 Vera Bin San Chan
Vera Bin San Chan Mary Beth Johnstone
Mary Beth Johnstone A. P. Wheeler
A. P. Wheeler Andrew S. Mount
Andrew S. Mount- Department of Biological Sciences, Clemson University, Clemson, SC, United States
Chitin is often reported in molluscan shells, where it likely contributes to the mechanical strength of the biomineral. However, the role of this polysaccharide in relation to the process of shell formation is not well understood. We investigated the deposition of chitin during shell repair in the Eastern oyster, Crassostrea virginica, by inserting stainless steel and glass implants in a region of shell damage. This work documents the time course of deposition of both chitin fibrils and calcium carbonate layers. Chitin was detected by confocal laser scanning microscopy (CLSM) using a chitin-specific fluorescent probe that was produced from clones of a chitin-binding domain. The presence of fibrils was confirmed using electron microscopy of implants. The fibrils’ dimensions were reduced after treatment with both acid and bleach, suggesting that chitin interacts with inorganic minerals and other organic components such as proteins and lipids as early as 5 h after shell damage. With CLSM, it was shown that chitin co-localized with the cell membrane, suggesting the importance of cells located on the implants in the process of fibril formation. Using observations from this study as well as those from the literature on chitin synthase production, we propose two cellular models for chitin deposition related to shell formation.
Introduction
Chitin is the second most abundant naturally occurring polysaccharide after cellulose (Kumar, 2000; Cohen, 2010), and this biopolymer is also a subject of major interest in material science (Merzendorfer and Zimoch, 2003). It has a crystalline structure made up of straight chain polysaccharides and is typically used by organisms for structural support (Stern and Jedrzejas, 2008). There are three allomorphic forms of chitin described in the literature, designated α-, ß-, and γ- chitin (Carlström, 1957; Kaya et al., 2017). The more commonly known allomorphs are α- and ß-chitin, which are differentiated based on the relative position of the constituent microfibril units. α-chitin is the more stable structure because it contains alternating chains, a configuration that enhances the number of intermolecular hydrogen bonds between the polysaccharide chains. The α- polymorph is found in crustacean and insect cuticles and the cell walls of yeast and other fungi. On the other hand, ß-chitin, with a parallel arrangement of chains, has fewer inter-chain hydrogen bonds. The ß-form is commonly hydrated and associated with proteins, such as in the cell walls of diatoms, and the internal skeleton of cephalopods (Carlström, 1957; Brunner et al., 2009). Kaya et al. (2017) characterized γ-chitin extracted from the cocoon of a moth and the stomach of a squid and concluded that this rarer form of chitin is structurally more similar to α- than ß-chitin.
Chitin is broadly distributed in the shells of mollusks where it constitutes 3.5 wt% of the insoluble organic components of calcified shell layers and their outermost organic covering layer, the periostracum (Galtsoff, 1964; Peters, 1972; Goffinet and Jeuniaux, 1979; Heinemann, 2008). Peters (1972) observed the presence of microfibrils in the shell, which he interpreted as chitin because of the fibers’ resistance to treatments with alkaline hydroxides and dilute acids (Goffinet and Jeuniaux, 1979). Using X-ray diffraction, Weiner and Traub (1980) concluded that the insoluble extracellular matrix of nacre from some species contained ß-chitin. However, the prominence of chitin was not uniform among the species and mineral microstructures they examined, and its presence was especially ambiguous in the foliated layers of Crassostrea irridescens and Crassostrea gigas (alternatively known as Magallana gigas). A chitin-rich membrane between the myostracum and foliated shell layer appears to define the boundary of the two crystal polymorphs at the adductor muscle scar of C. gigas (Lee and Choi, 2007).
While chitin plays a role in increasing the strength and resilience of many structures, including perhaps mineralized shell layers (Heredia et al., 2007), the toughness of the molluscan shell has most typically been associated with the extracellular matrix proteins which are interspersed between layers of micro- to nano-sized crystals thus forming a bio ceramic (Currey, 1999). Some of these proteins have also been repeatedly associated with the formation of different crystal morphologies (Gotliv et al., 2003). The presence of chitin-binding domains in matrix proteins suggests that chitin is a protein-anchoring material and may thereby play a role through matrix proteins both in the mechanical properties of the shell as well as in mineral formation. For example, Pif 177, a shell matrix protein from the inner aragonitic nacreous layer of the pearl oyster, Pinctada fucata, is cleaved post-translationally into Pif 80 and Pif 97. While Pif 80 binds aragonitic crystals and mediates their growth orientation, Pif 97 binds chitin (Suzuki et al., 2009, 2016). Chitin-binding domains have been identified in both the organic periostracal covering and outer prismatic layer of the shell of P. fucata as well (Suzuki et al., 2007; Nakayama et al., 2013). A homolog of Pif 177, which contained the chitin-binding cleavage product Pif 97 only, was cloned from the mantle of the oyster, C. gigas, and was found to be selectively expressed in the mantle (Wang et al., 2013). These findings suggest that the gene plays an important role in calcitic foliated shell similar to that of the homologs in the nacre of Pinctada. In fact, when these workers injected siRNA of Pif 97 into the adductor muscle of the animal, the structure of the foliar laths became thinner and narrower. In earlier studies, Falini et al. (1996) demonstrated that soluble proteins isolated from the molluscan shell were able to control the mineralogy of calcium carbonate in vitro when absorbed to chitin and silk protein. Using cryo-transmission electron microscopy (TEM), Levi-Kalisman et al. (2001) demonstrated the presence of chitin in the extracellular matrix between layers of nacre (interlamellar sheets) of Atrina rigida and proposed a model, based also on in vitro assemblages of protein and chitin, in which chitin was the principal organizing element of these sheets. This model was reinforced by Nudelman et al. (2008) using environmental and cryo-scanning electron microscopy (SEM) of chitinase and protease treated samples of nacre from two species.
The presence of chitin synthase in mineralizing tissue also indicates the potential significance of chitin in shell formation. For example, Suzuki et al. (2007) cloned a cDNA from mantle and demonstrated its expression in this tissue. Zhang et al. (2012) demonstrated that the enzyme transcript was highly expressed during early stages of C. gigas development when larval shell formation occurs, and, among all tissues, it was almost exclusively expressed in the mantle of adult oysters. Furthermore, in vivo partial inhibition of chitin synthase of developing Mytilus galloprovincialis larvae resulted in structurally and likely functionally abnormal larval shells (Schönitzer and Weiss, 2007). Molluscan chitin synthase has a unique myosin domain structure (Weiss, 2012), suggesting that the mechanism of chitin synthesis is very different from the better-studied taxonomic groups, such as yeast and fungi (Cohen, 2010).
In other systems, the biological role of chitin has been expanded beyond that of a structural molecule to include its involvement in various biological cascades (Hasegawa et al., 2001; Ichinohe et al., 2007; Di Rosa et al., 2016). When combined with various chitin-binding properties of proteins, chitin has the potential to be a spatial framework for biological functions, which may include cell-to-cell recognition and oxidative stress protection (Wang et al., 2012), actin bundling and organization (Öztürk-Çolak et al., 2016), and various innate immune responses (Lee et al., 2011), including apoptosis and necrosis (Jeon et al., 2012). Molluscan shell repair is known to begin with cell-driven inflammation and wound-healing responses at the shell margin mediated by the nervous system and hemocytes (Liu et al., 2017). Considering these examples of biological processes that are influenced by chitin, it is plausible that chitin may interact with cells and tissues as part of the cellular responses during mineral formation.
An important step in determining the role of chitin in shell formation is to understand its cellular origins and delivery to the mineralization front. Processes involved in shell formation of the Eastern oyster, Crassostrea virginica, have been successfully observed by the authors using a patented method for inducing shell repair (Mount et al., 2013, 2016; Johnstone et al., 2015). By inserting sterile metal implants between the mantle margin and the shell near the adductor muscle, adherence of granulocytic hemocytes containing 100 nm-sized crystals and deposition of prismatic and foliated layers was effectively observed within hours (Johnstone et al., 2015). In these studies, the outer mantle epithelium (OME), which is the closest to the shell inner surface, serves as a source of extracellular matrices and appears to introduce the junctional boundaries for the prismatic and foliated minerals (Johnstone et al., 2015). Because of their involvement in the production of other components of regenerating shell, hemocytes and OME are likely sources of chitin in this process as well.
Using the same method in this study, we followed the temporal changes of chitin deposition during shell regeneration in the Eastern oyster, employing a correlative microscopy approach. To specifically identify chitin at the mineralization front, we applied confocal laser scanning microscopy (CLSM) to detect a fluorescent conjugated chitin-binding domain produced in Escherichia coli using a circular plasmid vector. Counter staining of nuclei and membrane structures was used to uncover the relationship of adhering hemocytes to chitin-rich areas. To observe structural changes in chitin during the regeneration process, SEM was used to observe the ultrastructure of materials deposited on the implants. At high resolution, the interaction of putative chitin structures with other components was determined by treatment with acid to remove calcium carbonate and treatment with bleach to remove proteins and lipids. By using the Eastern oyster C. virginica as a model, the overall aim of this study was to examine the presence of chitin in the early stages of shell repair and its interaction with minerals and other molecules as well as with hemocytes.
Materials and Methods
Animal Collection and Maintenance
Wild Eastern oysters, C. virginica, were obtained from their natural habitats and were immediately transferred via overnight shipping to the Okeanos Research Laboratory at Clemson University. The animals used for stainless steel implant experiments were obtained from Louisiana1 in October 2015, while animals used for coverslip glass implant experiments were obtained from the Baruch Marine Field Lab, South Carolina in March 2018.
Upon arrival of each collection, 60–100 oysters were brushed to remove any adhering macrofauna and were submerged in an aerated holding tank which contained 680 L of artificial seawater (Red Sea salt mix, United States) maintained at 24 ppt salinity and 18°C. The water of the tank was recirculated through two cartridge containing cotton string-wound filters to remove particulates and one cartridge containing biological media to remove ammonia and nitrate waste. For the 2018 collection, a more advanced water treatment system was installed, including a 50 L sump, a sock filter, a protein skimmer, and a moving-bed Bio-reactor (Bashsea, United States). Ammonia and nitrate levels in the tanks for both experiments were monitored every other day using commercially available aquarium test solutions (Aquarium Pharmaceuticals, API). If these levels exceeded 0.25 ppm, the water was changed. The filtration system was bypassed during feeding hours which lasted 4–6 h per day, 5 days a week. The animals were fed ad libitum with a mix of six marine microalgae–Isochrysis sp., Pavlova sp., Tetraselmis sp., Chaetoceros calcitrans, Thalassiosira weissflogii, and Thalassiosira pseudonana (Shellfish Diet 1800, Reed Mariculture). Oysters were used within a month of collection.
Preparation of Materials for Implantation
Two types of implantation materials were prepared for microscopic observation of shell regeneration processes. In experiment 1, 1 cm × 1 cm × 1 mm polished stainless steel foil was applied for an improved conductivity during SEM. In experiment 2, circular glass cover slips (#1.5) were applied for CLSM. Before the experiments, all implant materials were cleaned to remove organic deposits (sequentially, methanol, 5 min; acetone, 5 min; isopropyl alcohol, 5 min; and ethanol, 5 min) prior to autoclave sterilization and air-drying.
Shell Regeneration Protocol
Shell repairing events were observed using an implantation procedure described in previous studies (Johnstone et al., 2015), and in United States Patents 8,541,031 and 9,371,451 (Mount et al., 2013, 2016). Briefly, the posterior margin adjacent to the adductor muscles of the oysters (length, 7.5–8.5 cm) were carefully cut with a cement saw to allow an approximately 1 cm opening to the extrapallial cavity where the implants were introduced. Then, rubber bands were wrapped around the oysters, covering the opening. The tightness of the rubber bands was adjusted by wrapping around the oysters 1–2 times, in order to prevent the implants from being rejected and yet allow normal opening of the valve and mantle activity during shell repair. Stainless steel implants (experiment 1) were collected at 5, 15, and 27 h and 6 days of incubation and glass coverslip implants (experiment 2) were collected at 5, 18, and 27 h of incubation. These time points were selected to optimize the capture of cellular activities (5, 15, and 27 h) on CLSM and mineral ultrastructures (27 h and 6 days) on the SEM. Oysters were kept in the same tank but not fed during the implantation experiment to avoid accumulation of dead algal cells on the implants which would compromise these observation. Foliated mineral is commonly deposited at the 6 days time point. After incubation, the implants were retrieved from the opening using a tweezer when possible. When necessary, the shell valves were separated by cutting through the adductor muscles with a shucking knife on the anterior side of the shell to minimize the introduction of shell fragments to the implant surfaces.
Preparation of Chitin-Binding Domain From Engineered Bacteria
To localize the presence of chitin during shell repair, a SNAP-conjugated chitin-binding domain (SNAP-CBD) fusion protein was produced by incubation and inoculation. A stab culture of E. coli transfected with the plasmid vector pYZ205 hosted by NEB T7 Express cells was obtained from Yinghua Zhang (New England Biolabs). The expression of plasmid pYZ205 produces a conserved chitin binding domain originating from the bacteria Bacillus circulans (superfamily cluster cl00046; NCBI). The domain has the following amino acid sequence: AWQVNTAYTAGQLVTYNGKTYKCLQPHTSLAGWEPSNVPALWQLQ. In addition to the chitin-binding domain, the construct has a polyhistidine site for protein purification, and a conjugation site with a SNAP fluorescent probe (Tang et al., 2015). The workflow of preparing a probe for fluorescent chitin detection includes (1) isolating a single colony of transfected E. coli and inducing expression of SNAP-CBD, (2) extraction and purification of SNAP-CBD, and (3) conjugating SNAP-CBD with a SNAP reactive fluorescent probe. A SNAP site on the CBD allows the use of a great variety of fluorescent probes. Here, we have made CBD-546 from SNAP-Surface Alexa Fluor 546 (New England Biolabs).
Determination of Specificity for Chitin Probes
Treatment of hemocytes with chitinase was used to test the specificity of the CBD signals (Heath-Heckman and McFall-Ngai, 2011). Hemocytes were drawn at 24 h after shell damage using the same oysters which were incubated with implants for the 5 and 18 h time points. The hemolymph from four oysters was spread on different glass coverslips and the hemocytes were allowed to aggregate for 20 min before fixation. Fixation of hemocytes was performed with 4% paraformaldehyde buffered in PBS for 20 min. Fixed hemocytes were washed twice with 0.4% Triton X-100 for 15 min and then permeabilized with phosphate buffered saline at pH 6.0 (PBS) with 0.4% Triton X-100 for 1 h. The permeabilized samples were washed three times with PBS at room temperature, with the duration of each wash being 15 min. Permeabilized hemocytes were exposed to three different conditions for 0.5 h at 37°C: (1) PBS; (2) PBS containing 0.25 mg/mL each of two chitinases one from Streptomyces griseus (Sigma-Aldrich) and one from Trichoderma viride (Sigma-Aldrich) (T); and (3) PBS containing heat-inactivated chitinases (inactivated at 75°C for 20 min) (HT).
The treatment time of 0.5 h at 37°C was chosen according to a trial study in which 1 and 2 h of chitinase treatment resulted in the disaggregation of the hemocytes and a major loss of cells for sampling. At the end of incubation, hemocytes were washed three times for 10 min with PBS and were labeled with SNAP-546 and 4’,6-diamidino-2-phenylindole (DAPI) as described for the glass implants. Each hemocyte sample was observed using a z-stack image from three random regions. The mean values of intensity (range from 0 to 255) from 10 areas around a cell nuclei were measured with point measurement tools from the maximum projection image using ImageJ Fiji (Schindelin et al., 2012).
CLSM Observation of Implants
One circular glass implant from each of three oysters at each time point was either unstained, stained only with the chitin probe, or triple-stained for chitin, nuclei and membranes. After fixation in 4% paraformaldehyde buffered with 0.1 M cacodylate trihydrate at pH 8.0 for 1 h, the implants were washed in a solution of 0.1 M cacodylate trihydrate buffer at pH 8.0 and 0.4% Triton X-100 detergent three times for 10 min, and then for 2 h to increase the permeability of the adherent cells. The implants were then washed with 0.1 M cacodylate trihydrate buffer, pH 8.0, three times for 10 min, and subsequently with freshly made 1 mg/mL sodium borohydride in cacodylate trihydrate buffer at pH 8.0, three times for 5 min to quench autofluorescence from fixation. They were then washed with 0.1 M cacodylate trihydrate buffer, pH 8.0, three times for 5 min prior to washing and incubating with blocking buffer (PBS with 1% BSA) for 1 h at room temperature. The implants were labeled with CBD-546, at a 1:10 dilution of CBD-546 in 1% BSA PBS overnight at 4°C. In addition, implants were counterstained for nuclei with 1 μg/mL DAPI for 30 min and for cell membranes with 5 μg/mL CellMask Deep Red plasma membrane stain (Thermo Fisher Scientific) for 30 min. Prior to CLSM, all implants were washed three times with PBS, for 15 min between and after staining procedures. Following the final wash they were mounted on microscope slides with liquid mountant (ProLong Gold, Thermo Fisher Scientific), and dried overnight before imaging on a CLSM.
An untreated 18 h implant was visualized on CLSM for 2 h in order to examine any changes of fluorescence intensity of the chitin probe due to photobleaching. Additionally, an 18 h implant was visualized to examine any changes in fluorescence intensity of due to chitinase treatment.
SEM Observation of Implants
The stainless steel foil implants were fixed in 4% paraformaldehyde buffered with 0.1 M cacodylate trihydrate buffer at pH 8.0 for 1 h. Following fixation, they were washed three times and stored in 0.1 M cacodylate trihydrate buffer at pH 8.0 prior to SEM imaging. To determine the chemical composition of the fibrils and any associated materials, the implants were treated with either 0.1% acetic acid for 1 min, 0.5% commercial bleach for 30 min or PBS as a control. After the chemical treatments, the implants were washed for 5 min with distilled water to remove salts in the buffer, then fixed in 1% osmium tetroxide for 30 min and finally washed with distilled water for 5 min to stop the fixation process. To enhance conductivity of the cells on the implants, the specimens were serially dehydrated with ethanol (50% for 10 min, 70% for 10 min, 85% for 10 min, 95% for 10 min, and 100% three times for 10 min), and then washed with 50% hexamethyldisilazane (HMDS) in ethanol, followed by 100% HMDS. The implants were then placed in a fume hood to dry overnight, after which they were mounted onto an aluminum stub with carbon tape. The cells and fibrous deposits on the implant were observed using a Hitachi 4800 SEM. The diameter of the fibril structures was quantified at high magnification of 10,000–90,000 times. Mean fibril diameters (FD) were measured from 7 to 10 fibrils chosen randomly, from implants for each time points point (5, 15, and 27 h and 6 days) and each experimental treatment level (control, acid, and bleach). Fibril that were partially covered were avoided for FD measurement. The typical morphology of fibrils which FD was measured are labeled in Figures 4–6.
Statistical Analysis
The effect of chitinase on the mean fluorescence intensity of CBD-546 was assessed using one-factor analysis of variance (ANOVA) and the post hoc Tukey’s HSD tests. The effects of experimental treatments (control, acid, and bleach) on the mean FD at 5, 15, and 27 h and 6 days after shell repair were assessed using two-factor ANOVA and the post hoc Tukey’s HSD tests. Before the analysis, data were checked to ensure a good fit for ANOVA assumptions such as data normality and variance heterogeneity using the Shapiro–Wilk test and the O’Brien test, respectively.
Results
Chitinase Digestion Reduces Fluorescence Intensity
Following the protocol by Heath-Heckman and McFall-Ngai (2011), we tested the specificity of the CBD Alexa-546 fluorescence to enzymatic digestion of chitin. In the cases of 1 and 2 h digestion, total removal of chitin signals were found; therefore, no pixel values were quantifiable. As shown in Figure 1, 0.5 h of chitinase treatment significantly reduced CBD Alexa-546 fluorescence associated with hemocytes when compared with PBS control and heat treated chitinase preparations, thus confirming the presence of chitin in the hemocytes and suggesting a high specificity of the probe for chitin (one-way ANOVA p < 0.05, Tukey’s test: PBS = HT > T; p < 0.01; O’Brien test demonstrated variance was homogeneous: p > 0.05). Our result demonstrated the probe is sensitive to not only the presence of chitin, but also the abundance of chitin.
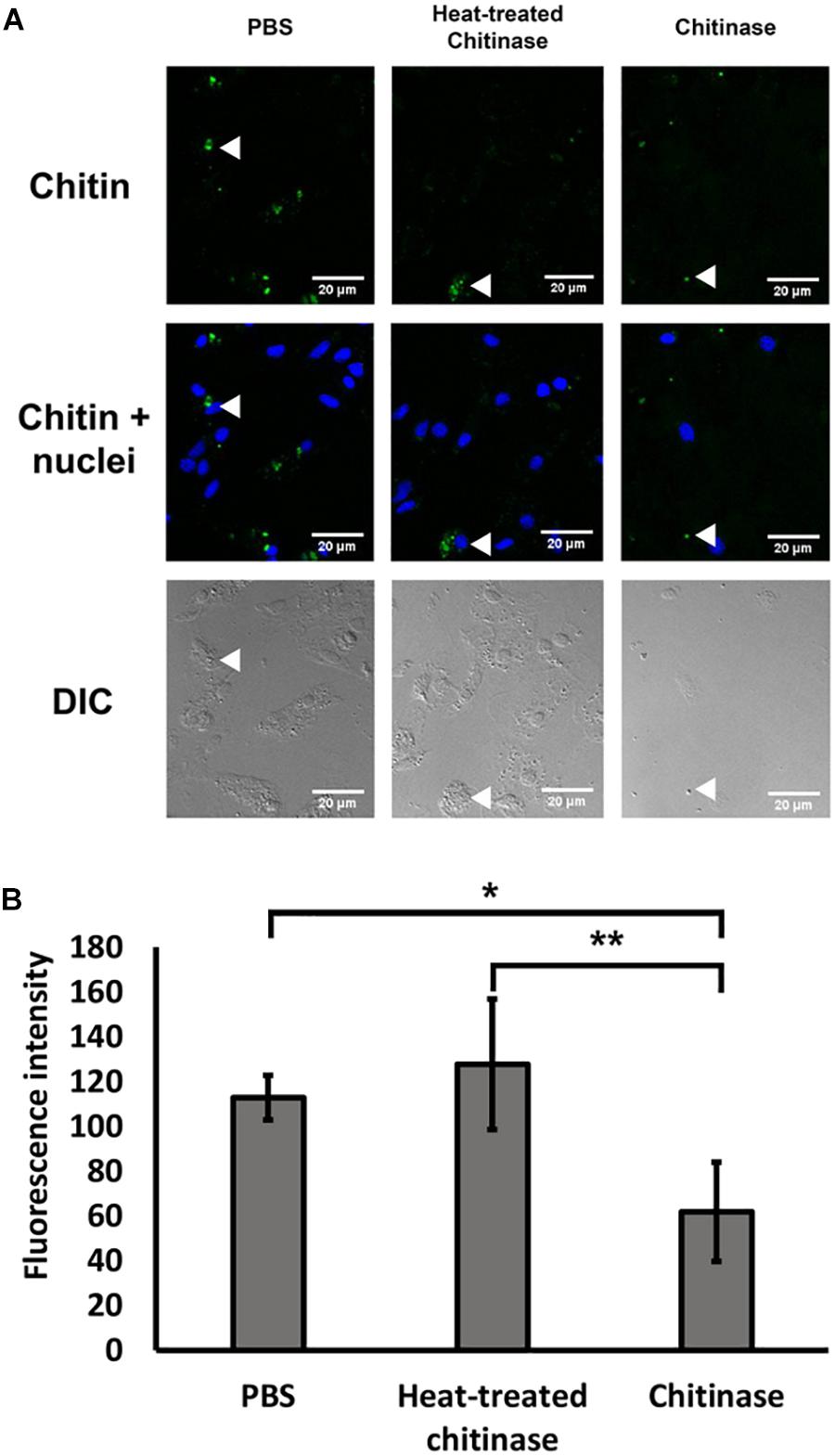
FIGURE 1. Specific detection of chitin by Alexa 546-labeled chitin-binding domain (CBD-546) in oyster hemocytes as demonstrated by chitinase digestion. (A) CLSM micrographs reveal that enzymatic digestion of chitin results in a reduction of fluorescence after treatment with chitinase, representative areas of the hemocytes by are quantified for comparison (white arrows). In this panel, fixed oyster hemocytes were treated with phosphate buffered saline (PBS) as a control (left column), or treated with heat inactivated chitinase in PBS (Heat-treated chitinase; middle column), or treated with active chitinase in PBS (Chitinase; right column) for 0.5 h. Chitin (in green, stained by CBD-546), cell nuclei (in blue, stained by DAPI). Differential interference contrast (DIC) microscopy (bottom row) shows the loss of cells after chitinase treatment. (B) Quantitation of fluorescence intensity (mean ± SD) from the CLSM images from the three experimental groups (PBS, Heat-treated Chitinase, and Chitinase; four replicates), demonstrates a reduction in pixel intensity with chitinase treatment. Statistical significance was determined by one-way ANOVA and Tukey’s post hoc test. p < 0.05 and p < 0.01 were denoted by ∗ and ∗∗, respectively.
To control for the possibility of photobleaching effects, the same CLSM setting that was used for a chitin treated implant was used to visualize an 18 h implant treated with PBS. The fluorescence intensity after 120 min in the control showed no reduction (intensity before: 222.7 ± 18.6; intensity after: 230.3 ± 18.0; mean ± SD), while the intensity for the chitinase-treated implant showed a 56% reduction in fluorescence intensity from 222.0 ± 44.8 to 96.4 ± 18.6.
CLSM Observation of Cells on the Glass Implants
Glass coverslips imaged by CLSM at 5, 18, and 27 h after implantation are shown in Figures 2, 3. Chitin structures, represented in green, appeared to be associated with cell nuclei at the early times of shell regeneration (5 and 18 h; Figure 2). These structures take the form of fibers (yellow arrows, Figure 3) and micron sized vesicles (magenta arrows, Figure 3). By a later stage of shell repair (27 h), the number of cells associated with the chitin materials was reduced while the coverage of chitin expanded on the implant surface. The 5, 18, and 27 h implants had no significant autofluorescence. Cell nuclei labeled with DAPI were represented in blue, and the membrane structures labeled with plasma membrane stain were represented in magenta (Figure 3 and Supplementary Figure S1). Since the complementary colors of green and magenta overlay to produce white pixels, structures where both chitin and membrane are present appear white. In addition to membrane associated with the expected fibrous structures, the presence of chitin-rich micron-sized spheres also looked white and appeared to be membrane-bound extracellular vesicles (white arrows, Figure 3 and Supplementary Figure S1).
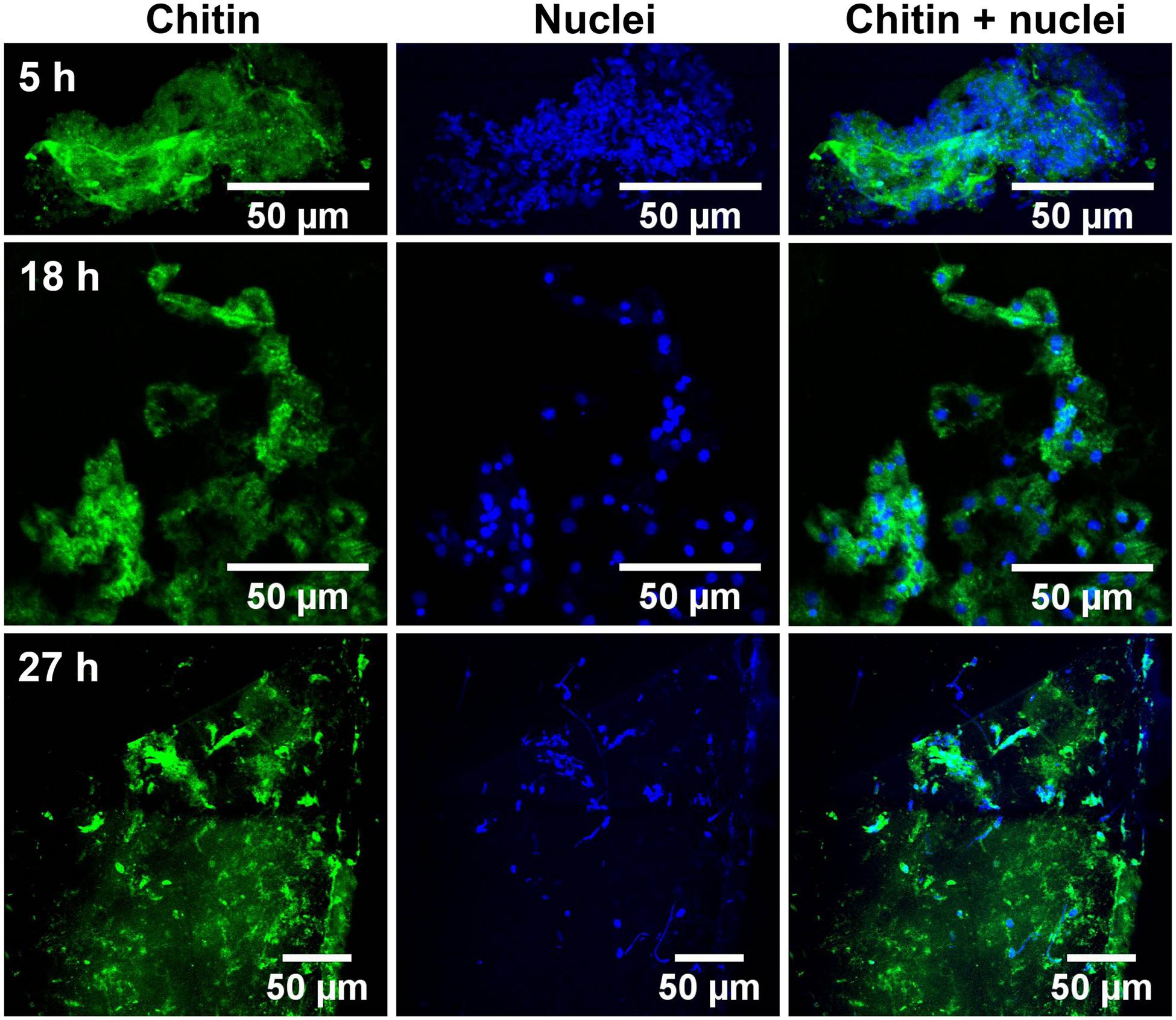
FIGURE 2. Succession of shell repair on glass implants, visualized at 5, 18, and 27 h after implantation. Maximum intensity projection of CLSM image at 5 (top row), 18 (middle row), and 27 h (bottom row). Chitin (in green, stained by CBD-Alexa 546 probe) and cell nuclei (in blue, stained by DAPI), merged image shows the distribution of nuclei and their relationship to chitin.
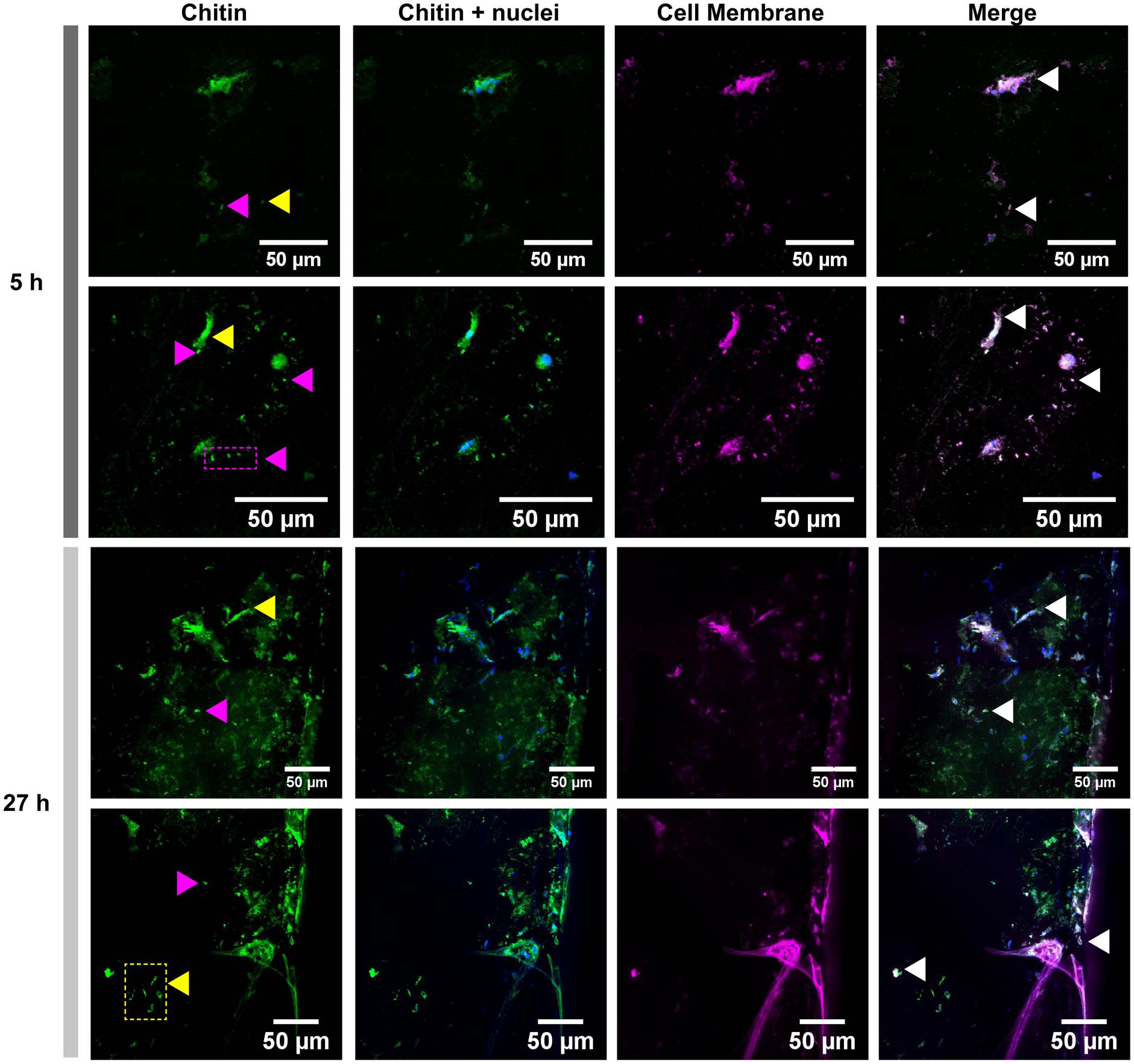
FIGURE 3. Cell membrane and chitin on glass implants visualized at 5 and 27 h after implantation. Maximum intensity projection of CLSM image shows the distribution of chitin (in green, stained by CBD-Alexa 546 probe), its location with respect to cell nuclei (in blue, stained by DAPI), and cell membrane (in magenta, stained by CellMask). A merge of all three channels shows a large overlapping of lipid membrane and chitin signals (white arrows). Fibrous chitin (yellow arrows) and micron sized particles (magenta arrows) are present. Two representative regions from 5 and 27 h implants are provided.
SEM Observation of Fibril Dimension on Metal Implants
With the benefit of the greater structural resolution provided by SEM, the fibrous features of the deposited chitin at 5, 15, and 27 h and 6 days after implantation were compared (Figure 4) and revealed (Figures 5–7). The deposition of fibril structures began as early as the time point of 5 h after shell repair, possibly as a result of cellular activities at the mineralization front. The thickness of these fibrous materials was reduced after this time point (Figure 4, two-way ANOVA: p < 0.0001, 5 h > 15 h = 27 h = 6 days). Overall thickness of the fibrous materials was reduced by treatment with acid and bleach (Figure 4, two-way ANOVA: p < 0.0001, Control > Acid etched > Bleached). These observations suggested that the fibril structures were likely conjugated to microcrystals in the first 5 h of shell repair (Figures 4, 5) and that these crystals were dissolved after treatment with dilute acid (Figures 4–6). Bleach reactivity agreed with the CLSM observation that some chitin structures are associated with membrane and possibly also with protein. We observed that the later stages (27 h and 6 days, Figures 6, 7) had more fibrils that were resistant to acid and bleach treatments, suggesting that these implants contained more chitin than in the earlier stages of shell repair (such as the 5 and 15 h time points, Figures 4, 5). In fact, the 5 h implants were devoid of fibers and the 15 h implants had nearly unobservable fibers (Figure 7). In general, we observed an increased level of structural complexity of fibers over the time course of shell repair (Figures 5–7). The deposition of fibril structures began as quickly as 5 h after the damage, with fibrils appearing tightly packed and in a random orientation. The fibrous structures later appeared to elaborate into a three-dimensional network by 27 h and 6 days (Figures 4, 7). In addition, a horizontally arranged foliated shell appeared by day 6, with the fibrous materials and the folia having an interconnected ultrastructure (Figure 6). Upon treatment with chitinase, the fibril structures remain, but appeared more membranous (Supplementary Figure S2).
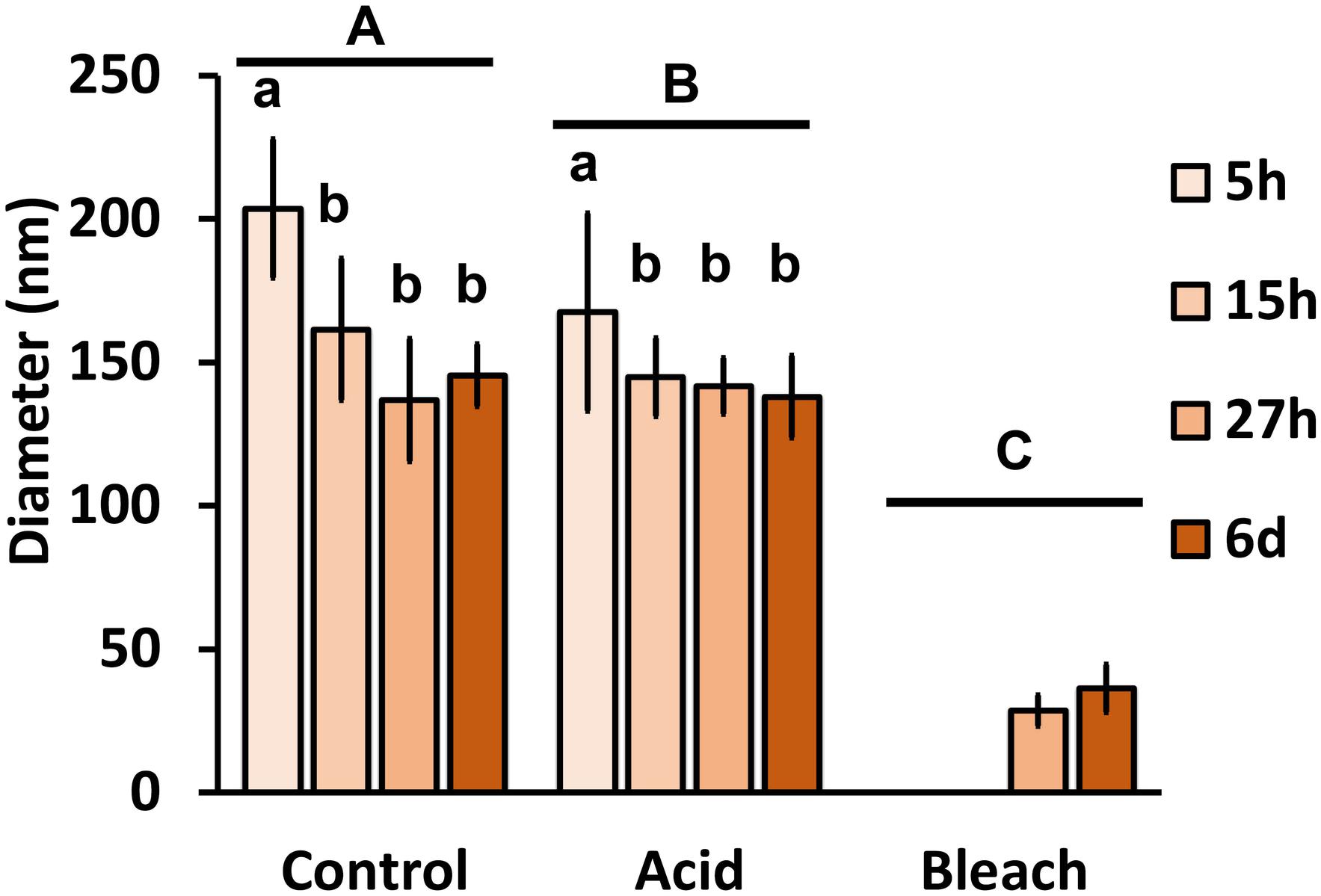
FIGURE 4. The effect of acid and bleach treatments on fibril diameter. PBS control fibril diameter (mean ± SD) from 5, 15, and 27 h and 6 days after implantation were compared to either acid (1 min incubation with 0.1% acetic acid) or bleach (30 min incubation with 0.5% bleach) treatment. Control fibril structures were the thickest at 5 h, and became thinner over time (two-way ANOVA: p < 0.0001, 5 h > 15 h = 27 h = 6 days). Acid treatment reduced fibril diameter (two-way ANOVA: p < 0.0001, Control > Acid etched > Bleached), suggesting the presence of mineral in these fibrils. Only fibrils from 27 h and 6 days remained after bleach treatment, indicating all of the fibrils are rich in organic components. Same letter denotes groups that are not significantly different. Lower case letters compare the different sampling time points; upper case letters compare the treatments.
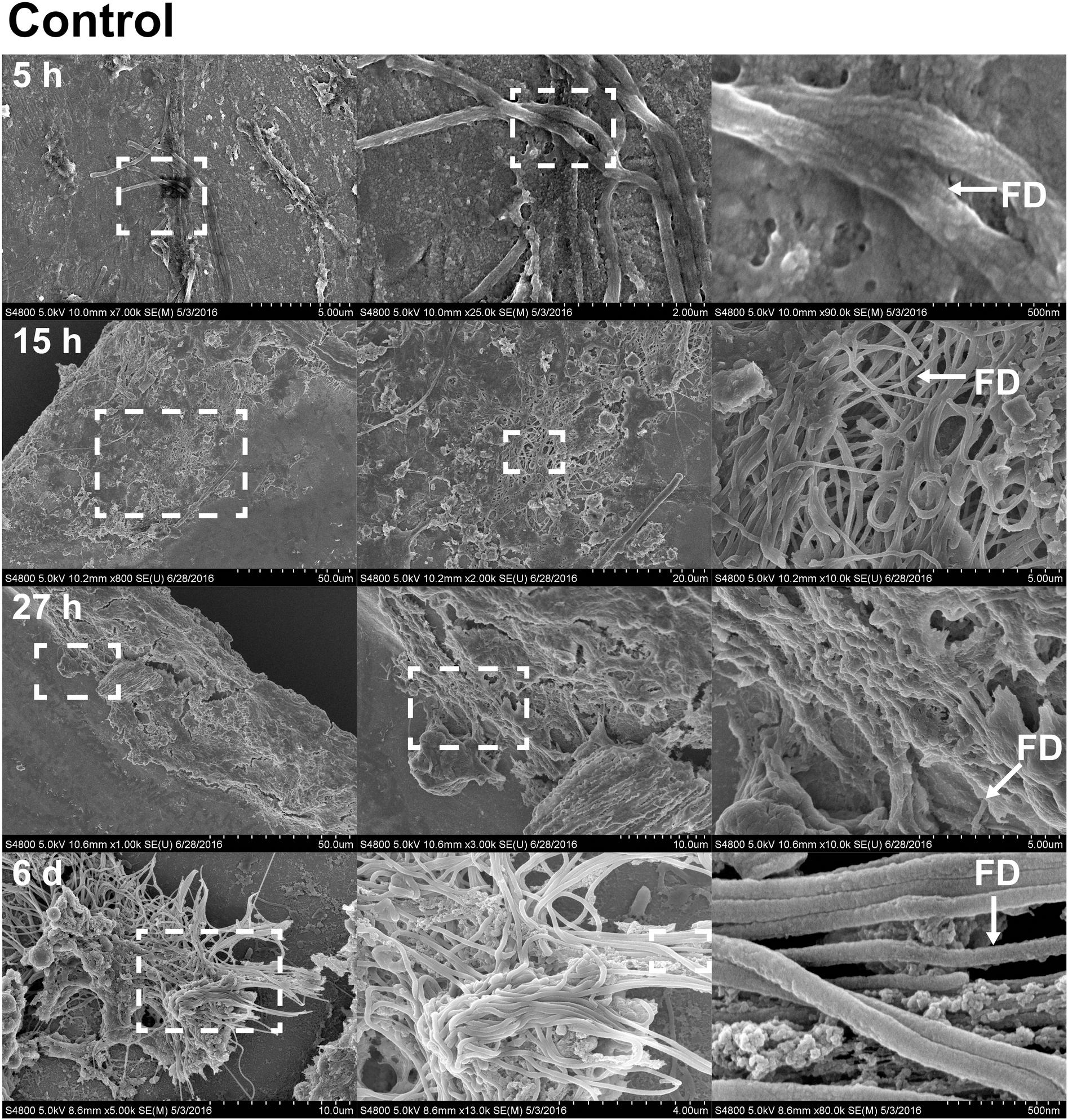
FIGURE 5. Fibril structures on stainless steel implants, 5, 15, and 27 h and 6 days after implantation on SEM. At 5 h (first row), chitin-like fibers are apparent over the entire surface of the implant. Enlarged view of the boxed region from A shows the interwoven chitin fibers. Scale bars from left to right = 5 μm, 2 μm, and 500 nm. At 15 h (second row), cell clusters are shown interacting with fibril structures. Scale bars from left to right = 50, 20, and 5 μm. At 27 h (third row), thick clusters of cells interacting with fibrils are found. Scale bars from left to right = 50, 10, and 5 μm. On sixth day (forth row), fibrils are organized to form a more complex structure. Scale bars from left to right = 10 μm, 4 μm, and 500 nm. Representative regions for the measurement of fibril diameter (FD) are shown on the right column (white arrows).
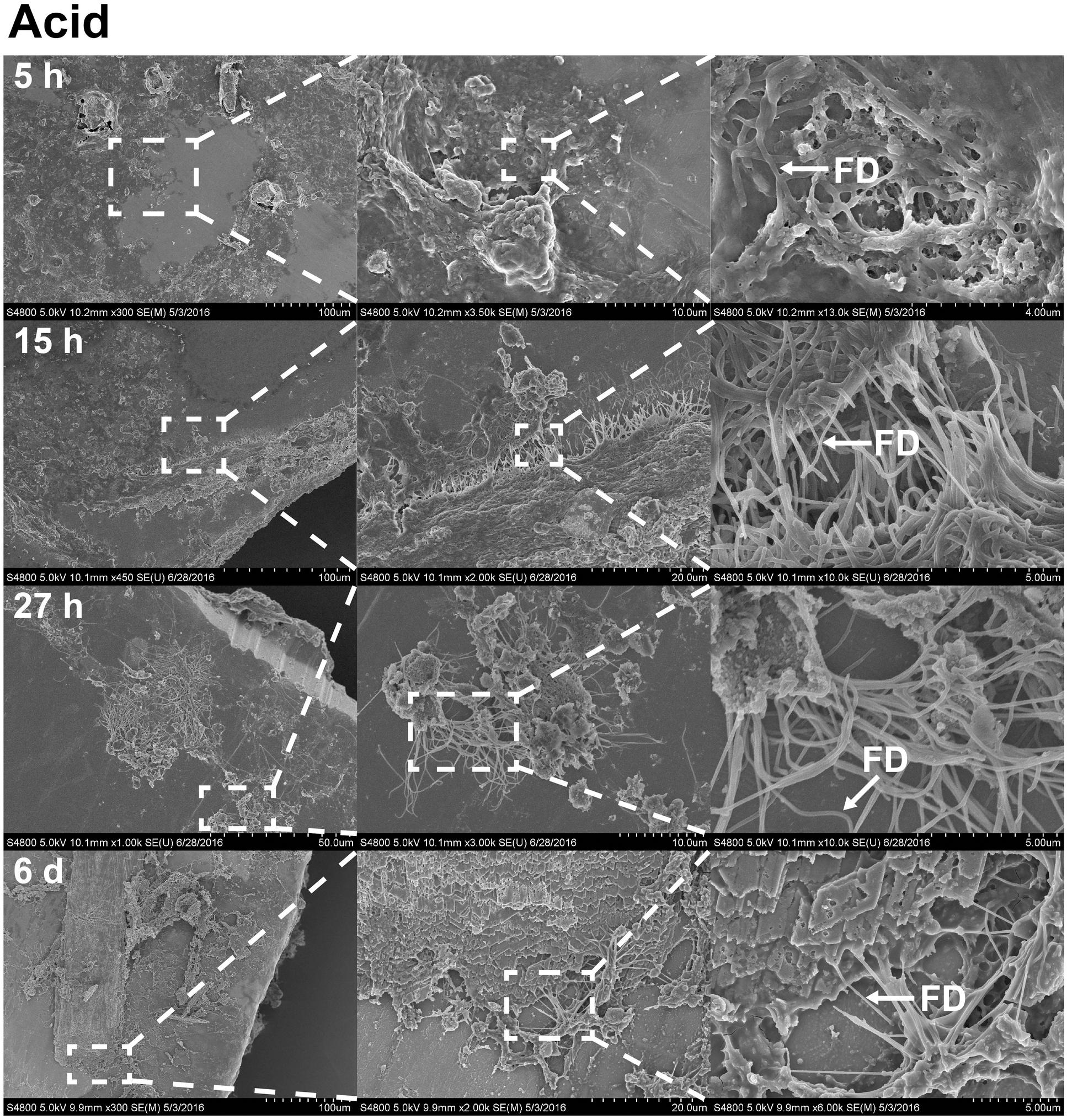
FIGURE 6. The effects of acetic acid on stainless steel implants, 5, 15, and 27 h and 6 days after implantation. 1 min incubation with 0.1% acetic acid treatment resulted in extracellular matrix becoming more open. The surface has become partially digested, revealing interacting fibrils. On sixth day (forth row), foliated shell structures and fibrils are organized as sheets across the surface of the implant. Scale bars from left to right, at 5 h (first row): 100, 10, and 4 μm; at 15 h (second row): 100, 20, and 5 μm; at 27 h (third row): 50, 10, and 5 μm; at sixth day (forth row): 100, 20, and 5 μm. Representative regions for the measurement of fibril diameter (FD) are shown on the right column (white arrows).

FIGURE 7. The effects of bleach on stainless steel implants, 5, 15, and 27 h and 6 days after implantation. Implant after 30 min incubation with 0.5% bleach treatment shows a total removal of fibril structures at 5 and 15 h (first and second rows). (right column) Enlarged view of boxed region from E indicates that fibril structures appeared to become thinner (arrow). Partial degradation of the implant surface and thinner fibril (arrow) were visible. Scale bars from left to right, at 5 h (first row): 5 and 2 μm; at 15 h (second row): 40 and 10 μm; at 27 h (third row): 10 and 2 μm; at sixth day (forth row): 10 and 2 μm. Representative regions for the measurement of fibril diameter (FD) are shown on the right column (white arrows).
Discussion
One of the goals of biomineralization research is to use approaches that capture the involvement of cells and molecules in this process as close to their native biological state as possible. Previously established work on chitin in mineral and biological samples were often treated using harsh chemicals such as NaOH or bleach (Peters, 1972; Ehrlich et al., 2010, 2013). Although these approaches were effective in revealing the chemical-resistant chitin fibers, cells and other molecules were degraded. The alternative approach of using advanced microscopy methods has greatly improved the biological relevance of observations on biominerals, including those related to chitin. Such methods would include cryo-preservation of the initial mineralization states in vitrified conditions (Levi-Kalisman et al., 2001; Addadi and Weiner, 2014). Previous studies demonstrated that shell repair can be observed on implants positioned between the shell and the mantle in C. virginica (Johnstone et al., 2015). In this study, when CLSM and SEM techniques were combined with the use of the implant system, we were able to reveal molecular and cellular aspects of chitin synthesis associated with shell deposition in real-time and under conditions which approximate in situ observations.
The use of recombinant green fluorescent protein-tagged chitin-binding protein has been used in earlier studies to perform fluorescent labeling of acid-etched shell structures (Nudelman et al., 2007), demineralized larvae (Schönitzer and Weiss, 2007), and squid hemocytes (Heath-Heckman and McFall-Ngai, 2011). The specificity of the chitin probe used in this study and the association of chitin with hemocytes was confirmed by subjecting fixed hemocytes to chitinase digestion and then observing the change in fluorescence by CLSM. Furthermore, chitin appeared to be involved in anchoring the hemocytes to the surface of the coverslips, as extended incubation with chitinase resulted in the loss of adhesive cells from the surface. This observation implied a role of chitin in cell adhesion and possibly in the molecular cascade of wound healing (Lee et al., 2011).
To address how cellular components are involved in the process of forming new biological minerals, glass implant surfaces were evaluated using various fluorescent stains under CLSM. In addition to the chitin probe, we counterstained cell nuclei and plasma membrane at the active sites of mineralization. In the early stages of regeneration, chitin fibers were found near where nuclei and thus cells were located. In addition, micron-sized structures that appeared to be membrane-bound stained for chitin, suggesting that exosomal-like structures are involved in chitin deposition.
Exosomes, called chitosomes, are well known for the production of chitin in yeast and fungi (Mills and Cantino, 1981; Gozalbo et al., 1993; Bartnicki-Garcia, 2006). In gastropod molluscs, chitin secretion appears to derive from Golgi-derived secretory granules in the columnar cells of mantle epithelia (Bevelander and Nakahara, 1970; Paillard and Le Pennec, 1993). In the process of periostracum formation, fine materials with a periodicity of 300 Å were abundant in these enlarged granules, and the materials are dispersed in the extracellular environment (Bevelander and Nakahara, 1970). Alternatively, the production of chitin may begin in the hemocytes where chitin synthase is abundantly expressed (Zhang et al., 2012; Ivanina et al., 2017). Furthermore, endogenous chitin production has been reported in a lysosomal compartment of hemocytes in squid (Heath-Heckman and McFall-Ngai, 2011). A recent study on molluscan shell formation examined the cooperative roles of mantle epithelial tissue and hemocytes (Ivanina et al., 2017), a relationship which may apply to chitin synthesis. The origin of the exosomes observed in this study, whether it is mantle or adherent hemocytes, or both tissues, remains unresolved.
In addition to the possibility that shell-matrix chitin may be made in vesicles, calcification sites are also often found within membrane-bound subcellular compartments. Examples of such sites include the microvesicles in magnetotactic bacteria (Hershey et al., 2016), the tubeworm (Neff, 1971), the Golgi in the coccolithophorids (Young and Henriksen, 2003), and the pinocytotic proton-transporting vesicles in the foraminifera (Bentov et al., 2009; de Nooijer et al., 2009; Toyofuku et al., 2017). Perhaps the most germane to this study, Mount et al. (2004) demonstrated the intracellular presence of calcite crystals in hemocytes of oysters during shell repair. In general, studies that target the cellular parameters required for biomineralization, such as intracellular or intraorganellar pH and calcium supply, are only emerging in the cases of multicellular organisms (Venn et al., 2009; Chan et al., 2015).
SEM observations of stainless steel implants revealed the presence of fibers. If these fibers contain chitin, a portion of them would be expected to be resistant to both bleach and acid treatments, as demonstrated in this study. Changes in the fiber dimensions after chemical treatments further suggest that the chitin does not exist as an isolated component, but is associated with other organics, such as proteins and lipids, as well as a calcium carbonate mineral phase. The presence of thicker fibrils found at 5 h of implantation compared to later stages could be a consequence of the inclusion of cellular products such as mineral, lipid membrane and proteins. In fact, after a 3 h chitinase treatment on an 18 h stainless steel implant, only morphological changes in fibers occurred without noticeable reduction in their diameter (Supplementary Figure S2). The complexity of the chitin structures increased over time, starting with a tightly packed random orientation and developing into a three dimensional structure. By the sixth day, the chitin became interconnected with mineral, which had an ultrastructure that appeared identical to the native foliated microstructure of the inner shell layer. The early appearance of chitin and its ultimate relationship to shell crystals suggests that chitin may play a significant role in shell regeneration, which resembles normal shell formation.
Based on previous studies and the work reported herein, we propose two hypothetical models of chitin involvement at the mineralization front in oysters (Figure 8). In both cases, chitin is produced in an intracellular compartment like the Golgi apparatus. The deposition of chitin at the site of mineral formation relies on an exosome-like microvesicle secretion process. These chitin-producing compartments could be generated from cell types different from those that produce mineral phase precursors and other organic matrix materials (model 1) or all these products could be produced by the same cell type (model 2). The latter products may be released in exosomes, or by more traditional secretion processes. The precise cellular origin of chitin exosomes putatively observed in this study and their cellular relationship of other shell components await additional study. Furthermore, the fact that hemocytes are present at the site of shell formation in conjunction with chitin, their role in secreting chitin or their use of it as a scaffold to advance mineralization events bears special scrutiny.
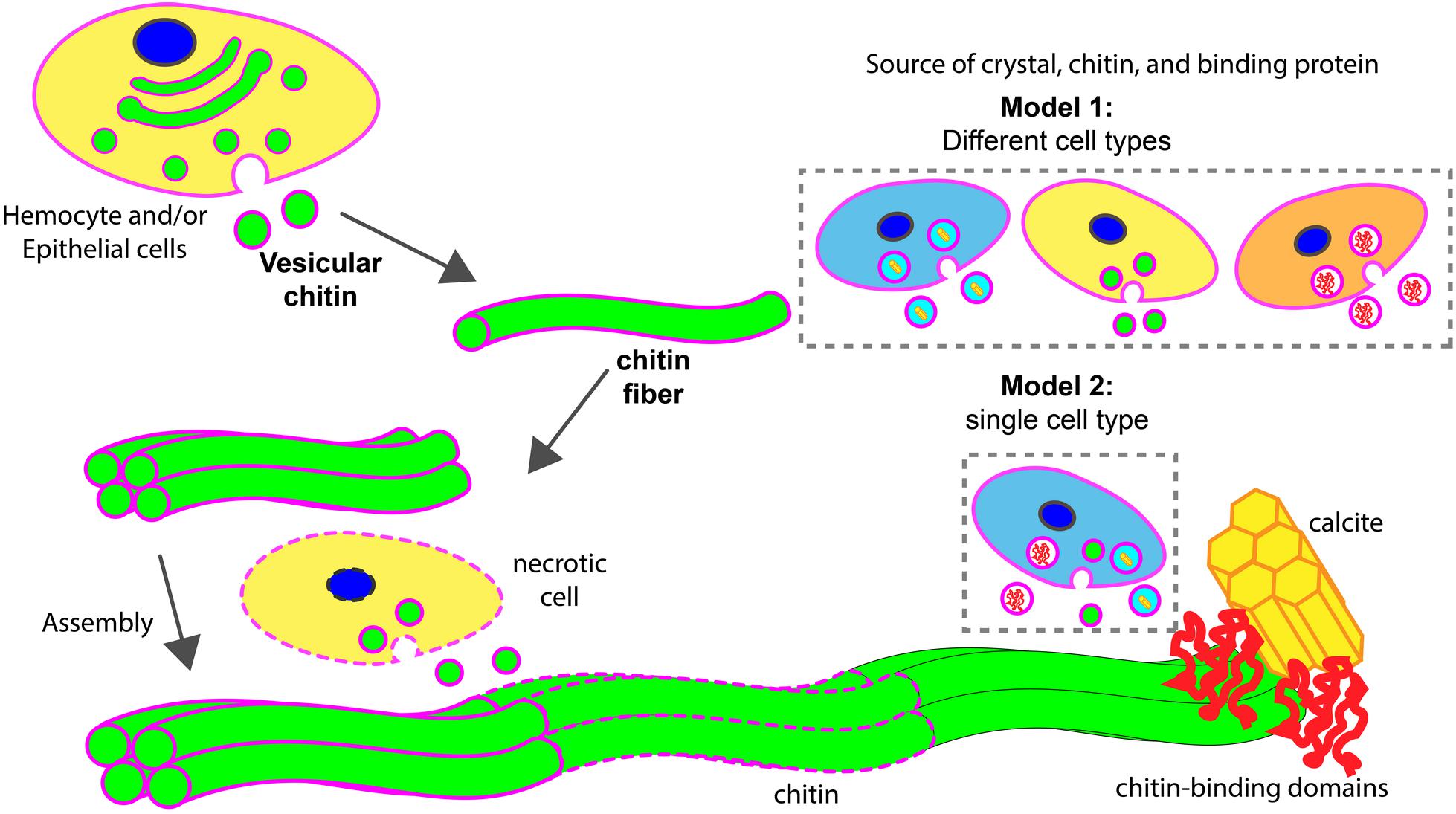
FIGURE 8. Models of chitin assisted mineralization. Cell secretes vesicular chitin, leading to chitin fibril maturation. These fibrils are assembled into a larger chitin fiber, often followed by multiple fiber assembly into a compact structural framework. This fiber assemblage interacts with chitin-binding domains of extracellular matrix proteins which in turn are associated with calcite. Upon maturation, lipid membrane (in magenta) disintegrates (dotted outlines). During the assembly process, necrotic cells (in dotted line) may serve as a supply of vesicular chitin. Two hypothetical models described the potential sources of chitin, mineral, and chitin-binding proteins for the assembly of minerals. In model 1, different cell types are involved. In model 2, a specialized cell type is responsible to supply all components for mineralization.
Author Contributions
VC carried out the experiments. VC and AW wrote the manuscript. MJ assisted VC with the protocol of expression and purification of the chitin-binding domain, and the design of the implant study. AM conceived and supervised the project and edited the manuscript.
Funding
This work was partially supported by ONR award # N00014-14-1-0488 and by the Clemson University Experimental Research Station.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank New England Biolabs for their supply of transfected E. coli. They thank Charles Rice, Amy Anderson, and Matthew Turnbull for their helpful discussion at the early stage of this work. They also thank Hermann Ehrlich, David Kisailus, Michiko Nemoto, and Michio Suzuki whose discussion with VC during the Biomin14 meeting greatly improved this project. They would also like to acknowledge Dr. S. Ramaswami for proof-reading of this manuscript, and Ms. Jan Lay for improving the quality of the figures.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2018.00347/full#supplementary-material
FIGURE S1 | Cell membrane and chitin on glass implants visualized at 18 h after implantation. Maximum intensity projection of CLSM image shows the distribution of chitin (in green, stained by CBD-Alexa 546 probe), its location with respect to cell nuclei (in blue, stained by DAPI), and cell membrane (in magenta, stained by CellMask). A merge of all three channels shows a large overlapping of lipid membrane and chitin signals (white arrows). Fibrous chitin (yellow arrows) and micron sized particles (magenta arrows) are present.
FIGURE S2 | The effects of chitinase treatment on stainless steel implants 18 h after implantation. Implant after 3 h of incubation treatment with chitinase shows a change in fibril morphology. (A–C) Untreated control fibril, (B) is the enlarged box region of (A), and (C) is the enlarged boxed region of (B). A representative fibril is indicated by a white arrow. (D–I) Chitinase treated fibril. (E) is the enlarged box region of D, and F is the enlarged boxed region of panel (E). Degradation by chitinase caused the implant surface to be more membranous. Panel (G) represents a mineralized region of the implant which appears to be free of fibrils after chitinase treatment. Panel (H) is a vesicle rich region and (I) is the enlarged box region of panel (H). Remaining fibril-like structures are indicated by white arrows in (F) and (I). Scale bars for (A–D): 10, 5, 3, and 20 μm; (E, F, G, I): 5 μm; (H): 1 μm.
Footnotes
References
Addadi, L., and Weiner, S. (2014). Biomineralization: mineral formation by organisms. Phys. Scr. 89:098003. doi: 10.1088/0031-8949/89/9/098003
Bartnicki-Garcia, S. (2006). Chitosomes: past, present and future. FEMS Yeast Res. 6, 957–965. doi: 10.1111/j.1567-1364.2006.00158.x
Bentov, S., Brownlee, C., and Erez, J. (2009). The role of seawater endocytosis in the biomineralization process in calcareous foraminifera. Proc. Natl. Acad. Sci. U.S.A. 106, 21500–21504. doi: 10.1073/pnas.0906636106
Bevelander, G., and Nakahara, H. (1970). An electron microscope study of the formation and structure of the periostracum of a gastropod, Littorina littorea. Calcif. Tissue Int. 5, 1–12. doi: 10.1007/bf02017528
Brunner, E., Richthammer, P., Ehrlich, H., Paasch, S., Simon, P., Ueberlein, S., et al. (2009). Chitin-based organic networks: an integral part of cell wall biosilica in the diatom Thalassiosira pseudonana. Angew. Chem. Int. Ed. 48, 9724–9727. doi: 10.1002/anie.200905028
Carlström, D. (1957). The crystal structure of α-chitin (poly-N-acetyl-D-glucosamine). J. Cell Biol. 3, 669–683. doi: 10.1083/jcb.3.5.669
Chan, V. B. S., Toyofuku, T., Wetzel, G., Saraf, L., Thiyagarajan, V., and Mount, A. S. (2015). Direct deposition of crystalline aragonite in the controlled biomineralization of the calcareous tubeworm. Front. Mar. Sci. 2:97. doi: 10.3389/fmars.2015.00097
Cohen, E. (2010). Chitin biochemistry: synthesis, hydrolysis and inhibition. Adv. Insect Physiol. 38, 5–74. doi: 10.1016/S0065-2806(10)38005-2
Currey, J. (1999). The design of mineralised hard tissues for their mechanical functions. J. Exp. Biol. 202, 3285–3294.
de Nooijer, L. J., Toyofuku, T., and Kitazato, H. (2009). Foraminifera promote calcification by elevating their intracellular pH. Proc. Natl. Acad. Sci. U.S.A. 106, 15374–15378. doi: 10.1073/pnas.0904306106
Di Rosa, M., Distefano, G., Zorena, K., and Malaguarnera, L. (2016). Chitinases and immunity: ancestral molecules with new functions. Immunobiology 221, 399–411. doi: 10.1016/j.imbio.2015.11.014
Ehrlich, H., Ilan, M., Maldonado, M., Muricy, G., Bavestrello, G., Kljajic, Z., et al. (2010). Three-dimensional chitin-based scaffolds from Verongida sponges (Demospongiae: Porifera). Part I. Isolation and identification of chitin. Int. J. Biol. Macromol. 47, 132–140. doi: 10.1016/j.ijbiomac.2010.05.007
Ehrlich, H., Kaluzhnaya, O. V., Brunner, E., Tsurkan, M. V., Ereskovsky, A., Ilan, M., et al. (2013). Identification and first insights into the structure and biosynthesis of chitin from the freshwater sponge Spongilla lacustris. J. Struct. Biol. 183, 474–483. doi: 10.1016/j.jsb.2013.06.015
Falini, G., Albeck, S., Weiner, S., and Addadi, L. (1996). Control of aragonite or calcite polymorphism by mollusk shell macromolecules. Science 271, 67–69. doi: 10.1126/science.271.5245.67
Goffinet, G., and Jeuniaux, C. (1979). Distribution et importance quantitative de la chitine dans les coquilles de mollusques. Cah. Biol. Mar. 20, 341–349.
Gotliv, B., Addadi, L., and Weiner, S. (2003). Mollusk shell acidic proteins: in search of individual functions. ChemBioChem 4, 522–529. doi: 10.1002/cbic.200200548
Gozalbo, D., Dubón, F., and Sentandreu, R. (1993). Effect of digitonin on membrane-bound and chitosomal chitin synthetase activity in protoplasts from yeast cells of Candida albicans. Antonie Van Leeuwenhoek 64, 67–74. doi: 10.1007/BF00870923
Hasegawa, M., Yagi, K., Iwakawa, S., and Hirai, M. (2001). Chitosan induces apoptosis via caspase-3 activation in bladder tumor cells. Jpn. J. Cancer Res. 92, 459–466. doi: 10.1111/j.1349-7006.2001.tb01116.x
Heath-Heckman, E. A., and McFall-Ngai, M. J. (2011). The occurrence of chitin in the hemocytes of invertebrates. Zoology 114, 191–198. doi: 10.1016/j.zool.2011.02.002
Heinemann, F. (2008). Investigation of Biopolymer- Mineral Interactions in the Natural Composite Material Nacre. Ph.D. thesis, University of Bremen, Bremen.
Heredia, A., Aguilar-Franco, M., Magaña, C., Flores, C., Piña, C., Velázquez, R., et al. (2007). Structure and interactions of calcite spherulites with α-chitin in the brown shrimp (Penaeus aztecus) shell. Mater. Sci. Eng. C 27, 8–13. doi: 10.1016/j.msec.2005.11.003
Hershey, D. M., Ren, X., Melnyk, R. A., Browne, P. J., Ozyamak, E., Jones, S. R., et al. (2016). MamO is a repurposed serine protease that promotes magnetite biomineralization through direct transition metal binding in Magnetotactic bacteria. PLoS Biol. 14:e1002402. doi: 10.1371/journal.pbio.1002402
Ichinohe, T., Nagata, N., Strong, P., Tamura, S. I., Takahashi, H., Ninomiya, A., et al. (2007). Prophylactic effects of chitin microparticles on highly pathogenic H5N1 influenza virus. J. Med. Virol. 79, 811–819. doi: 10.1002/jmv.20837
Ivanina, A. V., Falfushynska, H. I., Beniash, E., Piontkivska, H., and Sokolova, I. M. (2017). Biomineralization-related specialization of hemocytes and mantle tissues of the Pacific oysters Crassostrea gigas. J. Exp. Biol. 220, 3209–3221. doi: 10.1242/jeb.160861
Jeon, D.-W., Ahn, W. S., You, S. J., Chae, G. T., Shim, Y. B., and Chun, H. J. (2012). Induction of classical activation of macrophage in vitro by water soluble chitin. Appl. Surf. Sci. 262, 134–139. doi: 10.1016/j.apsusc.2012.03.078
Johnstone, M. B., Gohad, N. V., Falwell, E. P., Hansen, D. C., Hansen, K. M., and Mount, A. S. (2015). Cellular orchestrated biomineralization of crystalline composites on implant surfaces by the eastern oyster, Crassostrea virginica (Gmelin, 1791). J. Exp. Mar. Biol. Ecol. 463, 8–16. doi: 10.1016/j.jembe.2014.10.014
Kaya, M., Mujtaba, M., Ehrlich, H., Salaberria, A. M., Baran, T., Amemiya, C. T., et al. (2017). On chemistry of gamma-chitin. Carbohydr. Polym. 176, 177–186. doi: 10.1016/j.carbpol.2017.08.076
Kumar, M. N. R. (2000). A review of chitin and chitosan applications. React. Funct. Polym. 46, 1–27. doi: 10.1016/S1381-5148(00)00038-9
Lee, C. G., Da Silva, C. A., Dela Cruz, C. S., Ahangari, F., Ma, B., Kang, M. J., et al. (2011). Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 73, 479–501. doi: 10.1146/annurev-physiol-012110-142250
Lee, S., and Choi, C. (2007). The correlation between organic matrices and biominerals (myostracal prism and folia) of the adult oyster shell, Crassostrea gigas. Micron 38, 58–64. doi: 10.1016/j.micron.2006.03.018
Levi-Kalisman, Y., Falini, G., Addadi, L., and Weiner, S. (2001). Structure of the nacreous organic matrix of a bivalve mollusk shell examined in the hydrated state using cryo-TEM. J. Struct. Biol. 135, 8–17. doi: 10.1006/jsbi.2001.4372
Liu, Z., Zhou, Z., Jiang, Q., Wang, L., Yi, Q., Qiu, L., et al. (2017). The neuroendocrine immunomodulatory axis-like pathway mediated by circulating haemocytes in pacific oyster Crassostrea gigas. Open Biol. 7:160289. doi: 10.1098/rsob.160289
Merzendorfer, H., and Zimoch, L. (2003). Chitin metabolism in insects: structure, function and regulation of chitin synthases and chitinases. J. Exp. Biol. 206, 4393–4412. doi: 10.1242/jeb.00709
Mills, G. L., and Cantino, E. C. (1981). Chitosome-like vesicles from gamma particles of Blastocladiella emersonii synthesize chitin. Arch. Microbiol. 130, 72–77. doi: 10.1007/BF00527075
Mount, A., Wheeler, A., Paradkar, R., and Snider, D. (2004). Hemocyte-mediated shell mineralization in the eastern oyster. Science 304, 297–300. doi: 10.1126/science.1090506
Mount, A. S., Gohad, N. V., Hansen, D. C., Hansen, K. M., and Johnstone, M. B. (2013). Deposition of Nanocrystalline Calcite on Surfaces by a Tissue and Cellular Biomineralization. Available at: https://www.google.com/patents/US9371451
Mount, A. S., Gohad, N. V., Hansen, D. C., Hansen, K. M., and Johnstone, M. B. (2016). Deposition of Nanocrystalline Calcite on Surfaces by a Tissue and Cellular Biomineralization. Available at: https://www.google.com/patents/US9371451
Nakayama, S., Suzuki, M., Endo, H., Iimura, K., Kinoshita, S., Watabe, S., et al. (2013). Identification and characterization of a matrix protein (PPP-10) in the periostracum of the pearl oyster, Pinctada fucata. FEBS Open Bio 3, 421–427. doi: 10.1016/j.fob.2013.10.001
Neff, J. M. (1971). Ultrastructural studies of the secretion of calcium carbonate by the serpulid polychaete worm, Pomatoceros caeruleus. Cell Tissue Res. 120, 160–186. doi: 10.1007/BF00335534
Nudelman, F., Chen, H., Goldberg, H., Weiner, S., and Addadi, L. (2007). Spiers Memorial Lecture. Lessons from biomineralization: comparing the growth strategies of mollusc shell prismatic and nacreous layers in Atrina rigida. Faraday Discuss. 136, 9–25. doi: 10.1039/b704418f
Nudelman, F., Shimoni, E., Klein, E., Rousseau, M., Bourrat, X., Lopez, E., et al. (2008). Forming nacreous layer of the shells of the bivalves Atrina rigida and Pinctada margaritifera: an environmental- and cryo-scanning electron microscopy study. J. Struct. Biol. 162, 290–300. doi: 10.1016/j.jsb.2008.01.008
Öztürk-Çolak, A., Moussian, B., Araujo, S. J., and Casanova, J. (2016). A feedback mechanism converts individual cell features into a supracellular ECM structure in Drosophila trachea. eLife 5:e09373. doi: 10.7554/eLife.09373
Paillard, C., and Le Pennec, M. (1993). Ultrastructural studies of the mantle and the periostracal lamina in the manila clam, Ruditapes philippinarum. Tissue Cell 25, 183–194. doi: 10.1016/0040-8166(93)90018-G
Peters, W. (1972). Occurence of chitin in mollusca. Comp. Biochem. Physiol. B 41, 541–550. doi: 10.1016/0305-0491(72)90117-4
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schönitzer, V., and Weiss, I. M. (2007). The structure of mollusc larval shells formed in the presence of the chitin synthase inhibitor Nikkomycin Z. BMC Struct. Biol. 7:71. doi: 10.1186/1472-6807-7-71
Stern, R., and Jedrzejas, M. J. (2008). Carbohydrate polymers at the center of life’s origins: the importance of molecular processivity. Chem. Rev. 108, 5061–5085. doi: 10.1021/cr078240l
Suzuki, M., Mukai, H., Aoki, H., Yoshimura, E., Sakuda, S., Nagasawa, H., et al. (2016). Microstructure of iridescence-lacking pearl formed in Pinctada fucata. J. Cryst. Growth 433, 148–152. doi: 10.1016/j.jcrysgro.2015.10.014
Suzuki, M., Sakuda, S., and Nagasawa, H. (2007). Identification of chitin in the prismatic layer of the shell and a chitin synthase gene from the Japanese pearl oyster, Pinctada fucata. Biosci. Biotechnol. Biochem. 71, 1735–1744. doi: 10.1271/bbb.70140
Suzuki, M., Saruwatari, K., Kogure, T., Yamamoto, Y., Nishimura, T., Kato, T., et al. (2009). An acidic matrix protein, Pif, is a key macromolecule for nacre formation. Science 325, 1388–1390. doi: 10.1126/science.1173793
Tang, W. J., Fernandez, J. G., Sohn, J. J., and Amemiya, C. T. (2015). Chitin is endogenously produced in vertebrates. Curr. Biol. 25, 897–900. doi: 10.1016/j.cub.2015.01.058
Toyofuku, T., Matsuo, M. Y., De Nooijer, L. J., Nagai, Y., Kawada, S., Fujita, K., et al. (2017). Proton pumping accompanies calcification in foraminifera. Nat. Commun. 8:14145. doi: 10.1038/ncomms14145
Venn, A., Tambutté, E., Lotto, S., Zoccola, D., Allemand, D., and Tambutté, S. (2009). Imaging intracellular pH in a reef coral and symbiotic anemone. Proc. Natl. Acad. Sci. U.S.A. 106, 16574–16579. doi: 10.1073/pnas.0902894106
Wang, L., Li, F., Wang, B., and Xiang, J. (2012). Structure and partial protein profiles of the peritrophic membrane (PM) from the gut of the shrimp Litopenaeus vannamei. Fish Shellfish Immunol. 33, 1285–1291. doi: 10.1016/j.fsi.2012.09.014
Wang, X., Song, X., Wang, T., Zhu, Q., Miao, G., Chen, Y., et al. (2013). Evolution and functional analysis of the Pif97 gene of the Pacific oyster Crassostrea gigas. Curr. Zool. 59, 109–115. doi: 10.1093/czoolo/59.1.109
Weiner, S., and Traub, W. (1980). X-ray diffraction study of the insoluble organic matrix of mollusk shells. FEBS Lett. 111, 311–316. doi: 10.1016/0014-5793(80)80817-9
Weiss, I. (2012). Species-specific shells: chitin synthases and cell mechanics in molluscs. Z. Kristallogr. Cryst. Mater. 227, 723–738. doi: 10.1524/zkri.2012.1530
Young, J. R., and Henriksen, K. (2003). Biomineralization within vesicles: the calcite of coccoliths. Rev. Mineral. Geochem. 54, 189–215. doi: 10.2113/0540189
Keywords: biomineralization, chitin, oyster, chitin-binding domain, hemocytes, Crassostrea, shell formation, shell repair
Citation: Chan VBS, Johnstone MB, Wheeler AP and Mount AS (2018) Chitin Facilitated Mineralization in the Eastern Oyster. Front. Mar. Sci. 5:347. doi: 10.3389/fmars.2018.00347
Received: 17 April 2018; Accepted: 07 September 2018;
Published: 03 October 2018.
Edited by:
Pei-Yuan Qian, The Hong Kong University of Science and Technology, Hong KongReviewed by:
Gary H. Dickinson, The College of New Jersey, United StatesTing Xu, Hong Kong Baptist University, Hong Kong
Copyright © 2018 Chan, Johnstone, Wheeler and Mount. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vera Bin San Chan, dmJzY2hhbkBnLmNsZW1zb24uZWR1 Andrew S. Mount, bW91bnRAY2xlbXNvbi5lZHU=