 Alexandra Ehrens1,2†
Alexandra Ehrens1,2† Andrea Schiefer1,2†
Andrea Schiefer1,2† Anna K. Krome2,3
Anna K. Krome2,3 Tim Becker2,3Katharina Rox4,5Helene Neufeld1,2
Tim Becker2,3Katharina Rox4,5Helene Neufeld1,2 Tilman Aden1,2Karl G. Wagner2,3
Tilman Aden1,2Karl G. Wagner2,3 Rolf Müller5,6Miriam Grosse5,7Marc Stadler5,7Gabriele M. König2,8Stefan Kehraus2,8Silke Alt9Thomas Hesterkamp9
Rolf Müller5,6Miriam Grosse5,7Marc Stadler5,7Gabriele M. König2,8Stefan Kehraus2,8Silke Alt9Thomas Hesterkamp9 Marc Peter Hübner1,2
Marc Peter Hübner1,2 Kenneth Pfarr1,2*Achim Hoerauf1,2
Kenneth Pfarr1,2*Achim Hoerauf1,2- 1Institute for Medical Microbiology, Immunology and Parasitology, University Hospital Bonn, Bonn, Germany
- 2German Center for Infection Research (DZIF), Partner Site Bonn-Cologne, Bonn, Germany
- 3Department of Pharmaceutical Technology and Biopharmaceutics, University of Bonn, Bonn, Germany
- 4Department of Chemical Biology, Helmholtz Centre for Infection Research (HZI), Braunschweig, Germany
- 5German Center for Infection Research (DZIF), Partner Site, Hannover-Braunschweig, Germany
- 6Department of Microbial Natural Products, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Saarbrucken, Germany
- 7Department of Microbial Drugs, Helmholtz Centre for Infection Research, Braunschweig, Germany
- 8Institute for Pharmaceutical Biology, University of Bonn, Bonn, Germany
- 9Translational Project Management Office (TPMO), German Center for Infection Research (DZIF), Braunschweig, Germany
Corallopyronin A (CorA), a natural product antibiotic of Corallococcus coralloides, inhibits the bacterial DNA-dependent RNA polymerase. It is active against the essential Wolbachia endobacteria of filarial nematodes, preventing development, causing sterility and killing adult worms. CorA is being developed to treat the neglected tropical diseases onchocerciasis and lymphatic filariasis caused by Wolbachia-containing filariae. For this, we have completed standard Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) studies. In Caco-2 assays, CorA had good adsorption values, predicting good transport from the intestines, but may be subject to active efflux. In fed-state simulated human intestinal fluid (pH 5.0), CorA half-life was >139 minutes, equivalent to the stability in buffer (pH 7.4). CorA plasma-stability was >240 minutes, with plasma protein binding >98% in human, mouse, rat, dog, mini-pig and monkey plasma. Clearance in human and dog liver microsomes was low (35.2 and 42 µl/min/mg, respectively). CorA was mainly metabolized via phase I reactions, i.e., oxidation, and to a minimal extent via phase II reactions. In contrast to rifampicin, CorA does not induce CYP3A4 resulting in a lower drug-drug-interaction potential. Apart from inhibition of CYP2C9, no impact of CorA on enzymes of the CYP450 system was detected. Off-target profiling resulted in three hits (inhibition/activation) for the A3 and PPARγ receptors and COX1 enzyme; thus, potential drug-drug interactions could occur with antidiabetic medications, COX2 inhibitors, angiotensin AT1 receptor antagonists, vitamin K-antagonists, and antidepressants. In vivo pharmacokinetic studies in Mongolian gerbils and rats demonstrated excellent intraperitoneal and oral bioavailability (100%) with fast absorption and high distribution in plasma. No significant hERG inhibition was detected and no phototoxicity was seen. CorA did not induce gene mutations in bacteria (Ames test) nor chromosomal damage in human lymphocytes (micronucleus test). Thus, CorA possesses an acceptable in vitro early ADMET profile; supported by previous in vivo experiments in mice, rats and Mongolian gerbils in which all animals tolerated CorA daily administration for 7-28 days. The non-GLP package will guide selection and planning of regulatory-conform GLP models prior to a first-into-human study.
Introduction
Filarial nematodes are the causative agents of the devastating diseases lymphatic filariasis (LF) and onchocerciasis with approximately 68 million and 21 million people infected, respectively (1). LF is caused by the filarial nematodes Wuchereria bancrofti, Brugia malayi or Brugia timori and can lead to lymphedema in the lower extremities (elephantiasis) and in the scrotum of men (hydrocele). Onchocerca volvulus is the causative agent of onchocerciasis, which can cause severe dermatitis and vision impairment, including blindness, giving it the common name “river blindness” (2, 3). While LF is found in sub-Saharan Africa, South-East Asia and Central/South America, 99% of the onchocerciasis patients live in sub-Saharan Africa with additional foci in Yemen, Venezuela and Brazil (3–5). Due to the severity of these diseases and since they present a major public health burden in endemic countries, the WHO targeted the elimination of these diseases in the Roadmap for Neglected Tropical Diseases 2021-2030 (1, 2, 6–9). The goal is to eliminate LF as a public health problem and to eliminate onchocerciasis transmission by 2030 in most endemic countries. Currently, onchocerciasis is controlled by mass drug administration (MDA) using ivermectin (plus albendazole), while MDA with a triple therapy of ivermectin, diethylcarbamazine (DEC) and albendazole is recommended for LF (10, 11).
However, these treatment strategies face several problems that prevent the efficient elimination of LF and onchocerciasis. DEC used for the treatment of LF leads to a rapid killing of the first-stage larvae, the microfilariae (MF), which can cause severe adverse events (SAEs) if patients co-infected with onchocerciasis are inadvertently also treated (12, 13). Thus, the triple therapy against LF cannot be administered in areas co-endemic for onchocerciasis without major precautions, limiting the triple therapy to areas without onchocerciasis. In addition, ivermectin used for the treatment of onchocerciasis can lead to SAEs in highly microfilaremic loiasis patients, another filarial disease found in central Africa and often co-endemic with onchocerciasis (13–15). Current drugs used for MDA only target the MF stage and only temporarily inhibit embryogenesis in female adult worms, while macrofilaricidal activity, i.e., adult worm killing, is limited. Thus, MDA treatment has to be given on an annual or bi-annual basis for the reproductive life span of the female adult worms, which can be up to 15 years in the case of onchocerciasis (2).
To effectively achieve the goals set by the WHO, macrofilaricidal compounds are required. Many parasitic filarial nematode species harbor endosymbiotic Wolbachia bacteria that live in a mutualistic relationship with the filariae, providing the filariae with essential nutrients for growth, reproduction, and survival (16–20). Thus, targeting the endosymbiotic Wolbachia bacteria using antibiotics has several advantages. Antibiotics permanently sterilize adult worms, which leads to a slow decline in MF counts and concomitantly reduces the risk of SAEs in filariasis patients while inhibiting filarial transmission. The anti-Wolbachia drug doxycycline has macrofilaricidal activity (2, 21, 22). However, doxycycline is contraindicated for pregnant/breast feeding women and children below the age of 8. Moreover, the long treatment duration of at least 4 weeks makes doxycycline unsuitable for MDA. Thus, new macrofilaricidal drug candidates are required to effectively eliminate LF and onchocerciasis. Especially onchocerciasis elimination needs a new drug as ivermectin is, next to moxidectin (23), the only drug available and reduced responses to ivermectin have been described in different foci (24–27).
There are a few new macrofilaricidal drug candidates that have been developed for the treatment of filariasis, which are currently under investigation in different clinical trials for their efficacy and safety. Next to the direct acting drugs emodepside (28, 29) and oxfendazole (30), anti-wolbachial drugs such as high-dose rifampicin (31), AWZ1066S (32) and ABBV-4083 (33, 34) entered or have passed phase 1 clinical trials (35). However, due to high attrition rates in clinical studies, additional drug candidates are required.
Corallopyronin A (CorA) is a myxobacterial α-pyrone antibiotic which includes two side chains (Table 1). The eastern chain is structurally defined by a vinyl carbamate structure. The vinylogous carboxylic acid structure of the western chain leads to the acidic behavior of CorA (pKa: 3.7). However, long alkyl chains with several double bonds beginning from the pyrone ring cause the lipophilic character of CorA (logP: 5.4) (36, 37). It is an antibiotic naturally produced by the soil myxobacterium Corallococcus coralloides and inhibits the bacterial DNA-dependent RNA polymerase (RNAP) (38, 39). CorA is highly effective against Gram-positive bacteria, while it shows only low efficacy against Gram-negative bacteria (38, 40, 41). In contrast to rifampicin, which also inhibits the RNAP, CorA inhibits the RNAP at the switch region (40, 41), thus it is active against rifampicin-resistant Staphylococcus aureus (38, 42). Because CorA binds to a different RNAP target side, cross-resistance with rifampicin is not seen (38, 43).
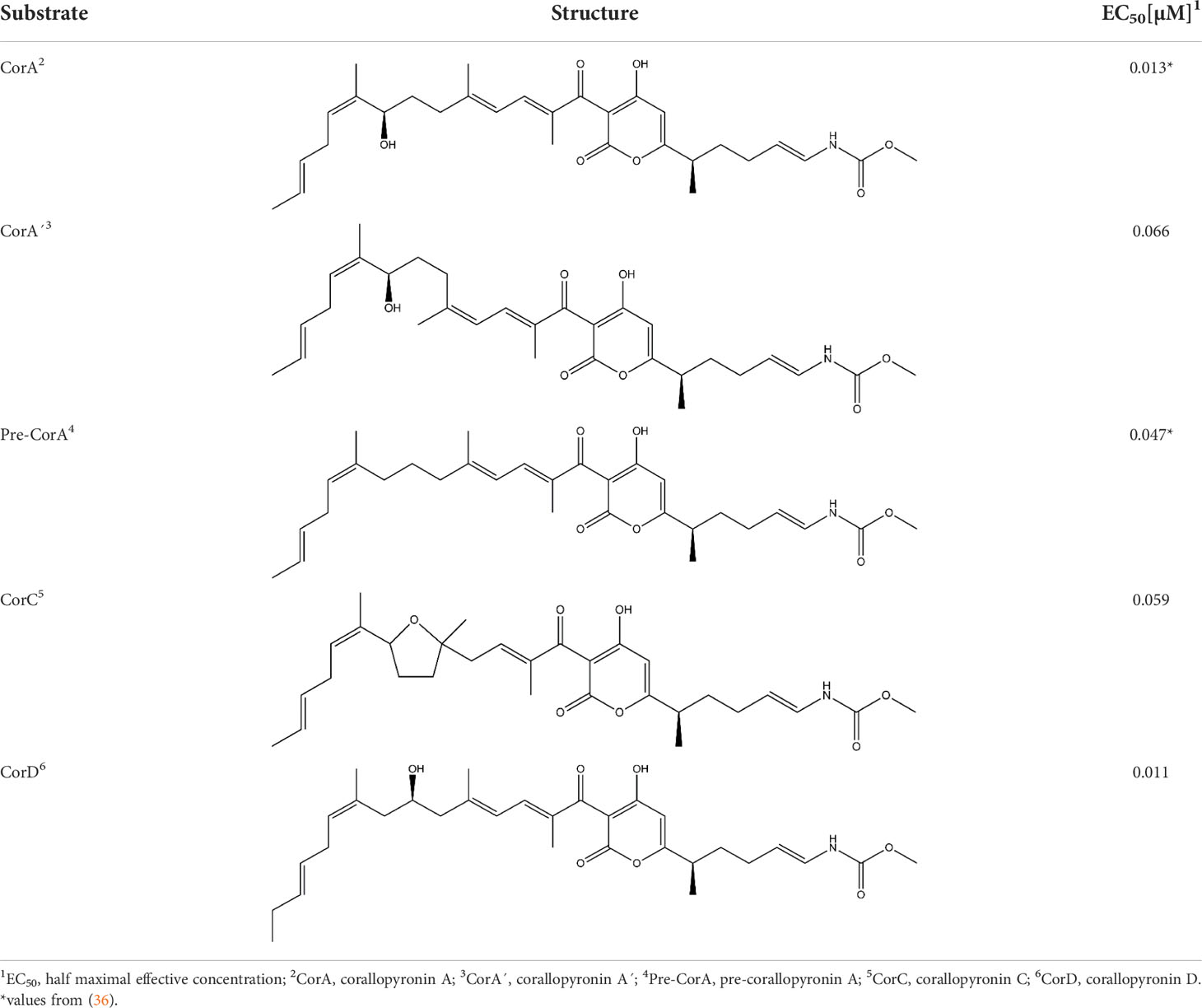
Table 1 In vitro potency of CorA and its derivatives against Wolbachia bacteria in C6/36 cells.
Even though endosymbiotic Wolbachia of filariae are Gram-negative bacteria, significant genome reduction has rendered them susceptible to CorA treatment in vitro and in vivo (16). In vitro CorA treatment reduced Wolbachia bacteria in infected insect cell cultures (39), while in vivo CorA treatment reduced Wolbachia bacteria in the Litomosoides sigmodontis rodent infection model (44). Administration of CorA one day after L. sigmodontis infection in mice and after adult worm development in mice and Mongolian gerbils for 14 days reduced Wolbachia from the female worms by more than 99% (44). Moreover, in L. sigmodontis-infected Mongolian gerbils, MF counts slowly declined beginning 6 weeks post CorA treatment and were eliminated 16 weeks post treatment through normal attrition of existing MF and inhibition of MF production/embryogenesis in the adult worms (44). In addition, other intracellular Gram-negative bacteria are depleted by CorA treatment: Rickettsia spp., Chlamydia trachomatis, and Orientia tsutsugamushi (45–47). Thus, CorA is a promising new drug candidate for treatment of onchocerciasis and LF, chlamydial infections, typhus, scrub typhus, and as a reserve antibiotic for MRSA.
To develop CorA as an antibiotic for human use, pre-clinical studies need to be conducted to understand its in vitro absorption, distribution, metabolism, excretion and toxicity (ADMET) to plan appropriate in vivo safety pharmacology and toxicity studies. Therefore, regulatory agencies require evaluating drug metabolic pathways in vitro, specific enzyme responses for elimination, drug-drug-interactions, pharmacological effects and drug metabolites. Herein we describe the in vitro ADME and toxicity profile of CorA as well as in vivo pharmacokinetic data. First, additional physiological and absorption properties of CorA were determined using FeSSIF and Caco-2 assays. CorA distribution and stability was assessed through protein stability and plasma protein binding experiments. Liver microsome and UGT metabolism assays were performed to analyze CorA metabolism and its metabolites. By using liver microsomes from six different species, interspecies metabolic comparisons were studied. Cytochrome P450 (CYP450) inhibition and CYP450 3A4 induction assays assessed drug-drug-interactions. Off-target effects were determined for CorA with a Cerep panel. As it is important to correlate in vitro and in vivo data, PK profiles in rats and Mongolian gerbils were determined. The data show that CorA is readily absorbed with a bioavailability in vitro and in vivo. Furthermore, the data indicate that CorA is primarily metabolized via phase I reactions and only slightly upregulated CYP450 3A4. Finally, toxicological analyses including hERG, Ames and micronucleus tests were carried out to define cardiotoxicity, genotoxicity and cytotoxicity potential, respectively, with no critical results. The completed non-GLP package will guide selection and planning of regulatory-conform GLP assays to be conducted prior to a first-into-human study.
Materials and methods
CorA
CorA with a purity of >91% was produced by the Helmholtz Centre for Infection Research, Department of Microbial Drugs using the heterologous producer strain Myxococcus xanthus carrying the CorA biosynthesis gene cluster and was purified as reported previously (37, 39).
In vitro potency
The in vitro activity of CorA against Wolbachia was determined in a Wolbachia-infected Aedes albopictus cell line (C6/36) as described previously (39). Briefly, 1 x 104 C6/36 cells infected with Wolbachia from A. albopictus B were seeded and cultured for 9 days with and without different CorA derivatives (CorA, CorA’, pre-CorA, CorC, CorD). The following concentrations were used to determine the IC50: 4, 2, 1 µM, 500, 125, 62.5, 31.25, 15.625, 7.8, 3.9 and 1.95 nM. The medium was exchanged every third day and the cells were harvested on day 9. Genomic DNA was extracted and Wolbachia depletion was determined via quantitative real-time polymerase chain reaction (qPCR) using primers targeting the 16S-rDNA gene of Wolbachia and the A. albopictus actin gene (39). Nonlinear fit curves were generated and the IC50 was determined.
In vitro metabolism
LC-MS/MS
Assays were performed and analyzed at Peakdale Molecular (Now part of Malvern Panalytical, Malvern, United Kingdom) and Pharmacelsus (Saarbrücken, Germany).
For the determination of CorA concentration at Peakdale Molecular, a Thermo Fisher Scientific (Waltham, USA) mass spectrometer TSQ Quantum Ultra was used with following settings: Spray Voltage: 3000 mV, Vaporiser Temperature: 350°C, Sheath Gas Pressure: 60, Ion Sweep Gas Pressure: 0.5, Auxillary Gas Pressure: 20, Capillary Temperature: 270°C. Detection was performed using a multiple reaction monitoring (MRM) method with QuickQuam software from Thermo Fisher Scientific (Waltham, USA).
The following LC-MS instruments were used at Pharmacelsus: LC-MS: Surveyor MS Plus HPLC (Thermo Electron, Waltham, USA) HPLC system connected to a TSQ Quantum Discovery Max (Thermo Electron) triple quadrupole mass spectrometer equipped with an electrospray (ESI) (Thermo Fisher Scientific); connected to a PC running the software Xcalibur 2.0.7. LC-MS: Accela U-HPLC pump and an Accela auto sampler (Thermo Fisher Scientific) connected to an Exactive mass spectrometer (Orbitrap with accurate mass (Thermo Fisher Scientific)); data handling with the software Xcalibur 2.1. LC-HRMS: Accela U-HPLC pump and an Accela Open auto sampler (Thermo Fisher Scientific) connected to an Q-Exactive mass spectrometer (Orbitrap); data handling with the software Xcalibur 2.2. LC-MS: Dionex UltiMate 3,000 RS pump (Thermo Fisher Scientific) and Dionex UltiMate 3000 RS column compartment and Accela Open Autosampler (Thermo Fisher Scientific) connected to an Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific), data handling with the software Xcalibur 4.0.27.19. The pump flow rate was set to 600 µl/min and the analytes were separated on a Kinetex Phenyl-Hexyl analytical column 2.6 µm, 50 x 2.1 mm (Phenomenex, Germany), on an Accucore RP-MS, 2.6 µm, 50 x 2.1 mm (Phenomenex, Germany) or a Luna Omega PS C18 5 µm, 100 x 2.1 mm (Phenomenex, Germany) with corresponding pre-columns using the following gradients (Suppl. Table 1).
Verapamil, diclofenac and propantheline were measured applying the triple quadrupole technology and full scan mass spectra acquired in the positive mode using syringe pump infusion to identify the protonated quasimolecular ions [M+H]+. Auto-tuning was carried out for maximizing ion abundance followed by the identification of characteristic fragment ions using a generic parameter set: ESI ion-transfer-capillary temperature 350°C, capillary voltage 3.8 kV, collision gas 0.8 mbar argon, sheath gas, ion sweep gas and auxiliary gas pressure were 40, 2 and 10 (arbitrary units), respectively. Ions with the highest S/N ratio were used to quantify the item in the selected reaction monitoring mode (SRM) and as qualifier, respectively. For warfarin, measurements were performed using an Orbitrap™ with accurate mass (Q-Exactive). As MS tune file, a generic tune file was used and as a lock mass for internal mass calibration the [M+H]+ ion of the Diisooctyl phthalate (m/z 391.28429), which is ubiquitously present in the solvent system, was used. Further analyzer settings were as follows: max. trap injection time 150 ms, sheath gas 40, aux gas 10, sweep gas 2, capillary voltage 4 kV for the positive and 2.8 kV for the negative mode, capillary temperature 350°C, H-ESI heater temperature 350°C.
For measurements applying the Q-Exactive Plus technology, as MS tune file, a generic tune file was used and as a lock mass for internal mass calibration the [M+H]+ ion of the Diisooctyl phthalate (m/z 391.28429), which is ubiquitously present in the solvent system, was used. For analysis of CorA, ß-estradiol and propofol, the MS was operated in the positive full scan mode, the accurate mass of the monitoring ions ±5 mDa were used for CorA and internal standard peak integration. Further analyzer settings were as follows: max. trap injection time 80 ms, sheath gas 40, aux gas 10, sweep gas 2, spray voltage 3.8 kV, capillary temperature 350°C, heater 350°C.
Absorption
Stability in fed-state simulated intestinal fluid and permeability in Caco-2 cells
FeSSIF assay
FeSSIF assay was performed and analyzed by Peakdale Molecular. To determine the stability in fed-state, CorA (1 µM) was incubated with fed state simulated intestinal fluid (FeSSIF, pH 5.0). For comparison CorA (1 µM) was incubated with PBS pH 7.4. Concentrations were measured at six time points (1, 5, 15, 30, 45 and 60 min) by using LC-MS/MS and the half-life for the three fluids were determined.
Caco-2 assay
Permeability in Caco-2 cells was performed and analyzed by Peakdale Molecular. Briefly, Caco-2 cells were cultured for 21 days in a 24-well plate and monolayer was confirmed using a Lucifer yellow test. CorA was added at a concentration of 10 µM and cells were placed on an orbital shaker for 2h at 37°C. Unidirectional transporter assay was performed by adding CorA to the apical well and measuring the concentration in the basolateral well after 2h (A→B). A bidirectional transporter assay was performed where CorA was added to the basolateral well and measured in the apical well after 2h (B→A). Efflux ratio was calculated by comparing the rates in both directions using LC-MS/MS. Cimetidine and propranolol were used as controls.
CorA plasma protein binding and stability in plasma from six different species
Plasma stability assay and plasma protein binding for dog, mini-pig and monkey was done by Pharmacelsus. Plasma protein binding for mouse, rat and human was done by Peakdale Molecular.
Plasma stability assay
Plasma stability assay was done by Pharmacelsus. CorA was tested at a final concentration of 10 µM in plasma from CD-1 mice, Sprague Dawley rats, beagle dogs, Goettingen mini-pig, Cynomolgus monkey and human. Propantheline was included as positive control. Samples were prepared at different time points (0, 30, 60, 120, and 240 minutes). Afterwards, plasma was inactivated by addition of acetonitrile (ACN) containing an internal standard and analyzed by LC-MS/MS. The pump flow rate was set to 600 µl/min and the analytes were separated on a Kinetex Phenyl-Hexyl analytical column 2.6 µm, 50 x 2.1 mm (Phenomenex, Germany), on an Accucore RP-MS, 2.6 µm, 50 x 2.1 mm (Phenomenex) or a Luna Omega PS C18 5 µm, 100 x 2.1 mm (Phenomenex) with corresponding pre-columns. Half-life (t1/2) estimates for the test item were determined using the rate of parent disappearance.
Plasma protein binding assay (PPB) at Pharmacelsus
Plasma protein binding for dog, mini-pig and monkey was determined by Pharmacelsus. Plasma samples and control plasma samples were incubated with 10 µM CorA or 10 µM warfarin as positive control at 37°C for 60 min in the dark. Afterwards, a modified ultrafiltration technique was applied. In parallel with each experimental plasma sample, a control plasma sample was additionally processed by ultrafiltration. The retentate from experimental and control plasma samples were mixed back into the filtrate of the respective partner sample. Therefore, plasma samples were added to sample reservoirs of Microcon® centrifugal filter units (UltracelYM30, MWCO 30,000 Da; Millipore, USA). All ultrafiltration units were centrifuged at 6,500 x g for 12 minutes at room temperature. The sample reservoirs containing plasma retentate were then inverted and placed on the filtrate collection tubes of the partner ultrafiltration unit. The ultrafiltration units were centrifuged a second time at 700 x g for 20 seconds, such that the retentate was mixed with the filtrate of the partner sample. Thus, the CorA concentration in the experimental filtrate represents the unbound fraction, whereas the experimental retentate contains the bound fraction (fb). Plasma samples were inactivated by the addition of ACN containing an internal standard (ISTD, 1 µM), precipitated and supernatant was used for LC-MS. The PPB was expressed as mean value of experimentally determined fb in the retentate and fb calculated from experimentally determined fraction unbound (fu) in the filtrate. The CorA concentration in the 60 min negative control sample was compared to the non-incubated negative control sample concentration (=100%) to determine CorA plasma stability. The percentage of compound bound to plasma proteins (%PPB) was calculated.
Plasma protein binding assay (PPB) at Peakdale Molecular
Plasma protein binding for mouse, rat and human was done by Peakdale Molecular. Pooled plasma (50% in buffer) was warmed to 37°C for 10 min and CorA was added at concentration of 5 µM. Five hundred microliters of dialyzed buffer was added to one side of the chamber of the RED device (Thermo Fischer) and 300 µl of the plasma-CorA-mixture was added to the other side of the chamber. The RED device was sealed with tape and shook at 500 rpm at 37°C for 4h. Afterwards, 50 µl from each chamber was collected and mixed with 50 µl buffer. Three hundred microliters of ice-cold ACN containing an internal standard was added to precipitate proteins. Samples were centrifuged at 3,800 rpm at 4°C for 20 min. As controls, warfarin was used. The supernatant was analyzed using LC-MS/MS.
Metabolism
In vitro metabolic stability
Metabolic stability in liver microsomes and identification of metabolites
Metabolic stability in liver microsomes was done at Pharmacelsus. Metabolic stability of CorA was tested using mouse, rat, dog, mini-pig, monkey and human liver microsomes fortified with the Phase I and II metabolism cofactors NADPH, UDPGA and alamethicin. For this, 0.25 mg/ml of microsomal protein in phosphate buffer (100 mM pH 7.4 supplemented with 2 mM MgCl2) were incubated with 1 µM CorA or 1 µM of the positive controls verapamil and diclofenac. The experiment was initiated by the addition of 22 mM UDPGA, 2.5 mg/ml alamethicin and 10 mM NADPH. After 0, 2, 15, 30, and 45 minutes, samples were drawn and precipitated with ACN containing internal standards. Microsomal metabolic activity was assessed in terms of verapamil or diclofenac turnover. The same instruments as for the plasma stability and plasma protein binding assay were used. The amount of compound in the samples was expressed as percentage of remaining compound compared to time zero (=100%). These percentages were plotted against the corresponding time points. In vitro intrinsic clearance (CLint) and half-life (t1/2) were determined using the rate of precursor disappearance with the following formula, based on the well-stirred liver mode:
In vitro CorA turnover in the presence of human recombinant UGT isoenzymes
UGT phenotyping was performed by Pharmacelsus using UGT-expressing Supersomes™ (Corning Life Sciences, Tewkbury, MA, USA) expressing human UGT1A1, UGT1A4, UGT1A6, UGT1A9, UGT2B7 and UGT2B10 at a standardized concentration of 0.25 mg/ml. The UGT suspension in 50 mM Tris buffer (pH 7.5, supplemented with 10 mM MgCl2) was incubated with 10 µM CorA or positive controls (15 µM β-estradiol, 20 µM trifluoperazine, 50 µM OH-trifluoromethylcoumarin, 10 µM propofol or 3 µM amitryptiline). The experiment was initiated by the addition of a solution consisting of 20 mM UDPGA and 0.25 mg/ml alamethicin in 50 mM Tris buffer to the UGT Supersomes™. Samples were removed after 0 and 60 minutes and precipitated by ACN containing the internal standards (1 or 10 µM). Samples and calibration standards containing trifluoperazine were diluted 1:20 (v/v) in a mixture of H2O/ACN containing the internal standard (1 µM) 1:3 (v/v). Dilutions of trifluoroperazine and all other supernatants were further diluted in H2O prior to LC-MS measurement. The conversion of test item over time as well as the formation of metabolites was assessed by LC-HRMS. The amount of test item in the samples after 60 min was expressed as percentage of remaining compound compared to time point zero (=100%).
Influence of CorA on human recombinant cytochrome P450 (CYP450) isoforms
CYP450 inhibition was analyzed by Eurofins (Fontenilles, France). CorA was pre-incubated with substrates as listed in Suppl. Table 2 and 0.1 mg/ml human liver microsomes (mixed gender, pool of 50 donors) in phosphate buffer (pH 7.4) for 5 min in a 37°C shaking water bath. The following CorA concentrations were used: 0.03, 0.1, 0.3, 1, 3, 10, 30, and 100 µM. The reaction was initiated by adding NADPH-generating system. The reaction was allowed for 10 min and stopped by transferring the reaction mixture to ACN/methanol. Samples were mixed and centrifuged. Supernatants were used for HPLC-MS/MS of the respective metabolite. The reference inhibitor was tested in each assay at multiple concentrations to obtain an IC50 value. Peak areas corresponding to the metabolite were recorded. The percent of control activity was calculated by comparing the peak area in the presence of CorA to the control samples containing the same solvent. Subsequently, the percent inhibition was calculated by subtracting the percent control activity from 100. The IC50 value (concentration causing a half-maximal inhibition of the control value) was determined by non-linear regression analysis of the concentration-response curve using the Hill equation.
CYP450 induction by PXR
CYP450 induction by PXR was performed and analyzed by Eurofins. Indigo Reporter Cells expressing a hybrid form of human PXR were used: The N-terminal sequence encoding the nuclear receptor DNA binding domain (DBD) has been substituted with that of the yeast GAL4-DBD. Ligand interaction activates the receptor, causing it to bind to the GAL4 DNA binding sequence, which is functionally linked to a resident luciferase reporter gene. Thus, changes in luciferase activity in the treated reporter cells provides a sensitive surrogate measure of the changes in nuclear receptor activity. The nuclear receptor reporter cells were seeded in 96-well plates. CorA (0.003, 0.01, 0.03, 0.1, 0.3, 1, 3, 10 µM), rifampicin and vehicle control (0.1% DMSO) were incubated with the cells at 37°C for 24 hours. Subsequently Luciferase Detection Reagent was added. The intensity of light emission from each sample was quantified using a luminometer.
The percent of control agonist effect was calculated using the following equation:
The EC50 value was determined by non-linear regression analysis of the concentration-response curve using the Hill equation. Compound is the individual reading in the presence of CorA. Rifampicin agonist is the highest nuclear receptor activity induced by the rifampicin. Background is the mean reading of the vehicle control (0.1% DMSO).
Off-targets of CorA significant and weak-moderate alerts
Off-targets of CorA were performed and analyzed by Eurofins (Fontenilles, France).
COX1: CorA (10 µM), reference compound or water (control) were pre-incubated for 15 min at room temperature with the enzyme (about 80 ng) in a buffer containing 80 mM Tris-HCl (pH 7.4), 4 mM EDTA/Tris and 0.8 mM hematin. Thereafter, the reaction was initiated by adding 3 µM arachidonic acid and 25 µM ADHP, then the mixture was incubated for 3 min at room temperature. The fluorescence of resofurin (oxydazed and N-deacetylated ADHP) was measured at λex=530 nm and λem=590 nm using a microplate reader (Envision, Perkin Elmer). An identical plate without enzyme (NSP) was prepared at the same time to verify compound interference with the fluorimetric detection method at these wavelengths. The enzyme activity was determined by subtracting the signal measured without enzyme (NSP) from that measured with enzyme. The results were expressed as a percent inhibition of the control enzyme activity. The standard inhibitory reference compound was diclofenac, which was tested in each experiment at several concentrations to obtain an inhibition curve from which its IC50 value was calculated.
Radioligand binding assay (A3, PPARγ, Cl-channel (GABA gated), I2)
A3: Cell membrane homogenates of HEK-293 cells transfected with human adenosine A3 receptor (32 µg protein) were incubated for 120 min at 22°C with 0.15 nM [125I]AB-MECA in the absence or presence of 10 µM CorA in a buffer containing 50 mM Tris-HCl (pH 7.4), 5 mM MgCl, 1 mM EDTA and 2 UI/ml ADA. Nonspecific binding was determined in the presence of 1 µM IB-MECA (standard reference compound).
PPARγ: Purified recombinant nuclear receptor (8 µg protein) was incubated for 120 min at 4°C with 5 nM [3H]rosiglitazone in the absence or presence of 10 µM CorA in a buffer containing 10 mM Tris-HCl (pH 8.2), 50 mM KCl and 1 mM DTT. Nonspecific binding was determined in the presence of 10 µM rosiglitazone (standard reference compound).
Cl-channel (GABA gated): Membrane homogenates of cerebral cortex (120 µg protein) were incubated for 120 min at 22°C with 3 nM [35S]TBPS in the absence or presence of 10 µM CorA in a buffer containing 50 mM Na2HPO4/KH2PO4 (pH 7.4) and 500 mM NaCl. Nonspecific binding was determined in the presence of 20 µM picrotoxinin (standard reference compound).
I2: Membrane homogenates of cerebral cortex (1 mg protein) were incubated for 30 min at 22°C with 2 nM [3H]idazoxan in the absence or presence of 10 µM CorA in a buffer containing 50 mM Tris-HCl (pH 7.4), 0.5 mM EDTA and 3 µM yohimbine. Nonspecific binding was determined in the presence of 10 µM cirazoline (standard reference compound).
Following incubation (A3, PPARγ, Cl-channel (GABA gated), I2 assays), the samples were filtered rapidly under vacuum through glass fiber filters (Filtermat B, Wallac) presoaked with 0.3% PEI and rinsed several times with an ice-cold buffer containing 50 mM Tris-HCl and 150 mM NaCl using a 48- or 96- sample cell harvester (Mach II, Tomtec). The filters were dried then counted for radioactivity in a scintillation counter (Betaplate 1204, Wallac) using a solid scintillator (Meltilex B/HS, Wallac). The results are expressed as a percent inhibition of the control radioligand specific binding. The standard reference compound was tested in each experiment at several concentrations to obtain a competition curve from which its IC50 was calculated.
Toxicity
hERG, Ames, micronucleus, phototoxicity test
hERG test
hERG test was done by B’SYS (Witterswill, Switzerland) using a whole-cell patch-clamp technique on hERG potassium channels stably expressed in HEK 293 cells. Cells were continuously perfused (1 ml/min) with bath solution (137 mM sodium chloride, 4 mM potassium chloride, 1.8 mM calcium chloride, 1 mM magnesium chloride, 10 mM HEPES, 10 mM D-Glucose, pH 7.4). After formation of a Gigaohm seal between the patch electrodes and individual hERG stably transfected HEK 293 cells (pipette resistance range: 2.0 MΩ to 7.0 MΩ; seal resistance range: > 1 GΩ) the cell membrane across the pipette tip was ruptured to assure electrical access to the cell interior (whole-cell patch-configuration). As soon as a stable seal was established, hERG outward tail currents were measured upon depolarization of the cell membrane to +20 mV for 2s (activation of channels) from a holding potential of -80 mV and upon subsequent repolarization to -40 mV for 3s. Once control recordings were accomplished, cells were continuously perfused with a bath solution containing CorA (0.3 µM, 1.0 µM, 3.0 µM and 10 µM), DMSO as negative control or 100 nM E-4031 as positive control. During washing of CorA, the voltage protocol indicated above was run continuously again at 10s intervals until the steady-state level of block was reached.
Ames test
Mutagenic properties of CorA were tested with the Ames MPF assay provided by Pharmacelsus. Salmonella typhimurium strains TA100, TA1535 and the Escherichia coli strains wp2 [pKM101] and wp2 uvrA were used for the detection of base substitution mutations, whereas S. typhimurium strains TA98 and TA1537 were used for the detection of frameshift mutations. Each of the bacterial strains was incubated with DMSO or H2O (negative control), a corresponding positive control (2-NF, 4-NQO, N4-ACT, 9-AA, 2-AA, 2-AF) or 1000, 500, 100, 25, 1 and 0.1 µg/ml CorA (1895, 948, 190, 47, 1.9, 0.19 µM, respectively) in the presence and absence of S9 mix (mitochondrial fraction). E. coli cultures with S9 were incubated for 20 minutes, all other bacteria cultures for 90 min at 37°C on a horizontal shaker. After incubation, the medium was exchanged to an indicator medium without tryptophan and histidine and bacterial growth was determined.
Micronucleus test
Micronucleus tests with and without S9 fraction were conducted by LPT Laboratory (Pharmacology and Toxicology GmbH & Co. KG, Hamburg, Germany) as published (48–51). After initiation of the human peripheral lymphocyte culture, 6.25, 12.5, 25, 50, 100, 200 µg/ml of CorA (11.9, 23.7, 47, 190, 379 µM, respectively) in DMSO was added to the cell cultures. As positive controls, 0.1 and 0.2 µg/ml mitomycin C, and 10 and 20 µg/ml cyclophosphamide for the 4h exposure, and 0.01 and 0.02 µg/ml colchicine for the 24h exposure were used. DMSO served as negative control. Experiments were performed with and without rat S9 mix. After incubation, mitotic activity was arrested by the addition of cytochalasin B to each culture at a final concentration of 5 µg/ml and incubated for 20h, then processed for microscopy using 10% Giemsa. At least 500 cells per replicate cell culture were scored and classified as mononucleated, binucleated or multinucleated to calculate the proliferation index as a measure of toxicity.
Phototoxicity test
Phototoxicity tests were conducted in BALB/c 3T3 cells by LPT Laboratory as published (52, 53). BALB/c 3T3, clone 31 cells were obtained from ATCC (American Type Culture Collection, Manassas, USA), and grown in DMEM containing 4 mM glutamine supplemented with 50 ml neonatal mouse brain-derived growth inhibitor and penicillin/streptomycin at 37°C and 5% CO2. Cells from the 24th or 25th passage were used for the test, CorA was dissolved in DMSO and further diluted to achieve final concentrations of 0.156, 0.313, 0.625, 1.25, 2.5, 5, 10 and 20 µg/ml (0.3, 0.59, 1.18, 2.37, 4.74, 9.48, 18.95, 37.91 µM, respectively). DMSO was used as the negative control and chlorpromazine was used as the positive reference. The cells were irradiated for 9.24 min with 9.01 mW/cm2 UVA (= 5 J/cm2). Duplicate plates (-UVA) were kept at room temperature in a dark box for 9.24 min. The test solution was decanted and the cells were washed twice with the buffered solution used for incubation. The buffer was replaced with culture medium and the cells were incubated overnight (18 - 22h) at 5% CO2, 37°C and a relative humidity of 95% ± 5%.
The cells were examined under a phase-contrast microscope. Changes in the morphology of the cells and effects on cell growth and integrity were recorded. Then the cells were stained with neutral red solution (1 ml 0.33% 3-amino-7-dimethylamino-2-methylphenazine hydrochloride were diluted with DMEM to a volume of 60 ml) without serum and incubated at 37°C with 95% ± 5% humidity and 5% CO2 for 3h. The stain was removed and the cells were washed. Exactly 150 µl neutral red desorb solution (freshly prepared 49 parts highly purified water + 50 parts ethanol + 1 part acetic acid) were added and the microtiter plate was gently shaken for 10 min until the neutral red had been extracted from the cells and had formed a homogeneous solution. The optical density of the neutral red extract was measured at 540 nm in a spectrophotometer (Tecan Sunrise Magellan Version 7.2, (Männedorf, Switzerland)), using blanks as a reference. Phototoxicity was determined by the IC50 in the absence (-UVA) divided by the IC50 in the presence (+UVA) of light.
Antiproliferation effect and cytotoxicity test
Antiproliferation effects were determined at the Leibniz-Institut für Naturstoff-Forschung und Infektionsbiologie e.V. in HUVEC- (ATCC CRL-1730), CHO-K1 (ACC-110), THP-1 (ACC-16) and K-562-cells (ACC-10). Cells were incubated with different concentrations of CorA (0.2, 0.39, 0.78, 1.6, 3.13, 6.25, 12.5, 25, 50 µg/ml (0.38, 0.74, 1.48, 3.03, 5.93, 11.85, 23.7, 47.38, 94.77 µM, espectively)) for 3 days. After fixation with glutaraldehyde, adherent live HUVEC or CHO-K1 cells were stained with 0.05% methylene blue (SERVA 29198). Non-adherent live K-562 or THP-1 cells were stained with CellTiter-Blue (Promega (Fitchburg, USA)). The absorbance of all wells of the microtiter plate was measured at 660 nm (Methylene Blue) or 570 nm (CellTiter-Blue, reference wavelength: 600 nm) with a microplate reader. Half-maximal antiproliferation effect (GI50) was determined.
For cytotoxicity tests, HeLa cell cultures (DSMZ, ACC-57) were incubated for 3 days with concentration series of CorA (0.2, 0.39, 0.78, 1.6, 3.13, 6.25, 12.5, 25, 50 µg/ml (0.38, 0.74, 1.48, 3.03, 5.93, 11.85, 23.7, 47.38, 94.77 µM, respectively)). After glutaraldehyde fixation and methylene blue staining, the absorbance of all wells at 660 nm was measured with a microplate reader. The half-maximal cytotoxic potency (CC50) was determined by analyzing cell cytolysis.
To determine changes in proliferation, the xCELLigence system (Roche Innovatis (Little Falls, USA)) and the Cedex HiRes I by Roche were used. Therefore, 50000 and 7500 Hep G2 cells were seeded into a 24-well plate or in 96-well plates, respectively. Hep G2 cells were used (passage number two to twelve) and cultured at 37°C and 5% CO2 in RPMI 1640 containing 5% fetal calf serum (FCS), 2 μM L-glutamine, 100 U/ml penicillin/streptomycin. After 24h cells were treated with CorA (200, 20, 2, 0.2, 0.02 µg/ml (379, 37.9, 3.79, 0.38, 0.04 µM, respectively)) for 5 days. Cells were counted with the Cedex HiRes I system and changes in proliferation were determined using the xCELLigence system (54).
In vivo PK studies
Polyvinylpyrrolidone (PVP, Kollidon® 30 LP) was kindly provided by BASF SE (Ludwigshafen, Germany). Polyethylene glycol 400 (PEG 400) was purchased from Carl Roth GmbH + Co.KG (Karlsruhe, Germany). Phosphate buffered saline pH 7.4 (PBS) was purchased from Thermo Fisher Inc. (Dreieich, Germany).
Female Mongolian gerbils were obtained from Janvier (Le Genest-Saint-Isle, France). All experimental procedures were conducted in accordance to the European Union animal welfare guidelines and all protocols were approved by the Landesamt für Natur Umwelt und Verbraucherschutz in North Rhine-Westphalia under the file #81-02.04.2020.A244. Animals were kept in individual ventilated cages at a 12h light/dark cycle with water and food ad libitum. Animal wellbeing was assessed daily. Mongolian gerbils were treated perorally (PO) (5 ml/kg dosing) with 30 and 60 mg/kg CorA-Povidone in PBS or intraperitoneally (IP) (5 ml/kg dosing) with 43 mg/kg CorA in PEG400/PBS (1:1). Blood was drawn from the tail vein after 5, 10, 15, 30, 60, 180 and 360 min. Blood was collected in EDTA tubes, stored on ice for<1h, centrifuged at 4°C at 3,220 x g for 10 min and plasma was collected. Samples were frozen at -20°C until analyzed.
Plasma PK analysis was done using male Sprague Dawley rats at Cyprotex Discovery Limited (Cheshire, United Kingdom). Animals were kept in individual ventilated cages at a 12h light/dark cycle with water and food ad libitum. Rats were treated intravenously (IV) via the tail vein with 0.48 mg/kg (2 ml/kg dosing), PO with 5 mg/kg (5 ml/kg dosing) or IP with 5 mg/kg (2.5 ml/kg dosing). CorA was dissolved in PBS and DMSO in a ratio 90% and 10%. Blood was collected into heparinized tubes with a polyurethane catheter inserted into the jugular vein after 5, 15, 30, 60, 120, 240 and 480 min.
CorA was detected using HPLC-DAD. Plasma (20 µL) was mixed 1:3 with ice-cold ACN, vortexed and then centrifuged for 25 min at 4°C and 11,600 x g. The supernatant was transferred into a HPLC-vial and analyzed using a Waters Alliance e2695 Separator module and Waters 2998 PDA Detector (Waters, Eschborn, Germany) with the Waters XBridge Shield RP18; 3.5 µm; 2.1 x 100 mm; 130 A column. Mobile phase was ACN/H2O 5/95 + 5 mM NH4Ac + 40 µl CH3COOH/L (A) and ACN/H2O 95/5 + 5 mM NH4Ac + 40µl CH3COOH/L (B) and a flow rate of 0.3 ml/min. A gradient from 70% A/30% B to 20% A/80% B within 30 min was used. CorA was quantified via an external reference standard measured at 300 nm. No major matrix effects were seen when mixing ACN containing CorA with mouse plasma (Suppl. Figure 1).
Results
In vitro potency of CorA
To determine CorA potency in vitro, Wolbachia-infected insect cell lines were treated with different CorA concentrations and Wolbachia 16S rDNA copy number normalized to insect cell actin copy number was measured to determine EC50 values. CorA and its derivatives had good in vitro potency against Wolbachia endobacteria (Table 1). CorA had the best activity with an EC50 of 0.013 µM. Two isoforms that are co-purified with CorA at low levels, pre-corallopyronin A (pre-CorA), a precursor of CorA, and CorA’ had an EC50 of 0.047 µM and 0.066 µM, respectively. Two other derivatives that are usually only formed in the presence of acid or oxygen (37), CorC and CorD, had an EC50 of 0.059 µM and 0.011 µM, respectively. Although CorD has an equivalent EC50 as CorA, it is almost never found during the production process of CorA (55).
In vitro metabolism
Next, the physiochemical properties of CorA as well as in vitro absorption, distribution, metabolism and excretion were measured.
Physiochemical characterization of CorA/absorption
Krome et al. determined that CorA has a pKa value of 3.7 and is highly lipophilic with poor solubility at pH values equivalent to the pH found in the human stomach (solubility: 0.1 µg/ml (0.19 µM) and Log D: 5.42) (37). With increasing pH, ionization of CorA increased and resulted in enhanced solubility. Thus, at a pH 6, equivalent to the pH found in the human small intestine, the solubility increased to 29 µg/ml. Despite the high degree of ionization, CorA remained highly lipophilic (Log D, pH 6: 3). At physiological pH (pH 7.4), a solubility of 723 µg/ml (1300 µM) and a lipophilicity of 1.81 was measured (37). Biphasic dissolution tests indicated slow and poor dissolution properties of neat CorA (37). The low amount of CorA distributed into the organic phase (absorption sink) predicted poor bioavailability of neat, unformulated CorA.
The stability of CorA in simulated intestinal fluid was measured for fed-state (FeSSIF, pH 5.0) to assess potential food and pH impact on the in vivo pharmacokinetic properties. The half-life of CorA under FeSSIF (>139 min) was equivalent to the half-life of CorA in physiological buffer (pH 7.4) (Table 2).

Table 2 Stability of CorA in fed-state simulated intestinal fluid.
CorA had good permeability in a Caco-2 cell line (Table 3). The permeability of CorA was analyzed and compared to cimetidine and propranolol. The absorption of CorA was comparable to propranolol with a Papp A to B of 20-6 cm/S vs. 29-6 cm/S, respectively. However, the efflux ratio of CorA was slightly higher than the one for cimetidine, a control compound known to exhibit active efflux. Thus, this suggests active efflux from the intestine for CorA. Based on these data, CorA is a Biopharmaceutical Classification System class II compound (56).

Table 3 Permeability of CorA in Caco-2 cells.
Metabolic stability and metabolites of CorA – Distribution, metabolism and excretion
To determine the distribution and metabolic stability of CorA, plasma protein binding and stability in plasma from six species (human, mouse, rat, dog, mini-pig, monkey) were determined. CorA had a high plasma protein binding and high plasma stability (Table 4). In all species, >98% of CorA was bound to plasma proteins, equivalent to plasma binding of the warfarin control. Moreover, CorA was stable in plasma of all tested six species with a half-life of >240 min.
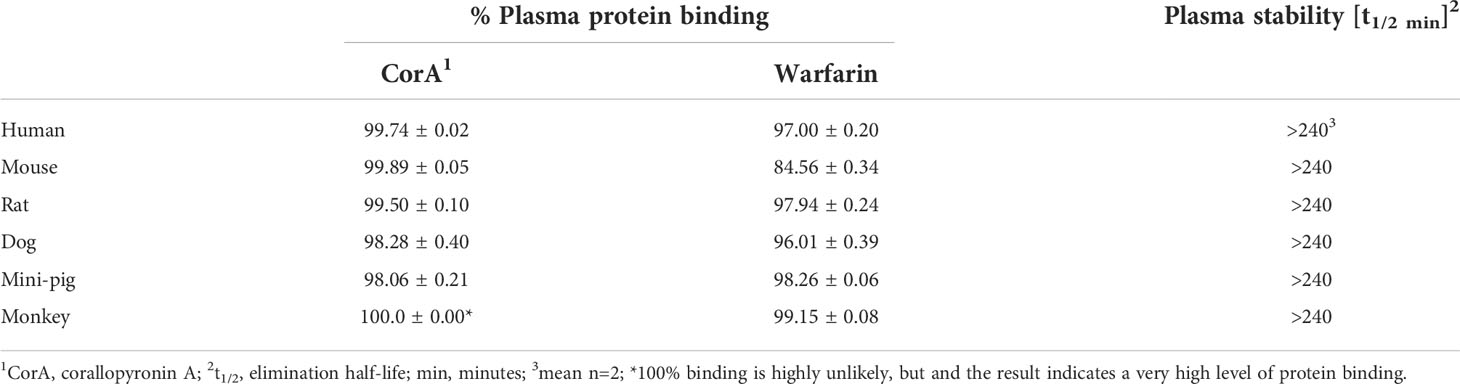
Table 4 CorA plasma protein binding and stability in plasma from six species.
The intrinsic clearance of CorA in liver microsomes from six species and the metabolites produced by liver microsomes were assessed (Table 5). Cofactors to assess phase I and phase II reactions were used. The intrinsic clearance of CorA was lowest in human and dog liver microsomes, followed by mouse, monkey, rat and mini-pig. In the time of the assay, almost no phase II metabolites were found. CorA was metabolized by phase I reactions resulting in four different metabolites (M1-M4) in liver microsomes due to oxidation within the “eastern” carbon chain of the molecule (Figure 1). CorA M1 metabolites were detected in all species, but human and dog microsomes produced the fewest. CorA M2 metabolites were detected in mouse, rat, monkey and mini-pig microsomes. M3 and M4 metabolites were only detected in mini-pig microsomes.

Table 5 In vitro metabolic stability of CorA in liver microsomes of six species.
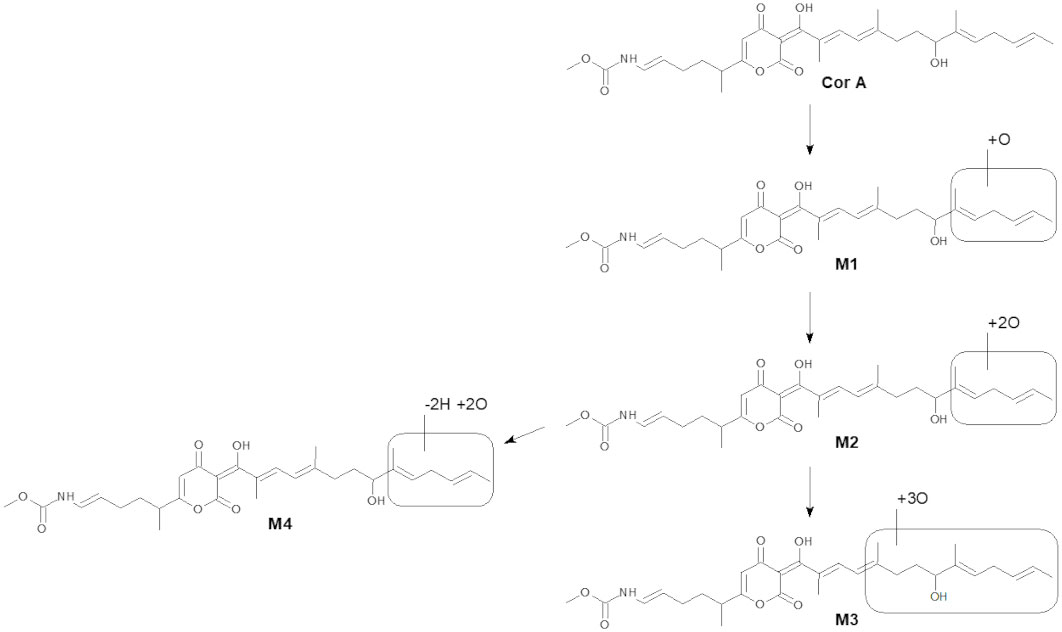
Figure 1 Scheme of CorA in vitro metabolism in liver microsomes. The accurate mass was used to propose the given structures.
Metabolism of CorA by uridine diphospho-glucuronosyltransferases (UGTs) was also measured. UGT enzymes are responsible for the vast majority of phase II metabolic reactions, leading to the attachment of hydrophilic glucuronic acid moieties to phase I drug metabolites, thus, facilitating faster excretion (57). CorA was poorly metabolized by 3 of the 6 tested UGTs as the turnover rate was <25% (Table 6), the cutoff for a UGT substrate defined by the FDA (58). Only the UGTs 1A1, 1A4 and 2B7 were able to measurably metabolize CorA. CorA concentrations remained stable within 60 min of incubation with the isoforms 1A6, 1A9 and 2B10, no glucuronide formation was observed.

Table 6 In vitro CorA turnover in the presence of human recombinant UGT isoenzymes.
Drug-drug interactions
To determine the drug-drug-interaction potential, CYP450 assays were performed to identify CorA-mediated inhibition of seven major human CYP450 enzymes (Table 7). CorA did not inhibit CYP1A2, 2B6, 2D6 and 3A4 (BFC substrate), but weakly inhibited CYP2C8, 2C19 and 3A4 (BzRes substrate). However, CorA strongly inhibited CYP2C9.
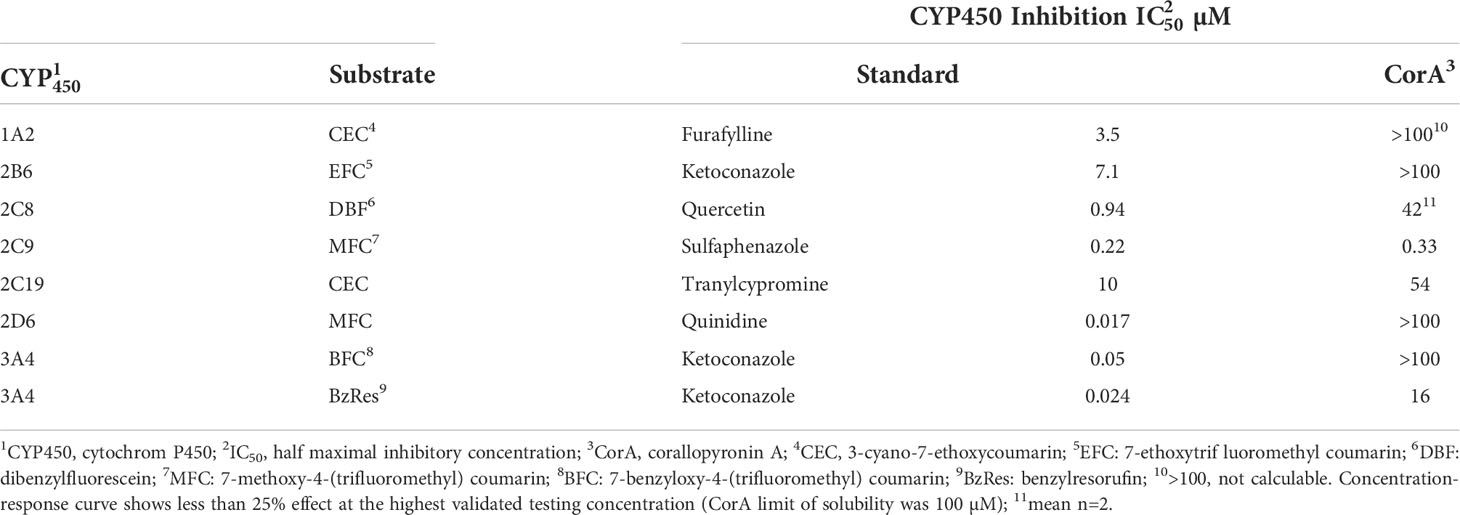
Table 7 Influence of CorA to human recombinant cytochrome P450 (CYP450) isoforms.
Induction of CYP3A4 via PXR was determined and resulted in a minimal induction of CYP3A4 (Figure 2). CorA did not activate the reporter gene above 10% until a concentration of ~3 µM. The EC50 of CorA was 12 µM, while rifampicin, used as positive control, had an EC50 of 1.5 µM. Taking into account that CorA is highly plasma protein bound, the unbound CorA is unlikely to reach sufficiently high concentrations to induce CYP3A4 in vivo. Therefore, CorA is not considered to induce CYP3A4 in vivo.
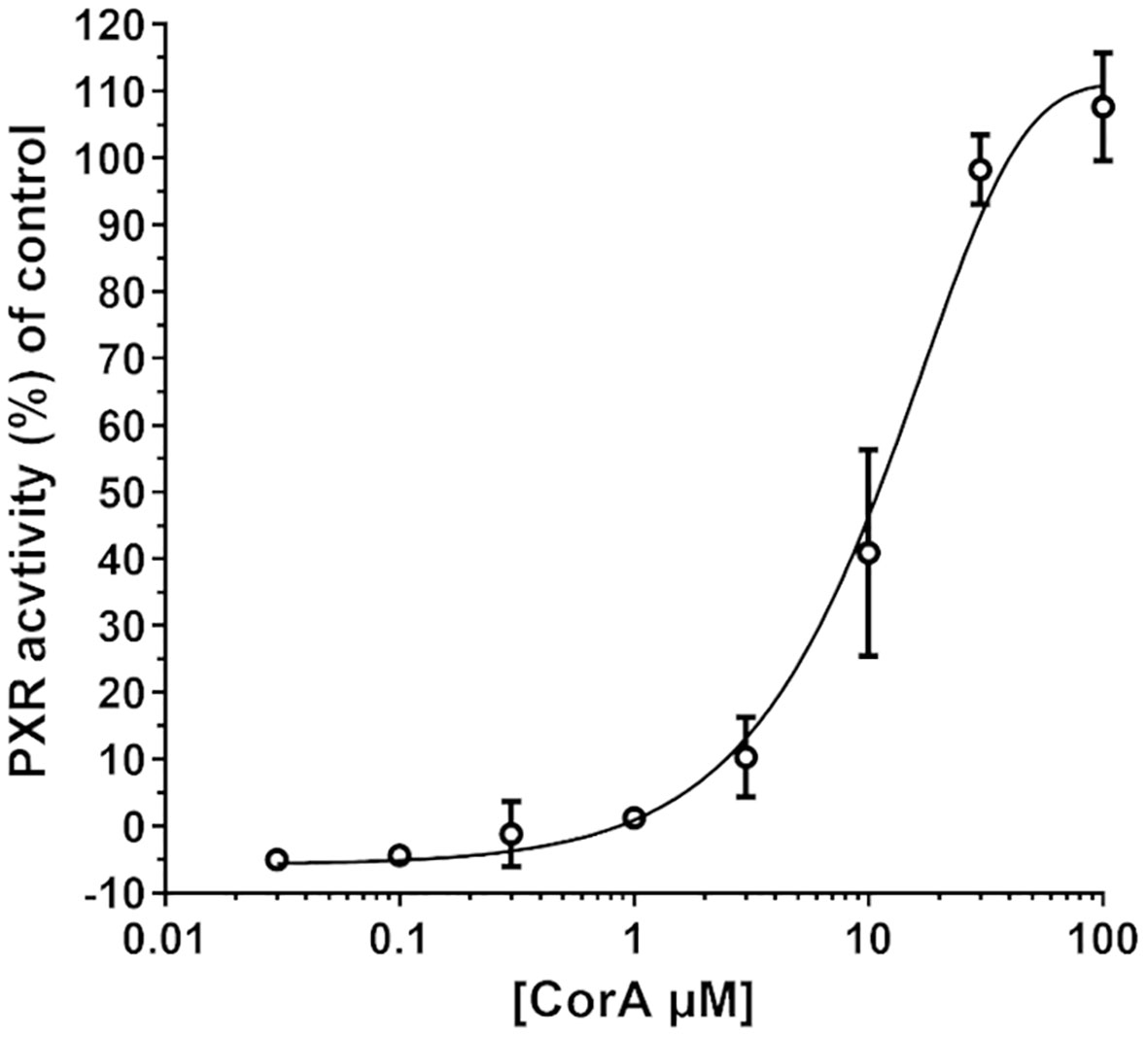
Figure 2 CYP3A4 is minimally induced by CorA. DPX2 cells transformed with human CYP3A4 were incubated with different concentrations of CorA. Activation of CYP3A4 was measured using the human pregnane X receptor (PXR)-linked luciferase reporter assay. Mean (N=3) ± 95% CI are shown.
To predict secondary pharmacologies, off-target effects of CorA were determined with the Eurofins Cerep panel of human receptors and enzymes. CorA had no effect on the ligand binding or activity of 93 receptors and enzymes (Suppl. Table 3). Weak to moderate off-target effects were found for the GABA-gated Cl-channel and the imidazoline receptor 2 (I2). Significant off-target effects were seen with adenosine receptor 3 (A3), peroxisome proliferator-activator receptor-γ (PPARγ) and cyclooxygenase 1 (COX1) (Table 8).
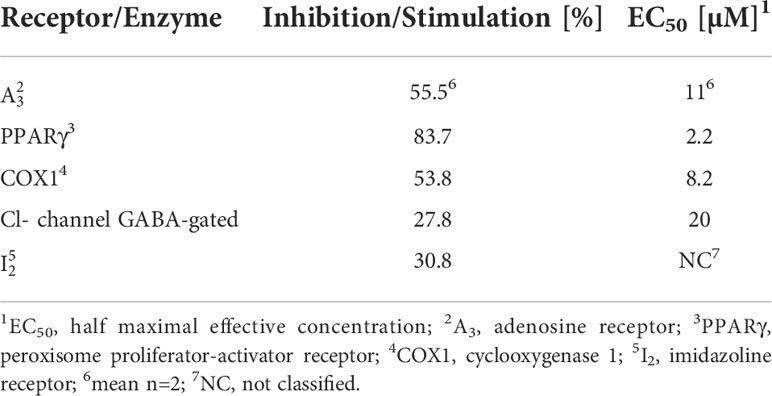
Table 8 CorA off-targets with significant and weak-moderate alerts.
In vitro toxicology
hERG safety assay was performed to identify the impact of CorA on ventricular repolarization. Different CorA concentrations maintained a mean relative tail current above 94%. Even the highest CorA concentration of 10 µM tested (the maximum concentration possible before precipitation of CorA in the test medium), resulted in a mean relative tail current of 94.12 ± 1.38%. Thus, CorA did not inhibit hERG potassium channels (Table 9).

Table 9 Toxicology profile results for CorA.
Genotoxicity was assessed by the Ames and micronucleus tests. Ames test using S. typhimurium strains TA100, TA1535, TA1537 and TA98 was performed with CorA dosed up to 1000 µg/ml (1895 µM). No mutagenic potential of CorA was observed (Table 9 and Suppl. Table 4). No phototoxicity occurred up to non-precipitating concentrations of 20 µg/ml (37.91 µM) (Table 9). Thus, in vitro, CorA showed no cardiotoxicity, genotoxicity and phototoxicity.
The micronucleus test was performed with and without S9 mix for 4 and 24h. Micronucleus formation due to CorA was negative (Table 9; Suppl. Table 5). The high CorA concentration (100 µg/ml (190 µM)) used for stimulation without S9 resulted in cytotoxicity after 24h (Suppl. Table 5).
Antiproliferation effects were determined and the half-maximal antiproliferation effect (Gl50) was above 50 µg/ml (94.77 µM) for HUVEC, K-562 and CHO-K1 cells, while for THP-1 a Gl50 of 39.1 µg/ml (74.1 µM) was observed. Half-maximal cytotoxicity effect on HeLa cells was above 50 µg/ml (94.77 µM) (Table 9).
The cytotoxicity xCell and Cedex tests showed no CorA toxicity within the therapeutic range of 1 µg/ml (1.9 µM). Up to 20 µg/ml (37.91 µM) CorA had no significant toxicity, while concentrations of 200 µg/ml (379 µM) had toxic effects on cells (Table 10).
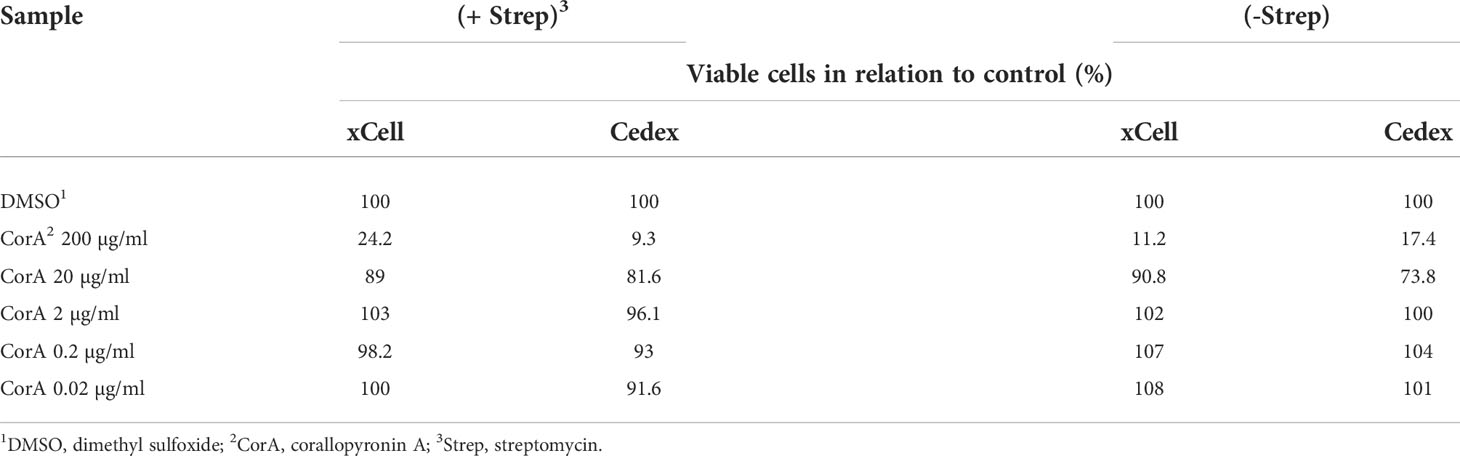
Table 10 CorA is non-toxic in therapeutic range.
In vivo pharmacokinetics
To assess CorA absorption and distribution in vivo, Mongolian gerbils and rats were treated with CorA solutions (PBS/10% DMSO) IV, IP or PO and plasma concentrations were determined over time. PK profiles for rats were determined from rats treated IV with 0.48 mg/kg CorA, or 5 mg/kg CorA IP and PO (Figure 3). Tmax was reached after 5 min, indicating fast absorption in rats. Cmax for IV application was 0.23 µg/ml (0.46 µM), while PO and IP application reached comparable Cmax of 1.87 and 1.44 µg/ml (3.54 and 2.723 µM), respectively. However, AUC0-inf after IP administration was almost twice as high as for PO administration (6.94 µg*h/ml (IP) and 3.70 µg*h/ml (PO)). After IV administration, a clearance of 1.688 l/h/kg was found. A volume of distribution of ~8.248 l/kg was measured, accounting for good distribution. The steady state volume of distribution after IP and PO administration was similar (Table 11). Finally, bioavailability for PO and IP application was ≥100%.
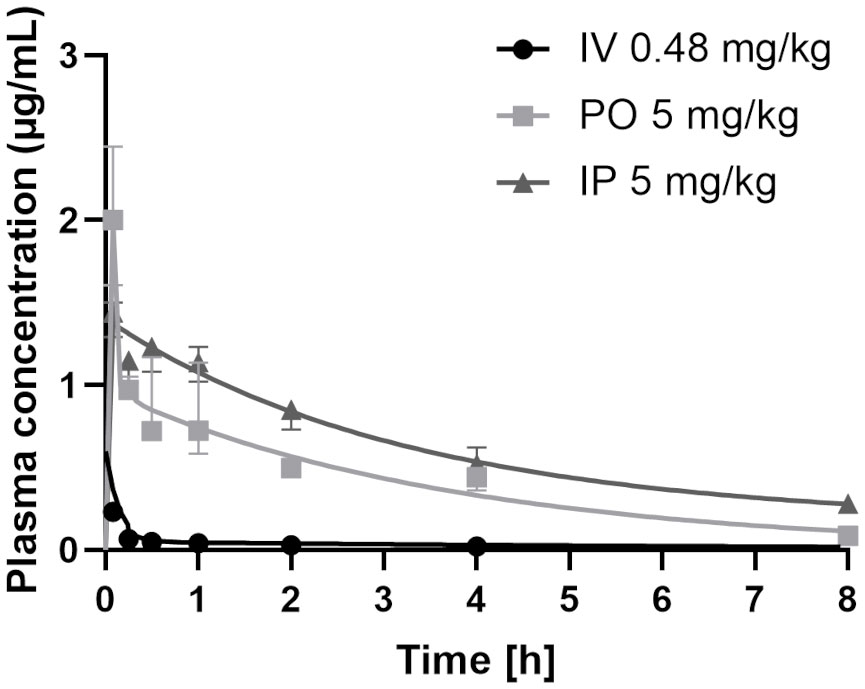
Figure 3 Plasma concentrations of CorA after intravenous (IV), intraperitoneal (IP) and oral (PO) administration in rats. 0.48 mg/kg CorA were administered in PBS/10% DMSO for IV or 5 mg/kg CorA in PBS/10% DMSO for PO and IP application. Plasma was prepared from blood drawn after 5, 15, 30, 60 min, 2h, 4h and 8h and CorA concentrations were quantified by LC-MS. Symbols indicate median ± IQR.
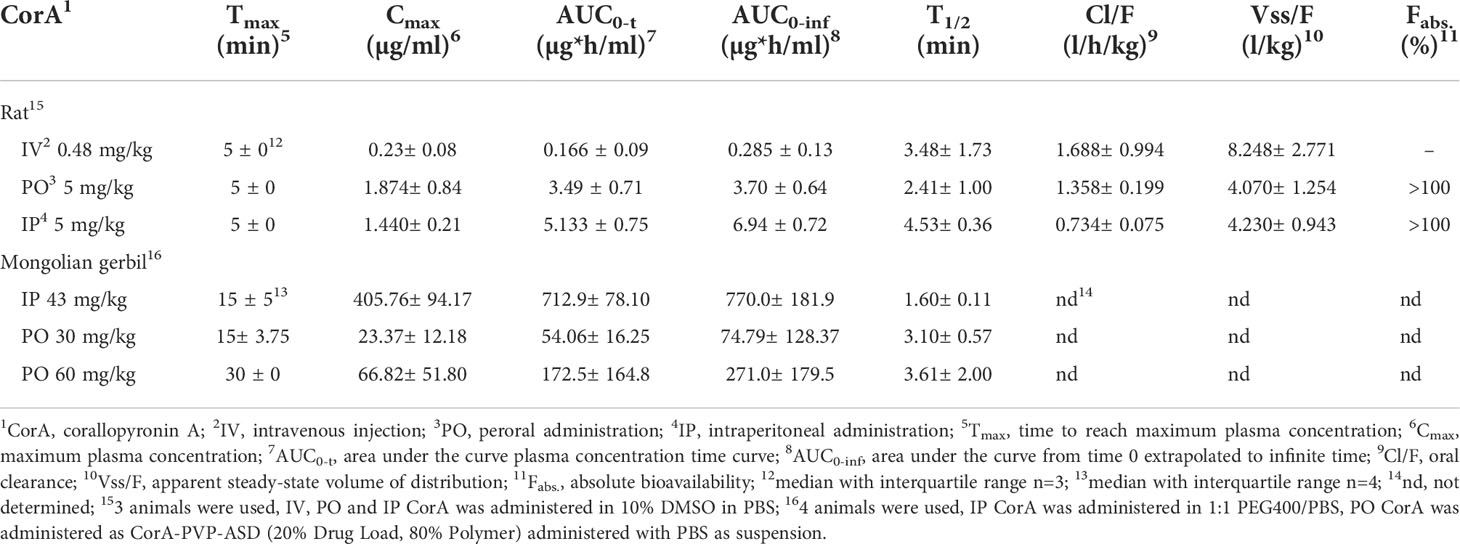
Table 11 Pharmacokinetics of CorA after intraperitoneal (IP), intravenous (IV) and peroral (PO) administration in Mongolian gerbils and rats.
Since oral administration in rats resulted in poor absorption of neat CorA (given as suspension in 10% DMSO, PBS), it was formulated in a povidone-based amorphous solid dispersion (ASD) (37) and administered orally suspended in PBS to Mongolian gerbils. IP administration of CorA resulted in a rapid increase of plasma CorA concentration and Tmax was reached after 15 min with a Cmax of 405.76 µg/ml (769.07 µM). In contrast, PO administration of 30 and 60 mg/kg (56.86 and 113.72 µM, respectively) CorA reached a Cmax of 23.27 and 66.82 µg/ml (44.1 and 126.65 µM), respectively. The higher dose reached Tmax after 30 min, while 30 mg/kg reached Tmax after 15 min. AUC0-inf was highest for IP administration (770.0 µg*h/ml), while PO administration reached an AUC0-inf of 74.79 and 271 µg*h/ml for 30 and 60 mg/kg, respectively (Table 11 and Figure 4).
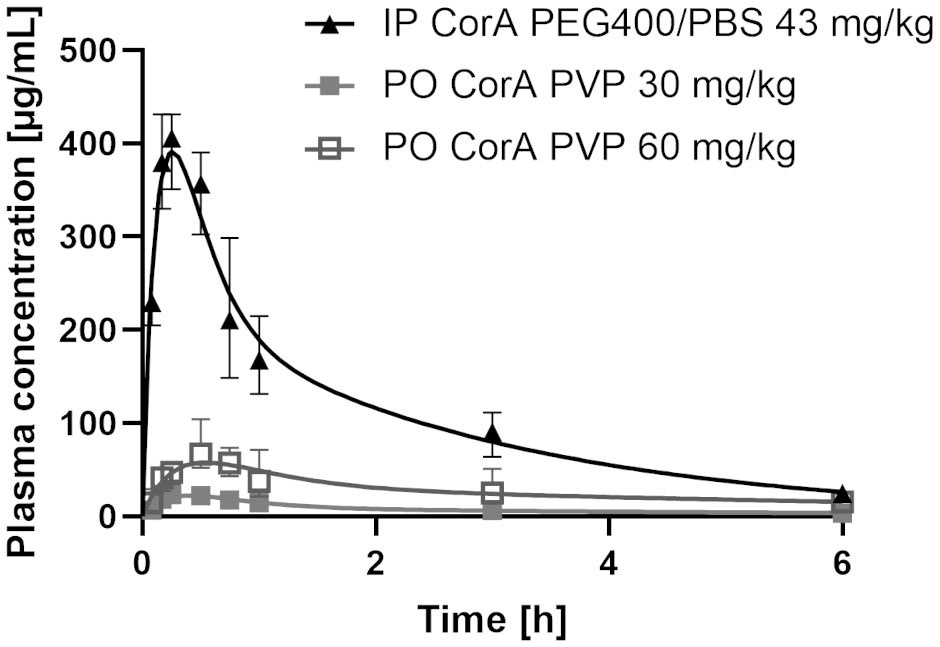
Figure 4 Plasma concentrations of CorA after intraperitoneal (IP) and oral (PO) administration in Mongolian gerbils. 43 mg/kg CorA were administered in PEG400/PBS for IP or 30 and 60 mg/kg CorA were administered in Povidone-PBS for PO application. Plasma was prepared from blood drawn after 5, 10, 15, 30, 60min, 3h and 6h and CorA concentrations were quantified by LC-MS. Symbols indicate median ± IQR.
Discussion
In the present study, the in vitro potency of CorA was confirmed. All CorA derivatives had good potency against Wolbachia in insect cells with CorA having the best-suited activity (EC50 = 0.013 µM). The derivatives CorC and CorD, which are only produced at low amounts under acidic or oxidative conditions during the purification, also had low EC50. Thus, CorA is the best corallopyronin derivate for the treatment of human filarial diseases by targeting Wolbachia endobacteria of filariae. To further assess the safety of CorA and to proceed into human clinical trials, non-GLP pharmacological and in vitro toxicological assays of CorA were conducted.
Drug absorption in the small intestine is dependent on several different factors of drug properties such as solubility, pH and food intake. The in vitro permeation of CorA was determined in a FeSSIF (pH 5.0) assay and in Caco-2 cells. CorA demonstrated a high permeability, which was comparable to the permeability of propranolol, a reference compound for passive transcellular transport with a high human absorption of 90% (59). Thus, CorA uptake from the apical to the basolateral site appears to be greatly facilitated. However, the efflux ratio of CorA was higher than for propranolol and slightly higher compared to cimetidine, which is known for its active efflux by an ATP-binding cassette transporter breast cancer resistance protein (BCRP) (60). Thus, CorA might be subject to active efflux into the lumen in a range similar to cyclosporine A, resulting in reduced absorption and oral bioavailability (61). Based on the previous and the current performed pharmacokinetic in vivo studies, CorA demonstrated high bioavailability (39, 44). Peroral and IP administration resulted in ≥100% bioavailability in rats, which could be a result of the different concentration used for the IV administration (0.48 mg/kg) in comparison to PO and IP administration (5 mg/kg). Successful application of enhanced formulation strategies overcame the detected poor dissolution and solubility properties of CorA when given orally (37). However, it has to be noted that the in vivo efficacy requires higher concentrations than in vitro in gerbils. Unpublished data show that 60 mg/kg CorA given bi-daily for 14 days are required to reduce Wolbachia from L. sigmodontis-infected gerbils by 2-3 logs. Thus, while in vitro an IC50 of 0.01µM is required, much higher doses for Wolbachia depletion are needed in vivo. This may be due to the low absorption of CorA in gerbils.
In summary, CorA has good permeability, and, although subject to active efflux in vitro, it has excellent bioavailability.
Next, CorA distribution and metabolism was determined. In all species, >98% of CorA was bound to plasma proteins, equivalent to plasma binding of the warfarin control and published values for ibuprofen (62). In parallel, CorA had a high stability and resulted in a long half-life. High plasma protein binding capacity might raise the concern of reduced efficacy in vivo. However, CorA has been shown to have good in vivo efficacy (44). We hypothesize that the plasma protein binding may protect CorA from rapid clearance, allowing it to reach the sites of infection, e.g., pleural cavity, of L. sigmodontis worms. In vivo PK data in rats and Mongolian gerbils show a fast absorption and distribution of CorA. Low volume of distribution values in rats suggest a high distribution in plasma and may reflect the high plasma binding capacity of CorA. A solubility enhanced oral formulation based on a povidone ASD as formulation principle (37) increased the plasma levels in Mongolian gerbils (63). Further biodistribution studies will show to which compartments CorA is distributed and how this might enable further indications.
The liver is the most important site of drug metabolism and most drugs are cleared by hepatic metabolism. CorA was analyzed for its half-life and intrinsic clearance in microsomes of different species. The metabolites resulting from the microsomes were also analyzed. Metabolic stability analyses using liver microsomes revealed that CorA is primarily converted to phase I metabolites, leading to the generation of four CorA metabolites (M1-M4) because of oxidation. M1 was detected in liver microsomal incubations across all tested species. In incubates with mouse, rat, mini-pig, and monkey liver microsomes, a two-fold hydroxylated metabolite (M2) was also observed. Further metabolites resulting from oxidative reactions were only observed with mini-pig liver microsomes, yielding M3 (threefold hydroxylation) and M4 (twofold oxidation combined with loss of two hydrogen atoms, most probably due subsequent insertion of two hydroxyl moieties and further oxidation of one of the hydroxyl groups to the corresponding aldehyde). CorA turnover by recombinant human UGT isoenzymes is low and only traces (far below the FDA defined cutoff of 25%) of CorA-glucuronide were observed as metabolite after incubation with 3 out of 6 isoforms. Human and dog microsomes had a moderate to low clearance of CorA, while liver microsomes from mice had a moderate to high intrinsic clearance of CorA. All other tested species exhibited a high intrinsic clearance of CorA. Thus, with the exception of human, dog and mouse microsomes, CorA is rapidly metabolized in vitro by microsomes. In accordance, the higher the intrinsic clearance in the microsomes, the more M1-M4 metabolites were observed after 45 min. Especially mini-pig microsomes had an extremely fast metabolism with no original drug, only M1 and M2 metabolites, detectable after 45 min. In contrast, human microsomes had moderate to low clearance and resulted in a high percentage of the original CorA and only low levels of M1 after 45 min. Experiments analyzing later time points, including tissue specific assays, will be conducted in GLP-studies to determine CorA metabolization over time. The intrinsic clearance rate and metabolite profile in dog microsomes was similar to human microsome metabolism of CorA. Thus, dogs appear to be a suitable model as a non-rodent species for further metabolic studies of CorA. In contrast, rat microsomes metabolized CorA at a higher rate than mouse microsomes, suggesting that mice might be a better-suited rodent model for studying CorA metabolism, which needs to be confirmed in upcoming PK-studies. Even though liver microsomal assays suggested fast metabolism, PK data show a moderate clearance of CorA in rats. Especially for IP application, clearance is lower than for oral administration in rats. Moreover, higher and prolonged plasma levels contribute to the higher efficacy after IP administration.
CorA was primarily metabolized through phase I reactions, which are mainly carried out by CYP450 enzymes. These enzymes play the most important role in metabolizing drugs (64). A strong inhibition of CYP2C9 was observed for CorA. CYP2C9 is a phase I drug-metabolizing enzyme and its substrates include sulfonamides, COX2 selective inhibitors, vitamin-K-antagonists, nonsteroidal anti-inflammatory drugs (NSAID) such as ibuprofen, angiotensin-AT1 selective receptor antagonists, and some antidepressants, such as fluoxetine and amitriptyline. Thus, these drug classes might be prone to potential drug-drug interactions (65). However, it has to be noted that in several cases other drugs could be used to prevent these drug-drug-interactions: For example, vitamin-K-antagonists could be exchanged for novel factor-Xa-inhibitors, which are not metabolized via CYP2C9. In addition, certain off-targets were identified suggesting possible side effects when taken together with drugs for the treatment of rheumatoid arthritis, diabetes, obesity, atherosclerosis and cancer as well as NSAID. Weak CYP450 induction was observed for CYP3A4. CYP3A4 is the most abundant and active CYP450 isoform that metabolizes most drugs. CYP3A4 can be induced by barbiturates, glucocorticoids and rifampicin (66). For rifampicin, pharmacokinetic drug-drug interactions via CYP3A4 induction have been described for benzodiazepines such as midazolam and triazolam as well as calcium channel antagonists such as verapamil and the HMG-CorA reductase inhibitor simvastatin. Most importantly, rifampicin reduces plasma concentrations of the HIV protease inhibitors indinavir, nelfinavir and saquinavir (67). CorA only weakly induced CYP3A4 with an EC50 of 12 µM, while enzyme induction by rifampicin already occurs at a much lower EC50 of 1.5 µM. When administering the minimal efficacious dose of CorA of 60 mg/kg in Mongolian gerbils, a Cmax of 66.82 µg/ml is reached which equals 126.7 µM. However, for CYP3A4 inhibition only unbound concentrations should be considered. Bearing in mind that CorA is highly plasma protein bound, the relevant concentrations to induce CYP3A4 will not be reached. Thus, based on the in vitro data, it appears that CorA has a significant advantage compared to rifampicin as it is unlikely to induce CYP3A4 in vivo. Additional GLP toxicology studies designed with these data will further evaluate the CYP inhibitory and inducing potential of CorA.
Potential drug-drug interactions with other anti-filarial drugs have to be considered. Currently, ivermectin, albendazole and DEC are used for MDA. Since ivermectin is metabolized by CYP3A4, CYP3A5 and CYP2C9 and CorA has an inhibitory effect on CYP2C9 and can activate CYP3A4, ivermectin metabolism may be affected by CorA treatment (68, 69). However, the two drugs would not be combined to treat onchocerciasis. Albendazole is primarily metabolized by CYP1A1, CYP1A2, CYP1B1 and CYP2C19 and thus, drug-drug interactions should be negligible (68, 70). Albendazole is also and partly metabolized by CYP3A4. Thus, drug-drug interaction with CorA may occur. DEC is metabolized via CYP2B6 and CYP1A2, and no drug-drug interaction with CorA would be expected (68).
CorA was profiled for early toxicology by determining effects on hERG, Ames, micronucleus, phototoxicity and cytotoxicity tests. CorA had three flagged off-target effects towards the adenosine receptor A3, PPARγ and COX1 with EC50 values of 11, 2.2 and 8.2 µM, respectively. A3 is involved in intracellular signaling pathways in the CNS and its binding by CorA could have an effect on drugs for the treatment of rheumatoid arthritis (71). Thus, we will investigate in further studies if CorA is able to reach the brain, which would render this interaction also physiologically relevant. PPARγ plays a role in lipogenesis in different cells and is a major activator of adipocyte differentiation, regulating fatty acid storage and glucose metabolism, and its binding can impair drugs for treatment of diabetes, e.g., thiazolidinedione, obesity, atherosclerosis and cancer (72). COX1 is involved in prostaglandin biosynthesis and is inhibited by the NSAIDs acetyl salicylic acid and ibuprofen (73). The GLP toxicology and pharmaceutical safety studies will include bespoke studies to analyze the effect of CorA on these receptors and enzyme to ensure safety of the final product.
No CorA cardiotoxicity and genotoxicity were observed. Cytotoxicity was only observed at concentrations higher than CorA solubility and much higher than the therapeutic range of CorA. Phototoxicity is an important factor to be determined for drugs used for the treatment of filarial diseases since they primarily occur in tropical and sub-tropical countries. It has been shown that tetracyclines, such as doxycycline, are phototoxic, which can lead to exaggerated sunburn reactions (74). However, CorA did not induce phototoxicity up to the limit of solubility; thus, its application should be safe in areas with high UV radiation.
In summary, CorA, is a potent anti-wolbachial drug with no major toxicity. Despite poor water solubility, CorA is absorbed in the intestine and proved to have good bioavailability as a solubility enhanced formulation (37); a formulation that also enhances shelf-life stability. Despite high plasma protein binding, CorA bioavailability has not been affected in previous in vivo efficacy studies (39, 44). CorA is primarily subject to phase I metabolism. It strongly inhibited CYP2C9 and weakly induced CYP3A4, which is unlikely to be relevant in vivo. Thus, potential drug-drug interactions could occur with antidiabetic medications, COX2 inhibitors, angiotensin AT1 receptor antagonists, vitamin K-antagonists, and certain antidepressants.
Human filariae currently infect millions of people in low- and middle-income countries. However, MDA faces several problems that prevent elimination of onchocerciasis in endemic areas and macrofilaricidal compounds would help to effectively achieve elimination. Peroral CorA was shown to deplete Wolbachia and to be macrofilaricidal in the L. sigmodontis rodent model (44). The current study shows that CorA possesses a favorable early ADMET profile.
New macrofilaricidal drugs for the treatment of filariasis need to fulfill a target product profile (TPP), e.g., that of the DNDi, which includes an oral formulation with treatment duration of up to 14 days leading to elimination of skin MF and adult worms superior to current drugs (75). Moreover, the drug should be safe to treat all individuals at risk, show no major adverse events including Mazzotti reaction and ocular reactions, and it should be safe in patients co-infected with L. loa due (75). Even though CorA has not been tested in humans and thus, there is no information on human-efficacy and safety, preliminary conclusions based on the early ADMET profile and the pharmacodynamics results in L. sigmodontis-infected gerbils can be drawn. CorA can be administered in an oral form and the treatment duration is expected to be 14 days. Thus, CorA would fit the TTPs from the DNDi. The risks, side effects and effectiveness against onchocerciasis MF and adult worms in humans has still to be determined in clinical studies. However, based on the in vivo experiments in mice, gerbils and rats, no major toxicity has been observed and Wolbachia are depleted from L. sigmodontis-infected animals after a 14-day regimen. Moreover, based on the nature of CorA as an anti-wolbachial, no side effects and adverse events in L. loa-infected individuals and no Mazzoti reaction nor adverse ocular reactions are expected (76, 77). In comparison to direct-acting drugs, anti-wolbachials such as CorA minimize excessive inflammation and thus, pathology (78–80). Moreover, published results show that CorA has no direct effect on the MF but rather induces a slow decline in peripheral MF due to the inhibition of the embryogenesis (44). Therefore, CorA should be safe in L. loa co-infected patients as well. Taken together, CorA is a promising candidate to support onchocerciasis elimination.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by Landesamt für Natur Umwelt und Verbraucherschutz in North Rhine-Westphalia under the file #81-02.04.2020.A244.
Author contributions
Writing original draft: AE, KP, and AS. Writing review and editing: AE, KP, AS, AH, MH, AK, TB, KW, and KR. Experimental work: AE, HN, TA, and SK. Resources: MG, MS, GK and RM. Supervision: KP, AS, and MH. Project Administration: AH, AS, KP, TH, and SA. All authors contributed to the article and approved the submitted version.
Funding
AH and KP received support from the German Center for Infection Research (DZIF, www.dzif.de, Translational Thematic Unit: Novel Antibiotics grants #09.807, #09.806, #09.822 and #09.914, the Deutsche Forschungsgesellschaft (DFG grant PF673/3-1) and the Federal Ministry of Education and Research (BMBF, grant 16GW0227K). MS and KW get funding from DZIF (grants TTU 09.914, #09.807, TTU09.70) and BMBF (grant 16GW0227K). GK received funding from the DFG grant KO902/5-1. MH and AE are funded by the DZIF Translational Thematic Unit: Novel Antibiotics grants TTU 09.701. KR receives support from the German Center for Infection Research (DZIF, TTU 09.719).
Acknowledgments
We thank Drs Martin Roth and Gundela Peschel, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI), Jena, Germany, for their discussion and lab scale production of CorA. We thank Dr. Ingo Stammberger for his support and correction of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor JT declared a past co-authorship with the author(s) MH and AH.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fitd.2022.983107/full#supplementary-material
References
1. WHO. Ending the neglect to attain the sustainable development goals – a road map for neglected tropical diseases 2021–2030. Geneva: World Health Organization (2020). p. 177.
2. Taylor MJ, Hoerauf A, Bockarie M. Lymphatic filariasis and onchocerciasis. Lancet (2010) 376(9747):1175–85. doi: 10.1016/S0140-6736(10)60586-7
3. Simonsen PE. “Chapter 84 - Filariases,” in Manson's Tropical Diseases (Twenty-second Edition), eds. Cook GC, Zumla AI. (London: W.B. Saunders) (2009). pp. 1477–1513. ISBN 978-1-4160-4470-3
4. WHO. Progress report on the elimination of human onchocerciasis, 2016-2017. Wkly Epidemiol Rec (2017) 92(45):681–700.
5. WHO. Onchocerciasis: WHO (2018). Available at: https://www.who.int/news-room/fact-sheets/detail/onchocerciasis.
6. Addisu A, Adriaensen W, Balew A, Asfaw M, Diro E, Garba Djirmay A, et al. Neglected tropical diseases and the sustainable development goals: An urgent call for action from the front line. BMJ Glob Health (2019) 4(1):e001334. doi: 10.1136/bmjgh-2018-001334
7. WHO. Progress report on the elimination of human onchocerciasis, 2016–2017. Wkly Epidemiol Rec (2017) 92(45):681–94. doi: 10.1111/polp.12181
8. WHO. Lymphatic filariasis and onchocerciasis. Global programme to eliminate lymphatic filariasis: progress report, 2016. Wkly Epidemiol Rec (2017) 92(40):594–607.
9. United Nations “Transforming our world: The 2030 Agenda for Sustainable Development”. (United Nations). Geneva, Switzerland (2015).
10. WHO. Lymphatic filariasis and onchocerciasis. Global programme to eliminate lymphatic filariasis: progress report, 2016. Wkly Epidemiol Rec (2017) 91(40):589–608. doi: 10.3098/ah.2017.091.4.589
11. WHO. Progress report on the elimination of human onchocerciasis, 2016–2017. Wkly Epidemiol Rec (2017) 92(45):681–700.
12. Greene BM, Taylor HR, Cupp EW, Murphy RP, White AT, Aziz MA, et al. Comparison of ivermectin and diethylcarbamazine in the treatment of onchocerciasis. N Engl J Med (1985) 313(3):133–8. doi: 10.1056/NEJM198507183130301[doi
13. Lariviere M, Vingtain P, Aziz M, Beauvais B, Weimann D, Derouin F, et al. Double-blind study of ivermectin and diethylcarbamazine in African onchocerciasis patients with ocular involvement. Lancet (1985) 2(8448):174–7. doi: 10.1016/S0140-6736(85)91496-5
14. Irvine MA, Stolk WA, Smith ME, Subramanian S, Singh BK, Weil GJ, et al. Effectiveness of a triple-drug regimen for global elimination of lymphatic filariasis: A modelling study. Lancet Infect Dis (2017) 17(4):451–8. doi: 10.1016/s1473-3099(16)30467-4
15. Gardon J, Gardon-Wendel N, Demanga N, Kamgno J, Chippaux JP, Boussinesq M. Serious reactions after mass treatment of onchocerciasis with ivermectin in an area endemic for loa loa infection. Lancet (1997) 350(9070):18–22. doi: 10.1016/S0140-6736(96)11094-1[doi
16. Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N, et al. The Wolbachia genome of Brugia malayi: Endosymbiont evolution within a human pathogenic nematode. PloS Biol (2005) 3(4):e121. doi: 10.1371/journal.pbio.0030121
17. Lentz CS, Halls V, Hannam JS, Niebel B, Strübing U, Mayer G, et al. A selective inhibitor of heme biosynthesis in endosymbiotic bacteria elicits antifilarial activity in vitro. Chem Biol (2013) 20(2):177–87. doi: 10.1016/j.chembiol.2012.11.009
18. Strübing U, Lucius R, Hoerauf A, Pfarr KM. Mitochondrial genes for heme-dependent respiratory chain complexes are up-regulated after depletion of Wolbachia from filarial nematodes. Int J Parasitol (2010) 40(10):1193–202. doi: 10.1016/j.ijpara.2010.03.004
19. Wu B, Novelli J, Foster J, Vaisvila R, Conway L, Ingram J, et al. The heme biosynthetic pathway of the obligate wolbachia endosymbiont of brugia malayi as a potential anti-filarial drug target. PloS Negl Trop Dis (2009) 3(7):e475. doi: 10.1371/journal.pntd.0000475
20. Hoerauf A, Volkmann L, Hamelmann C, Adjei O, Autenrieth IB, Fleischer B, et al. Endosymbiotic bacteria in worms as targets for a novel chemotherapy in filariasis. Lancet (2000) 355(9211):1242–3. doi: 10.1016/S0140-6736(00)02095-X[pii]10.1016/S0140-6736(00)02095-X [doi].
21. Debrah AY, Mand S, Marfo-Debrekyei Y, Batsa L, Pfarr K, Buttner M, et al. Macrofilaricidal effect of 4 weeks of treatment with doxycycline on wuchereria bancrofti. Trop Med Int Health (2007) 12(12):1433–41. doi: 10.1111/j.1365-3156.2007.01949.x
22. Debrah AY, Specht S, Klarmann-Schulz U, Batsa L, Mand S, Marfo-Debrekyei Y, et al. Doxycycline leads to sterility and enhanced killing of female onchocerca volvulus worms in an area with persistent microfilaridermia after repeated ivermectin treatment: A randomized, placebo-controlled, double-blind trial. Clin Infect Dis (2015) 61(4):517–26. doi: 10.1093/cid/civ363
23. Awadzi K, Opoku NO, Attah SK, Lazdins-Helds J, Kuesel AC. A randomized, single-Ascending-Dose, ivermectin-controlled, double-blind study of moxidectin in onchocerca volvulus infection. PloS Negl Trop Dis (2014) 8(6):e2953. doi: 10.1371/journal.pntd.0002953
24. Churcher TS, Pion SD, Osei-Atweneboana MY, Prichard RK, Awadzi K, Boussinesq M, et al. Identifying Sub-optimal responses to ivermectin in the treatment of river blindness. Proc Natl Acad Sci USA (2009) 106(39):16716–21. doi: 10.1073/pnas.0906176106
25. Osei-Atweneboana MY, Awadzi K, Attah SK, Boakye DA, Gyapong JO, Prichard RK. Phenotypic evidence of emerging ivermectin resistance in onchocerca volvulus. PloS Negl Trop Dis (2011) 5(3):e998. doi: 10.1371/journal.pntd.0000998[doi
26. Osei-Atweneboana MY, Eng JK, Boakye DA, Gyapong JO, Prichard RK. Prevalence and intensity of Onchocerca volvulus infection and efficacy of ivermectin in endemic communities in Ghana: A two-phase epidemiological study. Lancet (2007) 369(9578):2021–9. doi: 10.1016/S0140-6736(07)60942-8
27. Wanji S, Kengne-Ouafo JA, Esum ME, Chounna PW, Tendongfor N, Adzemye BF, et al. Situation analysis of parasitological and entomological indices of onchocerciasis transmission in three drainage basins of the rain forest of south West Cameroon after a decade of ivermectin treatment. Parasit Vectors (2015) 8:202. doi: 10.1186/s13071-015-0817-2
28. Krücken J, Holden-Dye L, Keiser J, Prichard RK, Townson S, Makepeace BL, et al. Development of emodepside as the first safe, short-course adulticidal treatment for human onchocerciasis – the fruit of a successful industrial–academic collaboration. PloS Pathog. (2021). 17(7):e1009682. doi: 10.1371/journal.ppat.1009682.
29. Townson S, Freeman A, Harris A, Harder A. Activity of the cyclooctadepsipeptide emodepside against onchocerca guttorosa, onchocerca lienalis and brugia pahangi. Am Soc Trop Med Hygiene 54th Annu Meeting (2005) 73(6 suppl):93. doi: 10.4269/ajtmh.2005.73.0
30. Hübner MP, Martin C, Specht S, Koschel M, Dubben B, Frohberger SJ, et al. Oxfendazole mediates macrofilaricidal efficacy against the filarial nematode litomosoides sigmodontis in vivo and inhibits onchocerca spec. Motil Vitro. PloS Negl Trop Dis (2020) 14(7):e0008427. doi: 10.1371/journal.pntd.0008427
31. Aljayyoussi G, Tyrer HE, Ford L, Sjoberg H, Pionnier N, Waterhouse D, et al. Short-course, high-dose rifampicin achieves wolbachia depletion predictive of curative outcomes in preclinical models of lymphatic filariasis and onchocerciasis. Sci Rep (2017) 7(1):210. doi: 10.1038/s41598-017-00322-5
32. Hong WD, Benayoud F, Nixon GL, Ford L, Johnston KL, Clare RH, et al. Awz1066s, a highly specific anti-wolbachia drug candidate for a short-course treatment of filariasis. Proc Natl Acad Sci U.S.A. (2019) 116(4):1414–9. doi: 10.1073/pnas.1816585116
33. Hübner MP, Koschel M, Struever D, Nikolov V, Frohberger SJ, Ehrens A, et al. In vivo kinetics of wolbachia depletion by abbv-4083 in l. Sigmodontis Adult Worms Microfilariae. PloS Negl Trop Dis (2019) 13(8):e0007636. doi: 10.1371/journal.pntd.0007636
34. von Geldern TW, Morton HE, Clark RF, Brown BS, Johnston KL, Ford L, et al. Discovery of abbv-4083, a novel analog of tylosin a that has potent anti-wolbachia and anti-filarial activity. PloS Negl Trop Dis (2019) 13(2):e0007159. doi: 10.1371/journal.pntd.0007159
35. Alami NN, Carter DC, Kwatra NV, Zhao W, Snodgrass L, Porcalla AR, et al. Anti-wolbachia candidate abbv-4083: Phase 1 safety and pharmacokinetics clinical trial in healthy adults. Am J Trop Med Hyg (2019) 101(5 suppl):392.
36. Schaberle TF, Schmitz A, Zocher G, Schiefer A, Kehraus S, Neu E, et al. Insights into structure-activity relationships of bacterial rna polymerase inhibiting corallopyronin derivatives. J Natural products (2015) 78(10):2505–9. doi: 10.1021/acs.jnatprod.5b00175
37. Krome A, Becker T, Kehraus S, Schiefer A, Steinebach C, Aden T, et al. Solubility and stability enhanced oral formulations for the anti-infective corallopyronin a. Pharmaceutics (2020) 12:1105. doi: 10.3390/pharmaceutics12111105
38. Irschik H, Jansen R, Hofle G, Gerth K, Reichenbach H. The corallopyronins, new inhibitors of bacterial rna synthesis from myxobacteria. J antibiotics (1985) 38(2):145–52. doi: 10.7164/antibiotics.38.145
39. Schiefer A, Schmitz A, Schäberle TF, Specht S, Lämmer C, Johnston KL, et al. Corallopyronin a specifically targets and depletes essential obligate Wolbachia endobacteria from filarial nematodes ii. J Infect Dis (2012) 206(2):249–57. doi: 10.1093/infdis/jis341
40. Belogurov GA, Vassylyeva MN, Sevostyanova A, Appleman JR, Xiang AX, Lira R, et al. Transcription inactivation through local refolding of the rna polymerase structure. Nature (2009) 457(7227):332–5. doi: 10.1038/nature07510
41. Mukhopadhyay J, Das K, Ismail S, Koppstein D, Jang M, Hudson B, et al. The rna polymerase "Switch region" is a target for inhibitors. Cell (2008) 135(2):295–307. doi: 10.1016/j.cell.2008.09.033
42. Krome AK, Becker T, Kehraus S, Schiefer A, Gutschow M, Chaverra-Munoz L, et al. Corallopyronin a: Antimicrobial discovery to preclinical development. Natural product Rep. doi: 10.1039/d2np00012a
43. O'Neill A, Oliva B, Storey C, Hoyle A, Fishwick C, Chopra I. Rna polymerase inhibitors with activity against rifampin-resistant mutants of staphylococcus aureus. Antimicrob Agents Chemother (2000) 44(11):3163–6. doi: 10.1128/aac.44.11.3163-3166.2000
44. Schiefer A, Hübner MP, Krome A, Lämmer C, Ehrens A, Aden T, et al. Corallopyronin a for short-course anti-wolbachial, macrofilaricidal treatment of filarial infections. PloS Negl Trop Dis (2020) 14(12):e0008930. doi: 10.1371/journal.pntd.0008930
45. Kock F, Hauptmann M, Osterloh A, Schäberle TF, Poppert S, Frickmann H, et al. Orientia tsutsugamushi is highly susceptible to the rna polymerase switch region inhibitor corallopyronin a in vitro and in vivo. Antimicrob Agents Chemother (2018) 62(4):e01732–17. doi: 10.1128/AAC.01732-17
46. Loeper N, Graspeuntner S, Ledig S, Kaufhold I, Hoellen F, Schiefer A, et al. Elaborations on corallopyronin a as a novel treatment strategy against genital chlamydial infections. Front Microbiol (2019) 10:943. doi: 10.3389/fmicb.2019.00943
47. Shima K, Ledig S, Loeper N, Schiefer A, Pfarr K, Hoerauf A, et al. Effective inhibition of rifampin-resistant Chlamydia trachomatis by the novel DNA-dependent rna-polymerase inhibitor corallopyronin a. Int J Antimicrob Agents (2018) 52:523–4. doi: 10.1016/j.ijantimicag.2018.07.025
48. Kirsch-Volders M, Elhajouji A, Cundari E, Van Hummelen P. The in vitro micronucleus test: A multi-endpoint assay to detect simultaneously mitotic delay, apoptosis, chromosome breakage, chromosome loss and non-disjunction. Mutat Res (1997) 392(1-2):19–30. doi: 10.1016/s0165-1218(97)00042-6
49. Maron DM, Ames BN. Revised methods for the salmonella mutagenicity test. Mutat Res (1983) 113(3-4):173–215. doi: 10.1016/0165-1161(83)90010-9
50. Wakata A, Matsuoka A, Yamakage K, Yoshida J, Kubo K, Kobayashi K, et al. Sftg international collaborative study on in vitro micronucleus test iv. using chl cells. Mutat Res (2006) 607(1):88–124. doi: 10.1016/j.mrgentox.2006.04.003
52. Spielmann H, Balls M, Brand M, Doring B, Holzhutter HG, Kalweit S, et al. Eec/Colipa project on in vitro phototoxicity testing: First results obtained with a Balb/C 3t3 cell phototoxicity assay. Toxicol In Vitro (1994) 8(4):793–6. doi: 10.1016/0887-2333(94)90069-8
53. Spielmann H, Balls M, Dupuis J, Pape WJ, Pechovitch G, de Silva O, et al. The international Eu/Colipa in vitro phototoxicity validation study: Results of phase ii (Blind trial). part 1: The 3t3 nru phototoxicity test. Toxicol In Vitro (1998) 12(3):305–27. doi: 10.1016/s0887-2333(98)00006-x
54. Schmitz A. The antibiotic corallopyronin a and the corallorazines from the myxobacterium corallococcus coralloides. B035: Rheinischen Friedrich-Wilhelms-Universität Bonn, Bonn, Germany (2013).
55. Pogorevc D, Panter F, Schillinger C, Jansen R, Wenzel SC, Muller R. Production optimization and biosynthesis revision of corallopyronin a, a potent anti-filarial antibiotic. Metab Eng (2019) 55:201–11. doi: 10.1016/j.ymben.2019.07.010
56. Tsume Y, Mudie DM, Langguth P, Amidon GE, Amidon GL. The biopharmaceutics classification system: Subclasses for in vivo predictive dissolution (Ipd) methodology and ivivc. Eur J Pharm Sci Off J Eur Fed Pharm Sci (2014) 57:152–63. doi: 10.1016/j.ejps.2014.01.009
57. King CD, Rios GR, Green MD, Tephly TR. Udp-glucuronosyltransferases. Curr Drug Metab (2000) 1(2):143–61. doi: 10.2174/1389200003339171
58. FDA. In vitro drug interaction studies —cytochrome P450 enzyme- and transporter-mediated drug interactions: Guidance for industry. Silver Spring, MD, USA: U.S. Department of Health and Human Services (2020). p. 46.
59. Borgstrom L, Johansson CG, Larsson H, Lenander R. Pharmacokinetics of propranolol. J Pharmacokinet Biopharm (1981) 9(4):419–29. doi: 10.1007/BF01060886
60. Mao Q, Unadkat JD. Role of the breast cancer resistance protein (Bcrp/Abcg2) in drug transport–an update. AAPS J (2015) 17(1):65–82. doi: 10.1208/s12248-014-9668-6
61. Wang K, Qi J, Weng T, Tian Z, Lu Y, Hu K, et al. Enhancement of oral bioavailability of cyclosporine a: Comparison of various nanoscale drug-delivery systems. Int J Nanomedicine (2014) 9:4991–9. doi: 10.2147/IJN.S72560
62. Whitlam JB, Crooks MJ, Brown KF, Pedersen PV. Binding of nonsteroidal anti-inflammatory agents to proteins–i. ibuprofen-serum albumin interaction. Biochem Pharmacol (1979) 28(5):675–8. doi: 10.1016/0006-2952(79)90154-0
63. Becker T, Krome AK, Vahdati S, Schiefer A, Pfarr K, Ehrens A, et al. In vitro-In vivo relationship in mini-ccale-Enabling formulations of corallopyronin A. Pharmaceutics (2022) 14, 1657.
64. Guengerich FP. Cytochrome P450s and other enzymes in drug metabolism and toxicity. AAPS J (2006) 8(1):E101–11. doi: 10.1208/aapsj080112
65. Van Booven D, Marsh S, McLeod H, Carrillo MW, Sangkuhl K, Klein TE, et al. Cytochrome P450 2c9-Cyp2c9. Pharmacogenet Genomics (2010) 20(4):277–81. doi: 10.1097/FPC.0b013e3283349e84
66. Guengerich FP. Cytochrome p-450 3a4: Regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol (1999) 39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1
67. Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT. Pharmacokinetic interactions with rifampicin : Clinical relevance. Clin Pharmacokinet (2003) 42(9):819–50. doi: 10.2165/00003088-200342090-00003
68. Li XQ, Bjorkman A, Andersson TB, Gustafsson LL, Masimirembwa CM. Identification of human cytochrome P(450)S that metabolise anti-parasitic drugs and predictions of in vivo drug hepatic clearance from in vitro data. Eur J Clin Pharmacol (2003) 59(5-6):429–42. doi: 10.1007/s00228-003-0636-9
69. Rendic SP. Metabolism and interactions of ivermectin with human cytochrome P450 enzymes and drug transporters, possible adverse and toxic effects. Arch Toxicol (2021) 95(5):1535–46. doi: 10.1007/s00204-021-03025-z
70. Asteinza J, Camacho-Carranza R, Reyes-Reyes RE, Dorado-Gonzalez VV, Espinosa-Aguirre JJ. Induction of cytochrome P450 enzymes by albendazole treatment in the rat. Environ Toxicol Pharmacol (2000) 9(1-2):31–7. doi: 10.1016/s1382-6689(00)00059-4
71. Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, et al. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase ii clinical trial. J Rheumatol (2008) 35(1):41–8.
72. Varga T, Czimmerer Z, Nagy L. Ppars are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta (2011) 1812(8):1007–22. doi: 10.1016/j.bbadis.2011.02.014
73. Tomić M, Micov A, Pecikoza U, Stepanović-Petrović R. Chapter 1 - clinical uses of nonsteroidal anti-inflammatory drugs (Nsaids) and potential benefits of nsaids modified-release preparations. Microsized Nanosized Carriers Nonsteroidal Anti-Inflammatory Drugs (2017)p:1–29.
74. Goetze S, Hiernickel C, Elsner P. Phototoxicity of doxycycline: A systematic review on clinical manifestations, frequency, cofactors, and prevention. Skin Pharmacol Physiol (2017) 30(2):76–80. doi: 10.1159/000458761
75. Drugs for Neglected Diseases Initiative. Target product profile for river blindness [Online]. (2022) Available: https://dndi.org/diseases/filaria-river-blindness/target-product-profile/#:~:text=DNDi%20aims%20to%20develop%20a,co%2Dinfected%20with%20Loa%20Loa. [Accessed 2022].
76. Ehrens A, Hoerauf A, Hubner MP. Current perspective of new anti-wolbachial and direct-acting macrofilaricidal drugs as treatment strategies for human filariasis. GMS Infect Dis (2022) 10:Doc02. doi: 10.3205/id000079
77. Hoerauf A, Specht S, Buttner M, Pfarr K, Mand S, Fimmers R, et al. Wolbachia endobacteria depletion by doxycycline as antifilarial therapy has macrofilaricidal activity in onchocerciasis: A randomized placebo-controlled study. Med Microbiol Immunol (2008) 197(3):295–311. doi: 10.1007/s00430-007-0062-1[doi
78. Brattig NW, Buttner DW, Hoerauf A. Neutrophil accumulation around onchocerca worms and chemotaxis of neutrophils are dependent on wolbachia endobacteria. Microbes Infect (2001) 3(6):439–46. doi: 10.1016/S1286-4579(01)01399-5
79. Gillette-Ferguson I, Hise AG, McGarry HF, Turner J, Esposito A, Sun Y, et al. Wolbachia-induced neutrophil activation in a mouse model of ocular onchocerciasis (River blindness). Infect Immun (2004) 72(10):5687–92. doi: 10.1128/IAI.72.10.5687-5692.2004
Keywords: Onchocerciasis, ADMET, filaria, macrofilaricidal, corallopyronin A, anti-Wolbachia, Onchocerca volvulus
Citation: Ehrens A, Schiefer A, Krome AK, Becker T, Rox K, Neufeld H, Aden T, Wagner KG, Müller R, Grosse M, Stadler M, König GM, Kehraus S, Alt S, Hesterkamp T, Hübner MP, Pfarr K and Hoerauf A (2022) Pharmacology and early ADMET data of corallopyronin A, a natural product with macrofilaricidal anti-wolbachial activity in filarial nematodes. Front. Trop. Dis 3:983107. doi: 10.3389/fitd.2022.983107
Received: 30 June 2022; Accepted: 31 August 2022;
Published: 30 September 2022.
Edited by:
Joseph Daniel Turner, Liverpool School of Tropical Medicine, United KingdomReviewed by:
Gert-Jan Wijnant, Université catholique de Louvain, BelgiumW. David Hong, University of Liverpool, United Kingdom
Copyright © 2022 Ehrens, Schiefer, Krome, Becker, Rox, Neufeld, Aden, Wagner, Müller, Grosse, Stadler, König, Kehraus, Alt, Hesterkamp, Hübner, Pfarr and Hoerauf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth Pfarr, a2VubmV0aC5wZmFyckB1a2Jvbm4uZGU=
†These authors have contributed equally to this work