 Juline Marta Walter1,2†
Juline Marta Walter1,2† Louisi Souza de Oliveira1,2†
Louisi Souza de Oliveira1,2† Diogo Antonio Tschoeke1,2
Diogo Antonio Tschoeke1,2 Pedro Milet Meirelles3,4Maria Helena Campos Baeta Neves5
Pedro Milet Meirelles3,4Maria Helena Campos Baeta Neves5 Daniela Batista5Ana Polycarpa Carvalho5
Daniela Batista5Ana Polycarpa Carvalho5 Rafaela Dos Santos Costa5
Rafaela Dos Santos Costa5 Sergey Dobretsov6,7
Sergey Dobretsov6,7 Ricardo Coutinho5
Ricardo Coutinho5 Jean Swings1,8
Jean Swings1,8 Cristiane Carneiro Thompson1,2
Cristiane Carneiro Thompson1,2 Fabiano L. Thompson1,2*
Fabiano L. Thompson1,2*- 1Laboratory of Microbiology, Institute of Biology, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 2Center of Technology – CT2, SAGE-COPPE, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 3Institute of Biology, Federal University of Bahia, Salvador, Brazil
- 4Instituto Nacional de Estudos Interdisciplinares e Transdisciplinares em Ecologia e Evolução, Salvador, Brazil
- 5Institute for Marine Studies Admiral Paulo Moreira, Rio de Janeiro, Brazil
- 6Department of Marine Science and Fisheries, College of Agricultural and Marine Sciences, Sultan Qaboos University, Muscat, Oman
- 7Center of Excellence in Marine Biotechnology, Sultan Qaboos University, Muscat, Oman
- 8Department of Biochemistry and Microbiology, Ghent University, Ghent, Belgium
The hypersaline lagoon system of Araruama (HLSA) is one of the largest in the world and one of the most important sources of evaporative salt in Brazil. The biogeochemical characteristics of this lagoon system led it to be considered a Precambrian relic. The HLSA also harbors extensive microbial mats, but the taxonomic and metabolic attributes of these mats are poorly understood. Our high-throughput metagenomics analyses demonstrated that the HLSA microbial mats are dominated by Proteobacteria, Cyanobacteria, and Bacteroidetes. Among Proteobacteria, Deltaproteobacteria comprises approximately 40% of the total population and it includes sulfate-reducing bacteria such as Desulfobacterales, Desulfuromonadales, and Desulfovibrionales. Differing in composition and function of their reaction centers, other phylogenetic diverse anoxygenic phototrophic bacteria were detected in the HLSA microbial mats metagenomes. The presence of photolithoautotrophs, sulfate reducers, sulfide oxidizers, and aerobic heterotrophs suggests the existence of numerous cooperative niches that are coupled and regulated by microbial interactions. We suggest that the HLSA microbial mats hold microorganisms and the necessary machinery (genomic repertoire to sustain metabolic pathways) to promote favorable conditions (i.e., create an alkaline pH microenvironment) for microbially mediated calcium carbonate precipitation process. Metagenome-assembled genomes (Ca. Thiohalocapsa araruaensis HLSAbin6 sp. nov. and Ca. Araruabacter turfae HLSAbin9 gen. nov. sp. nov.) obtained support the relevance of Sulfur metabolism and they are enriched with genes involved in the osmoadaptive networks, hinting at possible strategies to withstand osmotic stress. Metabolically versatile bacteria populations, able to use multiple nutrient sources and osmolytes, seem to be a relevant attribute to survive under such stressful conditions.
Introduction
Microbial mats are one of the oldest known ecosystems on Earth. They support complex consortia of many interdependent species belonging to different functional groups (van Gemerden, 1993; Bolhuis et al., 2014). Fossil records (dated > 3.5 Ga) and modern microbial mats have been investigated extensively from geological, biochemical, and microbiological perspectives (Walter et al., 1980; Vasconcelos et al., 2006; Nutman et al., 2016), raising interesting questions about interactions of microorganisms and its environment. Although modern microbial mats hold taxa that likely arose relatively recently, most metabolic pathways processed by them emerged early in Earth’s history and are likely retained at the community level (Bolhuis et al., 2014; Louca et al., 2018). Physicochemical microgradients and taxonomic stratification (Harris et al., 2013) are thus generated to accommodate the coexistence of a wide range of complementary metabolic strategies such as photosynthesis, chemosynthesis, and heterotrophy (Fullmer et al., 2015). Exopolymeric substances (EPSs) excreted by these complex microbial communities protect them against environmental stressors such as desiccation and excessive light. These substances also represent an important source of Organic Carbon under oligotrophic conditions and can serve as nucleation centers for carbonate precipitation processes (Rossi and De Philippis, 2015; Cangemi et al., 2016). These characteristics allow microbial mats to thrive in a variety of harsh environments around the world including hypersaline ecosystems, where intense evaporation and low levels of freshwater input lead to high salt concentrations in the water.
In aquatic ecosystems, the total concentration of inorganic ions such as NaCl (i.e., salinity) is a key environmental factor affecting the distribution of microbial communities (Lozupone and Knight, 2007; Schapira et al., 2009; Dupont et al., 2014). Hypersaline microbial mats are usually composed of extremophile bacteria and archaea that actively regulate cytoplasmic osmotic pressure, thereby maintaining protein integrity under hyperosmotic stress (Oren, 1994; Das et al., 2015). Under high salinity (>10% NaCl), cells tend to lose water to the environment through osmosis, causing dehydration and ultimately cell death. To survive and maintain cell turgor, acclimation processes are needed, including compatible solute accumulation and the expression of channel proteins and osmosensitive enzymes (Das et al., 2015).
The hypersaline lagoon system of Araruama (HLSA) is the largest complex of coastal hypersaline lagoons and salty ponds in Brazil, and one of the largest and commercially most important hypersaline sources of evaporative salt in the world (Kjerfve et al., 1996; Clementino et al., 2008; Laut et al., 2017). HLSA is a rare biogeochemical system, representing an analog for Precambrian environments (Vasconcelos et al., 2006). An excess of evaporation over precipitation maintains the lagoons hypersaline (approximately 52 g/L–1 salinity), although some annual unbalance might happen (Moreira-Turcq, 2000). High salinity, together with strong daily fluctuations of temperature, light intensity, UV radiation, and desiccation make this shallow system a harsh environment for any organism. These shifts impose important challenges for the ecosystem function, such as how microbial communities adapt to stay active, while maintaining the characteristic structure of vertically stratified groups of microorganisms. Anthropogenic activities also impose pressure on water quality in these lagoons. Understanding how the HLSA microbial mats are characterized is crucial to provide insights of the system and allow to monitor changes of microbial diversity in these unique ecosystems.
The set of hypersaline lagoons of Araruama has been a subject of study for over 30 years, and the examination of microbial communities inhabiting such environment has been performed through classical cultivation methods and 16S rRNA sequencing (Baeta Neves, 1983; Clementino et al., 2008; Ramos et al., 2017). Clementino et al. (2008) detected a high number of novel prokaryotic phylotypes in the HSLA water column, whereas a better understanding of the cyanobacterial composition in the HLSA microbial mats was given by Ramos et al. (2017). However, little is known about the metabolic diversity of the taxa composing the microbial mats of this lagoon system. In addition, a microbial-induced carbonate precipitation model has been described for the microbial mats of Lagoa Vermelha (L. Vermelha) in Araruama (Vasconcelos et al., 2006, 1995); however, it is not entirely clear what species of bacteria are involved in the carbonate formation in the HLSA mats. The main microbial diversity studies of HLSA lack information about the microorganisms involved in the Sulfur cycling, a key metabolism connected to the calcium carbonate precipitation and dissolution in microbial mats. The aim of the present study was to analyze the taxonomic and metabolic potential of the HLSA microbial mats using shotgun metagenomic sequencing, to avoid the taxonomic primer bias of the 16S rRNA sequencing approach (Jovel et al., 2016). We also sought to investigate the metabolic pathways enabling these microbes to thrive in such a unique environment, by shedding light on the genomic repertoire related to osmoadaptation.
Materials and Methods
Study Site and Sample Collection
Microbial mats were sampled in eight salty ponds across the HLSA (16°40′, 19°40′S–39°10′, 37°20′W) (Figures 1A,B). This shallow hypersaline lagoon system covers an area of approximately 300 km2 and is located on the coast 150 km east of Rio de Janeiro where it is subject to a semi-arid climate, an upwelling zone (Kjerfve et al., 1996; Spadafora et al., 2010), and northeast trade winds that promote strong daily fluctuations of temperature, light intensity, and desiccation (Vasconcelos et al., 2006). The low rainfall (annual evaporation rate of 1,390 mm) (Kjerfve et al., 1996) and high evaporation rates in this region result in high salt content in the lagoons (>5.2% total salts) (Kjerfve et al., 1996; Clementino et al., 2008).
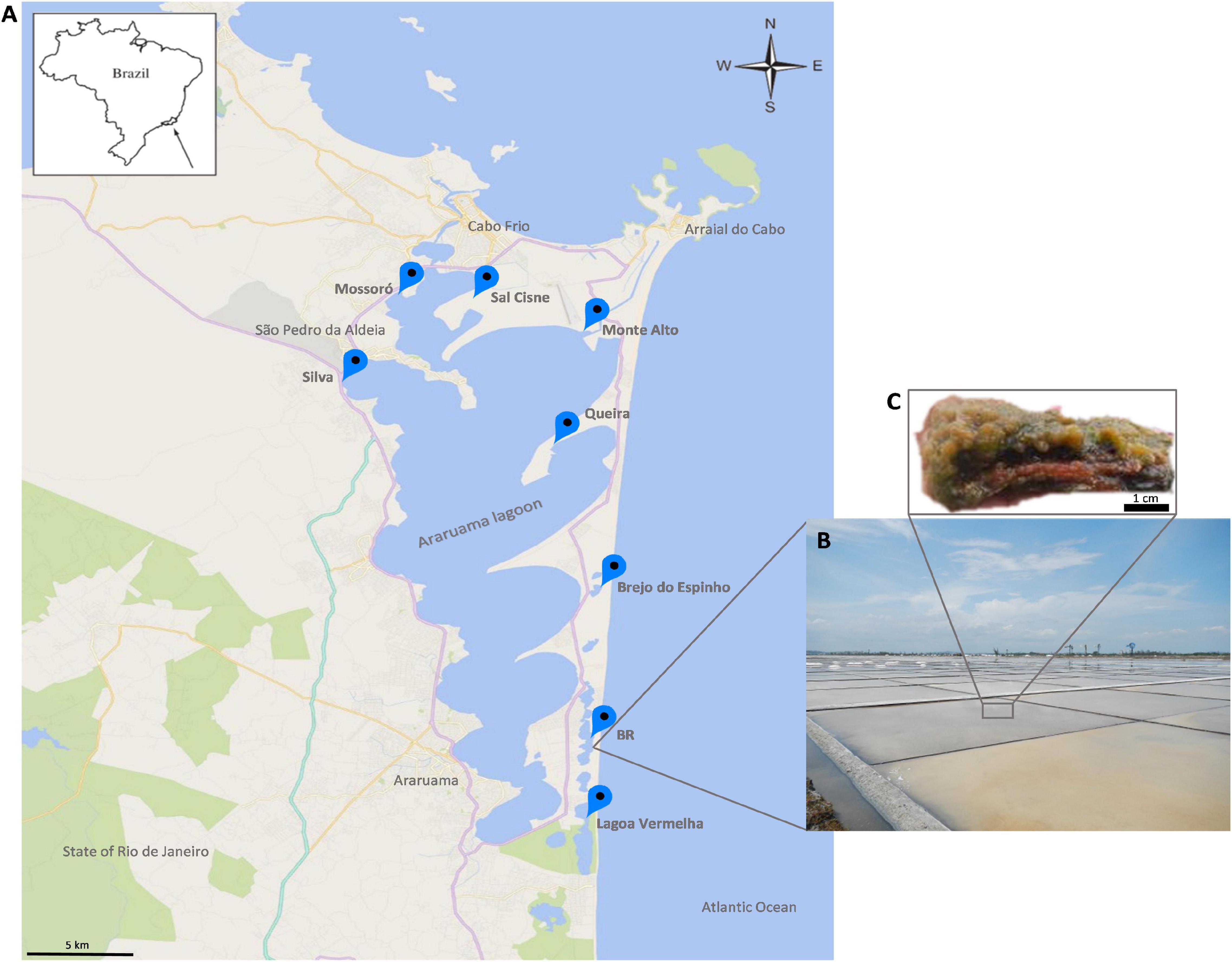
Figure 1. (A) Map of the study area. (B) Overview of the hypersaline lagoons. (C) An excised fraction of the microbial mat ecosystem from the hypersaline lagoon system of Araruama. A stratified structure is observed.
The abundant microbial mats are small and organized in stratified (stacked) layers (Figure 1C). To obtain a broad representation of the microbial taxonomic composition and metabolic potential, mat samples were collected in two seasons: summer (January 2013) at Brejo do Espinho (Br. Espinho), Monte Alto (M. Alto), Mossoró, Queira, Sal Cisne (S. Cisne), and Silva; and winter (June 2013) at BR, Br. Espinho, L. Vermelha, M. Alto, Mossoró, S. Cisne, and Silva (Figure 1A). The microbial mats were taken at different stages of maturity or stratification. Samples were collected with a small shovel and sterile metal spatulas, which were sterilized with ethanol and flame between samples. Approximately 75 g samples were collected, transferred to polypropylene tubes in the field and stored in liquid nitrogen. Samples comprised a mixture of the different layers.
DNA Extraction and Sequencing
The samples were separately ground in liquid nitrogen using ceramic mortars and pestles that were washed with SDS detergent, soaked in 10% bleach for 30 min, and autoclaved between samples. Approximately 200 mg of each sample were used for DNA extraction with the DNeasy PowerSoil Kit (Qiagen, Germantown, MD, United States). DNA integrity was evaluated by 1% agarose gel electrophoresis (GelRedTM, Biotium, Inc., Hayward, CA, United States), and DNA purity was assessed with a NanoDrop spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, United States). The DNA was quantified with a Qubit® 3.0 Fluorometer (Life Technologies-Invitrogen, Carlsbad, CA, United States). Metagenomic libraries were prepared with the Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, United States). Library size distribution was evaluated with a 2100 Bioanalyzer (Agilent, Santa Clara, CA, United States), and library quantification was carried out with a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, United States) and KAPA Library Quantification Kit (Kapa Biosystems, Wilmington, MA, United States). Paired-end sequencing (2 × 300 bp) was performed on a MiSeq System (Illumina).
Bioinformatics and Statistical Analysis
The fastq files generated by Illumina sequencing were qualitatively evaluated with FASTQC v.0.11.2 (Andrews, 2010). The sequences were preprocessed with PRINSEQ v0.20.4 (Schmieder and Edwards, 2011) to remove low-quality DNA sequences (Phred score < 20), duplicates, and short sequences (<35 bp). The resulting sequences were assembled using MIRA software (Chevreux et al., 1999) with default parameters. The assembled sequences (contigs) were then annotated via Metagenome Rapid Annotation using the Subsystem Technology (MG-RAST) (Meyer et al., 2008) with the following cut-off parameters: e-value ≤ 1e–5, 60% minimum sequence identity, and alignment length ≥ 15 bp. Taxonomic annotation was performed using the GenBank database (Benson et al., 2008), the largest (Strasser, 2008; Porter and Hajibabaei, 2018) and reliable (Leray et al., 2019) repository of genetic data for biodiversity; whereas the functional annotation was performed using the SEED Subsystems database (Overbeek et al., 2005), an accurate collection of functionally related protein families.
We compared the HLSA microbial mat metagenomic datasets from the present study (n = 13) with microbial mat metagenomic datasets from the following sources: Diamond Fork, Utah, United States (hot spring, n = 2) (Gomez-Alvarez et al., 2012), Yellowstone National Park, United States (Mushroom Springs, n = 4; Octopus Springs, n = 2) (Bhaya et al., 2007; Bolhuis et al., 2014), Guerrero Negro, Mexico (n = 10) (Kunin et al., 2008; Harris et al., 2013), Shark Bay, Australia (n = 6) (Ruvindy et al., 2016; Wong et al., 2018), Lake Meyghan, Iran (n = 3) (Naghoni et al., 2017), Clinton Creek, Canada (n = 2) (Unpublished, McCormick, M.)1, Schiermonnikoog, Netherlands (tidal and intermediate zone mats, n = 2) (Bolhuis and Stal, 2011), Abrolhos Bank, Brazil (n = 19) (Walter et al., 2016), Neutral Zone, Norway (n = 1) (Stokke et al., 2015), Cuatro Cienegas, Mexico (lithifying and non-lithifying microbialites, n = 2) (Breitbart et al., 2009; Peimbert et al., 2012), and Highbourne Cay, The Bahamas (stromatolites, n = 1) (Khodadad and Foster, 2012). All metagenomes were annotated using the same pipelines and settings, and they are publicly available on the MG-RAST website under the ID provided in Supplementary Table 1.
Statistical analyses were performed with R version 3.0.3 (R Core Team, 2011) with the vegan package (Oksanen et al., 2012). One-Way Analysis of Similarities (ANOSIM) were used to test differences between sampling locations at genera, phyla, and SEED Level 1 levels using Bray–Curtis distances and 999 permutations. Non-metric multidimensional scaling (nMDS) analyses were used to display the sampling locations based on Bray–Curtis dissimilarity matrices. The hierarchical clusters were built using Euclidian distances and Ward’s clustering method. The relative abundance of microbial taxa and the nMDS results were plotted with the ggplot2 (Wickham, 2009) and reshape (Wickham, 2007) packages.
Metagenome-Assembled Genomes
Cross-assembly of reads from all metagenomes was performed by metaSPAdes v.3.6.2 (Nurk et al., 2017), using the default parameters. Protein sequences were predicted from assembled scaffolds with Prodigal (Hyatt et al., 2010). The predicted protein sequences were searched against the NCBI nr database for functional and taxonomic annotation with DIAMOND (Buchfink et al., 2015) setting an e-value cut-off of 10–5. The assembled contigs were binned together using the super-specific configuration of MetaBAT (Kang et al., 2015) to obtain partial or complete microbial genomes. Genome quality was assessed by CheckM (Parks et al., 2015). The cross-assembly of reads between metagenomes approach is a central feature in most automated binning algorithms and often implemented in different studies (Sharon et al., 2013; Parks et al., 2017; Stewart et al., 2018). The cross-assembly of reads among the HLSA metagenomes aimed to increase the chances of full-length recovery of genomes from the metagenomes as long as the taxonomic profiles of the individual metagenomes seem to be similar. These data have been deposited in GenBank, https://www.ncbi.nlm.nih.gov, under the BioProject accession number PRJNA675017: BioSample SAMN16710161 and SAMN16710317.
Phylogenomic Analysis of the Reconstructed MAGs
Average amino acid identity (AAI), average nucleotide identity (ANI), and genome-to-genome distance (GGD) were used for genomic taxonomy (species cutoff of 95% AAI/ANI and 70% GGD) (Konstantinidis and Tiedje, 2005). Phylogenomic trees were generated for the two metagenome-assembled genomes (MAGs). Clustal Omega (Sievers and Higgins, 2014) was used to align the 43 phylogenetic markers used by CheckM (Parks et al., 2015) and that were identified in the bins 6 and 9, and in a set of bacterial genomes publicly available in the RefSeq database (O’Leary et al., 2016). These alignments were concatenated and used as input for phylogenomic reconstruction with FastTree 2.0 using default parameters (Price et al., 2010). One thousand bootstrap replications were calculated to evaluate the relative support of the branches.
Results
Overview of the Metagenomic Sequencing Dataset
We sequenced a total of 13 microbial mat samples (corresponding to 12.69 million reads) from eight different HLSA locations in summer (n = 6) and winter (n = 7) (Table 1). After quality control, the number of metagenome sequences pairs per sample ranged from 153,231 to 1,856,780, and the total number of contigs ranged from 36,860 to 1,140,943 (Table 1).
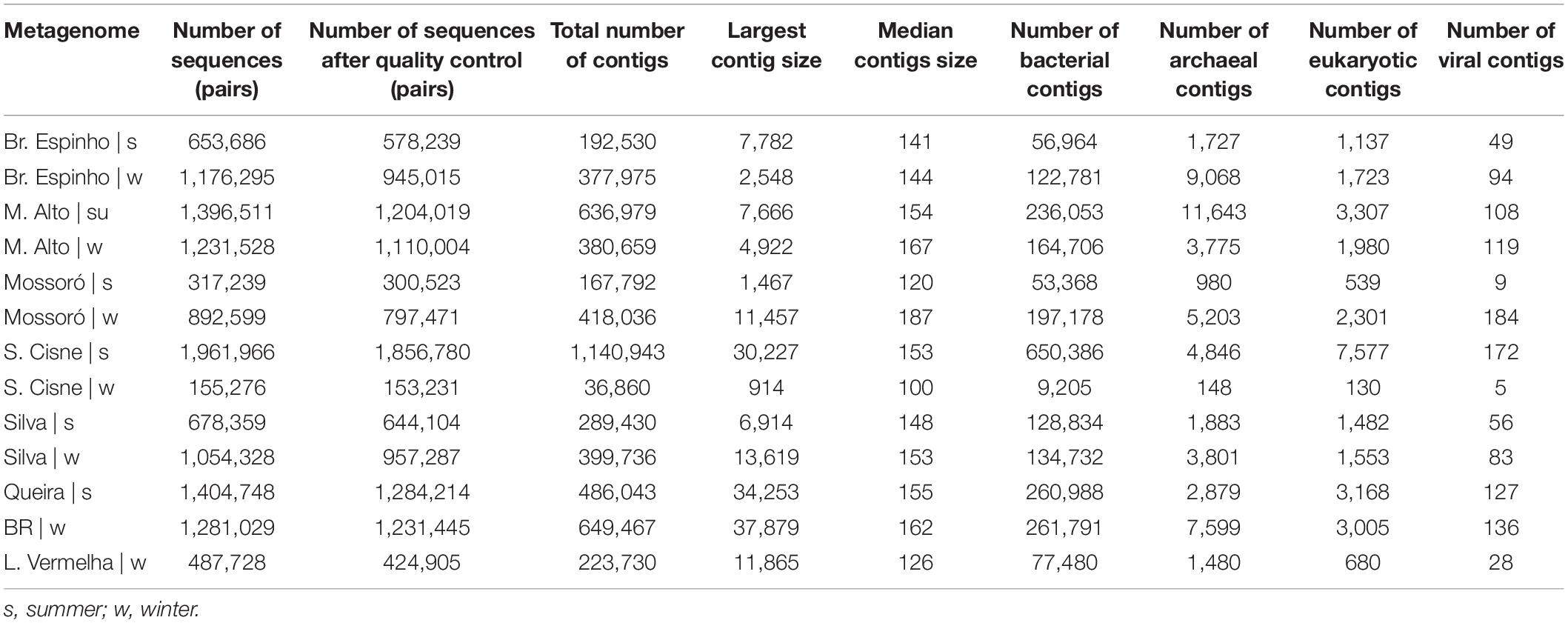
Table 1. Summary statistics of quality filtering and metagenomic assembly for the hypersaline lagoon system of Araruama.
Taxonomic Composition of HLSA Microbial Mats
The HLSA microbial mats sustain a taxonomically diverse assemblage of microorganisms (Figures 2A,B). A total of 32 phyla and 806 different genera were detected belonging to the prokaryotic fraction in the metagenomes of the HLSA microbial mats. The sequences were predominantly bacterial (95.9% on average) with a relatively minor proportion of Archaea (2.2% on average) (Supplementary Figure 1). The most abundant phylum was Proteobacteria (30.9–53.6%), followed by Cyanobacteria (9.7–33.0%), and Bacteroidetes (8.8–22.6%) (Figure 2A).
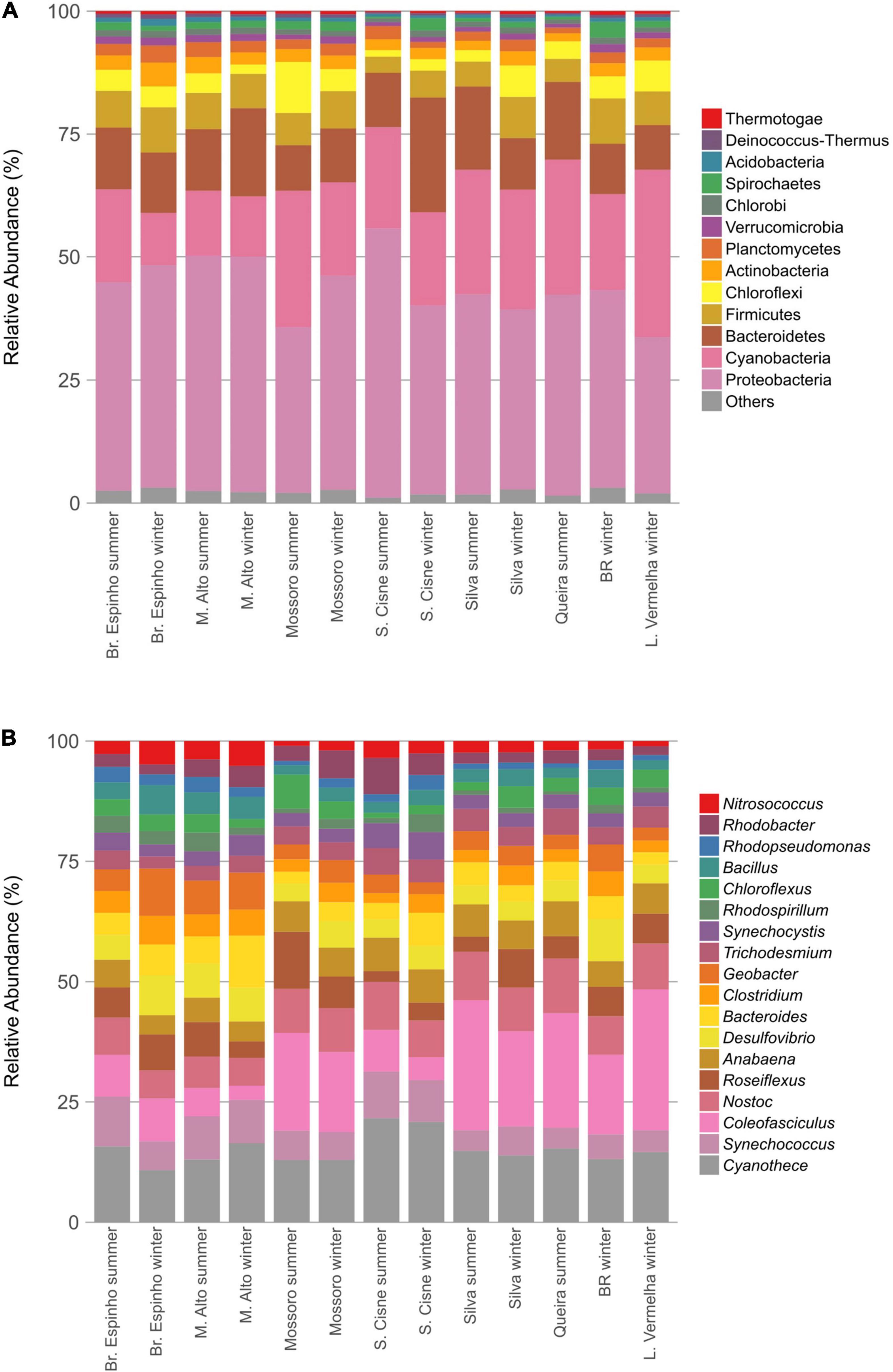
Figure 2. Taxonomic composition of the microbial mats from the hypersaline lagoon system of Araruama. Bar plots depict the relative abundances of bacterial phyla (A) and genera (B) detected in the metagenomes normalized to 100%. Only the most abundant taxa are shown.
Among Proteobacteria, the most abundant groups included the orders Desulfobacterales (1.3–4.6%), Desulfovibrionales (1.5–4.1%), and Desulfuromonadales (1.0–2.8%) and the genera Rhodobacter (0.4–1.8%), Rhodopseudomonas (0.3–0.8%), and Nitrosococcus (0.4–1.0%) (Figure 2B), likely because of the importance of their metabolic roles. Among Cyanobacteria, the difference in the profile abundance was attributed to an increase in reads associated with the orders Chroococcales (4.2–10.4%) and Oscillatoriales (2.1–14.6%). The cyanobacterium genus Coleofasciculus (formerly Microcoleus) was the most abundant genus in most metagenomes (0.6–11.6%), and the species Coleofasciculus chthonoplastes alone represented 23.6% of the total abundance of Cyanobacteria (ranging from 6.4 to 39.9%; n = 95,995). Reads related to Cyanothece sp. PCC 7425 (2.0% in Br. Espinho winter to 5.8% in L. Vermelha winter) were detected in all HLSA metagenomes. Most of the recovered archaeal sequences were assigned to the phylum Euryarchaeota (ranging from 0.7% in S. Cisne summer to 6.4% in Br. Espinho winter), with high relative abundances of the genera Halobacterium (0.1% in S. Cisne summer to 4.1% in Br. Espinho winter) and Methanomicrobia (0.3% in S. Cisne summer to 1.1% in BR winter).
Metabolic Potential of HLSA Microbial Mats
The metagenomic sequences were classified into 28 SEED subsystems (Supplementary Figure 2), a wide range of metabolic pathways which allows microbes to detect changes in the environment conditions to survive. Metabolisms related to Carbohydrates, Protein, and Amino Acids and Derivatives subsystems accounted for 35% of all identified sequences. Cyanobacteria were found to be a key component of this system as the main group responsible for Photosynthesis (50.9–82.4%), and the order Chroococcales alone was the main contributor of genes related to Nitrogen Fixation (6.1–27.3%) and Ammonia Assimilation (5.5–36.4%) metabolisms. Proteobacteria was the main contributor of genes related to Respiration (45.3–64.4%) and Fermentation (32.4–50.0%) subsystems. Genes related to Sulfur metabolism (0.5–0.8%) were attributed mainly to Proteobacteria, whereas genes related to Methanogenesis metabolism were attributed mainly to the bacterial phyla Actinobacteria (0.0–61.54%) and Proteobacteria (7.69–100%), and also to the archaeal orders Methanosarcinales (0.0–30.0%) and Methanopyrales (0.0–18.18%).
When examining the taxonomic profile of the HLSA metagenomes, we see that five out of the six most abundant phyla (Proteobacteria, Cyanobacteria, Bacteroidetes, Firmicutes, and Chloroflexi) (Figure 2A) contain members that are capable of oxygenic and anoxygenic reaction center-based phototrophy. Metabolic versatility of specific taxa present in the HLSA metagenomes is discussed below. Furthermore, genes related to Osmotic Stress were detected in all HLSA microbial mat metagenomes, following: L-ectoine synthesis (0.01–0.05%), L-proline transport, glycine betaine (0.1–0.2%), hyperosmotic potassium uptake (0.02–0.1%), glutathione-regulated potassium-efflux system (0.05–0.2%), and voltage-gated potassium efflux systems (0.01–0.1%).
Taxonomic and Metabolic Profiles Across Microbial Mats Metagenomic Samples
We compared the HLSA microbial mat metagenomes (n = 13) with metagenomes from 11 other microbial communities (n = 55). Notwithstanding the similar nature of the structures used for comparison, general bacterial composition differed by sample origin (ANOSIM: R = 0.706, P = 0.001; Figure 3). Cyanobacteria diversity and abundance accounted for much of the variation between samples. The HLSA metagenomes clustered together, without distinction between seasons or samples (Figures 3, 4). The HLSA metagenomes were more closely related to the hypersaline microbial mat metagenomes from Mexico (Guerrero Negro, in Baja California Sur, and Cuatro Cienegas green mat) (Figure 4 and Supplementary Figure 3).
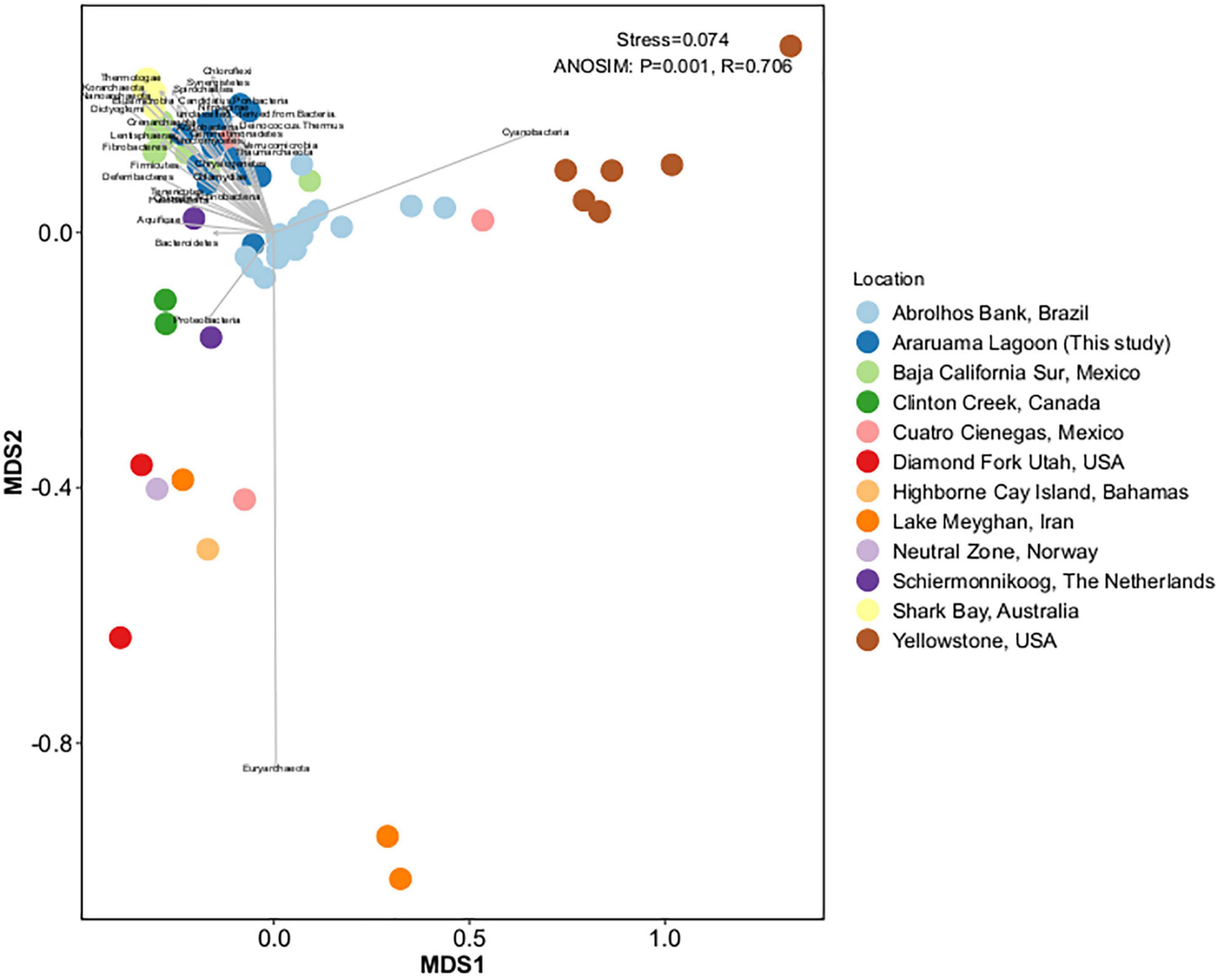
Figure 3. Non-metric multidimensional scaling (nMDS) plot representing the Bray–Curtis similarity of general microbial community structure composition of 68 metagenomes of microbial mats from different habitats. Each dot represents a sample, color represents the sampling location, and the arrows represent the nMDS phyla scores.
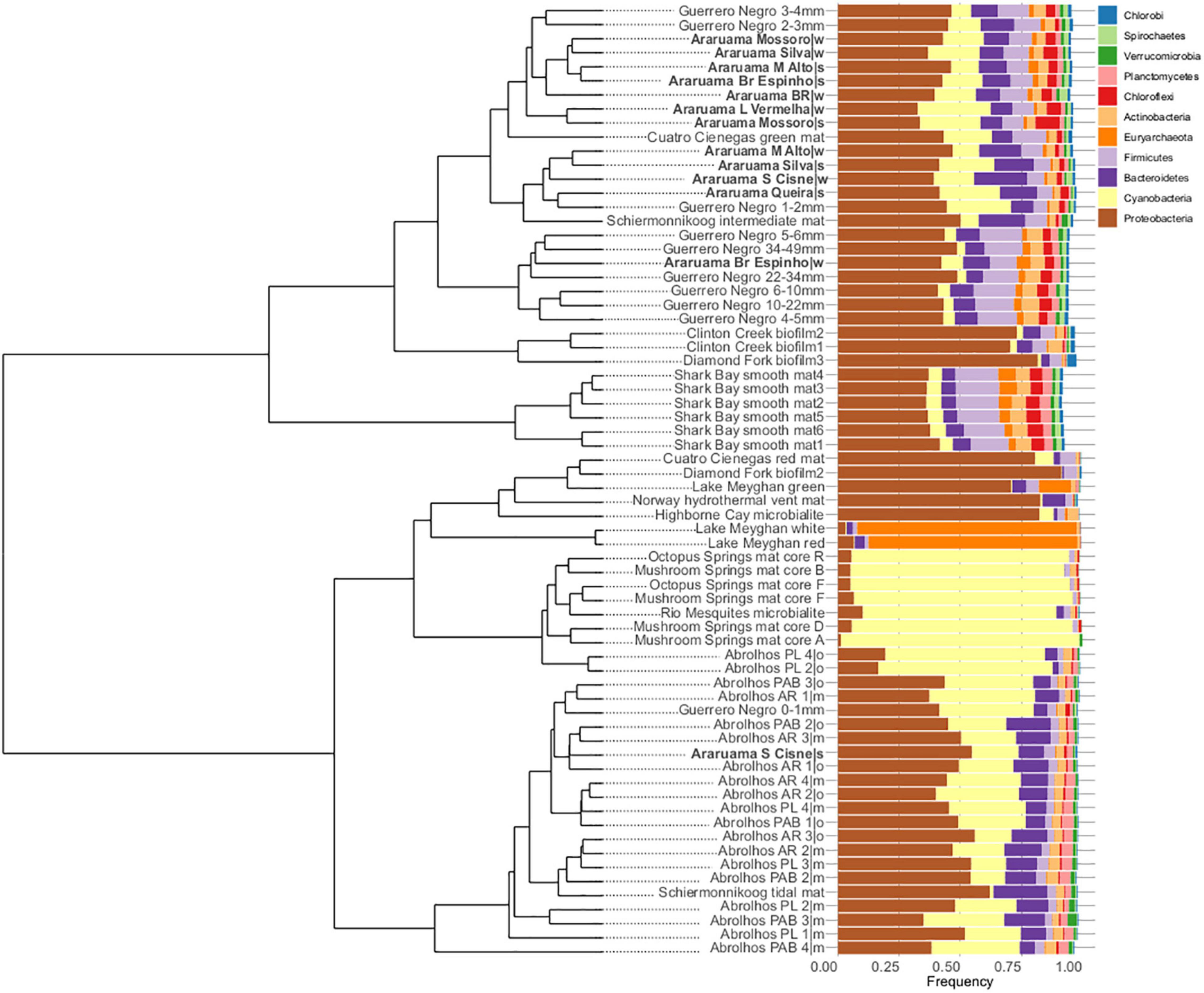
Figure 4. Hierarchical clustering and bar plots of relative abundances of the major microbial phyla found within 68 metagenomes of microbial mats from different habitats. Clustering was based on Euclidian distances and Ward’s clustering method.
Comparative Genomics and Functional Complexity of Recovered MAGs
To provide insights into the genomic context of microorganisms interacting within the HLSA microbial mats, we recovered genomes from the metagenomes and explored their taxonomic and functional diversity. Bacterial genomes with completeness >87% and presenting genome sizes of approximately 4 Mbp were obtained from the HLSA metagenomic dataset (Supplementary Table 2). Because of the relatively low sequencing depths obtained for the HLSA metagenomes, only the most abundant sequences were binned into individual genomes. Here, we highlighted the annotated taxonomic and functional genes of two reconstructed genomes. Following the standards suggested by Bowers et al. (2017), Bin6 is referred as a high-quality draft (>90% complete, <5% contamination), while Bin9 is a medium-quality draft (>50% complete, <10% contamination). A comparison of both reconstructed genomes with their most closely related reference genomes showed that AAI, ANI, and GGD values were much lower than the species cutoff, indicating the novelty of these microorganisms (Table 2). Bin6 represents a new species of Thiohalocapsa that is closely related to Thiohalocapsa sp. ML1 (70.2% AAI), a Gammaproteobacteria belonging to the order Chromatiales (Table 2 and Supplementary Figure 4). Bin9 represents a new genus, and the closest reference genome belongs to a member of Bacteroidetes, Phaeodactylibacter xiamenensis (47.6% AAI) (Table 2 and Supplementary Figure 5). To further identify these two reconstructed genomes, phylogenomic analysis were performed. Whereas Bin6 was placed closely with Thiohalocapsa sp. ML1 (Supplementary Figure 4), the phylogenomic placement of Bin9 shown a relatively distant evolutionary relationship with P. xiamenensis (Supplementary Figure 5). The novel species were named Ca. Thiohalocapsa araruaensis HLSAbin6 sp. nov. (Bin6), and Ca. Araruabacter turfae HLSAbin9 gen. nov. sp. nov. (Bin9).
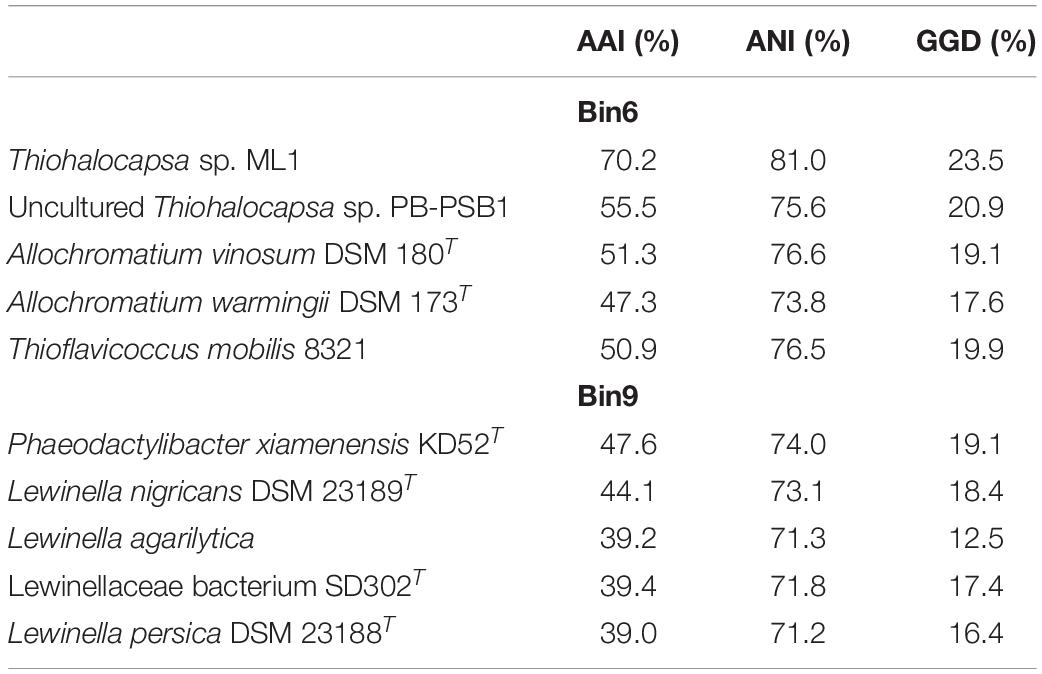
Table 2. Genetic relatedness between the metagenome-assembled genomes and the most closely related reference genomes based on average amino acid identity (AAI), average nucleotide identity (ANI), and genome-to-genome distance (GGD).
The closest relatives of these two recovered genomes are salt tolerant; therefore, we tested for the presence of genes related to osmoregulation. We identified genes encoding glycine betaine/proline transport systems (ABC transport systems, e.g., proV, proW, and proX), high-affinity choline uptake protein (betT), carnitine/choline transporter (opuCB), betaine uptake/biosynthesis systems, genes involved in glucan synthesis, an aquaporin Z, and another outer membrane protein (ompA) (Table 3).

Table 3. Osmoprotectant and osmoregulation profile in the two reconstructed genomes.
Key genes related to Carbon, Nitrogen, and Sulfur biogeochemical cycling were compiled and allowed delineation of the functional role of the taxa associated to the bins (Table 4). For instance, both bins encode complete and partial sulfate-reduction pathways (Table 4), potentially indicating the importance of Sulfur cycling in the HLSA microbial mats. Partial recovery of a determined pathway might be a result of lack of coverage in both not complete MAGs. Annotations for major metabolisms such as Sulfur (dsr, apr genes, and the soxABHWYZ complex), Nitrogen (nif and nar genes), and bacteriochlorophyll-based Photoautotrophy (e.g., bch, psb, puf, rbc, ccm, apcc, coo) are found in Bin6, which is associated with purple sulfur bacteria.

Table 4. Functional genetic diversity of biogeochemical cycling (C, N, S) in the two reconstructed genomes.
Discussion
This study aimed at gaining insights into the diversity of microorganisms in the HLSA microbial mats, expanding the knowledge generated by previous studies that employed operational taxonomic unit-based approach to focus on the cyanobacterial populations of these microbial mats (Ramos et al., 2017), and the prokaryotic diversity of the HLSA water column (Clementino et al., 2008). The use of shotgun metagenomics allowed us to delineate a broader characterization of the microbial mats coping with extreme environmental conditions in the coastal Araruama lagoon system. This approach circumvents the use of culture-dependent methodology as part of the polyphasic strategy employed previously (Clementino et al., 2008; Ramos et al., 2017). Although bacterial isolation technique has yielded valuable biodiversity information in the past, currently it provides little information which limits substantial characterization. For the first time in the HLSA ecosystem, shotgun metagenomics was generated, and near-complete genome bins were retrieved from the metagenomic data.
On the southeastern Brazilian continental margin, structures that dominate the HLSA have been extensively studied with focus on the mineralogical and biogeochemical features (Vasconcelos and McKenzie, 1997; van Lith et al., 2002; Delfino et al., 2012; Bahniuk et al., 2015), giving insights into stromatolite genesis. These laminated structures produced by the successive deposition of layers of microbial mat are found in proximity with their modern counterparts. Comprehensive fossil record found worldwide indicates that ancient microbial mat structures are the oldest biological communities known, dating back to 3.7 billion years for structures found in Greenland (Nutman et al., 2016) and 3.5 billion years for Australian stromatolites (Walter et al., 1980; Allwood et al., 2006).
Evaporation, flooding, and salinity fluctuations processes contribute to the dynamic of the naturally occurring shallow HLSA ecosystem. Modern microbial mats descended from stromatolites and are likely to harbor microorganisms adapted to such stressful conditions. The new sequences generated in the present study substantially increase the representation of all phyla described previously for the HLSA ecosystem (e.g., Proteobacteria and Cyanobacteria) (Clementino et al., 2008; Ramos et al., 2017). The HLSA microbial mats sustain taxonomically diverse assemblage of microorganisms, which exhibit high metabolic diversity.
In contrast to microbial mats in other extreme environments such as hot springs (e.g., Mushroom Springs and Octopus Spring, Yellowstone samples), which are dominated by Cyanobacteria (Bhaya et al., 2007; Bolhuis et al., 2014), the HLSA microbial mats are dominated by Proteobacteria. The prevalence and relative abundances of the three prevailing phyla in the HLSA microbial mats metagenomes were similar to other hypersaline microbial mats from Mexico, such as Guerrero Negro (Kunin et al., 2008; Harris et al., 2013), and Cuatro Cienegas green mat (Breitbart et al., 2009; Peimbert et al., 2012). The dominant proteobacterial groups in the HLSA microbial mats are similar to those in the Mexican microbial mats. Besides, the taxonomic similarity between both locations has been observed previously for the cyanobacterial community (Ramos et al., 2017). Taken together, a taxonomic signature for hypersaline environments may exists. Cyanobacteria were found to be a key component of this system as the main group responsible for photosynthesis and nitrogen fixation. Moreover, the relatively high abundance of the cyanobacterial orders Chroococcales and Oscillatoriales is in agreement with a previous study combining morphology and molecular-based tools to characterize the diversity of Cyanobacteria in the HLSA (Ramos et al., 2017). The abundance of the cyanobacterial genus Coleofasciculus in the HLSA microbial mats may be explained by its tolerance to high saline levels and its metabolic flexibility (i.e., ability to perform both photosynthesis and anoxic fermentation) (Burow et al., 2013). OTUs related to this halophilic Cyanobacteria was reported previously in the HLSA ecosystem (Clementino et al., 2008; Ramos et al., 2017), whereas microbial mats dominated by Coleofasciculus are found in hypersaline ponds of Guerrero Negro, Mexico (Garcia-Pichel et al., 1996; Marais, 2010; Harris et al., 2013). Acting as the primary producer in the mat, this microorganism maintains high numbers of metabolically active heterotrophs which hold catabolic and transport capabilities, for instance.
When examining the taxonomic profile of the HLSA metagenomes, we see that five out of the six most abundant phyla contain members that are capable of oxygenic and anoxygenic reaction center-based phototrophy. High taxa heterogeneity and metabolic versatility occurs in the HLSA mats, particularly considering the diverse taxa of anoxygenic phototrophic bacteria and oxygenic cyanobacterial communities driving the energetic flow. The utilization of different electron donors is well represented by the photoheterotrophic purple non-sulfur Rhodobacter spp., capable of anoxygenic photosynthesis, as well as aerobic and anaerobic respiration (Pérez et al., 2017). Another abundant genus, Rhodopseudomonas, is capable to switch among photoautotrophic, photoheterotrophic, chemoautotrophic, and chemoheterotrophic metabolisms (Larimer et al., 2004). Nitrosococcus, the most abundant genus of Chromatiales found in the HLSA, is a widespread chemolithoautotrophic ammonia-oxidizing bacterium that possesses monovalent cation transporters that confer salt tolerance (Klotz et al., 2006). Notably, Deltaproteobacteria make up to 37.5% of the Proteobacteria population and include sulfate-reducing bacteria such as Desulfobacterales, Desulfuromonadales, and Desulfovibrionales that obtain energy reducing sulfates to sulfides (Wasmund et al., 2017). Also, very abundant (up to 40.5%) and more diverse is Gammaproteobacteria, which contain anoxygenic phototrophic sulfide-oxidizing members that provide the heterotrophic sulfate reducers with some Organic Carbon, hence closing the Sulfur cycle within the HLSA mats. In correspondence to that, several metabolically versatile microorganisms were identified in the HLSA mats, including a high-quality reconstructed genome related to Thiohalocapsa sp. (Bin6), a purple bacterium, which indicates its involvement in the Sulfur metabolism. Bin6 contains annotations for both Sulfur (dsr, apr genes, and the soxABHWYZ complex) and Nitrogen (nif and nar genes) metabolisms. These, together with the annotations for bacteriochlorophyll-based Photoautotrophy (e.g., bch, psb, puf, rbc, ccm, apcc, coo) suggest a dynamic role of Bin6 in the HLSA microbial mats. This purple sulfur bacteria contain a puf operon encoding a type-2 photochemical reaction center (subunits PufL, PufM, and PufH) for aerobic anoxygenic metabolism. Another indication of its anoxygenic metabolism is the presence of BchF, which is exclusively found in those groups of bacteria that can synthetize bacteriochlorophyll a (Bryant et al., 2012). Observations of active Sulfur and Nitrogen metabolisms in other purple sulfur bacteria have been shown elsewhere (Bebout et al., 1993; Yurkov et al., 1994).
Another genome recovered from the metagenomes, Bin9, is related to Bacteroidetes. Members of this phylum act as specialists for the degradation of high molecular weight organic matter and complex polysaccharides (Fernandez-Gomez et al., 2013). They have been detected in high abundance and diversity in several hypersaline microbial mats (e.g., Guerrero Negro, Shark Bay), and the occurrence of specialists have been hypothesized. A strain specialized on the scavenging of Cyanobacteria was found in a hypersaline microbial mat (Hania et al., 2017). Complex cyanobacterial exudates become available to the general microbial community thought those bacteria. Therefore, it is likely that Bacteroidetes play a key role in the degradation and cycling of mat compounds.
Altogether, they indicate the importance of the energy flow (e.g., Carbon and Sulfur) (Canfield and Marais, 1993; Baumgartner et al., 2006) in the HLSA microbial mats. The major role of sulfur-bacteria to calcium mineralization has been demonstrated (Visscher et al., 1998; Braissant et al., 2007; Dupraz et al., 2009; Saghai et al., 2015). Previous Oxygen and Sulfur profiles taken at L. Vermelha demonstrated oxygen peak (oxygen-producing Cyanobacteria) and decrease (oxygen consuming heterotrophs), followed by sulfide consumption (anaerobic sulfide-oxidizing purple bacteria), and sulfide increase (sulfide producing sulfate- and sulfur reducers) (Warthmann et al., 2011). Notably, high sulfate reduction rates coincided with zones of carbonate precipitation in oxygenated zones of another hypersaline microbial mats (Highborne Cay, Bahamas) (Visscher et al., 2000; Dupraz et al., 2004). Different sulfur-reducing bacteria display different tolerances for oxygen exposure and, hence, may present a broad distribution in the microbial mat. Like in the hypersaline mats from Guerrero Negro (Minz et al., 1999), different genera of sulfur-reducing bacteria most likely populate different depths within the HLSA mats, and the oxic zone near the mat surface may present the highest rates of sulfate reduction (Canfield and Marais, 1991). Key genomic repertoire related to calcium carbonate was identified in the HLSA microbial mats metagenomes (Table 5). Microbial mats found across the HLSA seem to present thin and discontinuous calcium carbonate deposition, and two lagoons (L. Vermelha and Br. do Espinho) are well-known for containing Ca–Mg carbonate formations alternating with non-lithified organic layers (Vasconcelos et al., 2006; Nascimento et al., 2019). In addition to the cyanobacterial contribution to the precipitation of calcium carbonate, Cyanothece sp. PCC 7425 and Thermosynechococcus elongatus BP-1 strains are known to accumulate calcium carbonate inclusions in their cytoplasm (Benzerara et al., 2014), and both were abundantly present in the HLSA metagenomes.
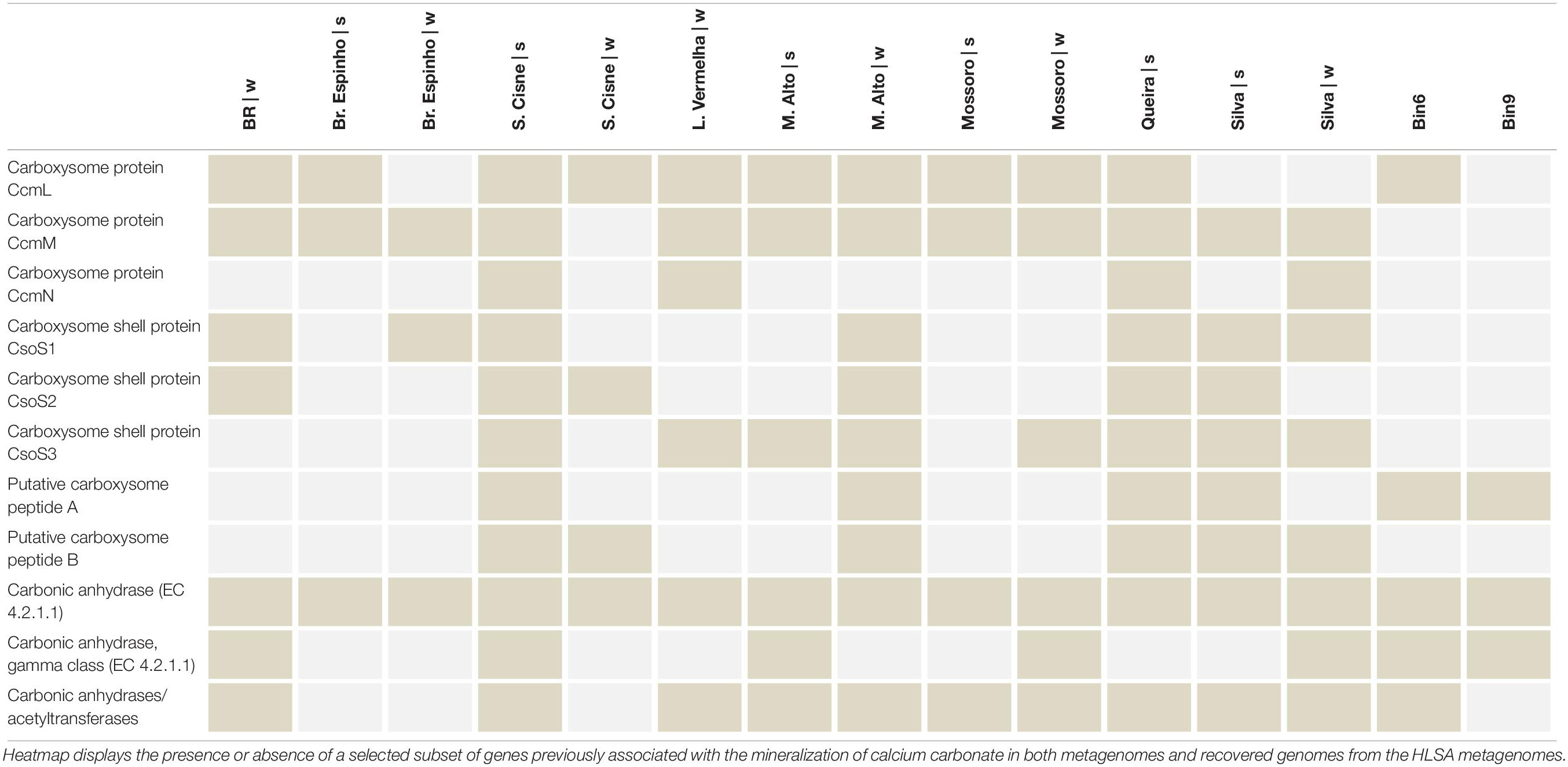
Table 5. Key genes related to microbial-induced calcium carbonate precipitation.
Interestingly, the final net production of carbonates depends on the balance of different microbial metabolisms. Metabolisms such as oxygenic and anoxygenic photosynthesis (Dupraz and Visscher, 2005; Bundeleva et al., 2012), sulfate reduction (Visscher et al., 2000; Gallagher et al., 2014), and anaerobic methane oxidation coupled to sulfate reduction (Michaelis et al., 2002) contribute to a state of carbonate saturation, in an alkaline pH, promoted by a matrix of EPS that leads to calcium ions to precipitate as calcium carbonate (Baumgartner et al., 2006; Zhu and Dittrich, 2016). On the other hand, aerobic respiration, sulfide oxidation, and fermentation (Dupraz and Visscher, 2005) tend to promote dissolution by acidification.
Microorganisms adapted to saline and hypersaline environments display different strategies to cope with high osmotic pressure. These microorganisms may use two main strategies to maintain osmotic balance: (1) accumulate (biosynthesize and/or import) organic compatible solutes (osmoprotectants) that do not interfere with enzymatic activity (e.g., L-ectoine, L-proline, sucrose, trehalose, glucosylglycerol, and glycine betaine) and (2) control ion flow across cellular membranes through regulated potassium uptake and efflux pumps (Martinac et al., 1987). Efflux pumps are not sufficient to cope with high osmolarity (Roberts, 2005), because microorganisms may only transiently accumulate potassium ions. Thus most halotolerant organisms use multiple osmolyte strategies to cope with hypersaline environments (Yaakop et al., 2016). The two recovered genomes contain salt tolerant genomic repertoire. Bin6, associated with purple sulfur bacteria, has genes that encode aquaporin Z water channels that may enhance the flux of water across the cellular membrane in response to abrupt changes in osmotic pressure (Calamita, 2000). This species may also achieve osmotolerance by importing proline, glycine, and betaine through the proU operon (proV, proW, and proX). Whereas Bin9, related to the family Saprospiraceae, also possesses osmoregulation genes encoding proteins involved in the biosynthesis of trehalose and proline and in the uptake and biosynthesis of choline and betaine (Chen et al., 2014). In addition, this species has a gene that encodes the OmpA outer membrane protein, which has multiple functions, including osmoprotection (Hong et al., 2006). Osmoprotectant compounds can be used as Carbon and Nitrogen sources and for energy storage (Welsh, 2000), which may help microorganisms, including the novel candidate species identified in this study, to survive under stressful conditions, such as a sudden temperature increase, desiccation, and UV radiation. Indeed, the new candidate species exemplify different strategies for halotolerance as mentioned before.
Although detected in all HLSA metagenomes, the reconstruction of a particular bin associated with a Cyanobacteria representative (the most closely related reference genome was C. chthonoplastes) did not pass the binning thresholds (Bowers et al., 2017) due to high level of sequences contamination. Despite that, annotation of the cyanobacterial bin could provide some interesting information (results not shown). This bin contains a genetic repertoire for compatible solute metabolism (e.g., trehalose biosynthesis and glycine betaine uptake and biosynthesis). Interestingly, most mat-forming filamentous Cyanobacteria accumulate trehalose, and the combination of EPS with trehalose protects against desiccation (Potts, 1994). Another compound that may be used by Coleofasciculus as an osmoprotectant is carnitine, which can protect against fluctuations in salinity, water content, and temperature (Meadows and Wargo, 2015). Also, we identified sequences related to the permease proteins involved in carnitine transport. Although many bacteria can generate carnitine from direct precursors, these metabolic pathways are not completely understood. Coleofasciculus sp. may also use Na+/H+ antiporters for ion exclusion (e.g., Na+) under hypersaline conditions, as described for other Cyanobacteria (Waditee et al., 2002). Na+ is the main inorganic cation in saline environments and thus, active sodium ion export mechanisms exist in these cells. Clusters of genes encoding the Mrp operon system were also found in all HLSA metagenomes. The Mrp cluster is a monovalent cation/proton antiporter system also involved in Na+ extrusion (Hagemann, 2011). MAGs allow to disentangle the drivers of functional complexity in other microbial mats (Saghai et al., 2015; Wong et al., 2020), where the genomic repertoire of such candidate microbial taxa was investigated. The genomes recovered from the HLSA metagenomes support the environmental relevance of the microorganisms represented by the assemblies described in this study.
Conclusion
Hypersaline lagoon system of Araruama microbial mats have evolved to encompass high taxonomic and metabolic diversity, illustrated by the autotrophic and heterotrophic guilds found in their metagenomes. The similarity between HLSA, Cuatro Cienegas and Guerrero Negro hint to possible adaptative mechanisms to thrive in hypersaline environments. High metabolic flexibility and the production of osmoprotectant compounds appear to be important for survival in the HLSA microbial mats. Halotolerance, phototrophy, and chemosynthesis pathways by bacterial representatives in both the HLSA microbial mats metagenomes and the recovered genomes are indicative of a diverse metabolic repertoire needed to sustain life in the HLSA. A high proportion of sulfur bacteria is remarkable. Deltaproteobacteria, which includes sulfate-reducing bacteria such as Desulfobacterales, Desulfuromonadales, and Desulfovibrionales, comprise approximately 40% of the Proteobacteria population, the most abundant phylum in the HLSA microbial mat metagenomes. This result supports the relevance of sulfate-reducing bacteria in the hypersaline microbial mats of HLSA, where versatile populations in synergy with other taxa cover most of the metabolic activities within the mat, including the precipitation of calcium carbonate in these unique microbial structures.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
SD, RC, FT, CT, MN, DB, and AC conceived the study and designed the experiments. LO and DB processed the samples and performed DNA sequencing. JW, LO, DT, and PM analyzed the data. JW, LO, and FT wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Support offered by the Foundation Carlos Chagas Filho Research Support of the State of Rio de Janeiro (FAPERJ), National Counsel of Technological and Scientific Development (CNPq), and Coordination for the Improvement of Higher Education Personnel (CAPES).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.715335/full#supplementary-material
Footnotes
- ^ McCormick, M. Shotgun Metagenome of Clinton Creek Biofilm, Canada. Clinton, NY: Hamilton College. Available online at: https://www.mg-rast.org/linkin.cgi?project=mgp15973
References
Allwood, A. C., Walter, M. R., Kamber, B. S., Marshall, C. P., and Burch, I. W. (2006). Stromatolite reef from the early Archaean era of Australia. Nature 441, 714–718. doi: 10.1038/nature04764
Andrews, S. (2010). FastQC: a Quality Control Tool for High Throughput Sequence Data. Babraham: Babraham Institute.
Baeta Neves, M. H. C. (1983). Flora ficológica da lagoa hipersalina da Araruama (Estado do Rio de Janeiro-Brasil) 2. Cyanophyceae. Inst. Pesq. Mar. 149, 1–18.
Bahniuk, A., McKenzie, J. A., Perri, E., Bontognali, T. R. R., Vögeli, N., Rezende, C. E., et al. (2015). Characterization of environmental conditions during microbial Mg-carbonate precipitation and early diagenetic dolomite crust formation: Brejo do Espinho, Rio de Janeiro, Brazil. Geol. Soc. Lond. Spec. Publ. 418, 243–259. doi: 10.1144/SP418.11
Baumgartner, L. K., Reid, R. P., Dupraz, C., Decho, A. W., Buckley, D. H., et al. (2006). Sulfate reducing bacteria in microbial mats: changing paradigms, new discoveries. Sedimentary Geol. 185, 131–145. doi: 10.1016/j.sedgeo.2005.12.008
Bebout, B. M., Fitzpatrick, M. W., and Paerl, H. W. (1993). Identification of the sources of energy for nitrogen fixation and physiological characterization of nitrogen-fixing members of a marine microbial mat community. Appl. Environ. Microbiol. 59, 1495–1503. doi: 10.1128/aem.59.5.1495-1503
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Wheeler, D. L. (2008). GenBank. Nucleic Acids Res. 36, D25–D30.
Benzerara, K., Skouri-Panet, F., Li, J., Férard, C., Gugger, M., Laurent, T., et al. (2014). Intracellular Ca-carbonate biomineralization is widespread in cyanobacteria. Proc. Natl. Acad. Sci. U S A. 111, 10933–10938. doi: 10.1073/pnas.1403510111
Bhaya, D., Grossman, A. R., Steunou, A.-S., Khuri, N., Cohan, F. M., Hamamura, N., et al. (2007). Population level functional diversity in a microbial community revealed by comparative genomic and metagenomic analyses. ISME J. 1, 703–713. doi: 10.1038/ismej.2007.46
Bolhuis, H., Cretoiu, M. S., and Stal, L. J. (2014). Molecular ecology of microbial mats. FEMS Microbiol. Ecol. 90, 335–350. doi: 10.1111/1574-6941.12408
Bolhuis, H., and Stal, L. J. (2011). Analysis of bacterial and archaeal diversity in coastal microbial mats using massive parallel 16S rRNA gene tag sequencing. ISME J. 5, 1701–1712. doi: 10.1038/ismej.2011.52
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731. doi: 10.1038/nbt.3893
Braissant, O., Decho, A. W., Dupraz, C., Glunk, C., Przekop, K. M., and Visscher, P. T. (2007). Exopolymeric substances of sulfate-reducing bacteria: interactions with calcium at alkaline pH and implication for formation of carbonate minerals. Geobiology 5, 401–411. doi: 10.1111/j.1472-4669.2007.00117.x
Breitbart, M., Hoare, A., Nitti, A., Siefert, J., Haynes, M., Dinsdale, E., et al. (2009). Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Cienegas. Mexico. Environ. Microbiol. 11, 16–34. doi: 10.1111/j.1462-2920.2008.01725.x
Bryant, D., Liu, Z., Li, T., Zhao, F., Klatt, C. G., Ward, D., et al. (2012). “Comparative and functional genomics of anoxygenic green bacteria from the taxa Chlorobi, Chloroflexi, and Acidobacteria,” in Functional Genomics and Evolution of Photosynthetic Systems. Advances in Photosynthesis and Respiration, eds R. L. Burnap and W. Vermaas (Dordrecht: Springer), 47–102.
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Bundeleva, I. A., Shirokova, L. S., Bénézeth, P., Pokrovsky, O. S., Kompantseva, E. I., and Balor, S. (2012). Calcium carbonate precipitation by anoxygenic phototrophic bacteria. Chem. Geol. 291, 116–131. doi: 10.1016/j.chemgeo.2011.10.003
Burow, L. C., Woebken, D., Marshall, I. P., Lindquist, E. A., Bebout, B. M., Prufert-Bebout, L., et al. (2013). Anoxic carbon flux in photosynthetic microbial mats as revealed by metatranscriptomics. ISME J. 7, 817–829. doi: 10.1038/ismej.2012.150
Calamita, G. (2000). The Escherichia coli aquaporin-Z water channel. Mol. Microbiol. 37, 254–262. doi: 10.1046/j.1365-2958.2000.02016.x
Canfield, D. E., and Marais, D. J. D. (1991). Aerobic sulfate reduction in microbial mats. Science 251, 1471–1473.
Canfield, D. E., and Marais, D. J. D. (1993). Biogeochemical cycles of carbon, sulfur, and free oxygen in a microbial mat. Geochim. Cosmochim. Acta 57, 3971–3984. doi: 10.1016/0016-7037(93)90347-Y
Cangemi, M., Censi, P., Reimer, A., D’Alessandro, W., Hause-Reitner, D., Madonia, P., et al. (2016). Carbonate precipitation in the alkaline lake Specchio di Venere (Pantelleria Island, Italy) and the possible role of microbial mats. Appl. Geochem. 67, 168–176. doi: 10.1016/J.APGEOCHEM.2016.02.012
Chen, Z., Lei, X., Lai, Q., Li, Y., Zhang, B., Zhang, J., et al. (2014). Phaeodactylibacter xiamenensis gen. nov., sp. nov., a member of the family Saprospiraceae isolated from the marine alga Phaeodactylum tricornutum. Int. J. Syst. Evol. Microbiol. 64, 3496–3502. doi: 10.1099/ijs.0.063909-63900
Chevreux, B., Wetter, T., and Suhai, S. (1999). “Genome sequence assembly using trace signals and additional sequence information. in computer science and biology,” in Proceedings of the German Conference on Bioinformatics (GCB). (Accra).
Clementino, M. M., Vieira, R. P., Cardoso, A. M., Nascimento, A. P. A., Silveira, C. B., Riva, T. C., et al. (2008). Prokaryotic diversity in one of the largest hypersaline coastal lagoons in the world. Extremophiles 12, 595–604. doi: 10.1007/s00792-008-0162-x
Das, P., Kumar Behera, B., Kumar Meena, D., Afrin Azmi, S., Chatterjee, S., Meena, K., et al. (2015). Salt stress tolerant genes in halophilic and halotolerant bacteria: paradigm for salt stress adaptation and osmoprotection. Int. J. Curr. Microbiol. App. Sci. 4, 642–658.
Delfino, D. O., Wanderley, M. D., da Silva e Silva, L. H., Feder, F., and Lopes, F. A. S. (2012). Sedimentology and temporal distribution of microbial mats from Brejo do Espinho, Rio de Janeiro, Brazil. Sediment. Geol. 263–264, 85–95. doi: 10.1016/j.sedgeo.2011.08.009
Dupont, C. L., Larsson, J., Yooseph, S., Ininbergs, K., Goll, J., et al. (2014). Functional tradeoffs underpin salinity-driven divergence in microbial community composition. PLoS One 9:e89549. doi: 10.1371/journal.pone.0089549
Dupraz, C., Reid, R. P., Braissant, O., Decho, A. W., Norman, R. S., and Visscher, P. T. (2009). Processes of carbonate precipitation in modern microbial mats. Earth Sci. Rev. 96, 141–162.
Dupraz, C., and Visscher, P. T. (2005). Microbial lithification in marine stromatolites and hypersaline mats. Trends Microbiol. 13, 429–438. doi: 10.1016/j.tim.2005.07.008
Dupraz, C., Visscher, P. T., Baumgartner, L. K., and Reid, R. P. (2004). Microbe–mineral interactions: early carbonate precipitation in a hypersaline lake (Eleuthera Island, Bahamas). Sedimentology 51, 745–765.
Fernandez-Gomez, B., Richter, M., Schuler, M., Pinhassi, J., Acinas, S. G., Gonzalez, J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/ismej.2012.169
Fullmer, M. S., Soucy, S. M., and Gogarten, J. P. (2015). The pan-genome as a shared genomic resource: mutual cheating, cooperation and the black queen hypothesis. Front. Microbiol. 6:728. doi: 10.3389/fmicb.2015.00728
Gallagher, K. L., Dupraz, C., and Visscher, P. T. (2014). Two opposing effects of sulfate reduction on carbonate precipitation in normal marine, hypersaline, and alkaline environments. Geology 42:e313. doi: 10.1130/G34639C.1
Garcia-Pichel, F., Prufert-Bebout, L., and Muyzer, G. (1996). Phenotypic and phylogenetic analyses show Microcoleus chthonoplastes to be a cosmopolitan cyanobacterium. Appl. Environ. Microbiol. 62, 3284–3291.
Gomez-Alvarez, V., Revetta, R. P., and Domingo, J. W. S. (2012). Metagenomic analyses of drinking water receiving different disinfection treatments. Appl. Environ. Microbiol. 78, 6095–6102. doi: 10.1128/AEM.01018-1012
Hagemann, M. (2011). Molecular biology of cyanobacterial salt acclimation. FEMS Microbiol. Rev. 35, 87–123. doi: 10.1111/j.1574-6976.2010.00234.x
Hania, W. B., Joseph, M., Bunk, B., Cathrin, S., Klenk, H.-P., Fardeau, M.-L., et al. (2017). Characterization of the first cultured representative of a bacteroidetes clade specialized on the scavenging of cyanobacteria. Environ. Microbiol. 19, 1134–1148.
Harris, J. K., Caporaso, J. G., Walker, J. J., Spear, J. R., Gold, N. J., Robertson, C. E., et al. (2013). Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. ISME J. 7, 50–60. doi: 10.1038/ismej.2012.79
Hong, H., Szabo, G., and Tamm, L. K. (2006). Electrostatic couplings in OmpA ion-channel gating suggest a mechanism for pore opening. Nat. Chem. Biol. 2, 627–635. doi: 10.1038/nchembio827
Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Jovel, J., Patterson, J., Wang, W., Hotte, N., O’Keefe, S., Mitchel, T., et al. (2016). Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 7:459. doi: 10.3389/fmicb.2016.00459
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Khodadad, C. L., and Foster, J. S. (2012). Metagenomic and metabolic profiling of nonlithifying and lithifying stromatolitic mats of Highborne Cay. The Bahamas. PLoS One 7:e38229. doi: 10.1371/journal.pone.0038229
Kjerfve, B., Schettini, C. A. F., Knoppers, B., Lessa, G., and Ferreira, H. O. (1996). Hydrology and salt balance in a large, hypersaline coastal lagoon: lagoa de araruama, Brazil. Estuar. Coast Shelf. Sci. 42, 701–725. doi: 10.1006/ECSS.1996.0045
Klotz, M. G., Arp, D. J., Chain, P. S. G., El-Sheikh, A. F., Hauser, L. J., Hommes, N. G., et al. (2006). Complete genome sequence of the marine, chemolithoautotrophic, ammonia-oxidizing bacterium Nitrosococcus oceani ATCC 19707. Appl. Environ. Microbiol. 72, 6299–6315. doi: 10.1128/AEM.00463-466
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Kunin, V., Raes, J., Harris, J. K., Spear, J. R., Walker, J. J., Ivanova, N., et al. (2008). Millimeter-scale genetic gradients and community-level molecular convergence in a hypersaline microbial mat. Mol. Syst. Biol. 4:198. doi: 10.1038/msb.2008.35
Larimer, F. W., Chain, P., Hauser, L., Lamerdin, J., Malfatti, S., Do, L., et al. (2004). Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 22, 55–61. doi: 10.1038/nbt923
Laut, L., Martins, M. V. A., Frontalini, F., Ballalai, J. M., Belart, P., Habib, R., et al. (2017). Assessment of the trophic state of a hypersaline-carbonatic environment: vermelha Lagoon (Brazil). PLoS One 12:e0184819. doi: 10.1371/journal.pone.0184819
Leray, M., Knowlton, N., Ho, S.-L., Nguyen, B. N., and Machida, R. J. (2019). GenBank is a reliable resource for 21st century biodiversity research. Proc. Natl. Acad. Sci. U S A. 116, 22651–22656. doi: 10.1073/pnas.1911714116
Louca, S., Polz, M. F., Mazel, F., Albright, M. B. N., Huber, J. A., O’Connor, M. I., et al. (2018). Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2, 936–943.
Lozupone, C. A., and Knight, R. (2007). Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U S A. 104, 11436–11440.
Marais, D. J. D. (2010). “Marine hypersaline microcoleus-dominated cyanobacterial mats in the saltern at guerrero negro, baja california sur, mexico: a system-level perspective,” in Microbial Mats. Cellular Origin, Life in Extreme Habitats and Astrobiology, Vol. 14, eds J. Seckbach and A. Oren (Dordrecht: Springer).
Martinac, B., Buechner, M., Delcour, A. H., Adler, J., and Kung, C. (1987). Pressure-sensitive ion channel in Escherichia coli. Proc. Natl. Acad. Sci. U S A. 84, 2297–2301. doi: 10.1073/PNAS.84.8.2297
Meadows, J. A., and Wargo, M. J. (2015). Carnitine in bacterial physiology and metabolism. Microbiology 161, 1161–1174. doi: 10.1099/mic.0.000080
Meyer, F., Paarmann, D., D’Souza, M., Olson, R., Glass, E., Kubal, M., et al. (2008). The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 9:386. doi: 10.1186/1471-2105-9-386
Michaelis, W., Seifert, R., Nauhaus, K., Treude, T., Thiel, V., Blumenberg, M., et al. (2002). Microbial reefs in the black sea fueled by anaerobic oxidation of methane. Science 297, 1013–1015. doi: 10.1126/science.1072502
Minz, D., Fishbain, S., Green, S. J., Muyzer, G., Cohen, Y., Rittmann, B. E., et al. (1999). Unexpected population distribution in a microbial mat community: sulfate-reducing bacteria localized to the highly oxic chemocline in contrast to a eukaryotic preference for anoxia. Appl. Environ. Microbiol. 65, 4659–4665.
Moreira-Turcq, P. F. (2000). Impact of a low salinity year on the metabolism of a hypersaline coastal lagoon (Brazil). Hydrobiologia 429, 133–140. doi: 10.1023/A:1004037624787
Naghoni, A., Emtiazi, G., Amoozegar, M. A., Cretoiu, M. S., Stal, L. J., Etemadifar, Z., et al. (2017). Microbial diversity in the hypersaline Lake Meyghan. Iran. Sci Rep. 7:11522. doi: 10.1038/s41598-017-11585-11583
Nascimento, G. S., Eglinton, T. I., Haghipour, N., Albuquerque, A. L., Bahniuk, A., McKenzie, J. A., et al. (2019). Oceanographic and sedimentological influences on carbonate geochemistry and mineralogy in hypersaline coastal lagoons, Rio de Janeiro state, Brazil. Limnol. Oceanogr. 64, 2605–2620.
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Nutman, A. P., Bennett, V. C., Friend, C. R., Van Kranendonk, M. J., and Chivas, A. R. (2016). Rapid emergence of life shown by discovery of 3,700-million-year-old microbial structures. Nature 537, 535–538. doi: 10.1038/nature19355
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., O’Hara, R. B., Simpson, G. L., et al. (2012). vegan: Community Ecology Package. R Package version 1: R Package Version 2.0–4.
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Oren, A. (1994). The ecology of the extremely halophilic archaea. FEMS Microbiol. Rev. 13, 415–439. doi: 10.1016/0168-6445(94)90063-9
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H.-Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P. A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-17
Peimbert, M., Alcaraz, L. D., Bonilla-Rosso, G., Olmedo-Alvarez, G., Garcia-Oliva, F., Segovia, L., et al. (2012). Comparative metagenomics of two microbial mats at Cuatro Cienegas basin I: ancient lessons on how to cope with an environment under severe nutrient stress. Astrobiology 12, 648–658. doi: 10.1089/ast.2011.0694
Pérez, V., Hengst, M., Kurte, L., Dorador, C., Jeffrey, W. H., Wattiez, R., et al. (2017). Bacterial survival under extreme UV radiation: a comparative proteomics study of Rhodobacter sp., isolated from high altitude wetlands in chile. Front. Microbiol. 8:1173. doi: 10.3389/fmicb.2017.01173
Porter, T. M., and Hajibabaei, M. (2018). Over 2.5 million COI sequences in GenBank and growing. PLoS One 13:e0200177. doi: 10.1371/journal.pone.0200177
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
R Core Team (2011). R: A Language and Environment for Statistical Computing. Vienna: The R Foundation for Statistical Computing.
Ramos, V. M. C., Castelo-Branco, R., Leão, P. N., Martins, J., Carvalhal-Gomes, S., and Sobrinho (2017). Cyanobacterial diversity in microbial mats from the hypersaline lagoon system of Araruama, Brazil: an in-depth polyphasic study. Front. Microbiol. 8:1233. doi: 10.3389/fmicb.2017.01233
Roberts, M. F. (2005). Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Systems 1:5. doi: 10.1186/1746-1448-1-5
Rossi, F., and De Philippis, R. (2015). Role of cyanobacterial exopolysaccharides in phototrophic biofilms and in complex microbial mats. Life 5, 1218–1238. doi: 10.3390/life5021218
Ruvindy, R., White, R. A. I. I. I., Neilan, B. A., and Burns, B. P. (2016). Unravelling core microbial metabolisms in the hypersaline microbial mats of Shark Bay using high-throughput metagenomics. ISME J. 10, 183–196. doi: 10.1038/ismej.2015.87
Saghai, A., Zivanovic, Y., Zeyen, N., Moreira, D., Benzerara, K., Deschamps, P., et al. (2015). Metagenome-based diversity analyses suggest a significant contribution of non-cyanobacterial lineages to carbonate precipitation in modern microbialites. Front. Microbiol. 6:797. doi: 10.3389/fmicb.2015.00797
Schapira, M., Buscot, M. J., Leterme, S. C., Pollet, T., Chapperon, C., et al. (2009). Distribution of heterotrophic bacteria and virus-like particles along a salinity gradient in a hypersaline coastal lagoon. Aquatic Microbial Ecol. 54, 171–183.
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Sharon, I., Morowitz, M. J., Thomas, B. C., Costello, E. K., Relman, D. A., and Banfield, J. F. (2013). Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res. 23, 111–120. doi: 10.1101/gr.142315.112
Sievers, F., and Higgins, D. G. (2014). Clustal omega. Curr. Protocols Bioinform. 48, 3.13.1–16. doi: 10.1002/0471250953.bi0313s48
Spadafora, A., Perri, E., McKenzie, J. A., and Vasconcelos, C. (2010). Microbial biomineralization processes forming modern Ca:Mg carbonate stromatolites. Sedimentology 57, 27–40. doi: 10.1111/j.1365-3091.2009.01083.x
Stewart, R. D., Auffret, M. D., Warr, A., et al. (2018). Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 9:870. doi: 10.1038/s41467-018-03317-3316
Stokke, R., Dahle, H., Roalkvam, I., Wissuwa, J., Daae, F. L., Tooming-Klunderud, A., et al. (2015). Functional interactions among filamentous Epsilonproteobacteria and Bacteroidetes in a deep-sea hydrothermal vent biofilm. Environ. Microbiol. 17, 4063–4077. doi: 10.1111/1462-2920.12970
Strasser, B. J. (2008). GenBank - natural history in the 21st century? Science 322, 537–538. doi: 10.1126/science.1163399
van Gemerden, H. (1993). Microbial mats: a joint venture. Mar. Geol. 113, 3–25. doi: 10.1016/0025-3227(93)90146-M
van Lith, Y., Vasconcelos, C., Warthmann, R., Martins, J. C. F., and McKenzie, J. A. (2002). Bacterial sulfate reduction and salinity: two controls on dolomite precipitation in Lagoa Vermelha and Brejo do Espinho (Brazil). Hydrobiologia 485, 35–49. doi: 10.1023/A:1021323425591
Vasconcelos, C., and McKenzie, J. A. (1997). Microbial mediation of modern dolomite precipitation and diagenesis under anoxic conditions (Lagoa Vermelha, Rio de Janeiro, Brazil). J. Sediment Res. 67, 378–390.
Vasconcelos, C., McKenzie, J. A., Bernasconi, S., Grujic, D., and Tien, A. J. (1995). Microbial mediation as a possible mechanism for natural dolomite formation at low temperatures. Nature 377, 220–222. doi: 10.1038/377220a0
Vasconcelos, C., Warthmann, R., McKenzie, J. A., Visscher, P. T., Bittermann, A. G., and van Lith, Y. (2006). Lithifying microbial mats in Lagoa Vermelha, Brazil: modern precambrian relics? Sediment Geol. 185, 175–183. doi: 10.1016/J.SEDGEO.2005.12.022
Visscher, P. T., Reid, R. P., and Bebout, B. M. (2000). Microscale observations of sulfate reduction: correlation of microbial activity with lithified micritic laminae in modern marine stromatolites. Geology 28, 919–922.
Visscher, P. T., Reid, R. P., Bebout, B. M., Hoeft, S. E., Macintyre, I. G., and Thompson, J. A. (1998). Formation of lithified micritic laminae in modern marine stromatolites (Bahamas): the role of sulfur cycling. Am. Mineral 83, 1482–1493. doi: 10.2138/am-1998-11-1236
Waditee, R., Hibino, T., Nakamura, T., Incharoensakdi, A., and Takabe, T. (2002). Overexpression of a Na+/H+ antiporter confers salt tolerance on a freshwater cyanobacterium, making it capable of growth in sea water. Proc. Natl. Acad. Sci. U S A. 99, 4109–4114. doi: 10.1073/pnas.052576899
Walter, J. M., Tschoeke, D. A., Meirelles, P. M., de Oliveira, L., Leomil, L., Tenório, M., et al. (2016). Taxonomic and functional metagenomic signature of turfs in the Abrolhos reef system (Brazil). PLoS One 11:e0161168. doi: 10.1371/journal.pone.0161168
Walter, M. R., Buick, R., and Dunlop, S. R. (1980). Stromatolites 3,400–3,500 Myr old from the North Pole area. West. Aust. Nat. (Perth) 284, 443–445. doi: 10.1038/284443a0
Warthmann, R., Vasconcelos, C., Bittermann, A. G., and McKenzie, J. A. (2011). “The role of purple sulphur bacteria in carbonate precipitation of modern and possibly early precambrian stromatolites,” in Advances in Stromatolite Geobiology. Lecture Notes in Earth Sciences, (Berlin: Springer), doi: 10.1007/978-3-642-10415-2_9
Wasmund, K., Mußmann, M., and Loy, A. (2017). The life sulfuric: microbial ecology of sulfur cycling in marine sediments. Environ. Microbiol. Rep. 9, 323–344. doi: 10.1111/1758-2229.12538
Welsh, D. T. (2000). Ecological significance of compatible solute accumulation by micro-organisms: from single cells to global climate. FEMS Microbiol. Rev. 24, 263–290. doi: 10.1111/j.1574-6976.2000.tb00542.x
Wong, H. L., MacLeod, F. I., White, R. A., Visscher, P. T., and Burns, B. P. (2020). Microbial dark matter filling the niche in hypersaline microbial mats. Microbiome 8, 1–14. doi: 10.1186/s40168-020-00910-910
Wong, H. L., White, R. A. I. I. I., Visscher, P. T., Charlesworth, J. C., Vázquez-Campos, X., and Burns, B. P. (2018). Disentangling the drivers of functional complexity at the metagenomic level in Shark Bay microbial mat microbiomes. ISME J. 12, 2619–2639. doi: 10.1038/s41396-018-0208-208
Yaakop, A. S., Chan, K.-G., Ee, R., Lim, Y. L., Lee, S.-K., Manan, F. A., et al. (2016). Characterization of the mechanism of prolonged adaptation to osmotic stress of Jeotgalibacillus malaysiensis via genome and transcriptome sequencing analyses. Sci. Rep. 6:33660. doi: 10.1038/srep33660
Yurkov, V., Stackebrandt, E., Holmes, A., Fuerst, J. A., Hugenholtz, P., Golecki, J., et al. (1994). Phylogenetic positions of novel aerobic, bacteriochlorophyll a-containing bacteria and description of roseococcus Thiosulfatophilus gen. nov., sp. nov., Erythromicrobium ramosum gen. nov., sp. nov., and Erythrobacter litoralis sp. nov. Int. J. Syst. Bacteriol. 44, 427–434. doi: 10.1099/00207713-44-3-427
Keywords: biofilms, microbiome, metagenome, metagenome-assembled genomes, sulfate-reducing bacteria, carbonatogenesis, calcium carbonate, compatible solutes
Citation: Walter JM, de Oliveira LS, Tschoeke DA, Meirelles PM, Neves MHCB, Batista D, Carvalho AP, Dos Santos Costa R, Dobretsov S, Coutinho R, Swings J, Thompson CC and Thompson FL (2021) Metagenomic Insights Into Ecosystem Function in the Microbial Mats of a Large Hypersaline Coastal Lagoon System. Front. Mar. Sci. 8:715335. doi: 10.3389/fmars.2021.715335
Received: 26 May 2021; Accepted: 16 July 2021;
Published: 17 August 2021.
Edited by:
Punyasloke Bhadury, Indian Institute of Science Education and Research Kolkata, IndiaReviewed by:
Brajogopal Samanta, Mount Allison University, CanadaTamara Lazic, University of Bari Aldo Moro, Italy
Copyright © 2021 Walter, de Oliveira, Tschoeke, Meirelles, Neves, Batista, Carvalho, Dos Santos Costa, Dobretsov, Coutinho, Swings, Thompson and Thompson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabiano L. Thompson, ZmFiaWFub3Rob21wc29uMUBnbWFpbC5jb20=
†These authors have contributed equally to this work