 Georg Steinert1*†
Georg Steinert1*† Bernd Wemheuer2†
Bernd Wemheuer2† Dorte Janussen3
Dorte Janussen3 Dirk Erpenbeck4
Dirk Erpenbeck4 Rolf Daniel2
Rolf Daniel2 Meinhard Simon1,5
Meinhard Simon1,5 Thorsten Brinkhoff1
Thorsten Brinkhoff1 Peter J. Schupp1,5*
Peter J. Schupp1,5*- 1Institute for Chemistry and Biology of the Marine Environment, Carl von Ossietzky University of Oldenburg, Oldenburg, Germany
- 2Genomic and Applied Microbiology and Göttingen Genomics Laboratory, Institute of Microbiology and Genetics, University of Göttingen, Göttingen, Germany
- 3Senckenberg Research Institute and Nature Museum, Frankfurt, Germany
- 4Department of Earth and Environmental Sciences and GeoBio-Center, Ludwig-Maximilians-Universität München, Munich, Germany
- 5Helmholtz Institute for Functional Marine Biodiversity, Carl von Ossietzky University of Oldenburg, Oldenburg, Germany
Marine sponges (Phylum Porifera) are globally distributed within marine and freshwater ecosystems. In addition, sponges host dense and diverse prokaryotic communities, which are potential sources of novel bioactive metabolites and other complex compounds. Those sponge-derived natural products can span a broad spectrum of bioactivities, from antibacterial and antifungal to antitumor and antiviral compounds. However, most analyses concerning sponge-associated prokaryotes have mainly focused on conveniently accessible relatively shallow sampling locations for sponges. Hence, knowledge of community composition, host-relatedness and biotechnological potential of prokaryotic associations in temperate and cold-water sponges from greater depths (mesophotic to mesopelagic zones) is still scarce. Therefore, we analyzed the prokaryotic community diversity of four phylogenetically divergent sponge taxa from mesophotic to mesopelagic depths of Antarctic shelf at different depths and locations in the region of the South Shetland Islands using 16S rRNA gene amplicon-based sequencing. In addition, we predicted functional profiles applying Tax4Fun from metagenomic 16S rRNA gene data to estimate their biotechnological capability and possible roles as sources of novel bioactive compounds. We found indications that cold and deep-water sponges exhibit host-specific prokaryotic communities, despite different sampling sites and depths. Functional prediction analysis suggests that the associated prokaryotes may enhance the roles of sponges in biodegradation processes of xenobiotics and their involvement in the biosynthesis of secondary metabolites.
Introduction
Marine sponges (Phylum Porifera) are taxonomically highly diverse with so far ∼ 9115 accepted species, which are distributed within marine and freshwater ecosystems (van Soest et al., 2012, 2018). Along with the vast phylogenetic diversity of the Porifera phylum, sponges are also known to host a dense and diverse microbiota, consisting of prokaryotes, uni- and multicellular eukaryotes (Taylor et al., 2007). Recent advances in sequencing technologies and the collaborative effort of a global sponge microbiome sequencing project found 63–72 prokaryotic phyla (including candidate phyla) among 268 sponge species (Moitinho-Silva et al., 2017b). However, most analyses concerning diversity and host-relatedness of sponge-associated microorganisms, such as bacteria or archaea, focused mainly on relative conveniently accessible sampling locations. Hence, most prokaryotic diversity studies account for sponges collected in relatively shallow coastal locations, with an additional tendency toward tropical and subtropical sponge species (Webster et al., 2010; Schmitt et al., 2012; Steinert et al., 2016; Thomas et al., 2016). Consequently, the knowledge of community composition, host-relatedness and potential functions of prokaryotic associations in temperate and cold-water sponges from greater depths is still scarce. The technical complexities and financial costs, which are related to the goal to reach the seafloor beyond technical diving depths, makes thorough sampling and methodological fieldwork either very challenging or even impossible. Therefore, the sparse existing sponge-associated prokaryotic diversity studies that encompass mesophotic to mesopelagic zones in various temperate and cold-water environments were often constrained by either only a few available sponge species, low or missing replication or the collection along large depth gradients and geographic locations (Webster et al., 2004; Xin et al., 2011; Jackson et al., 2013; Schöttner et al., 2013; Kennedy et al., 2014; Reveillaud et al., 2014). To the best of our knowledge, only two prokaryotic diversity characterization studies have been conducted in the Antarctic region with marine sponges collected from two coastal shallow sampling sites using high throughput sequencing (Rodríguez-Marconi et al., 2015; Cárdenas et al., 2018). Therefore, most of the prokaryotic diversity in Antarctic sponges from greater depths and the potential prokaryotic metabolic functions still remain unknown.
Closing this knowledge gap is crucial considering that sponges in general are prolific sources of secondary metabolites (Blunt et al., 2016; Indraningrat et al., 2016) and that the marine fauna in greater depths can also be considered as a rich source of novel natural products (Skropeta, 2008; Schupp et al., 2009; Skropeta and Wei, 2014). While results are still scarce emerging evidence indicates that North-Atlantic (Jackson et al., 2013; Kennedy et al., 2014) or Antarctic (Webster et al., 2004; Xin et al., 2011; Rodríguez-Marconi et al., 2015) cold- and/or deep-water sponges harbor diverse prokaryotic communities. Moreover, deep-sea sponges from the Atlantic exhibited novel secondary metabolite synthetic gene clusters of potential prokaryotic origin (Borchert et al., 2016), and Antarctic deep-water sponges proved to be rich sources of bioactive natural compounds (Turk et al., 2013; Berne et al., 2016; Botić et al., 2017; Angulo-Preckler et al., 2018; Mangano et al., 2018). Consequently, further studies on sponge-microbe associations from rarely accessed environments, such as the Antarctic shelf have the potential to lead to the discovery of novel metabolic mechanisms and bioactive natural compounds for a vast range of biotechnological applications (Sipkema, 2016).
Still overlooked are the xenobiotics degradation and metabolism capacities of the sponge-associated microbiota. The related pathway class comprises a set of metabolic pathways, which are involved in the metabolic breakdown of drugs and pollutants or the degradation of secondary metabolites foreign to the organisms degrading these compounds. Although research on marine sponges indicated already their capability in the biodegradation of xenobiotics, the presence of multixenobiotic resistance mechanisms or their ability to act as bioindicators for xenobiotic-related environmental stress has only been investigated for sponge-associated microbes to a very limited extent (Müller et al., 1996, 1998; Wagner et al., 1998; Krasko et al., 2001; Perez et al., 2002). The role of the sponge microbiota in some of these processes has mostly been restricted to reports on the presence of xenobiotics biodegradation pathways derived from the available sequence data (Lesser et al., 2016). However, there is a need for an initial evaluation of the bioremediation capabilities of marine sponges and their associated prokaryotic communities due to an increased awareness of a widespread marine pollution problem and its possible impact on the Antarctic marine environment (Goerke et al., 2004; Narayanaswamy et al., 2014; Courtene-Jones et al., 2017).
In the present study 16S rRNA gene amplicon data from sponge-associated prokaryotic communities was used to investigate two aims. First, we analyzed the diversity and composition of prokaryotic communities of four sponge taxa, Antarctotetilla leptoderma, Haliclona sp., Homaxinella balfourensis, and Isodictya bentarti, collected at the Antarctic shelf around the South Shetland Islands from various depths (96–469 m) (Figure 1). In addition, we investigated the influence of sponge-host identity on the prokaryotic community variance. Secondly, by applying the Tax4Fun functional gene profile prediction software with sponge prokaryotic community 16S rRNA gene data as input, we predicted functional profiles using 16S rRNA gene data to estimate the biotechnological capability and especially the potential roles of the associated prokaryotic communities as sources for novel bioactive compounds and in the degradation of xenobiotics. Corresponding with the present prokaryotic diversity and community, to our knowledge, only little or nothing is known about metabolic functions and secondary metabolites produced by the present sponges and / or their associated prokaryotic community. On the basis of the taxonomic status of the present Haliclona sp. specimens a comprehensive assessment regarding its known bioactivity is difficult. Nonetheless, different Haliclona taxa are already identified as hosts of several bioactive bacteria and sources of secondary metabolites (Erickson et al., 1997; Santalova et al., 2007; Kennedy et al., 2009; Khan et al., 2011; Turk et al., 2013; Viegelmann et al., 2014). In contrast, for the remaining three sponge taxa information is scarce, with only H. balfourensis known to contain stanols (Santalova et al., 2007). However, the here predicted metabolic functions remain to be validated by metagenomic or whole genome sequencing in future sponge microbiome studies. Nonetheless, predictive functional profiling from 16S rRNA gene sequence data can serve as a complementary and cost-effective metagenomic pre-study.
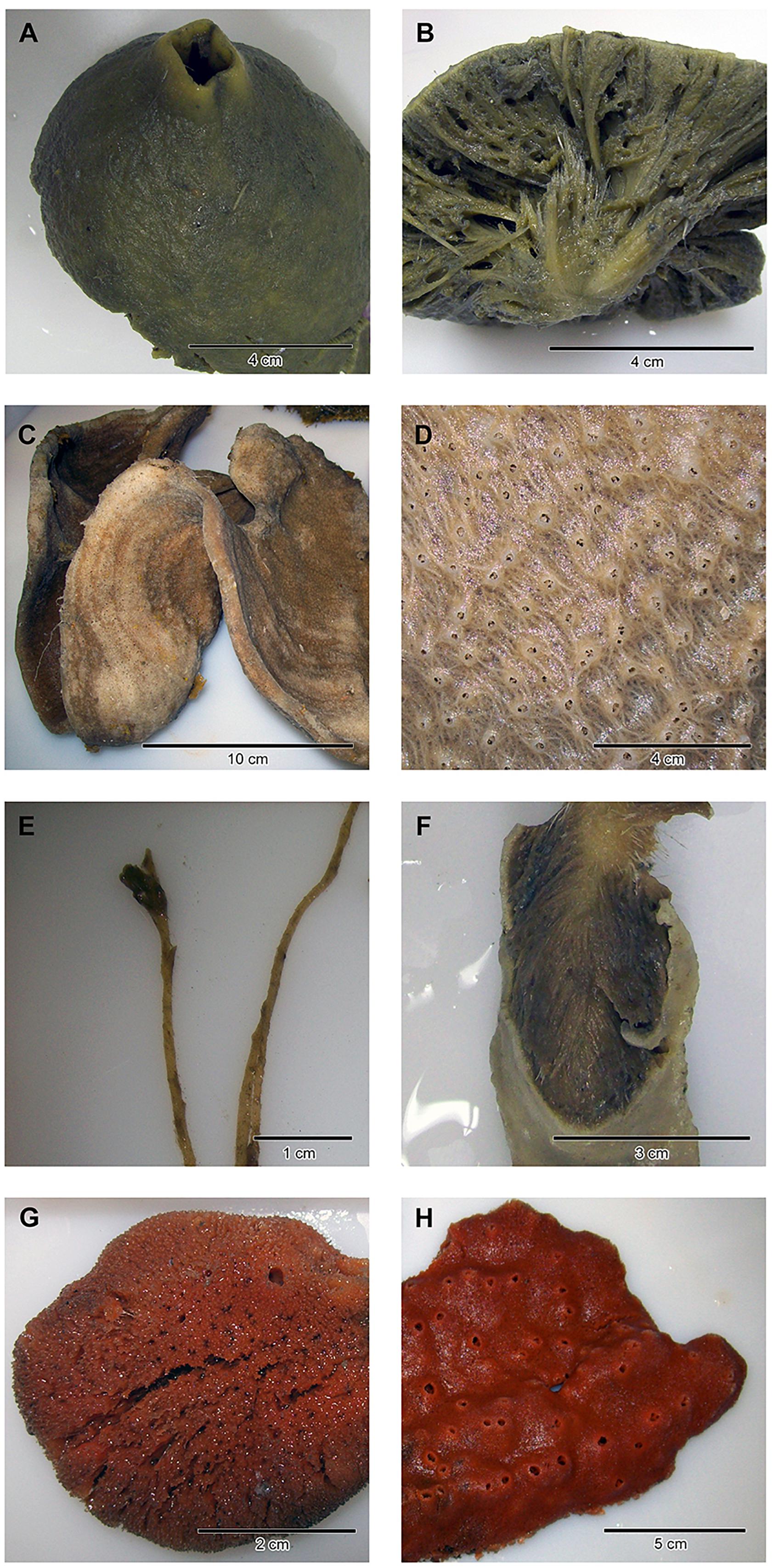
Figure 1. Close-ups and/or dissections of representatives of the four sponge taxa analyzed in the present study: (A,B) Antarctotetilla leptoderma (C,D) Haliclona sp. (E,F) Homaxinella balfourensis, and (G,H) Isodictya bentarti.
Materials and Methods
Sample Processing and Sponge Taxonomy
During the expedition of the research vessel “Polarstern” to the Southern Ocean in the Antarctic Peninsula in 2012 (ANT-XXVIII/4), 15 sponge specimens, comprising four sponge taxa, were collected by dredging from different depths (96–469 m) and sites located near the South Shetland Islands (Table 1). Collection of each specimen via dredging could have caused contamination with the surrounding sediments. To minimize the risk of such environmental contamination dissected specimens were rinsed with 0.2 μm filtered seawater. The sponges were photographed and subsequently dissected (Figure 1). Smaller tissue samples were stored in EtOH (70%) for molecular analyses, while larger tissue parts were stored in EtOH (70%) as vouchers for morphological identification. All samples were stored at -20°C until further processing. In addition, 12 seawater samples were collected at several stations from the same area at 20 m depth using Niskin bottles attached to a conductivity-temperature-depth sensor. Approximately 1000 ml of each seawater sample were filtered using 0.2 μm pore size filters and stored at -80°C until further processing. Sponge species were identified by morphological and molecular taxonomic methods (see Supplementary Material Section 1 and Supplementary Table 1). Specimens for the present study were selected and further processed if a minimum of two replicates per sponge taxon were collected. The research described refers to several Antarctic sponge species. It conforms to Antarctic Treaty legislation and to the SCAR Code of Conduct for the Use of Animals for Scientific Purposes in Antarctica (ATCM XXXIV, 2011).
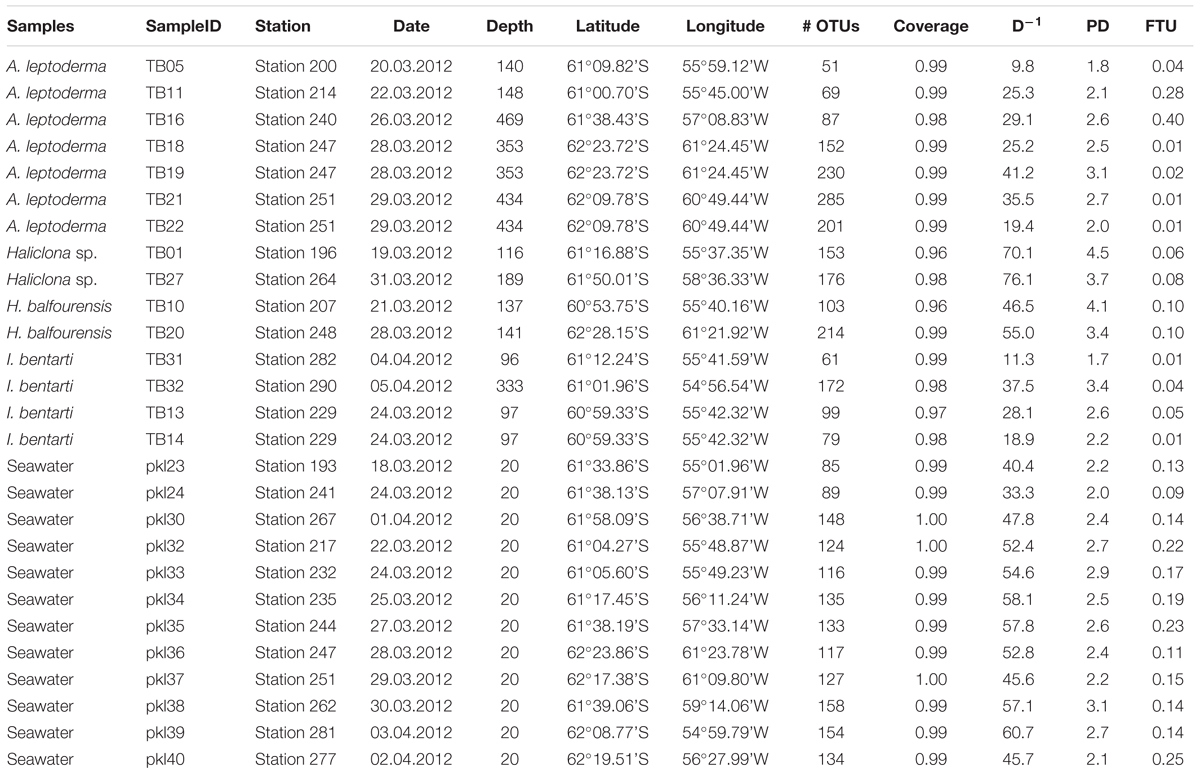
Table 1. Sample information with sample identifiers, station, date, depth, coordinates, and OTU statistics, such as number of OTUs per sample, OTU coverage, phylogenetic diversity (PD), and fraction of unmapped tax4fun OTUs (FTU).
Pyrosequencing and Prokaryote 16S rRNA Gene Amplicon Processing
Whole genomic DNA was extracted using a modified protocol by bead beating, phenol:chloroform:isoamyl alcohol purification, isopropanol/sodium acetate precipitation and ethanol washing (Zhou et al., 1996). Extracted DNA from all specimens was subjected to PCR of the variable regions V3 to V5 of bacterial 16S rRNA genes. Each reaction mixture (50 μl) contained 25 μl of Phusion High-Fidelity PCR Master Mix with HF buffer (Thermo Scientific, Waltham, MA), 0.5 μM of each primer, 3% dimethylsulfoxide, 30 μg BSA and 50 ng template. Primers used for V3 forward were 5′-MID-TACGGRAGGCAGCAG-3′ (Liu et al., 2007) and for V5 reverse 5′-CCGTCAATTCMTTTGAGT-3′ (Wang and Qian, 2009). Forward primer sequences included also the variable multiplex identifier (MID) consisting of 10 bases. DNA from each sample was amplified in three parallel PCR runs (95°C for 4 min; 30 cycles of 45 s at 95°C, 1 min at 58°C, and 45 s at 72°C; 5 min at 72°C). Products were electrophoresed for 1 h at 80 V and gel-purified using the PeqGold Gel Extraction Kit (Peqlab, Erlangen, Germany). PCR products from parallel runs were pooled and quantified using the Quant-iT dsDNA HS assay kit and a Qubit fluorometer (Invitrogen, Carlsbad, CA, United States). 16S rRNA gene amplicons were sequenced using the GS-FLX 454 pyrosequencer and titanium chemistry as recommended by the manufacturer (Roche, Branford, CT, United States). The raw data have been deposited in the Sequence Read Archive with BioProject number: PRJNA471833.
Sequences were processed using mothur v.1.37.6 (Schloss et al., 2009, 2011). Pyrosequencing flowgrams were filtered and denoised using the mothur implementation of AmpliconNoise (Quince et al., 2009). Sequences were removed from the analysis if they were <300 bp or contained ambiguous characters, homopolymers longer than 8 bp, more than one MID mismatch, or more than two mismatches to the primer sequence. After chimera removal with UCHIME (Edgar et al., 2011), unique sequences were aligned and identified using the SILVA128 taxonomy1. Non-target sequences (e.g., chloroplasts, mitochondria, eukaryotic 18S rRNA) were removed prior to operational taxonomic unit (OTU) clustering. After raw data processing, mothur was used to cluster the obtained sequences into OTUs at 97% genetic identity (species level).
Prokaryotic Community Analyses
The OTU statistics / alpha-diversity calculations and subsequent diversity analyses, if not stated otherwise, were performed in R v.3.3.3 using the picante v.1.6 package (Kembel et al., 2010; R Development Core Team, 2018). In order to compare the (dis)similarity of OTU compositions between communities the OTU abundance table was standardized using decostand (method = “hellinger”) and pairwise Bray–Curtis dissimilarities were calculated using vegdist (method = “bray”) (Bray and Curtis, 1957; Ramette, 2007; Anderson and Walsh, 2013). For univariate and multivariate analyses (e.g., Kruskal–Wallis rank sum test or analysis of variance using distance matrices – adonis) all samples were arranged into sample groups (i.e., host-identity or seawater). Please note that due to the uneven sample size and low number of replicates (n = 2) for two sponge species (i.e., Haliclona sp. and H. balfourensis) these statistical results should be interpreted cautiously. Univariate relationships between alpha-diversity indices (i.e., OTU richness, coverage, Simpson’s inverse index of diversity D-1, Faith’s phylogenetic diversity PD) and sample groups were analyzed by the Kruskal-Wallis rank sum test using kruskal test (R stats package v.3.3.3) followed by Dunn’s Kruskal-Wallis multiple comparisons test using dunnTest (FSA package v.0.8.21) and the Benjamini–Hochberg p-value correction for multiple testing (Benjamini and Hochberg, 1995; Ogle, 2018). Multivariate beta-diversity analysis based on the host-identity and non-metric multidimensional scaling (nMDS) was performed using adonis and metaMDS functions of the vegan package v.2.5.3 (Oksanen et al., 2012). Hierarchical cluster analysis was performed using hclust (method = “averag”e). The heatmap was created using JColorGrid v.1.86 (Joachimiak et al., 2006). NCBI blastn search was performed against two NCBI databases (i.e., “rRNA type strains/prokaryotic 16S ribosomal RNA” and “Nucleotide collection – nt”) using the standard settings. The 16S rRNA 97% OTU table including the SILVA128 taxonomy for each individual OTU can be accessed via the Figshare online repository2.
Functional profiles were predicted using Tax4Fun, which is a software package that predicts the functional capabilities of prokaryotic communities based on 16S rRNA datasets (Aßhauer et al., 2015). Analyses were performed with short read mode disabled. To assess the quality of the predictions, the Fraction of Taxonomic units Unexplained (FTU) value was obtained. The FTU measures the fraction of sequences assigned to taxonomic units that cannot be mapped to KEGG organisms using the Tax4Fun association matrix. The KEGG Orthology (KO) abundance profile was visualized using metaMDS in R. Subsequently, LEfSe v.1 was used to identify KEGG pathways as significant biomarkers for the individual sponge-hosts by calculating the linear discriminant analysis effect size (LDA) (Segata et al., 2011). Note that results from predictive functional profiling using 16S rRNA marker genes can deviate from metagenomic profiling and functional gene annotation. In addition, because of functional overlap, some KOs can be assigned to more than one pathway. The KEGG Orthologs relative abundance table can be accessed via the Figshare online repository3. The KEGG Orthologs pathway relative abundance table can be accessed via the Figshare online repository4.
Results
Sponge Taxonomy
We morphologically assigned four taxa to the 15 collected demosponge specimens: A. leptoderma (Order Tetractinellida), Haliclona sp. (Haplosclerida), H. balfourensis (Suberitida), and I. bentarti (Poecilosclerida) (Figure 1 and Table 1). The molecular taxonomic results corroborate the morphological results. The resulting phylogenetic tree (Supplementary Figure 1) reflects the phylogenetic distances of the four taxa.
Prokaryotic Diversity and Community Patterns
The sequencing of sponge-associated prokaryotic communities and additional seawater samples yielded 1287 OTUs (Table 1). Coverage for all samples was >0.98, with one exception (sample TB01; coverage >0.96). Sponge specimens belonging to A. leptoderma (OTUs n = 153.6 ± 88.9), Haliclona sp. (164.5 ± 16.3), and H. balfourensis (158.5 ± 78.5), exhibited a higher average OTU richness compared to seawater samples (126.7 ± 22.8). The I. bentarti OTU richness was on average lower (102.8 ± 48.7) than seawater samples and all other sponges. We measured the shared phylogenetic history among prokaryotic OTUs in all samples using Faith’s PD index (Faith, 1992). Obtained values almost reflect OTU richness with high PD for Haliclona sp. (4.1 ± 0.6) and H. balfourensis (n = 3.8 ± 0.5) and low PD for I. bentarti (2.5 ± 0.7) and seawater (2.5 ± 0.3). Only the A. leptoderma PD values (2.4 ± 0.4) do not correspond with the observed relatively high OTU richness.
In total, 31 prokaryotic phyla, including the candidate phylum Berkelbacteria and two archaeal phyla Euryarchaeota and Thaumarchaeota were found within the present four sponge taxa and seawater samples (Figure 2 and Supplementary Table 2). Phylum richness ranged from 14 phyla in Haliclona sp., H. balfourensis, and I. bentarti, over 15 phyla in seawater samples, to 22 phyla in A. leptoderma. The most abundant prokaryotic phyla among all sponges and seawater samples were Proteobacteria (mainly Alpha- and Gammaproteobacteria) and Bacteroidetes (Figure 2). Compared to all the samples, Actinobacteria contributed a substantial share to the A. leptoderma (65.6%) and seawater (18.2%) microbiota. In addition, Firmicutes were considerably abundant only in I. bentarti (50%), whereas Marinimicrobia was only relatively well represented in seawater (68.6%). Altogether, these phyla made up the most dominant taxonomic groups (93.1%) responsible for the characteristic differences between the present sponge- and seawater-associated prokaryotic community patterns (Figure 2). Noteworthy is also the presence of typical sponge-associated prokaryotic phyla such as Nitrospinae (2.1%, n = 75 reads), Planctomycetes (1.7%, n = 62), Acidobacteria (0.6%, n = 22), Chloroflexi (0.6%, n = 21), Verrucomicrobia (0.4%, n = 13), Gemmatimonadetes (0.3%, n = 11), and Spirochaetae (0.3%, n = 10), which were still found in higher numbers among all samples compared to the taxonomically rich (n = 19 phyla) but rare prokaryotic community that accounted only for minuscule parts of the individual sponge and seawater microbiota (reads n ≤ 9) in the present study (Supplementary Table 2).
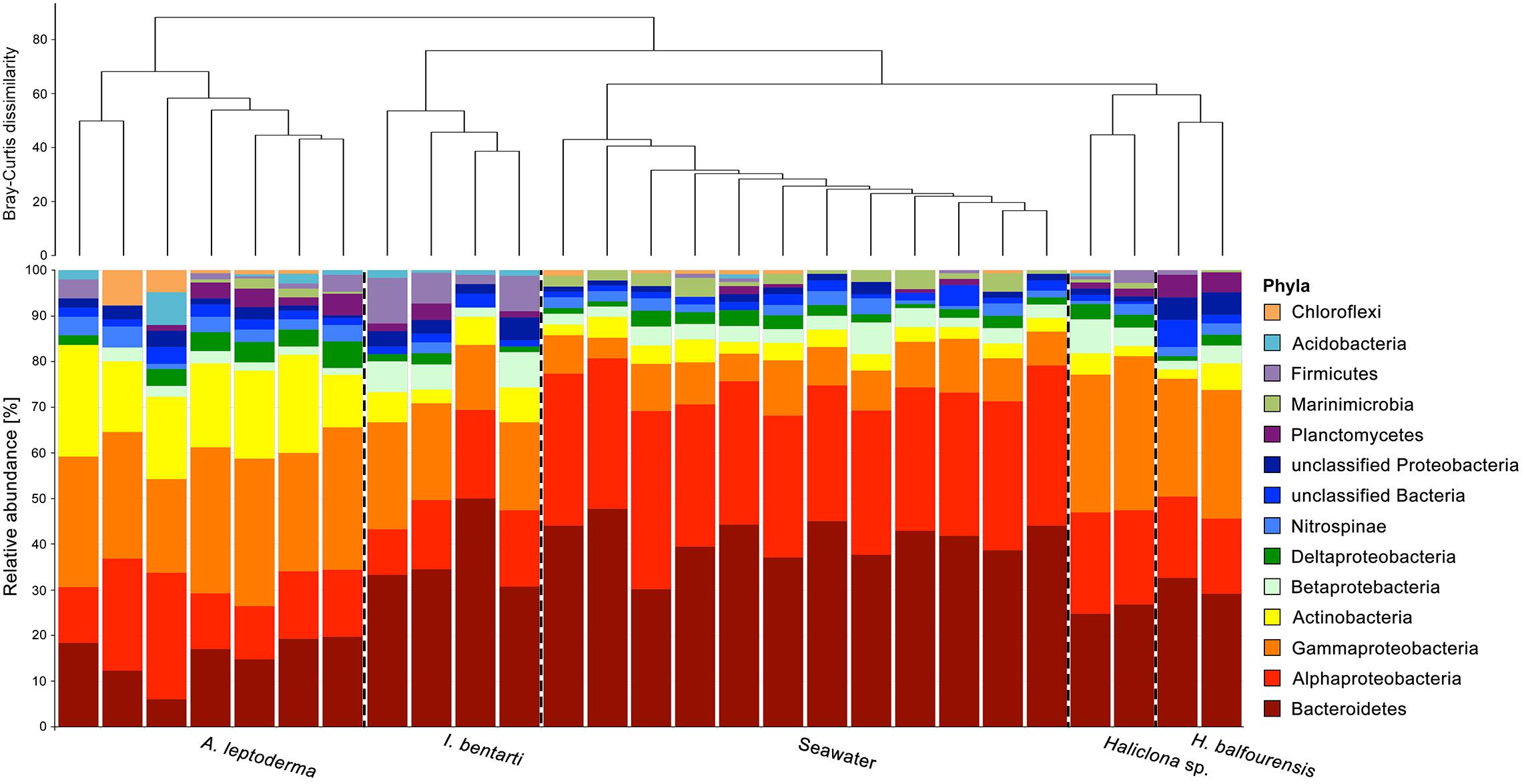
Figure 2. Relative abundance of the most abundant prokaryotic phyla, with Proteobacteria divided into classes, based on 16S rRNA gene sequences obtained from all individual sponge specimens. Only the most abundant taxa are shown. Rare phyla, below 0.5% among all samples, were excluded. The dendrogram is based on Bray–Curtis dissimilarities (relative abundances of 97%-OTUs).
The most abundant OTUs were predominantly present within particular sponge-hosts or seawater samples (Figure 3). The two most abundant OTUs belong to the Sva0996 marine group family and were almost exclusively present within A. leptoderma. Moreover, this sponge species also exhibited two additional rather host-specific OTUs, which belong to the E01-9C-26 marine group and Flavobacteriaceae NS3a marine group families (Figure 3). In contrast, the genus Candidatus Branchiomonas was only present in I. bentarti, and besides a second OTU (SAR11 clade), the only abundant bacterium among all top OTUs in this sponge taxon. In contrast to the rather sponge-host specific OTU patterns, Haliclona sp. and H. balfourensis shared many predominant OTUs with the abundant seawater microbiota (Figure 3). Especially the phylum Bacteroidetes with the order Flavobacteriales was well represented among those shared OTUs (i.e., OTUs 9, 10, 12, 16, 18, 24, 26, 28, 30, 36) belonging to seawater, Haliclona sp. and H. balfourensis samples. Finally, H. balfourensis exhibits one apparently abundant and host-specific OTU (OTU0011, unclassified gammaproteobacterium), which was only classified to class level using the SILVA128 taxonomy. The subsequent NCBI blastn search revealed that the closest similar sequence in the NCBI 16S bacterial / archaeal database belongs to the gammaproteobacterial Spongiispira norvegica species (sequence similarity 86%; GenBank accession number: NR_042453), which was isolated from the boreal sponge identified as Geodia phlegraei. An additional blastn search against the nucleotide collection yielded as best match (sequence similarity 94%; GenBank accession number: JQ435988) also a sponge-associated, yet uncultured bacterial clone that was isolated from the marine sponge identified as Hymeniacidon perlevis.
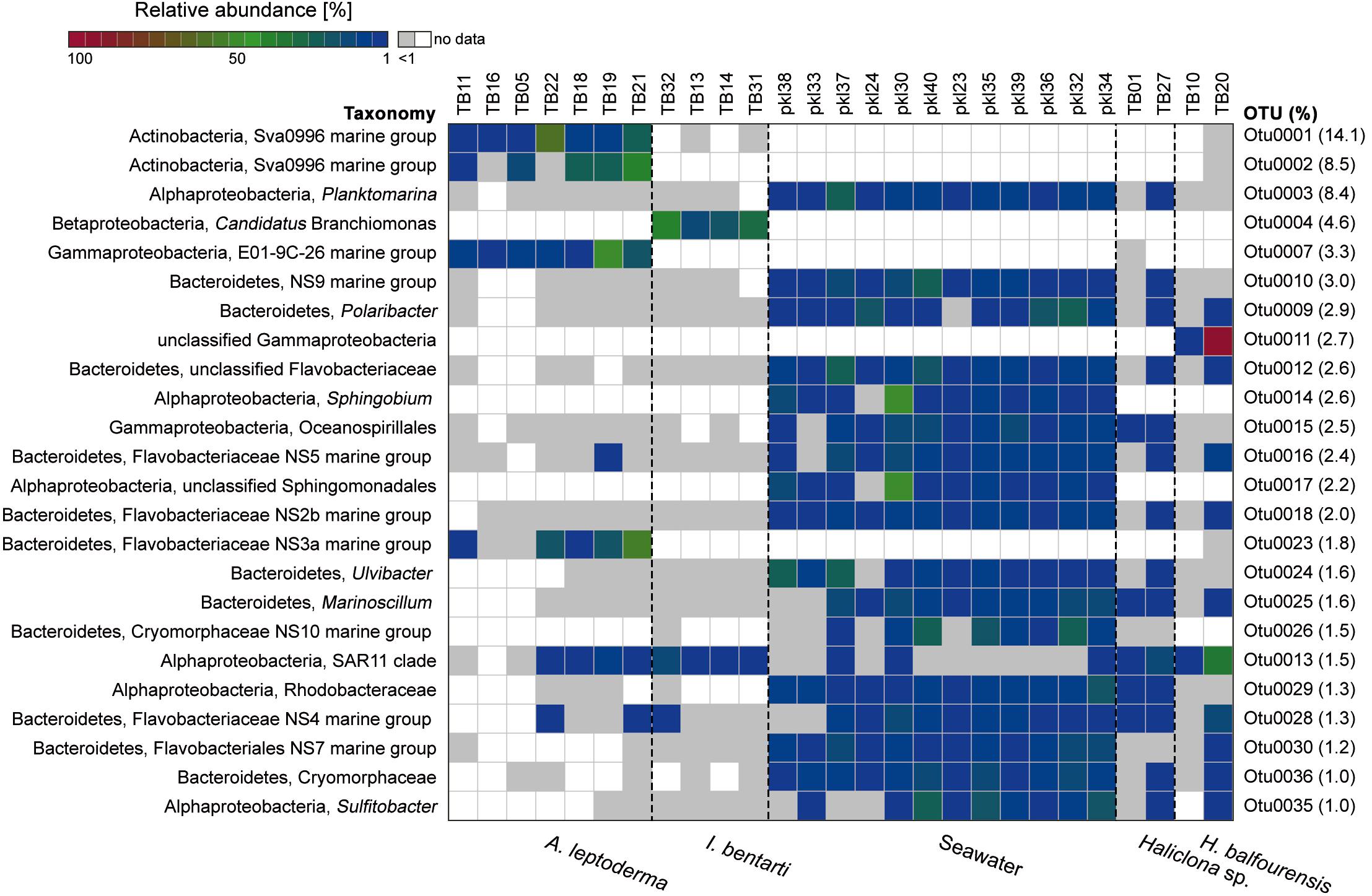
Figure 3. Relative abundance heatmap of the 24 most abundant (i.e., with >1600 reads or 1% relative abundance among all OTUs based on the sum of all available reads) 16S rRNA gene OTUs. The corresponding relative abundance of each OTU among all available OTUs is given on the right side, whereas the colorization depicts the sum of reads per individual OTU. The taxonomy consists of the highest and lowest available taxonomic rank available for each OTU. Low abundance occurrences of OTUs per sample (<1%) are colored in light gray. Samples are sorted using the cluster analysis shown in Figure 2.
Multivariate beta-diversity 16S rRNA gene community analyses using Bray–Curtis dissimilarities consolidated the preceding phylum and OTU patterns and underlined the influence of sponge-host identity on the prokaryotic community composition (Figures 2, 4A). The analysis of variance (i.e., adonis) revealed significant correlation (F4,22 = 11, R2= 0.67, p < 0.001) between community structure and host-identity. In addition, all seawater samples were clearly separated from the sponge specimens in the observed nMDS ordination and cluster analysis. Correspondingly, Simpson’s inverse alpha-diversity indices were also significantly different between seawater and A. leptoderma (p adjusted = 0.012) and I. bentarti (p adjusted = 0.017) sample groups, which are the two sponge taxa with the highest distances to seawater samples observed in the multivariate analyses (Supplementary Table 3 and Figures 2, 4A). Conversely, no significant correlation has been observed between the Haliclona sp. and H. balfourensis derived Simpson’s inverse diversity indices and seawater samples. Finally, no significant pairwise differences were found for the two remaining indices (observed OTUs and Phylogenetic Diversity PD) after corrections for multiple tests (Supplementary Table 3).
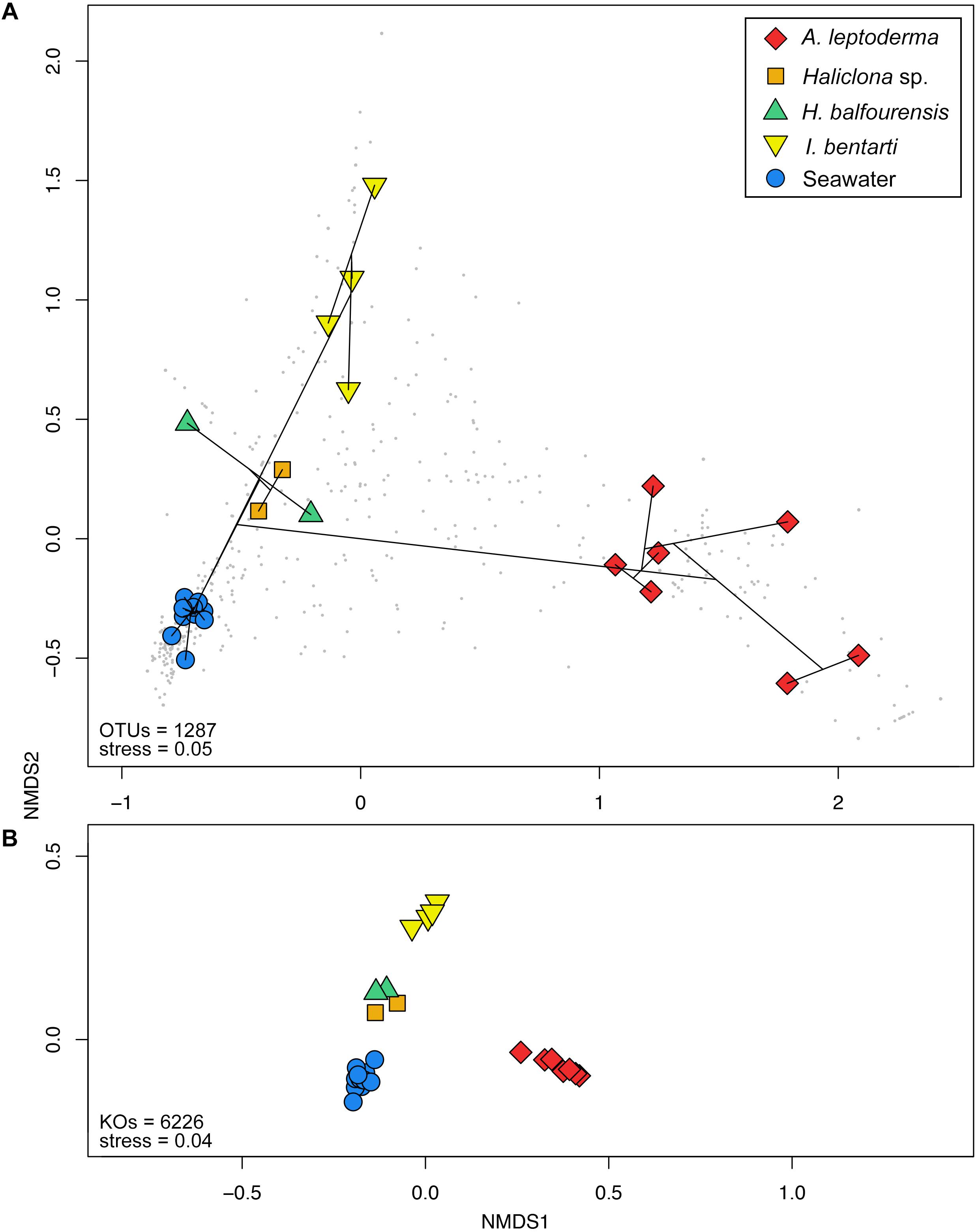
Figure 4. Non-metric multidimensional scaling plots based on Bray–Curtis distances calculated from the (A) standardized 97% OTU table and (B) predicted KEGG Orthologs for each specimen and seawater samples. Colorization is based on the corresponding sponge taxonomy. Cluster information based on Bray–Curtis dissimilarities is added to (A) via the ordispider vegan function in R, in addition, taxonomic variables are added as gray dots to the background.
Predicted Functional Profiles
The potential functional roles of the sponge-associated prokaryotic community were predicted using Tax4Fun, which is a software package that predicts the functional capabilities of prokaryotic communities based on 16S rRNA gene datasets (Aßhauer et al., 2015). Compared to the PICRUSt tool, which is also a software package that predicts functional profiles of prokaryotic communities using 16S rRNA gene sequences, Tax4Fun showed higher correlations of predicted profiles with corresponding functional profiles obtained from whole metagenome sequence data (Langille et al., 2013; Aßhauer et al., 2015). Additionally, in direct comparison Tax4Fun predicted almost 15% more KEGG Orthologs than PICRUSt (Koo et al., 2017). However, due to the predictive nature of 16S rRNA gene based functional profiling this approach cannot substitute whole metagenome profiling completely (Aßhauer et al., 2015; Weigel and Erwin, 2017). Nonetheless, in broad prokaryotic community ecology surveys, like the present study, predictive functional profiling from 16S rRNA gene sequence data can serve as a complementary and cost-effective metagenomic pre-study.
The FTU value, as a measure of the quality of the predictions, was relatively low for most of the samples and reached only >0.20 for five out of 27 samples (Table 1). A low FTU indicates that the majority of OTUs were included in the functional prediction, which further indicates that most OTUs were represented by at least one KEGG organism in the reference data of Tax4Fun. Hence, the FTU value can serve as an indirect measure of quality, because a low FTU value is usually associated with a higher correlation to real metagenomic data. For instance, FTU values for the Guerrero Negro microbial mat were up to 0.5 for some samples, but still a correlation coefficient of around 0.65 between functional prediction made with Tax4Fun and the functional profile derived from metagenome sequencing were obtained (Aßhauer et al., 2015). Moreover, for the HMP data, all FTU values were >0.1 and Spearman correlation coefficients were around 0.85 (for further details see5). As a consequence of habitat and ecosystem related differences in microbiome functions the quality of predicted functional profiles is influenced by the presence of marine derived prokaryotic genes and genomes in the provided pre-computed metabolic reference file (Arrigo, 2005; Sunagawa et al., 2015). Therefore, it is important to emphasize that the predicted metagenomic functional pathways and implied synthesis of metabolites can deviate from actual functional capabilities of the present sponge-associated microbiomes assessed by metagenomic shotgun sequencing.
In total, 6323 KEGG Orthologs (KOs; i.e., sets of homologous sequences) were predicted to be present among all samples. Using the relative KO abundances samples are arranged similar to the 16S rRNA gene profile (Figures 4A,B). Furthermore, the correlation between the KO abundance profile and the sample groups was even higher (F4,22 = 47.6, R2= 0.9, p < 0.001) compared to the 16S rRNA gene OTU profile. In total, 278 pathways were found, which resulted in 161 significantly different pathways using LEfSe (Supplementary Table 4).
In the sponge taxa A. leptoderma, H. balfourensis, and I bentarti three different predicted energy metabolism pathways, comprising the oxidation and reduction reactions of the respective pathway-associated compounds, were significantly enriched [i.e., methane (ko00680), sulfur (ko00920), and nitrogen (ko00910), respectively] (Table 2). Besides energy metabolism, we focused on specific metabolic pathway classes involved in the synthesis of bioactive compounds and in the degradation of xenobiotics, which were identified as significant biomarkers among the four present sponge taxa (Table 2). Especially, the two sponges A. leptoderma and I. bentarti featured many predicted metabolic pathways within the biosynthesis of other secondary metabolites, like terpenoids and polyketides metabolism, and xenobiotics biodegradation and metabolism classes (Table 2). For instance, in A. leptoderma the predicted secondary metabolite pathways streptomycin (ko00521), neomycin, kanamycin and gentamicin (ko00524), and phenylpropanoid (ko00940) were significantly enriched. Furthermore, in A. leptoderma certain xenobiotics biodegradation and metabolism pathways were enriched, inter alia, styrene (ko00643), caprolactam (ko00930), and steroid (ko00984) degradation (Table 2). The number of significantly enriched predicted xenobiotics biodegradation pathways was even higher in I. bentarti, which comprised a wide range of potential (chlorinated) hydrocarbon degradation capabilities: e.g., toluene (ko00623), ethylbenzene (ko00642), naphthalene (ko00626), chloroalkane and chloroalkene (ko00625), chlorocyclohexane and chlorobenzene (ko00361), dioxin (ko00621), and atrazine (ko00791) degradation (Table 2).
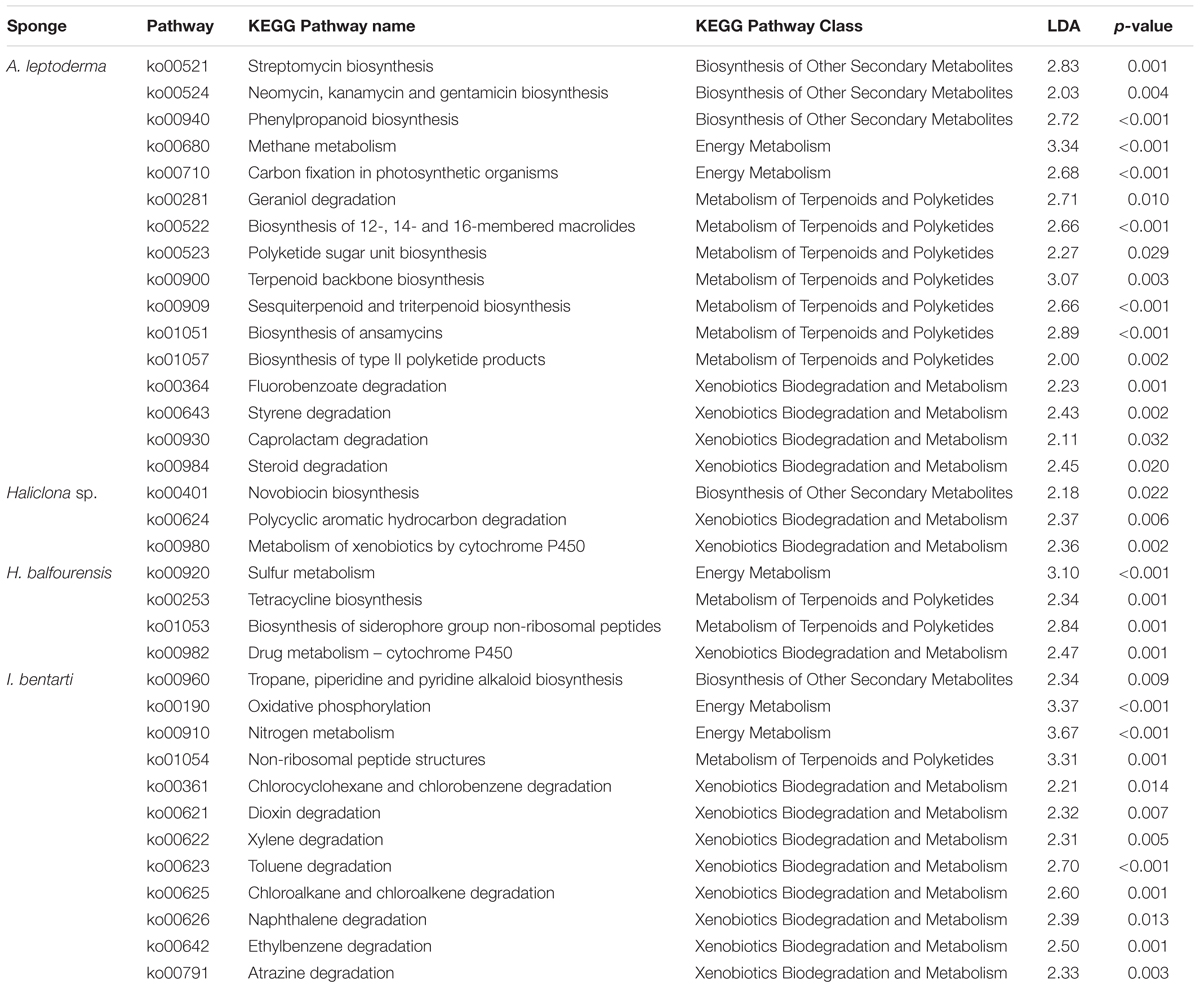
Table 2. KEGG pathways information sorted by sponge taxa with pathway identifier, pathway name and class, the linear discriminant analysis effect size (LDA), and corresponding p-values.
In contrast, the predicted bioactive potential of the associated prokaryotic community in Haliclona sp. and H. balfourensis seems to be rather low considering the number of significant predicted secondary metabolite and xenobiotics pathways. Both sponges exhibited significant P450 xenobiotics metabolism pathways, metabolism of xenobiotics by cytochrome P450 (ko00980) and drug metabolism – cytochrome P450 (ko00982), which are pathways involved in a multitude of metabolic processes. Furthermore, in Haliclona sp. only novobiocin secondary metabolite biosynthesis (ko00401), together with polycyclic aromatic hydrocarbon degradation (ko00624) pathways were enriched.
Discussion
The investigated cold-water sponges from the Antarctic shelf host diverse prokaryotic communities, which feature several bacterial phyla, such as Proteobacteria (Alpha-, Beta-, and Gamma-), Actinobacteria, or Bacteroidetes, that are usually enriched in marine sponges (Hentschel et al., 2012; Thomas et al., 2016; Moitinho-Silva et al., 2017c). In particular, most of these phyla are significantly enriched in low microbial abundance (LMA) sponges, whereas typical phyla of high microbial abundance (HMA) sponges, such as Chloroflexi, Acidobacteria, Gemmatimonadetes, are underrepresented in the present sponges (Moitinho-Silva et al., 2017c). While it is possible to utilize prokaryotic diversity patterns to predict the LMA or HMA status of sponge species, only one recent study confirmed the reliability of HMA-LMA dichotomy for cold-water sponges using microscopic cell counts (Schöttner et al., 2013). However, the present potential host-specific prokaryotic community patterns indicate that all four sponges belong to the LMA group.
Our results showed that the prokaryotic communities of the sponges were distinctly different from those in surface waters. A comparison to data of a study conducted during the same cruise, which assessed bacterial communities in mesopelagic and bathypelagic waters, showed that the sponge-associated bacterial communities are also different from bacterial communities in mesopelagic waters (Milici et al., 2017). Even though Gammaproteobacteria also dominated the mesopelagic bacterial communities, other taxa than those prevalent in the sponges were dominant, such as Pseudoalteromonas, SAR86 and ZD0417 (Milici et al., 2017). Furthermore, a plethora of prokaryotic community studies demonstrated consistent apparent differences between either various sponge-associated communities or individual sponge-specific bacteria and seawater communities, independent of spatial distances between sponge and seawater sampling locations (Blanquer et al., 2013; Taylor et al., 2013; Reveillaud et al., 2014; Thomas et al., 2016; Steinert et al., 2017). However, a recent study that utilized 98 sponge specimens and corresponding seawater samples suggested that horizontal acquisition is an important mechanism for the establishment and maintenance of sponge-species specific prokaryotic communities (Turon et al., 2018). Consequently, seawater samples should be collected close to the related sponges in order to improve the accuracy of sponge-seawater microbiota comparisons.
To our knowledge, of the present four sponge taxa, only the prokaryotic community associated with H. balfourensis has been investigated using culture-dependent and independent methods (Webster et al., 2004; Xin et al., 2011). Especially alphaproteobacterial clones, and cultured isolates belonging to several Gram-positive bacteria, such as Pseudonocardia sp., Bacillus sp., or Virgibacillus picturae, were part of the H. balfourensis microbiota in these two studies. For Haliclona sp. one Antarctic specimen from the same genus has been screened using high-throughput-sequencing and several phyla, such as Proteobacteria (Alpha-, Beta-, and Gamma-), Actinobacteria or Planctomycetes were present in relatively high abundances, which is similar to the results obtained with the specimens in the present study (Rodríguez-Marconi et al., 2015). Moreover, in different Haliclona taxa from other locations, these bacterial phyla were also found in high abundances (Kennedy et al., 2009; Sipkema et al., 2009; Thomas et al., 2016). However, the apparent large fraction of OTUs assigned to Bacteroidetes in the present study appears to be novel considering the available literature that studied Haliclona spp. specimens.
In general, sponges from deep-sea and/or cold-water habitats exhibit a diverse range of prokaryotic taxa, which are known to be associated with sponges to a greater or lesser extent. Prokaryotic phyla, such as Acidobacteria, Bacteroidetes, Chloroflexi, PAUC34f, Planctomycetes, Poribacteria, Proteobacteria (Alpha-, Beta-, Gamma-, and Delta-), Thaumarchaeota, or Verrucomicrobia, were detected in several phylogenetically divergent sponge species (Radax et al., 2012; Jackson et al., 2013; Kennedy et al., 2014; Rodríguez-Marconi et al., 2015). Correspondingly, the present study also identified most of these taxa as major part of the sponge-associated microbiota. However, on the basis of the differences in the abundance and composition of the individual communities, apparently host-identity significantly influences the prokaryotic composition; this is a defining pattern of the complex sponge-prokaryote and fungal relationships with strong supportive evidence from multiple sponge species inhabiting environmentally diverse marine habitats and climate zones (Pita et al., 2013; Easson and Thacker, 2014; Steinert et al., 2016; Thomas et al., 2016; Chaib De Mares et al., 2017).
Besides the here indicated host-specificity of prokaryotic communities, also the predominance of one single taxon, or even single OTUs, is an often observed circumstance in sponge-symbiont relationships (Croué et al., 2013; Giles et al., 2013; Jackson et al., 2013; Simister et al., 2013; Hardoim and Costa, 2014). Likewise, also the present sponge specimens possessed OTUs that made up large fractions of their potential host-specific microbiota. This includes the two most abundant bacterial OTUs, both belonging to the same taxon Sva0996 marine group (Phylum Actinobacteria), which has been recovered from various environments/organisms such as Red Sea sponges (Moitinho-Silva et al., 2014), tropical ascidians (Steinert et al., 2015), Arctic and Atlantic ocean bacterioplankton assemblages (Bano and Hollibaugh, 2002; Milici et al., 2016), or Arctic marine sediment (Ravenschlag et al., 1999). While this taxon seems to be a cosmopolitan, the enrichment in just one sponge species hints to a putative symbiotic relationship between the host and this bacterium. The presence of Candidatus Branchiomonas (order Nitrosomonadales) among the most abundant OTUs is also noteworthy. To the best of our knowledge this taxon has not been found to be present in sponges so far. However, recent research provided evidence that “ammonia oxidizing” Nitrosomonadales representatives might contribute to the nitrogen cycling in sponges and are often present in high abundances in several sponge taxa, such as Crambe crambe (Croué et al., 2013) or in one Clathria sp. individual from the Antarctic (Rodríguez-Marconi et al., 2015). In addition, the order Nitrosomonadales has also been detected in deep-sea coral and sponge communities, although at relatively low abundances (Kennedy et al., 2014; Simister et al., 2016). Finally, the presence of a potentially novel and sponge-specific bacterium (i.e., low BLAST similarity to known bacterial 16S rRNA gene sequences), which is predominant in H. balfourensis, suggests that certain bacterial symbionts are potentially highly adapted to their specific sponge-host and its metabolic as well as chemical environment. In fact, in recent years ‘omics-related analyses started to reveal the complexity of metabolic interactions between symbionts and sponge-hosts (Thomas et al., 2010; Liu et al., 2012; Radax et al., 2012; Moitinho-Silva et al., 2017a; Slaby et al., 2017).
Functional metabolic profiling by a predictive approach using PICRUSt (Langille et al., 2013) has been successfully applied in several recent sponge-microbiota studies either by investigating individual sponges and their functional potential (de Voogd et al., 2015; Cleary et al., 2017), comparing predicted profiles with shotgun sequenced metagenomic data (Weigel and Erwin, 2017), correlating predicted functions with ocean acidification and elevated seawater temperature (Lesser et al., 2016), or focusing on predicted functions of individual key symbionts (Morrow et al., 2015). We applied a similar approach with a different predictive program, Tax4fun (Aßhauer et al., 2015), which was successfully applied for bacterioplankton communities and providing valid correlations of functional profiles to microbial and environmental parameters (Wemheuer et al., 2017). The present study demonstrates that this approach can provide first insights into potential functional aspects of sponge-associated prokaryotic communities. Similar to the differences observed here in community composition among different sponge and seawater samples, the predicted functional KO profiles also resulted in a clear distinction between the four sponge-hosts and/or seawater samples. So far, comparative metagenomic functional analyses between different sponge species are scarce (Pita et al., 2016). Only one metagenomic study, which investigated the microbiomes of six phylogenetically divergent sponge species, has shown that host-specific symbiont communities exhibited apparent core functions despite maintaining sponge species-specific gene composition patterns (Fan et al., 2012). Apart from the presence of energy metabolism classes (e.g., carbon or nitrogen metabolism) or symbiosis factors (e.g., eukaryotic-like proteins, ankyrin repeats, or tetratricopeptide repeats), potential prokaryotic symbionts often also synthesize secondary metabolites and bioactive natural products for which sponges are widely acknowledged (Hentschel et al., 2012; Reynolds and Thomas, 2016; Bhushan et al., 2017; Lackner et al., 2017). Here, the predicted pathway analysis found first indications that the four Antarctic sponge species may possess significantly different secondary metabolite biosynthesis pathways. Predicted significant secondary metabolism biosynthesis pathways of known antibiotics, such as streptomycin, butirosin and neomycin in A. leptoderma, and novobiocin in Haliclona sp. suggest that these sponge species may possess host-specific antimicrobial activity. So far, a broad range of antibacterial compounds and activities has been reported for phylogenetically divergent sponges from different marine habitats (Laport et al., 2009; Indraningrat et al., 2016), including the Southern Ocean (Berne et al., 2016; Angulo-Preckler et al., 2018). In the present study, certain predicted xenobiotics-related degradation pathways are significantly enriched in all four sponges, especially in A. leptoderma and I. bentarti. For instance, styrene and its derivatives are aromatic compounds that pose environmental risks due to their toxicity (O’Leary et al., 2002; Mooney et al., 2006). In addition, the degradation process of steroids, a rich class of organic molecules in all natural environments, can utilize steroids as growth substrates for prokaryotes (Bergstrand et al., 2016; Holert et al., 2018). Hence, the predicted presence of the styrene and steroid degradation pathways may indicate a potential role in bioremediation processes and the biotechnological potential of the A. leptoderma microbiota. Finally, the sponge I. bentarti exhibited a wide range of predicted xenobiotic degradation pathways that encompass several organic compound classes, such as alkylbenzenes, chlorinated alkenes, chloroarenes, organochlorides, or naphtalenes. Many derivatives of these compound classes are known contaminants and pollutants due to their (geno)toxic properties (Singh, 2012). Local enrichment of pollutants could explain the predicted differential xenobiotic degradation pathway patters. However, the sponge specimens that belonged to the same species were mostly collected at different sites and depths. Hence, it seems unlikely that certain predicted degradation pathways may reflect the local enrichment of pollutants. On the other hand, genomic analyses indicated that prokaryotes possess a broad range of detoxifying functions within the sponge-holobiont (Hoffmann et al., 2009; Konoki et al., 2010; Fan et al., 2012; Uriz et al., 2012; Tian et al., 2014; Karimi et al., 2017; Keren et al., 2017). Therefore, a host-specific microbiota that is adapted to the host-intrinsic chemistry and metabolism could have shaped the distribution patterns of the predicted xenobiotic degradation pathways.
However, it should be emphasized that the performed functional metabolic predictions are no substitute for metabolic gene and gene cluster analyses. For instance, a direct comparison between predicted and shotgun sequenced metagenome derived nitrogen cycling genes revealed relatively similar KO counts and abundances, though with noticeable deviations (Weigel and Erwin, 2017). Moreover, prokaryotic secondary metabolism can be highly strain and environment-specific (Jensen et al., 2007; Romano et al., 2018), and 90% of the marine prokaryotic genes detected by the TARA Oceans project had no match in public databases at the time when the survey was conducted (Sunagawa et al., 2015). Therefore, it remains to be validated by metagenomic or whole genome sequencing if specific sponge-associated prokaryotic taxa are responsible for the metabolic functions predicted in the present study.
Conclusion
We could find indications that cold and deep-water sponges exhibit host-specific prokaryotic communities, despite different sampling sites and depths. This is similar to the conclusions regarding host-specificity from studies of sponge-prokaryotic communities from several (sub)tropical and temperate regions (Pita et al., 2013; Easson and Thacker, 2014; Steinert et al., 2016, 2017). Finally, predictive functional analysis suggests that sponge-associated prokaryotes may also enhance the roles of sponges in bioremediation processes of environmental pollutants, besides their acknowledged involvement in the biosynthesis of secondary metabolites and detoxification of metabolic waste products.
Author Contributions
PS and GS designed and conceived the study. MS and TB collected samples during the ANT-XXVIII/4 Polarstern cruise. DJ and DE classified the sponge samples and wrote the Supplementary Section 1: Sponge barcoding and morphological taxonomy. BW undertook the laboratory work and sequencing, and generated data. RD contributed reagents and materials for sequencing. GS analyzed and interpreted the data and wrote the first draft of the manuscript. All authors contributed to manuscript corrections and improved the final version. The authors greatly appreciate the thoughtful and insightful comments by the reviewers.
Funding
TB, RD, MS, and BW were supported by the Transregional Collaborative Research Center Roseobacter (TRR51) funded by Deutsche Forschungsgemeinschaft (DFG). DJ thanks the Deutsche Forschungsgemeinschaft (DFG) for financial support to our Antarctic Porifera research (JA-1063/17 1). DE thanks Gabriele Büttner, Simone Schätzle, and Gert Wörheide, LMU Munich for support in various aspects of the molecular investigations. Additionally, we acknowledge support by DFG and the Open Access Publication Funds of the Carl-von-Ossietzky University of Oldenburg.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful for the excellent support of captain Thomas Wunderlich and his crew on board RV Polarstern.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00297/full#supplementary-material
Footnotes
- ^https://www.mothur.org/wiki/Silva_reference_filese
- ^https://doi.org/10.6084/m9.figshare.6671720
- ^https://doi.org/10.6084/m9.figshare.6671735
- ^https://doi.org/10.6084/m9.figshare.6671726
- ^http://tax4fun.gobics.de/
References
Anderson, M. J., and Walsh, D. C. I. (2013). PERMANOVA, ANOSIM, and the mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 83, 557–574. doi: 10.1890/12-2010.1
Angulo-Preckler, C., San Miguel, O., García-Aljaro, C., and Avila, C. (2018). Antibacterial defenses and palatability of shallow-water Antarctic sponges. Hydrobiologia 806, 123–138. doi: 10.1007/s10750-017-3346-5
Arrigo, K. R. (2005). Marine microorganisms and global nutrient cycles. Nature 437, 349–355. doi: 10.1038/nature04159
ATCM XXXIV. (2011). “SCAR’s Code of Conduct for the Use of Animals for Scientific Purposes in Antarctica,” in Proceedings of the IP53, Agenda Item CEP 8c, presented by SCAR during XXXIV Antarctic Treaty Consultative Meeting, (Buenos Aires).
Aßhauer, K. P., Wemheuer, B., Daniel, R., and Meinicke, P. (2015). Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31, 2882–2884. doi: 10.1093/bioinformatics/btv287
Bano, N., and Hollibaugh, J. T. (2002). Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl. Environ. Microbiol. 68, 505–518. doi: 10.1128/AEM.68.2.505-518.2002
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300.
Bergstrand, L. H., Cardenas, E., Holert, J., van Hamme, J. D., and Mohn, W. W. (2016). Delineation of steroid-degrading microorganisms through comparative genomic analysis. mBio 7, :e00166. doi: 10.1128/mBio.00166-16
Berne, S., Kalauz, M., Lapat, M., Savin, L., Janussen, D., Kersken, D., et al. (2016). Screening of the Antarctic marine sponges (Porifera) as a source of bioactive compounds. Polar Biol. 39, 947–959. doi: 10.1007/s00300-015-1835-4
Bhushan, A., Peters, E. E., and Piel, J. (2017). “Entotheonella Bacteria as Source of Sponge-Derived Natural Products: Opportunities for Biotechnological Production,” in Blue Biotechnology: From Gene to Bioactive Product, eds W. E. G. Müller, H. C. Schröder, and X. Wang (Cham: Springer International Publishing), 291–314. doi: 10.1007/978-3-319-51284-6_9
Blanquer, A., Uriz, M. J., and Galand, P. E. (2013). Removing environmental sources of variation to gain insight on symbionts vs. transient microbes in high and low microbial abundance sponges. Environ. Microbiol. 15, 3008–3019. doi: 10.1111/1462-2920.12261
Blunt, J. W., Copp, B. R., Munro, M. H. G., Northcote, P. T., and Prinsep, M. R. (2016). Marine natural products. Nat. Prod. Rep. 27, 165–237. doi: 10.1039/b906091j
Borchert, E., Jackson, S. A., O’Gara, F., and Dobson, A. D. W. (2016). Diversity of natural product biosynthetic genes in the microbiome of the Deep Sea sponges Inflatella pellicula, Poecillastra compressa, and Stelletta normani. Front. Microbiol. 07:1027. doi: 10.3389/fmicb.2016.01027
Botić, T., Defant, A., Zanini, P., Žužek, M. C., Frangež, R., Janussen, D., et al. (2017). Discorhabdin alkaloids from Antarctic Latrunculia spp. sponges as a new class of cholinesterase inhibitors. Eur. J. Med. Chem. 136, 294–304. doi: 10.1016/j.ejmech.2017.05.019
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Cárdenas, C. A., González-aravena, M., Font, A., Hestetun, J. T., Hajdu, E., Trefault, N., et al. (2018). High similarity in the microbiota of cold- water sponges of the genus mycale from two different geographical areas. PeerJ 6, :e4965. doi: 10.7717/peerj.4935
Chaib De Mares, M., Sipkema, D., Huang, S., Bunk, B., Overmann, J., and van Elsas, J. D. (2017). Host specificity for bacterial, archaeal and fungal communities determined for high- and low-microbial abundance sponge species in two genera. Front. Microbiol. 8:2560. doi: 10.3389/fmicb.2017.02560
Cleary, D. F. R., Polónia, A. R. M., and de Voogd, N. J. (2017). Prokaryote composition and predicted metagenomic content of two cinachyrella morphospecies and water from West Papuan Marine Lakes. FEMS Microbiol Ecol. 94, :fix175. doi: 10.1093/femsec/fix175
Courtene-Jones, W., Quinn, B., Gary, S. F., Mogg, A. O. M., and Narayanaswamy, B. E. (2017). Microplastic pollution identified in deep-sea water and ingested by benthic invertebrates in the Rockall Trough, North Atlantic Ocean. Environ. Pollut. 231, 271–280. doi: 10.1016/j.envpol.2017.08.026
Croué, J., West, N. J., Escande, M.-L., Intertaglia, L., Lebaron, P., and Suzuki, M. T. (2013). A single betaproteobacterium dominates the microbial community of the crambescidine-containing sponge Crambe crambe. Sci. Rep. 3, :2583. doi: 10.1038/srep02583
de Voogd, N. J., Cleary, D. F. R., Polónia, A. R. M., and Gomes, N. C. M. (2015). Bacterial community composition and predicted functional ecology of sponges, sediment and seawater from the thousand islands reef complex, West Java, Indonesia. FEMS Microbiol. Ecol. 91, 1–12. doi: 10.1093/femsec/fiv019
Easson, C. G., and Thacker, R. W. (2014). Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Front. Microbiol. 5:532. doi: 10.3389/fmicb.2014.00532
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Erickson, K. L., Beutler, J. A., Cardellina, J. H., and Boyd, M. R. (1997). Salicylihalamides A and B, novel cytotoxic macrolides from the marine sponge haliclona sp. J. Org. Chem. 62, 8188–8192. doi: 10.1021/jo971556g
Faith, D. P. (1992). Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10. doi: 10.1016/0006-3207(92)91201-3
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. 109, E1878–E1887. doi: 10.1073/pnas.1203287109
Giles, E. C., Kamke, J., Moitinho-Silva, L., Taylor, M. W., Hentschel, U., Ravasi, T., et al. (2013). Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol. Ecol. 83, 232–241. doi: 10.1111/j.1574-6941.2012.01467.x
Goerke, H., Weber, K., Bornemann, H., Ramdohr, S., and Plötz, J. (2004). Increasing levels and biomagnification of persistent organic pollutants (POPs) in Antarctic biota. Mar. Pollut. Bull. 48, 295–302. doi: 10.1016/j.marpolbul.2003.08.004
Hardoim, C. C. P., and Costa, R. (2014). Temporal dynamics of prokaryotic communities in the marine sponge Sarcotragus spinosulus. Mol. Ecol. 23, 3097–3112. doi: 10.1111/mec.12789
Hentschel, U., Piel, J., Degnan, S. M., and Taylor, M. W. (2012). Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 10, 641–654. doi: 10.1038/nrmicro2839
Hoffmann, F., Radax, R., Woebken, D., Holtappels, M., Lavik, G., Rapp, H. T., et al. (2009). Complex nitrogen cycling in the sponge Geodia barretti. Environ. Microbiol. 11, 2228–2243. doi: 10.1111/j.1462-2920.2009.01944.x
Holert, J., Cardenas, E., Bergstrand, L. H., Zaikova, E., Hahn, A. S., Hallam, S. J., et al. (2018). Metagenomes reveal global distribution of bacterial steroid catabolism in natural, engineered, and host environments. mBio 9, e02345—e2317. doi: 10.1128/mBio.02345-17
Indraningrat, A., Smidt, H., and Sipkema, D. (2016). Bioprospecting sponge-associated microbes for antimicrobial compounds. Mar. Drugs 14, :87. doi: 10.3390/md14050087
Jackson, S. A., Flemer, B., McCann, A., Kennedy, J., Morrissey, J. P., O’Gara, F., et al. (2013). Archaea appear to dominate the microbiome of Inflatella pellicula deep sea sponges. PLoS One 8:e84438. doi: 10.1371/journal.pone.0084438
Jensen, P. R., Williams, P. G., Oh, D. C., Zeigler, L., and Fenical, W. (2007). Species-specific secondary metabolite production in marine actinomycetes of the genus Salinispora. Appl. Environ. Microbiol. 73, 1146–1152. doi: 10.1128/aem.01891-06
Joachimiak, M. P., Weisman, J. L., and May, B. C. (2006). JColorGrid: software for the visualization of biological measurements. BMC Bioinformatics 7:225. doi: 10.1186/1471-2105-7-225
Karimi, E., Ramos, M., Gonçalves, J. M. S., Xavier, J. R., Reis, M. P., and Costa, R. (2017). Comparative metagenomics reveals the distinctive adaptive features of the Spongia officinalis endosymbiotic consortium. Front. Microbiol. 8:2499. doi: 10.3389/fmicb.2017.02499
Kembel, S. W., Cowan, P. D., Helmus, M. R., Cornwell, W. K., Morlon, H., Ackerly, D. D., et al. (2010). Picante: {R} tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Kennedy, J., Baker, P., Piper, C., Cotter, P. D., Walsh, M., Mooij, M. J., et al. (2009). Isolation and analysis of bacteria with antimicrobial activities from the marine sponge Haliclona simulans collected from irish waters. Mar. Biotechnol. 11, 384–396. doi: 10.1007/s10126-008-9154-1
Kennedy, J., Flemer, B., Jackson, S. A., Morrissey, J. P., O’Gara, F., and Dobson, A. D. W. (2014). Evidence of a putative deep sea specific microbiome in marine sponges. PLoS One 9:e91092. doi: 10.1371/journal.pone.0091092
Keren, R., Mayzel, B., Lavy, A., Polishchuk, I., Levy, D., Fakra, S. C., et al. (2017). Sponge-associated bacteria mineralize arsenic and barium on intracellular vesicles. Nat. Commun. 8, :14393. doi: 10.1038/ncomms14393
Khan, S. T., Komaki, H., Motohashi, K., Kozone, I., Mukai, A., Takagi, M., et al. (2011). Streptomyces associated with a marine sponge Haliclona sp.; biosynthetic genes for secondary metabolites and products. Environ. Microbiol. 13, 391–403. doi: 10.1111/j.1462-2920.2010.02337.x
Konoki, K., Saito, K., Matsuura, H., Sugiyama, N., Cho, Y., Yotsu-Yamashita, M., et al. (2010). Binding of diarrheic shellfish poisoning toxins to okadaic acid binding proteins purified from the sponge Halichondria okadai. Bioorg.anic Med. Chem. 18, 7607–7610. doi: 10.1016/j.bmc.2010.08.043
Koo, H., Hakim, J. A., Morrow, C. D., Eipers, P. G., Davila, A., Andersen, D. T., et al. (2017). Comparison of two bioinformatics tools used to characterize the microbial diversity and predictive functional attributes of microbial mats from Lake Obersee, Antarctica. J. Microbiol. Methods 140, 15–22. doi: 10.1016/j.mimet.2017.06.017
Krasko, A., Kurelec, B., Batel, R., Müller, I. M., and Müller, W. E. G. (2001). Potential multidrug resistance gene POHL: an ecologically relevant indicator in marine sponges. Environ. Toxicol. Chem. 20, 198–204. doi: 10.1002/etc.5620200123
Lackner, G., Peters, E. E., Helfrich, E. J. N., and Piel, J. (2017). Insights into the lifestyle of uncultured bacterial natural product factories associated with marine sponges. Proc. Natl. Acad. Sci. U.S.A. 114, E347–E356. doi: 10.1073/pnas.1616234114
Langille, M., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Laport, M., Santos, O., and Muricy, G. (2009). Marine sponges: potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. 10, 86–105. doi: 10.2174/138920109787048625
Lesser, M. P., Fiore, C., Slattery, M., and Zaneveld, J. (2016). Climate change stressors destabilize the microbiome of the Caribbean barrel sponge, Xestospongia muta. J. Exp. Mar. Bio. Ecol. 475, 11–18. doi: 10.1016/j.jembe.2015.11.004
Liu, M., Fan, L., Zhong, L., Kjelleberg, S., and Thomas, T. (2012). Metaproteogenomic analysis of a community of sponge symbionts. ISME J. 6, 1515–1525. doi: 10.1038/ismej.2012.1
Liu, Z., Lozupone, C., Hamady, M., Bushman, F. D., and Knight, R. (2007). Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 35, :e120. doi: 10.1093/nar/gkm541
Mangano, S., Caruso, C., Michaud, L., and Lo Giudice, A. (2018). First evidence of quorum sensing activity in bacteria associated with Antarctic sponges. Polar Biol. 41, 1435–1445. doi: 10.1007/s00300-018-2296-3
Milici, M., Deng, Z. L., Tomasch, J., Decelle, J., Wos-Oxley, M. L., Wang, H., et al. (2016). Co-occurrence analysis of microbial taxa in the Atlantic ocean reveals high connectivity in the free-living bacterioplankton. Front. Microbiol. 7:649. doi: 10.3389/fmicb.2016.00649
Milici, M., Vital, M., Tomasch, J., Badewien, T. H., Giebel, H. A., Plumeier, I., et al. (2017). Diversity and community composition of particle-associated and free-living bacteria in mesopelagic and bathypelagic Southern Ocean water masses: evidence of dispersal limitation in the Bransfield Strait. Limnol. Oceanogr. 62, 1080–1095. doi: 10.1002/lno.10487
Moitinho-Silva, L., Bayer, K., Cannistraci, C. V., Giles, E. C., Ryu, T., Seridi, L., et al. (2014). Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol. Ecol. 23, 1348–1363. doi: 10.1111/mec.12365
Moitinho-Silva, L., Díez-Vives, C., Batani, G., Esteves, A. I., Jahn, M. T., and Thomas, T. (2017a). Integrated metabolism in sponge–microbe symbiosis revealed by genome-centered metatranscriptomics. ISME J. 11, 1651–1666. doi: 10.1038/ismej.2017.25
Moitinho-Silva, L., Nielsen, S., Amir, A., Gonzalez, A., Ackermann, G. L., Cerrano, C., et al. (2017b). The sponge microbiome project. Gigascience 6, 1–7. doi: 10.1093/gigascience/gix077
Moitinho-Silva, L., Steinert, G., Nielsen, S., Hardoim, C. C. P., Wu, Y.-C., McCormack, G. P., et al. (2017c). Predicting the HMA-LMA status in marine sponges by machine learning. Front. Microbiol. 8:752. doi: 10.3389/fmicb.2017.00752
Mooney, A., Ward, P. G., and O’Connor, K. E. (2006). Microbial degradation of styrene: biochemistry, molecular genetics, and perspectives for biotechnological applications. Appl. Microbiol. Biotechnol. 72, 1–10. doi: 10.1007/s00253-006-0443-1
Morrow, K. M., Bourne, D. G., Humphrey, C., Botté, E. S., Laffy, P., Zaneveld, J., et al. (2015). Natural volcanic CO2 seeps reveal future trajectories for host-microbial associations in corals and sponges. ISME J. 9, 894–908. doi: 10.1038/ismej.2014.188
Müller, W. E. G., Batel, R., Lacorn, M., Steinhart, H., Simat, T., Lauenroth, S., et al. (1998). Accumulation of cadmium and zinc in the marine sponge Suberites domuncula and its potential consequences on single-strand breaks and on expression of heat-shock protein: a natural field study. Mar. Ecol. Prog. Ser. 167, 127–135. doi: 10.3354/meps167127
Müller, W. E. G., Steffen, R., Rinkevich, B., Matranga, V., and Kurelec, B. (1996). The multixenobiotic resistance mechanism in the marine sponge Suberites domuncula: its potential applicability for the evaluation of environmental pollution by toxic compounds. Mar. Biol. 125, 165–170. doi: 10.1007/BF00350770
Narayanaswamy, B. E., Paterson, G. L. J., Calafat, A., Canals, M., Thompson, R. C., Sanchez-Vidal, A., et al. (2014). The deep sea is a major sink for microplastic debris. R. Soc. Open Sci. 1, :140317–140317. doi: 10.1098/rsos.140317
Ogle, D. H. (2018). FSA: Fisheries Stock Analysis. Available at: https://github.com/droglenc/FSA (accessed October 22, 2018).
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., and Minchin, P. R. (2012). Vegan: Community Ecology Package. R Packag. version 2.0-2.
O’Leary, N. D., O’Connor, K. E., and Dobson, A. D. W. (2002). Biochemistry, genetics and physiology of microbial styrene degradation. FEMS Microbiol. Rev. 26, 403–417. doi: 10.1016/s0168-6445(02)00126-2
Perez, T., Sarrazin, L., Rebouillon, P., and Vacelet, J. (2002). First evidences of surfactant biodegradation by marine sponges (Porifera): an experimental study with a linear alkylbenzenesulfonate. Hydrobiologia 489, 225–233. doi: 10.1023/A:1023217218585
Pita, L., Fraune, S., and Hentschel, U. (2016). Emerging sponge models of animal-microbe symbioses. Front. Microbiol. 7:2102. doi: 10.3389/fmicb.2016.02102
Pita, L., Turon, X., López-Legentil, S., and Erwin, P. M. (2013). Host rules: Spatial stability of bacterial communities associated with marine sponges (Ircinia spp.) in the western mediterranean sea. FEMS Microbiol. Ecol. 86, 268–276. doi: 10.1111/1574-6941.12159
Quince, C., Lanzén, A., Curtis, T. P., Davenport, R. J., Hall, N., Head, I. M., et al. (2009). Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods 6, 639–641. doi: 10.1038/nmeth.1361
R Development Core Team. (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Radax, R., Rattei, T., Lanzen, A., Bayer, C., Rapp, H. T., Urich, T., et al. (2012). Metatranscriptomics of the marine sponge Geodia barretti: tackling phylogeny and function of its microbial community. Environ. Microbiol. 14, 1308–1324. doi: 10.1111/j.1462-2920.2012.02714.x
Ramette, A. (2007). Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 62, 142–160. doi: 10.1111/j.1574-6941.2007.00375.x
Ravenschlag, K., Sahm, K., and Pernthaler, J. (1999). High bacterial diversity in permanently cold marine sediments High Bacterial Diversity in Permanently Cold Marine Sediments. Appl. Environ. Microbiol. 65, 3982–3989.
Reveillaud, J., Maignien, L., Eren, M. A., Huber, J. A., Apprill, A., and Sogin, M. L. (2014). Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 8, 1198–1209. doi: 10.1038/ismej.2013.227
Reynolds, D., and Thomas, T. (2016). Evolution and function of eukaryotic-like proteins from sponge symbionts. Mol. Ecol. 25, 5242–5253. doi: 10.1111/mec.13812
Rodríguez-Marconi, S., De la Iglesia, R., Díez, B., Fonseca, C. A., Hajdu, E., and Trefault, N. (2015). Characterization of bacterial, archaeal and eukaryote symbionts from antarctic sponges reveals a high diversity at a three-domain level and a particular signature for this ecosystem. PLoS One 10:e0138837. doi: 10.1371/journal.pone.0138837
Romano, S., Jackson, S., Patry, S., and Dobson, A. (2018). Extending the “one strain many compounds” (OSMAC) principle to marine microorganisms. Mar. Drugs 16, :244. doi: 10.3390/md16070244
Santalova, E. A., Makarieva, T. N., Ponomarenko, L. P., Denisenko, V. A., Krasokhin, V. B., Mollo, E., et al. (2007). Sterols and related metabolites from five species of sponges. Biochem. Syst. Ecol. 35, 439–446. doi: 10.1016/j.bse.2006.10.020
Schloss, P. D., Gevers, D., and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing Artifacts on 16s rRNA-based studies. PLoS One 6:e47075. doi: 10.1371/journal.pone.0027310
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-1549
Schmitt, S., Tsai, P., Bell, J., Fromont, J., Ilan, M., Lindquist, N., et al. (2012). Assessing the complex sponge microbiota: Core, variable and species-specific bacterial communities in marine sponges. ISME J. 6, 564–576. doi: 10.1038/ismej.2011.116
Schöttner, S., Hoffmann, F., Cárdenas, P., Rapp, H. T., Boetius, A., and Ramette, A. (2013). Relationships between host phylogeny, host type and bacterial community diversity in cold-water coral reef sponges. PLoS One 8:1–11. doi: 10.1371/journal.pone.0055505
Schupp, P. J., Kohlert-Schupp, C., Whitefield, S., Engemann, A., Rohde, S., Hemscheidt, T., et al. (2009). Cancer chemopreventive and anticancer evaluation of extracts and fractions from marine macro- and microorganisms collected from twilight zone waters around guam. Nat. Prod. Commun. 4, 1717–1728.
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, :R60. doi: 10.1186/gb-2011-12-6-r60
Simister, R., Taylor, M. W., Rogers, K. M., Schupp, P. J., and Deines, P. (2013). Temporal molecular and isotopic analysis of active bacterial communities in two New Zealand sponges. FEMS Microbiol. Ecol. 85, 195–205. doi: 10.1111/1574-6941.12109
Simister, R. L., Antzis, E. W., and White, H. K. (2016). Examining the diversity of microbes in a deep-sea coral community impacted by the Deepwater Horizon oil spill. Deep. Res. Part II Top. Stud. Oceanogr. 129, 157–166. doi: 10.1016/j.dsr2.2015.01.010
Singh, N. S. (2012). in Microbial Degradation of Xenobiotics, ed. S. N. Singh (Berlin: Springer). doi: 10.1016/j.dsr2.2015.01.010
Sipkema, D. (2016). Marine biotechnology: diving deeper for drugs. Microb. Biotechnol. 10, 7–8. doi: 10.1111/1751-7915.12410
Sipkema, D., Holmes, B., Nichols, S. A., and Blanch, H. W. (2009). Biological characterisation of Haliclona (?gellius) sp.: Sponge and associated microorganisms. Microb. Ecol. 58, 903–920. doi: 10.1007/s00248-009-9534-8
Skropeta, D. (2008). Deep-sea natural products. Nat. Prod. Rep. 25, 1131–1166. doi: 10.1039/b808743a
Skropeta, D., and Wei, L. (2014). Recent advances in deep-sea natural products. Nat. Prod. Rep. 31, 999–1025. doi: 10.1039/c3np70118b
Slaby, B. M., Hackl, T., Horn, H., Bayer, K., and Hentschel, U. (2017). Metagenomic binning of a marine sponge microbiome reveals unity in defense but metabolic specialization. ISME J. 11, 2465–2478. doi: 10.1038/ismej.2017.101
Steinert, G., Rohde, S., Janussen, D., Blaurock, C., and Schupp, P. J. (2017). Host-specific assembly of sponge-associated prokaryotes at high taxonomic ranks. Sci. Rep. 7, :2542. doi: 10.1038/s41598-017-02656-6
Steinert, G., Taylor, M. W., Deines, P., Simister, R. L., de Voogd, N. J., Hoggard, M., et al. (2016). In four shallow and mesophotic tropical reef sponges from Guam the microbial community largely depends on host identity. PeerJ 4, :e1936. doi: 10.7717/peerj.1936
Steinert, G., Taylor, M. W., and Schupp, P. J. (2015). Diversity of actinobacteria associated with the marine ascidian Eudistoma toealensis. Mar. Biotechnol. 17, 377–385. doi: 10.1007/s10126-015-9622-3
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., et al. (2015). Structure and function of the global ocean microbiome. Science 348, :1261359. doi: 10.1126/science.1261359
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/mmbr.00040-06
Taylor, M. W., Tsai, P., Simister, R. L., Deines, P., Botte, E., Ericson, G., et al. (2013). ‘Sponge-specific’ bacteria are widespread (but rare) in diverse marine environments. ISME J. 7, 438–443. doi: 10.1038/ismej.2012.111
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7, :11870. doi: 10.1038/ncomms11870
Thomas, T., Rusch, D., DeMaere, M. Z., Yung, P. Y., Lewis, M., Halpern, A., et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4, 1557–1567. doi: 10.1038/ismej.2010.74
Tian, R. M., Wang, Y., Bougouffa, S., Gao, Z. M., Cai, L., Bajic, V., et al. (2014). Genomic analysis reveals versatile heterotrophic capacity of a potentially symbiotic sulfur-oxidizing bacterium in sponge. Environ. Microbiol. 16, 3548–3561. doi: 10.1111/1462-2920.12586
Turk, T., Avguštin, J. A., Batista, U., Strugar, G., Kosmina, R., Čivović, S., et al. (2013). Biological activities of ethanolic extracts from deep-sea antarctic marine sponges. Mar. Drugs 11, 1126–1139. doi: 10.3390/md11041126
Turon, M., Cáliz, J., Garate, L., Casamayor, E. O., and Uriz, M. J. (2018). Showcasing the role of seawater in bacteria recruitment and microbiome stability in sponges. Sci. Rep. 8, 1–10. doi: 10.1038/s41598-018-33545-33541
Uriz, M. J., Agell, G., Blanquer, A., Turon, X., and Casamayor, E. O. (2012). Endosymbiotic calcifying bacteria: a new cue to the origin of calcification in Metazoa? Evolution 66, 2993–2999. doi: 10.1111/j.1558-5646.2012.01676.
van Soest, R. W., Boury-Esnault, N., Hooper, J. N. A., Rützler, K., de Voogd, N. J., Alvarez, B., et al. (2018). World Porifera Database. Available at: http://www.marinespecies.org/porifera (accessed March 15, 2018).
van Soest, R. W. M., Boury-Esnault, N., Vacelet, J., Dohrmann, M., Erpenbeck, D., de Voogd, N. J., et al. (2012). Global diversity of sponges (Porifera). PLoS One 7:e35105. doi: 10.1371/journal.pone.0035105
Viegelmann, C., Margassery, L. M., Kennedy, J., Zhang, T., O’Brien, C., O’Gara, F., et al. (2014). Metabolomic profiling and genomic study of a marine sponge-associated Streptomyces sp. Mar. Drugs 12, 3323–3351. doi: 10.3390/md12063323
Wagner, M., Roger, A. J., Flax, J. L., Brusseau, G. A., and Stahl, D. A. (1998). Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J. Bacteriol. 180, 2975–2982.
Wang, Y., and Qian, P. Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4:e7401. doi: 10.1371/journal.pone.0007401
Webster, N. S., Negri, A. P., Munro, M. M. H. G., and Battershill, C. N. (2004). Diverse microbial communities inhabit Antarctic sponges. Environ. Microbiol. 6, 288–300. doi: 10.1111/j.1462-2920.2004.00570.x
Webster, N. S., Taylor, M. W., Behnam, F., Lücker, S., Rattei, T., Whalan, S., et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082. doi: 10.1111/j.1462-2920.2009.02065.x
Weigel, B. L., and Erwin, P. M. (2017). Effects of reciprocal transplantation on the microbiome and putative nitrogen cycling functions of the intertidal sponge, Hymeniacidon heliophila. Sci. Rep. 7, :43247. doi: 10.1038/srep43247
Wemheuer, B., Wemheuer, F., Meier, D., Billerbeck, S., Giebel, H.-A., Simon, M., et al. (2017). Linking compositional and functional predictions to decipher the biogeochemical significance in dfaa turnover of abundant bacterioplankton lineages in the North Sea. Microorganisms 5, :68. doi: 10.3390/microorganisms5040068
Xin, Y., Kanagasabhapathy, M., Janussen, D., Xue, S., and Zhang, W. (2011). Phylogenetic diversity of gram-positive bacteria cultured from Antarctic deep-sea sponges. Polar Biol. 34, 1501–1512. doi: 10.1007/s00300-011-1009-y
Keywords: 16S rRNA, Antarctic shelf, functional prediction, host-specificity, prokaryotic diversity, sponge microbiota
Citation: Steinert G, Wemheuer B, Janussen D, Erpenbeck D, Daniel R, Simon M, Brinkhoff T and Schupp PJ (2019) Prokaryotic Diversity and Community Patterns in Antarctic Continental Shelf Sponges. Front. Mar. Sci. 6:297. doi: 10.3389/fmars.2019.00297
Received: 08 February 2019; Accepted: 20 May 2019;
Published: 05 June 2019.
Edited by:
Zhiyong Li, Shanghai Jiao Tong University, ChinaReviewed by:
Angelina Lo Giudice, National Research Council (CNR), ItalyMarinella Silva Laport, Federal University of Rio de Janeiro, Brazil
Copyright © 2019 Steinert, Wemheuer, Janussen, Erpenbeck, Daniel, Simon, Brinkhoff and Schupp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georg Steinert, Z2Vvcmcuc3RlaW5lcnRAdW5pLW9sZGVuYnVyZy5kZQ==; Peter J. Schupp, cGV0ZXIuc2NodXBwQHVuaS1vbGRlbmJ1cmcuZGU=
† Present address: Georg Steinert, Marine Symbioses, GEOMAR – Helmholtz Centre for Ocean Research Kiel, Kiel, Germany Bernd Wemheuer, Centre for Marine Bio-Innovation, School of Biological, Earth and Environmental Sciences, University of New South Wales, Sydney, NSW, Australia