 Gowri Krishna Girija
Gowri Krishna Girija Li-Chun Tseng
Li-Chun Tseng Yu-Ling Chen1
Yu-Ling Chen1 Pei-Jie Meng
Pei-Jie Meng Jiang-Shiou Hwang
Jiang-Shiou Hwang Ying-Ning Ho
Ying-Ning Ho- 1Institute of Marine Biology, College of Life Science, National Taiwan Ocean University, Keelung, Taiwan
- 2Graduate Institute of Marine Biology, National Dong Hwa University, Pingtung, Taiwan
- 3Centre of Excellence for the Oceans, National Taiwan Ocean University, Keelung, Taiwan
The Indo-Pacific native azooxanthellate Tubastraea (Scleractinia) has been identified as an invasive marine species with substantial environmental, economic, and social implications worldwide. Despite their exceptional invasive capacity, our understanding of the role of their symbiotic microbiota in host resilience, as well as their response to ambient environmental conditions, remains limited. In this study, we analyzed the symbiotic bacterial communities found in the tissue and mucus of Tubastraea aurea from different habitats along the northeastern coast of Taiwan. These habitats included two extreme sites (a hydrothermal vent [HV] and a copper mining [CM] site) and two normal environments (inlet of a nuclear power plant [NPP] and a habitat adjacent to a conservation zone [CZ]). We employed full-length 16S rRNA sequencing (~1.5 kilobases) to determine coral-associated microbiome responses to local environments. Results showed significant variations in bacterial communities between corals from extreme and normal habitats. Chemoheterotrophic Endozoicomonas bacteria dominated the tissue samples from the HV and CM sites, whereas phototrophic Synechococcus cyanobacteria dominated the NPP and CZ sites. Hydrographic parameters such as pH, salinity, biological oxygen demand, turbidity, and concentration of heavy metals (e.g., Cu and Fe) increased at the HV and CM sites compared with those at the NPP and CZ sites. This difference created more stressful conditions at the HV and CM sites. The microbial assemblages associated with T. aurea exhibited a prevalence of diverse symbiotic bacteria that could potentially contribute to the host’s ability to adapt and survive in challenging ecological conditions. Therefore, these advantageous microorganisms, along with the host’s physiological mechanisms of dispersion, range expansion, and invasiveness, may enhance the resilience and ability of T. aurea to thrive in extreme environments.
1 Introduction
Healthy corals maintain a robust association with a wide range of microorganisms, including the symbiotic dinoflagellate Symbiodinium spp., as well as a diverse array of other protistan, bacterial, archaeal, fungal, and viral associates (Meron et al., 2011; Thompson et al., 2014; Ziegler et al., 2017). A coral host and its associated microbiome form a coral holobiont (Rohwer et al., 2002). Microbial communities on and within this holobiont play crucial roles in the fitness and life cycle of coral hosts. These roles include mediating an effective host metabolism, assisting in the defense against pathogens, and improving host resilience to environmental stressors (Peixoto et al., 2017). Because the associated microbiome is sensitive to environmental disturbances and host physiological conditions (Bourne et al., 2016), the coral holobiont is considered a dynamic system in which the microbiome needs to adjust its community composition and consortial physiology in response to changing environmental conditions. With the presumably mutualistic system of the holobiont, such responses are expected to be favorable to the needs of the host (Ainsworth et al., 2011).
Microbiome studies have focused on reef-building corals (Ziegler et al., 2017; Ziegler et al., 2019); conversely, few studies have explored the microbial communities of azooxanthellate corals although they comprise one-third of the scleractinian diversity (Engelen et al., 2018). As azooxanthellate corals represent the most ancestral extant scleractinians in terms of evolutionary history and phylogeny, a deeper understanding of their microbiome may help provide valuable insights into the development of coral holobiont structure and function (Kitahara et al., 2010; Engelen et al., 2018).
The azooxanthellate coral Tubastraea aurea (Quoy & Gaimard, 1833) grows naturally in various habitats along the northeastern coast of Taiwan. Shallow water hydrothermal vents on Kueishan Island (also known as Turtle Island or Kueishantao) are distinctive because they have the lowest recorded vent water pH worldwide (Chen et al., 2005a; Chen et al., 2005b). They consist of two types, namely, yellow and white vents (Dahms et al., 2018). The main gases released at these vent sites are carbon dioxide and low amounts of hydrogen sulfide (Tang et al., 2013). Yellow-spring fluids have a temperature range of 78°C to 116°C, while white-spring fluids have a temperature range of 30°C to 65°C (Kuo et al., 2001). Despite the challenging extreme physicochemical conditions, T. aurea thrives naturally near white hydrothermal vents.
T. aurea also grows along the northeast coast of Taiwan, specifically in Liang-Dong Bay, known as the Yin-Yang Sea (YYS). This area is affected by Cu pollution from abandoned mines in the catchment area (Chan et al., 2014). Effluents containing dissolved iron and aluminum reacts with estuarine water to form chips of ferric and aluminous hydroxide, which consequently absorbs heavy metals suspended in the clay and carries them to the YYS (Fang et al., 2003). T. aurea has been discovered in the vicinity of nuclear power plant II and the Chao-Jing Bay Resource Protected Area at the northeast coast of Taiwan. Chan et al. (2014) investigated metal accretion in the coral T. coccinea in response to varying levels of metal enrichment from the YYS, the Kueishan Island (KI) hydrothermal vent field, and the nearshore area of the remote Green Island (GI). However, studies have yet to investigate T. aurea from hydrothermal vents in the KI, YYS, the nuclear power plant II inlet, and areas outside the Chao-Jing Bay Resource Protected Area. Additionally, the microbial composition associated with T. aurea has yet to be explored even though the microbiomes associated with other Tubastraea species have been reported in a limited number of studies (Carlos et al., 2013; Engelen et al., 2018).
Several Tubastraea species are invasive in various parts of the world, particularly along the southwest Atlantic coasts (Precht et al., 2014; Creed et al., 2017). They can alter the benthic community structure by competing with native coral species and negatively affecting new coral recruits (Riul et al., 2013; Miranda et al., 2018). Invasive alien species can increase the vulnerability of natural habitats, making them more susceptible to the impacts of climate change (IUCN Brief Issues, 2021). Because of the combined effects of climate change, ocean acidification, marine pollution, and invasive species spread, the recovery of marine ecosystems, such as coral reefs, will be challenging (Hoegh-Guldberg, 2014; Van Oppen et al., 2017). However, these invasive species demonstrate a remarkable ability to survive in harsh environments amid acidity and marine pollution. Therefore, further studies should elucidate how invasive organisms respond to various extreme conditions.
In the present study, we hypothesized that the resilience of the azooxanthellate hard coral T. aurea in extremely acidified and polluted environments could be attributed to a highly flexible bacterial community. To test this hypothesis, we analyzed the structure of the microbiota in T. aurea collected from acidified (HV), polluted (CM), and normal environments (NPP and CZ) by using a third-generation DNA sequencing platform from Oxford Nanopore Technologies (ONT). Oxford nanopore sequencing provides long reads, which allow us to efficiently and cost-effectively cover the entire 16S rRNA gene (V1–V9 regions) with a high throughput; thus, we can rapidly monitor bacterial communities (Carradec et al., 2020; Ho et al., 2021). Utilizing ONT, we examined the variation in the bacterial communities of T. aurea. This pioneering study explored the changes in the microbiome of T. aurea in different environments.
2 Materials and methods
2.1 Sampling sites
Coral samples were collected from four sites along the northeastern coast of Taiwan from September to October 2020 (Supplementary Figure S1; Supplementary Table S1). The selection of the study sites was based on their direct exposure to environmental and anthropogenic stressors. The shallow hydrothermal vent (HV) site is situated on Kueishan Island, approximately 10 km off the northeast coast of Taiwan. Kueishan Island is known for accommodating numerous gaseohydrothermal vents, ranging from depths of 10–300 m, and it is home to the world’s most acidic vents, with pH of as low as 1.52 (Chen et al., 2005a; Chan et al., 2016).
Carbon dioxide, along with hydrogen sulfide, constitutes the majority of the gases released by these vents (92%) (Tang et al., 2013; Chen et al., 2018). The coral samples from the HV site were collected from a depth of 20 m near white vents. The nuclear power plant (NPP) inlet is characterized by minimal or no direct ecological and man-made disturbances. T. aurea colonies were found to be attached to wave breakers in the intertidal region of the NPP inlet. Similarly, the site adjacent to the conservation zone (CZ) is less susceptible to direct natural and anthropogenic stressors; the coral colonies were collected from a rock at a depth of 2–3 m. In contrast to the other sampling sites, the YYS is characterized by a mixture of seawater and polluted water from the copper refinery of the Taiwan Metal Mining Corporation. This sampling site was referred to as the copper mining site (CM). Effluents with dissolved iron and aluminum contain chips of ferric and aluminous hydroxide upon contact with estuarine water, which in turn absorbs heavy metals suspended in clay and carries them to the YYS (Fang et al., 2003). Coral samples were collected from the CM site at a depth of 8–9 m.
2.2 Sample collection
Three T. aurea colonies were collected from each of the HV, CM, and CZ sites by scuba diving. At the NPP site, colonies were collected from concrete wave blockers during the lowest low tide. The size of the colonies had a width of 5–10 cm. In addition to coral colonies, ambient water and sediment samples were collected in triplicate. Water samples were collected in 1 L sterile glass bottles, and sediment samples were collected in sterile 50 mL falcon tubes in triplicate. The coral colonies and the sediments were immediately preserved in liquid nitrogen upon reaching the surface. The collected water samples were transported to the laboratory in an icebox and filtered using 0.22 µm mixed cellulose ester membrane filter paper (ADVANTEC, Tokyo, Japan). All samples were stored in a refrigerator at −80°C in the laboratory until further processing or DNA extraction.
2.3 Heavy metal analysis
The test samples contained in dry filter papers were digested for 3 h at 120°C in Teflon tubes with 1 mL nitric acid (Merck Suprapur®, 65%, Darmstadt, Germany) and 3 mL hydrochloric acid (Merck Suprapur®, 33%). The resulting digested solutions were diluted with ultrapure water to a final volume of 10 mL and stored at 4°C until analysis. Graphite atomic absorption spectrophotometry (GAAS) was performed to determine the contents of heavy metals (Cd, Zn, Pb, Cu, Ni, Cr, Fe, and Hg) in triplicate (Hitachi Z-8000, Japan; Pai, 1988). Standard reference material samples (NASS-3), including bovine liver (NBSSRM-1577) and orchard leaf (NASS-3), were utilized to assess data quality (NBS-SRM-1571).
2.4 Chemical and physical parameter analysis
Physical and chemical parameters were analyzed in accordance with the procedure described by Meng et al. (2008) and Huang et al. (2012). Nutrients such as ammonia, nitrate, nitrite, phosphate, and silicate were measured using a flow injection analyzer (FIA) and a spectrophotometer (Hitachi Model U-3000, Japan; Pai et al., 2001). Ammonia was determined using an indophenol–blue-based spectrophotometric method. Nitrate was converted to nitrite through cadmium reduction, and nitrite was measured by diazoting with sulfanilamide and coupling with N-(1-naphtha)-ethylenediamine-dihydrochloride. Phosphorus was determined using the molybdate–antimony technique. Silicate was detected using the silicomolybdate method and subsequently reduced by a metal–oxalic acid solution. Ammonia, nitrite, nitrate, phosphate, and silicate had the following precision and minimum detection limits (MDLs): 3.8% and 0.71 mM; 0.9% and 0.03 mM; 5.0% and 0.10 mM; and 4.5% and 0.06 mM, respectively. Duplicate, standard sample spiking, and quality assurance (QA) sample analyses were performed to standardize each nutrient measurement. For example, the duplicate, standard sample spiking, and QA sample phosphate examination were 0.16%, 4.1%, and 4.03%, respectively. The Winkler method, with an average of three readings at each sampling, was applied to measure dissolved oxygen (DO) by using a multiparameter monitoring sensor (YSI model 600XLM, Yellow springs, Ohio, USA). The precision and accuracy of the measurements of biological oxygen demand for 5 days (BOD5) were ±2.38% and 98.3% ± 7.7%, respectively, which were verified using a control chart. pH was measured using a pH meter from Hanna Instruments (Smithfield, Rhode Island, USA). A spectrophotometric approach was applied to assess phytoplankton Chl-a. In this approach, water samples were filtered using a Whatman GF/C glass microfiber filter, and Chl-a was extracted in 90% aqueous acetone at 4°C in the dark for 24 h. The turbidity of the water samples was determined using a nephelometric approach.
2.5 DNA extraction
The tissue samples were thawed at room temperature, and the mucus was collected by gently swabbing a sterile cotton ball on the coral surface. The coral colony was then washed with sterile seawater to remove any remaining mucus and loosely attached bacteria. Subsequently, the tissue was carefully scraped off the skeleton by using a sterile scalpel. The scraped tissue and some skeletal particles were ground to a fine paste with a sterile mortar and pestle. For DNA extraction, 0.25 g of the ground tissue paste was used. The cotton balls containing coral mucus were cut into small pieces with sterile scissors and forceps. From the cut cotton balls, 0.25 g was used for DNA extraction. Similarly, the filter papers (0.22 µm) were cut into pieces for the seawater samples, and 0.25 g of the materials were taken for DNA extraction. DNA was extracted from the tissue, mucus, sediment (0.25 g), and water samples from all sites by using a DNeasy PowerSoil kit (QIAGEN, Hilden, Germany) in accordance with the manufacturer’s protocol.
2.6 Full-length 16S rRNA gene amplification
DNA was extracted from each sample and amplified with the V1–V9 full-length (~1.5 kbp) 16S rRNA gene by using 16S rRNA gene primer sets (Urban et al., 2021). The universal primers 27F and 1492R were utilized for bacterial 16S rRNA amplification. Both primers were combined with a unique barcode to identify different samples (Supplementary Table S2). In the full-length PCR of bacterial 16S rRNA, the following were used: 20.5 µL of sterilized water, 25 µL of PCR master mix (2x Master mix red), 4.0 µL of primer set (2 µL forward and 2 µL reverse) with barcode sequences (10 µM), and 0.5 µL of sample DNA extract in a total volume of 50 µL. The following PCR conditions were used for amplification: initial denaturation at 95°C for 3 min; 30 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 1.5 min; and a final extension at 72°C for 5 min.
2.7 Preparation of the nanopore library and nanopore sequencing
The amplicons were purified and the full-length 16S rRNA gene products were concentrated using an AMPure XP purification kit (Beckman Coulter Life Sciences, Indianapolis, USA). After the amplicons were mixed with the AMPure XP purification kit, the reactions were left at 25°C for 20 min. Subsequently, the samples were washed with 70% fresh ethanol for one round, air-dried at room temperature for 1 min, and eluted in 30 µL of sterilized ddH2O. Each purified sample was quantified using a Qubit 4 fluorometer dsDNA HS (Thermo Fisher Scientific, Waltham, United States). Forty-eight purified samples were pooled at equimolar ratios of approximately 2000 ng. Then, nanopore sequencing libraries were generated in accordance with the SQK-LSK109 protocol (Oxford Nanopore Technologies, Oxford, UK). The library (75 µL) was loaded into R9.4.1 MinION flow cells (Oxford Nanopore Technologies) and run for approximately 96 h. Afterward, the reads were base-called using Guppy (version 3.15) with a high-accuracy model (HAC), and the output DNA sequences with a quality score (Q) of >9 were saved as “.fastq” files for subsequent analyses.
2.8 Read data processing
The package “Porechop” (version 0.2.4, https://github.com/rrwick/porechop) was employed to demultiplex reads and trim adapters under the default parameters. Nanofilt (version 2.8.0, https://github.com/wdecoster/nanofilt) was used to remove any reads shorter than 1.4 kbp and longer than 1.6 kbp (De Coster et al., 2018). NanoStat (version 1.1.2, https://github.com/wdecoster/nanostat; De Coster et al., 2018) was utilized to examine read statistics, including quality scores and read lengths. Additionally, Pistis (https://github.com/mbhall88/pistis) was used to generate quality control plots. The k-mer-based read computation tool Centrifuge (version 1.0.4), which relies on a Burrows–Wheeler transform and the Ferragina–Manzini index, was utilized to obtain the taxonomic assignments of the reads (Kim et al., 2016). For this purpose, the Centrifuge database was constructed using 16s rRNA sequencing data of the type strain from the National Center for Biotechnology Information (NCBI; released on July 2022, https://ftp.ncbi.nlm.nih.gov). With the NCBI reference database, a more precise taxonomic classification than SILVA could be obtained, because the SILVA database lacks species-level information (Matsuo et al., 2021). The custom Centrifuge database has been uploaded to the Mendeley Data, an online repository (DOI: 10.17632/tt5ntb4zyv.1), along with the manufacturing process. The results were visualized using the Pavian package on the R language (Breitwieser and Salzberg, 2020). After the taxonomic assignments of the reads were obtained, the read data were rarefied by removing the following: (1) unclassified reads, (2) reads of cellular organisms, and (3) bacterial groups with reads comprising <0.05% of the total reads obtained. This procedure was accomplished using the Vegan package version 2.6.2. Rarefaction was set to 22,555 reads per sample (Supplementary Table S3) and conducted to mitigate the potential false discovery rate caused by significant disparities in bacterial read counts between samples (Weiss et al., 2017).
2.9 Statistical analysis
Bacterial read data were statistically analyzed using R version 4.2.0 and Past version 4.05. A rarefied taxon rank data table was used for statistical analysis. The Vegan package version 2.6.2 (https://CRAN.R-project.org/package=vegan) was used to calculate the alpha-diversity indices; for visualization, box plots were created using the ggplot2 package (Wickham, 2009). The Bray–Curtis dissimilarity index was calculated and visualized through principal coordinate analysis (PCoA) to assess the difference in bacterial community composition between sites. The significance of the dissimilarity was determined through two-way PERMANOVA with 999 permutations using the Adonis function of the Vegan R package (https://CRAN.R-project.org/package=vegan). PERMDIS was conducted using the Vegan R package to verify whether the observed variations in multivariate datasets are influenced by differences in the spread or variability of data points. ANOVA was performed to assess the significance of PERMDIS analysis. Similarity percentage analysis (SIMPER) based on the Bray–Curtis dissimilarity matrices was carried out in PAST 4.05 to identify the bacterial genera with a higher contribution to the dissimilarity between sites. A Wald chi-square test was conducted to identify the significantly expressed dominant bacterial groups between sites by using the DESeq2 package (Love et al., 2014). The Functional Annotation of the Prokaryotic Taxa (FAPROTAX) database was employed to predict the relevant function of the bacteria (Louca et al., 2016). FAPROTAX can transform taxonomic bacterial community profiles into possible functional profiles by considering the species present in a sample. Spearman’s rank correlation was performed using the microbiomeSeq package (Torondel et al., 2016) to understand the correlation of dominant bacterial taxa with hydrographic parameters and the environmental microbiome. A heatmap was generated using abundance data by ClustVis (Metsalu and Vilo, 2015) to examine the difference in the distribution of Endozoicomonas species between sites. All analyses for testing the significance and correlation were corrected for multiple comparisons by using the Benjamini–Hochberg false discovery rate (FDR) method.
3 Results
3.1 Quality metrics of nanopore sequencing analysis
The bacterial communities in the tissues and mucus of the coral collected from four different sites were subjected to nanopore sequencing to investigate the variation in the bacterial community structure associated with the tissue and mucus of T. aurea in various habitats; ambient seawater and sediment were also analyzed to examine any influence of the ambient environment on changes in the bacterial community composition. A total of 11.2 million reads were obtained from 48 collected samples. Among them, 79.1% (n = 8,889,171) of raw reads were taxonomically assigned to the genus level (Supplementary Figure S2). For statistical analysis, the overall rarefied taxonomy count data were used across different sites and among different sample types (Supplementary Table S3).
3.2 Bacterial diversity of the coral and environmental microbiota
Alpha-diversity was computed using the rarefied taxonomy count data to estimate the diversity of bacterial communities associated with not only the tissue and mucus samples of T. aurea but also the water and sediment samples within each habitat. The bacterial richness (Chao1), diversity (Shannon), and evenness (Pielou) of the sediment were significantly lower at the HV site than at the three other sites (p = 0.023, 0.024, and 0.054, respectively; Tukey’s post hoc test; Figure 1). The richness of the tissue samples at the HV site was also lower than that at the three other sites. The average alpha-diversity of the water samples from the HV site was lower than that from the other sites (Figure 1). However, the alpha-diversity of the mucus and water samples varied across the sites, and no significant differences were observed (Figure 1; Supplementary Table S4).
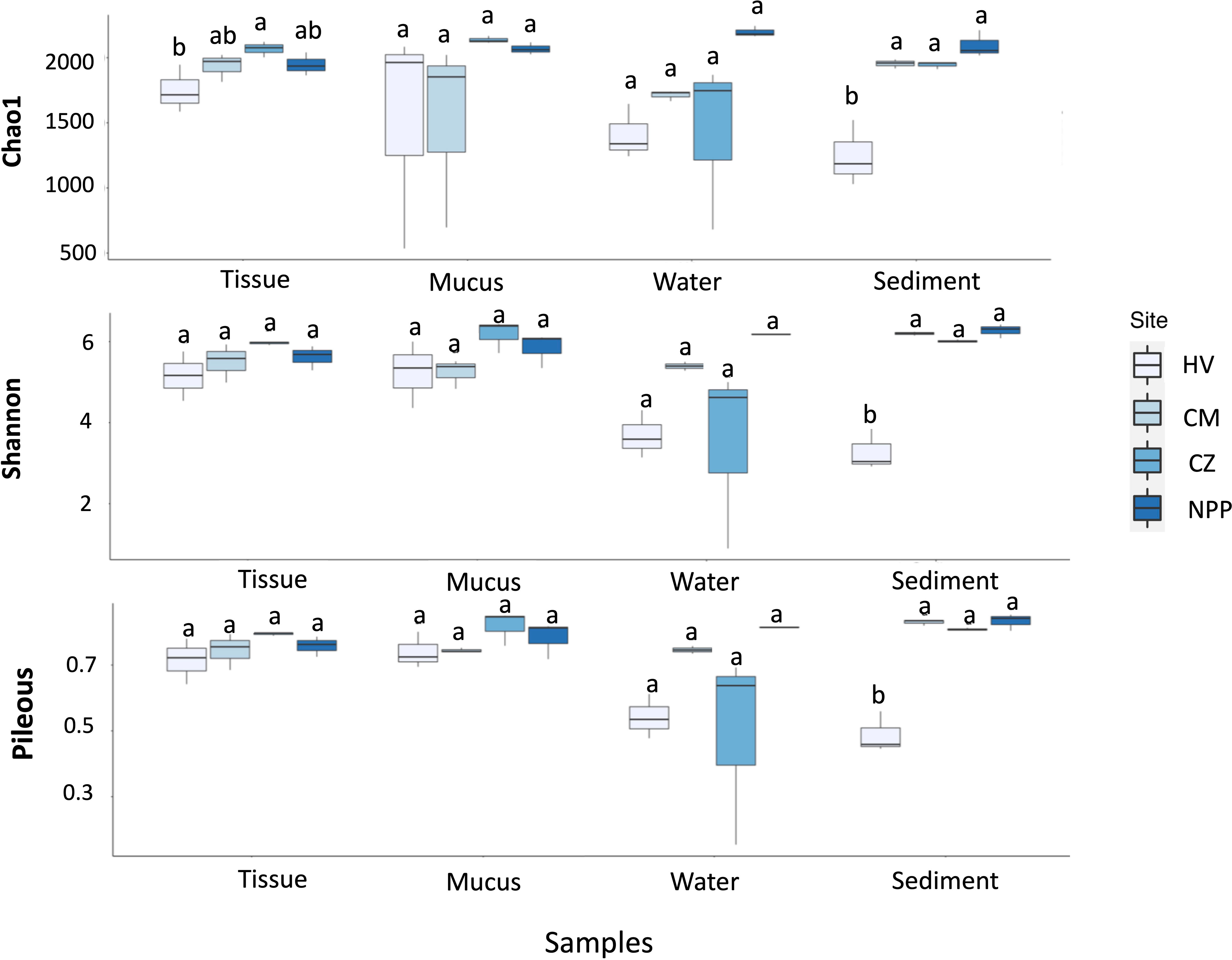
Figure 1 Box plots showing the indices of bacterial diversities (upper), evenness (middle) and richness (bottom) in different sample types collected from different habitats. The significances obtained by the Tukey post hoc test are indicated by different letters. HV, hydrothermal vent site; CM, copper mining site; CZ, conservation zone site; NPP, nuclear power plant inlet.
The variation in bacterial community composition among different samples was assessed using Bray–Curtis dissimilarity with PCoA based on the rarefied reads. The bacterial community structure of all samples, including the separated tissue, mucus, water, and sediment samples from the HV site, differed from that of the samples from the three other sites (Figure 2). PERMANOVA revealed that the sites significantly affected the bacterial community structure of the coral tissue, mucus, and environmental samples (Supplementary Table S5). The tissue and mucus samples respectively accounted for 57.4% and 50.3% of the variance in bacterial communities between sites. Conversely, the sediment sample contributed to 85.6% of the variance in the bacterial community (Figure 2; Supplementary Table S5). The CM site samples showed a significantly high variation of 76.1% in the bacterial community within the site (Supplementary Table S5; Supplementary Figure S3). These results indicated that different environmental sites could shape the distinct microbial communities of the coral tissues and mucus. At the CM site, the bacterial community structure significantly varied among different sample types, accounting for 76.1% of the variance (p < 0.001; Supplementary Table S5). According to the null hypothesis, dispersion did not differ among the groups. Considering that p > 0.05 (Supplementary Table S5), we accepted the null hypothesis, indicating that no significant differences in dispersion were observed among the groups. Therefore, our results confirmed the presence of position effects (observed by PERMANOVA results, Supplementary Table S5) in comparison with dispersion (indicated by PERMDISP results, Supplementary Table S5). This finding further supported the validity of our PERMANOVA analysis. However, no significant differences were found using the PERMDIS (beta-dispersion) test (Supplementary Table S5). Furthermore, no significant differences were observed when the samples were grouped by sites (HV, CM, CZ, and NPP) or sample types (tissue, mucus, water, and sediment). Nevertheless, the trend of the pseudo-F value in PERMDISP resembled that in PERMANOVA (Supplementary Table S5).

Figure 2 Habitat-dependent bacterial community structure based on principle co-ordinate analysis (PCoA) of Bray-Curtis dissimilarities associated with different type samples from different habitats. Symbol shapes represent the sample types and color represents different sampling sites. The PCoA results of all samples include tissue, mucus, water and sediment from different habitats (left panel), and separated sample types from different habitats (right panel). HV, hydrothermal vent site; CM, copper mining site; CZ, conservation zone site; NPP, nuclear power plant inlet.
SIMPER was performed to identify the bacterial genus that contributed the most to the difference in the total bacterial communities between sites in each sample type. Endozoicomonas contributed the most to the Bray–Curtis dissimilarity matrix of the tissue samples from different sites, followed by Synechococcus, Kistomonas, and Sulfurovum (Supplementary Table S6; Supplementary Figure S4). The bacterial phenotype that accounted for the dissimilarity in the bacterial communities of the mucus samples from different sites was Methylobacterium, followed by Sulfurovum and Brucella. Additionally, Thiomicrorhabdus, followed by Lebetimonas, Synechococcus, Marinifilum, and Candidatus Pelagibacter, played a crucial role in driving the differences in bacterial communities in the water samples from different sites. Sulfurovum and Woeseia were also responsible for community differences in sediments among different sampling sites (Figure S4).
3.3 Dominant bacterial communities in the coral and environmental samples
The abundance data showed that the coral tissue and mucus samples from different habitats had similar bacterial genera, while the relative abundance of the dominant genera varied across habitats (Figure 3A). For example, the dominant bacterial genera such as Synechococcus and Ruegeria were found in tissue samples from all sites. However, their relative abundance was comparatively higher in the tissue samples from CZ (5.8%) and NPP (8.3%) sites (Figure 3A) than in the other sites. The tissue samples from the HV and CM sites were dominated by the bacterial group Endozoicomonas (18.72% and 7.5%, respectively). Methylobacterium, Ralstonia, and Brucella were present in the mucus samples from all sites; however, their abundance was higher in the mucus samples from the HV (5%, 5.4%, and 3.3%) and CM (14.2%, 4.9%, and 7.7%) sites (Figure 3A). Similarly, Candidatus Pelagibacter and Synechococcus were observed in water samples from all sites. Although Candidatus Pelagibacter (7.3%) had higher dominance in the water samples from the CM (12.7%) site, Synechococcus was comparatively abundant in the water samples from CZ (13.3%) and NPP (6.5%) sites (Figure 3A). Likewise, Woeseia had higher dominance in the CZ (9%) sediment sample even though this genus was common at all sites (Figure 3A).
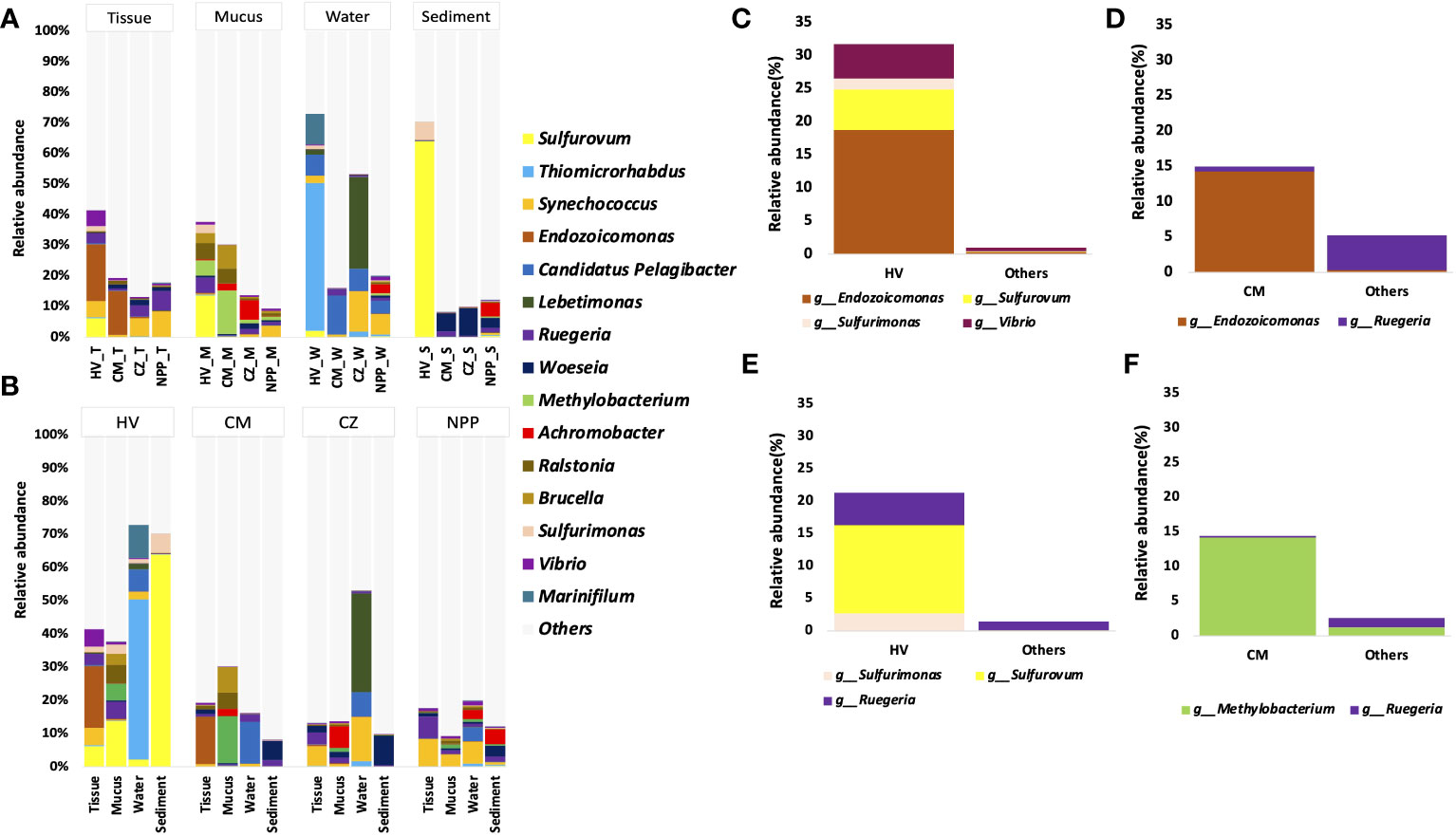
Figure 3 The relative abundance of dominant bacterial genera with T. aurea collected from four different habitats. The plot compares the relative abundance of top 15 bacterial genera in same sample type among different sites (A), in different samples from same collection site (B). The plot shows significantly dominant bacterial genera in HV tissue samples (C) and mucus samples (D) compared to other sites. The plot shows significantly dominant bacterial genera in CM tissue samples (E) and mucus samples (F) compared to other sites. Others sites represent the locations of CZ and NPP. The statistical method of significance based on the Wald chi-square test. HV, hydrothermal vent site; CM, copper mining site; CZ, conservation zone site; NPP, nuclear power plant inlet.
At the HV site, the bacterial community in the coral tissue sample was dominated by Endozoicomonas (18.73%), followed by Sulfurovum (6.12%), Synechococcus (5.19%), Vibrio (5.15%), and Ruegeria (3.38%). In the mucus sample at the HV site, Sulfurovum (13.64%), along with Ralstonia (5.38%), Methylobacterium (5%), Brucella (3.34%), and Sulfurimonas (2.72%; Figure 3B), showed a comparatively higher abundance. Among all bacterial genera obtained from the HV site, the most dominant was Sulfurovum, which was found in significantly higher numbers in the sediment samples (64%). At the metal-ion-enriched CM site, Endozoicomonas was dominant (14.30%) in tissue samples.
To understand the significantly different bacterial groups in terms of relative abundance, we performed the Wald chi-square test by using DEseq2. Our results represented only the bacterial groups that were significantly different among the top 15 genera (Figures 3C–F). In comparison with the two other sites (CZ and NPP), HV site tissue samples were significantly enriched with Sulfurovum, Sulfurimonas, and Vibrio (p < 0.001, Wald chi-square test), and the mucus sample was significantly enriched with Sulfurovum, Sulfurimonas, and Ruegeria (p < 0.05, Wald chi-square test; Figures 3C, D). Endozoicomonas and Methylobacterium were significantly enriched in CM site tissues and mucus samples (p < 0.001, Wald chi-square test; Figures 3E, F).
3.4 Bacterial functional prediction analyses
The metabolic and ecological functions of the bacterial groups in the coral host and the environmental samples were predicted by FAPROTAX. FAPROTAX results suggested that chemoheterotrophs and aerobic chemoheterotrophs were the predominant functional groups observed in all the collected samples except in the HV site sediment samples, which were dominated by nitrate reducers (13.22%), followed by bacteria involved in nitrogen respiration (13.08%) and nitrate respiration (13.03%; Supplementary Figure S5A). Furthermore, we analyzed the difference in the mean proportion of the relative abundance of the hypothesized functional groups from all samples between different sites and compared the bacterial functional groups between the samples from the HV sites without vent sites (CM, CZ, and NPP). We observed that the percentage mean proportion of sulfur-related functional groups (such as dark oxidation of sulfur compounds, dark sulfur oxidation, dark sulfide oxidation and sulfur respiration) were significantly higher in the HV site samples than outside the HV sites (Figure 4). However, the difference in the mean proportion of photoheterotrophy, nitrogen fixation, and predatory or ectoparasitic taxa was significantly higher in the CM site samples (p = 0.017, p < 0.001, and p < 0.001, respectively), and phototrophy was significantly higher in the CZ site samples (p = 0.045) than in the HV site samples (Figure 4B). Phototrophic (p < 0.001), photoautotrophic (p < 0.001), oxygenic photoautotrophic (p < 0.001), and photosynthetic cyanobacteria (p < 0.001) were significantly lower in the HV site samples than in the NPP site samples (Figure 4C).
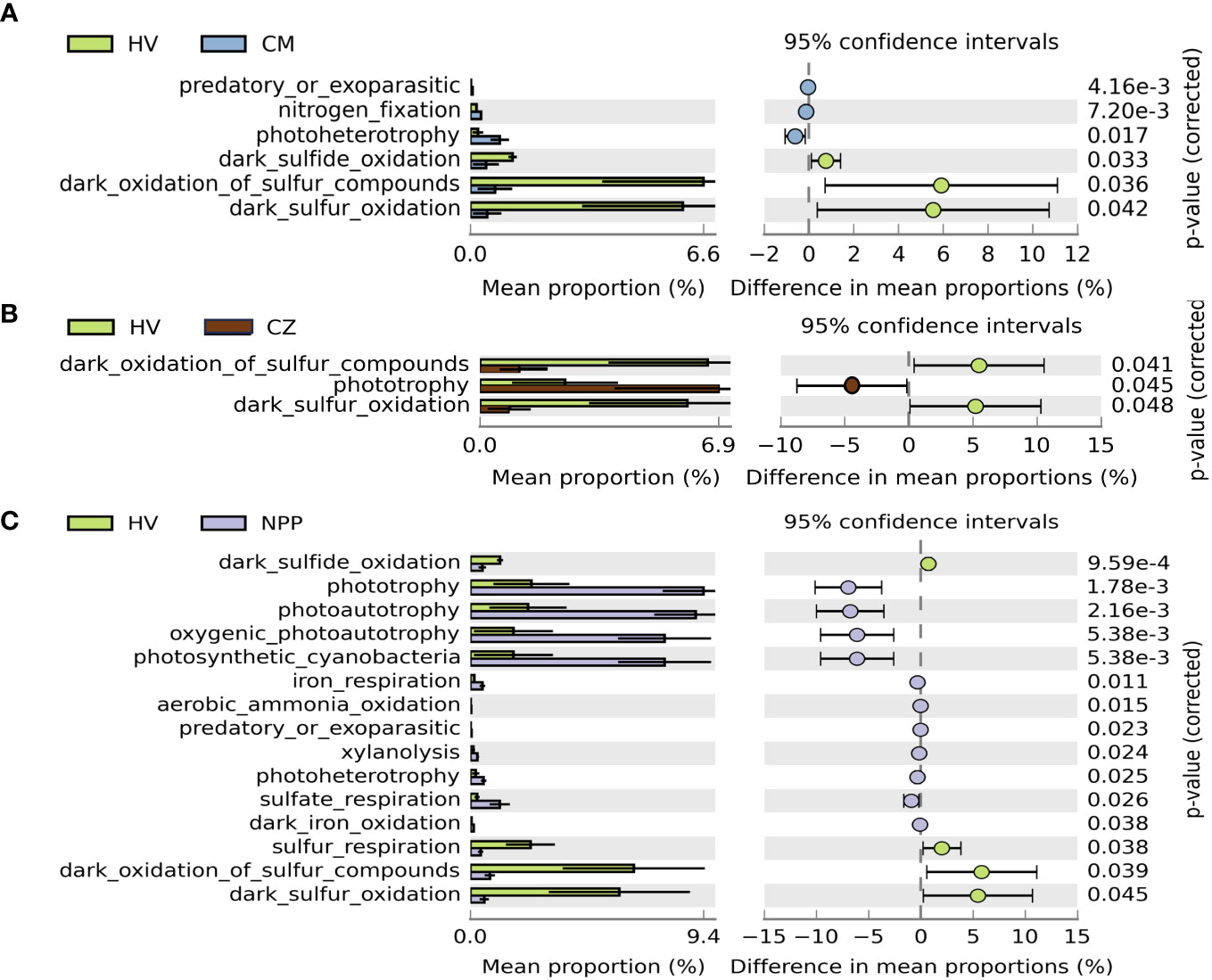
Figure 4 Extended error bar plot demonstrated the mean proportion of bacterial functional groups with significant difference among sampling sites. Difference of functional groups between HV and CM site (A), between HV and CZ site (B), and between HV and NPP site (C). Only functional groups with significant difference (p < 0.05, Welch’s t test) are shown in the figures. Error bars show 95% confidence intervals. HV, hydrothermal vent site; CM, copper mining site; CZ, conservation zone site; NPP, nuclear power plant inlet.
The abundance of the hypothesized functional groups in the tissue samples from the HV site was compared with the mean abundance of the functional groups in the tissue samples from the CZ and NPP sites. Phototrophic, photoautotrophic, oxygenic photoautotrophic, and photosynthetic cyanobacteria significantly decreased in the HV site tissue samples (Figure 5A). Similarly, in the HV site mucus samples, phototrophy, photoautotrophy, and aerobic chemoheterotrophy significantly decreased (Figure 5B). The functional groups in the tissue samples from the CM site showed no significant changes compared with those in the tissue samples from the CZ and NPP sites (Figure 5C). Conversely, sulfur respiration, sulfur compound respiration, phototrophy, photoautotrophy, fermentation, dark thiosulfate oxidation, dark sulfur oxidation, dark sulfur compound oxidation, anoxygenic phototrophic sulfur oxidizers, and anoxygenic phototrophs in the mucus samples from the CM site were significantly reduced compared with those in the mucus samples from the CZ and NPP sites (Figure 5D).

Figure 5 The difference of bacterial functional groups in tissue and mucus samples of coral, Tubastraea aurea. (A) Change values (Log2 fold) determined to show the differential dominant functional groups of tissue sample (A) and mucus sample (B) between the HV site and the other three sites. Change values (Log2 fold) determined to show the differential dominant functional groups from tissue samples (C) and mucus samples (D) between CM site and other three sites. * indicates significantly different functional groups (p < 0.05, T-Test). HV, hydrothermal vent site; CM, copper mining site; CZ, conservation zone site; NPP, nuclear power plant inlet.
3.5 Analysis of hydrographic parameters and heavy metals and their effects on coral microbiome
Seawater quality parameter analysis revealed that the NPP and CZ sites shared similar conditions (Table 1), while the HV and CM sites had different water quality parameters from those in the other sites. The HV site was comparatively more acidic, with the lowest pH (5.74), followed by CM (pH 7.57). CM was characterized by lower salinity (25.31) and higher concentration of heavy metals such as copper (104.05 µgL-1) and iron (183.11 µgL-1). The BOD was greater at the HV site, while turbidity was higher at the CM and HV sites than at the NPP and CZ sites (Table 1). The concentrations of heavy metals such as Cr, Ni, and Cd were extremely low at the four sites, while Zn had a higher concentration at the NPP and CZ sites with 6.65 and 3.80 µgL-1, respectively (Table 2). The concentrations of nutrients, such as NO3, NO2, PO4, and NH3, were relatively similar across all four sites, whereas the concentration of SiO2 was higher at the HV (1.52 mg/L) and CM (1.24 mg/L) sites than at the NPP (0.098 mg/L) and CZ (0.9 mg/L) sites.
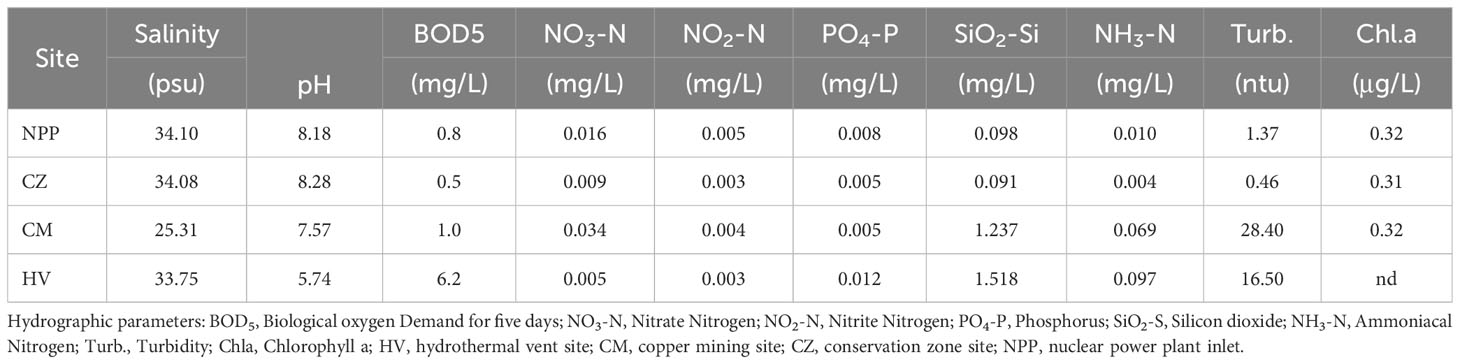
Table 1 Results of hydrographic parameters collected from four different environmental sites.

Table 2 Concentrations of heavy metals in seawater collected from four different environmental sites.
Spearman’s rank correlation analysis was performed to analyze the relation between the hydrographic parameters and bacterial communities of the tissue and mucus samples. Several bacterial genera were positively and negatively correlated with hydrographic parameters (Figure 6A; Supplementary Table S7). In tissue samples, Vallitalea showed the highest positive correlation with salinity (r = 0.96, p < 0.001; Figure 6A). Thermostilla, Roseivirga, and Denitrobaculum showed a higher negative correlation with BOD5, SiO2, and NH3 (r < -0.8, p < 0.001); they also had a higher positive correlation with pH (r > 0.8, p < 0.001; Figure 6A; Supplementary Table S7). Endozoicomonas was significantly and negatively correlated with pH and salinity, but it was significantly and positively correlated with BOD5, SiO2, NH3, and turbidity (Figure 6A) in the tissue samples. For environmental metal concentrations, Endozoicomonas had a highly negative correlation with Zn, Cd and Pb, whereas it was positively correlated with iron (Fe; Figure 6B). These results demonstrated that certain bacterial groups were highly positively correlated with some of the heavy metals. Therefore, these bacterial groups might be considered indicators of those elements. Vallitalea, Thermostilla, Methyloceanibacter, Kryptousia, Hoeflea, Denitrobaculum, Bythopirellula, Aliterella, and Actinomarinicola were significantly and positively correlated with heavy metals, such as Cd, Zn, and Pb (p < 0.05, Figure 6B; Supplementary Table S8).

Figure 6 Heatmap of Spearman’s correlation results between most abundant bacterial genera identified from mucus and tissue samples and hydrographic parameters (A) and heavy metals (B). The scale on the right side indicates the Spearman’s correlation coefficient (1 to -1). The color gradient in red indicates a positive correlation and in blue indicates a negative correlation. Significance levels (*p < 0.05; **p < 0.01; ***p < 0.001).
4 Discussion
A healthy halobiont may require changes in the microbiome makeup in response to changing environmental conditions (Epstein et al., 2019). The capacity of microbiome members to react to environmental changes is a crucial but frequently underappreciated characteristic affecting microbiome reorganization (Tandon et al., 2022). For the first time, we investigated the relative role of environmental heterogeneity in shaping the bacterial community structure associated with the tissue and mucus of the azooxanthellate coral T. aurea, supporting our initial hypothesis. We sequenced the full-length 16S rRNA amplicons obtained from the coral tissue, mucus, and environmental bacteria via Oxford nanopore sequencing (De Siqueira et al., 2021; Urban et al., 2021). The resulting 16S rRNA amplicon data supported our hypothesis that the highly flexible bacterial community associated with T. aurea helped the host adapt to different environmental conditions.
4.1 Environmental parameters play a crucial role in shaping the associated bacterial consortium in T. aurea
We observed that hydrographic parameters such as pH, turbidity, salinity, and BOD5 at the HV and CM sites differed from those at the two other sites. Changing environmental parameters, such as temperature, salinity, turbidity, reduced pH, and nutrient level loads, cause stressful conditions that can alter coral-associated bacterial communities (Thurber et al., 2009). Hydrographic parameters at the NPP inlet and CM sites did not show any extreme variations (Table 1). Hence, they are considered regular coastal habitats. Studies have not yet reported the environmental conditions prevailing at the CZ site. Lee et al. (2018) suggested that the nuclear power plant inlet has an inflow of natural seawater that cools power plant chambers. Since natural seawater with zooplankton continuously flows, the NPP inlet provides better conditions for the growth of T. aurea. This phenomenon could explain the observed variation in the dominance of bacterial communities and functional groups between sites (Figure 7).
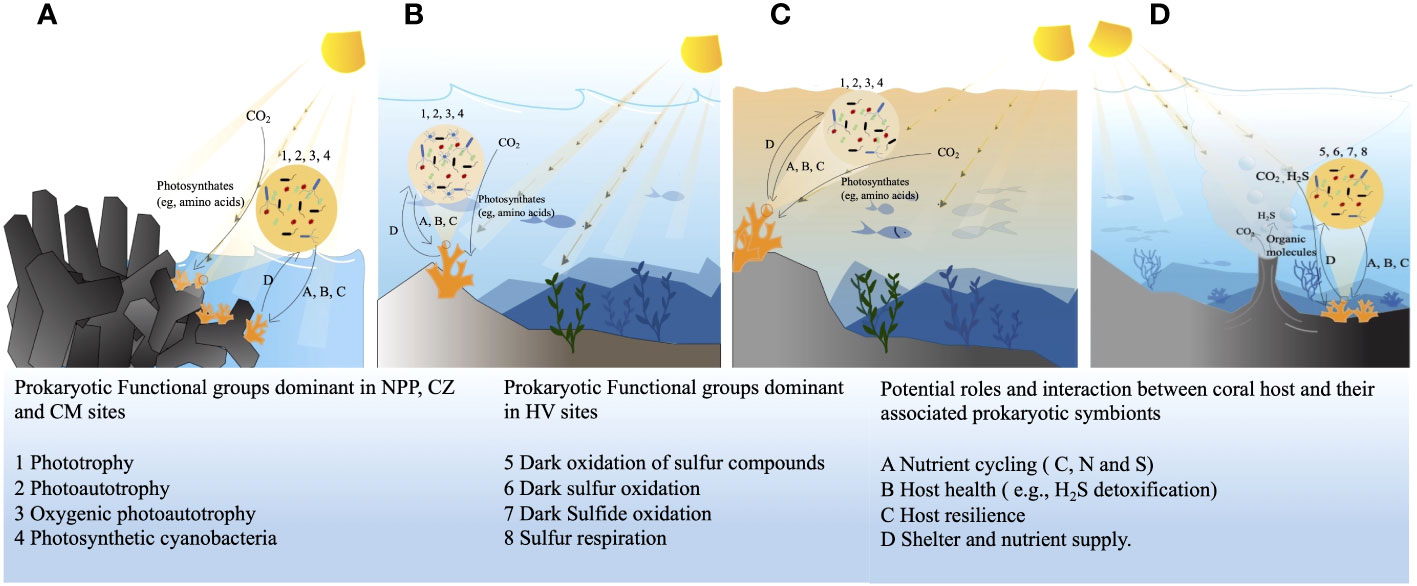
Figure 7 Dominant bacterial functional profiles and their possible relation and interaction with the host coral at four different study sites, in nuclear power plant site (A), in conservation zone site (B), at the copper mining site (C), at the hydrothermal vent site (D). HV, hydrothermal vent site; CM, copper mining site; CZ, conservation zone site; NPP, nuclear power plant inlet.
The relative abundance of the dominant bacterial communities in the tissue, mucus, sediment, and water samples varied among the different sites (Figure 3A). The dominant genera from the hydrothermal vent samples were Sulfurovum and Sulfurimonas of Epsilonproteobacteria; the dominant genus from the water sample was Thiomicrorhabdus of Gammaproteobacteria (Figure 3B). Previous reports showed that bacterial communities from the hydrothermal vent region are dominated by Epsilonproteobacteria and Gammaproteobacteria (Wang et al., 2015; Wang et al., 2017). Sequences related to Sulfurovum have also been found in shallow-water hydrothermal vents off the coast of Milos Island, Greece (Giovannelli et al., 2013). Recent research indicated that Epsilonproteobacteria are chemolithoautotrophs responsible for primary production (Hügler et al., 2005); among them, Sulfurovum can fix CO2 by using elemental sulfur or thiosulfate as the sole electron donor and oxygen or nitrate as the electron acceptor (Inagaki et al., 2004). Similarly, Thiomicrorhabdus can fix CO2 and oxidize sulfide to elemental sulfur (Magnuson et al., 2020). Therefore, the higher abundance of these bacteria in the samples from the HV site could be linked to vent fluids with a surplus of CO2 and H2S.
In the present study, Endozoicomonas was the predominant group in the coral tissue samples from the HV and CM sites (Figure 3A). The species-level classification of the bacterial groups revealed that nine Endozoicomonas species were present in the tissue samples from the HV and CM sites (Supplementary Figure S6). Endozoicomonas is a core microbiont of corals (Ding et al., 2016) since it commonly provides the dominant bacterial taxon in healthy coral colonies (Vezzulli et al., 2013; Meyer et al., 2014). Previous studies confirmed the occurrence of Endozoicomonas in the associated microbiota of T. coccinea (Engelen et al., 2018). Endozoicomonas has less specificity toward the sun coral because they are less tightly associated with the coral host. The present study showed a contrastingly higher abundance of Endozoicomonas in the tissue of T. aurea collected from the HV (18.73%) and CM (14.3%) sites with ambient water pH of 5.74 and 7.57, respectively. This finding was different from the NPP and CZ sites with high pH of 8.18 and 8.28, respectively (Table 1). Similar to our observation, previous findings showed that Endozoicomonas is enriched in two Porites species at low pH (Shore et al., 2021). Both results suggested that Endozoicomonas might help specific coral hosts survive under common pH conditions. Studies have shown that environmental factors can play a crucial role in shaping microbial communities in coral reefs, leading to variations in the composition of these communities (Zaneveld et al., 2016; Ziegler et al., 2019). T. aurea may use coral-mediated tolerance mechanisms, such as gene expression, genetic adaptation, or other metabolic/genetic processes, to withstand stressful conditions and consequently sustain symbiotic relationships with important microbes such as Endozoicomonas (Reigel et al., 2021).
The differential expression of Endozoicomonas species in extreme environments compared with that in more common habitats shows that they are more acclimated to extreme environmental conditions. The presence of Endozoicomonas may be a good indicator of healthy T. aurea in stressful environments, such as HV and CM sites. The different microbial profiles of the relative abundance of Endozoicomonas species and different major Endozoicomonas species varied between the HV and CM sites (Supplementary Figure S6). E. acroporae dominated the HV site tissue samples, and it is considered the dominant coral-associated Endozoicomonas species, which participates in sulfur cycling via DSMP metabolism (Tandon et al., 2020). E. elysicola and E. montiporae, which were dominant in the HV tissue samples, are involved in nitrogen cycling (Neave et al., 2017). E. euniceicola, E. atrinae, and E. numazuensis, which were dominant in the CM site, are closely related to E. elysicola and E. montiporae. E. eunceicola isolated from octocorals, E. numazuensis isolated from purple marine sponges, and E. atrinae isolated from the gut of a comb pen shell grow in seawater with an optimum NaCl concentration of 2%–3%, which could explain the higher dominance of these species in CM site tissue samples. The comparative analysis of the genomes of E. elysicola, E. atrinae, E. montiporae, E. eunceicola, and E. numazuensis revealed a high proportion of the substantial presence of transposable elements within the genomes of Endozoicomonas, suggesting that these elements likely facilitate rapid evolutionary adaptations inhosts or environmental niches (Neave et al., 2016a). The differential expression of Endozoicomonas species between the tissue samples from different sites could be attributed to the coral-mediated tolerance mechanisms and the presence of transposable elements within the bacterial genomes.
The functional role of Endozoicomonas in hosts is unknown (Neave et al., 2016b; Engelen et al., 2018). Various Endozoicomonas strains can provide various functions, such as producing vitamins and co-factors, cycling carbohydrates, and synthesizing amino acids (Neave et al., 2017). In addition, Endozoicomonas contributes to the health of their host by supplying macromolecular nutrients that are otherwise unavailable to the host, altering the associated microbial community through signaling molecules and antimicrobial action, and contributing to nitrogen and or sulfur cycling (Morrow et al., 2015).
Vibrio was found at the four sites, but its abundance was significantly high in the tissue and mucus samples of the HV site (Figure 3A). Several studies have reported an increased abundance of Vibrio in stressed corals (Ben-Haim et al., 2003; Rosenberg and Falkovitz, 2004; Geffen et al., 2009; Rypien et al., 2010; Tait et al., 2010). Meron et al. (2011) observed that the dominance of Vibrio sp. increases when the coral Acropora eurystoma is exposed to reduced pH conditions. This finding suggests that Vibrio can be a natural and common member of the coral-associated microbiome and act as an opportunistic pathogen (Bourne and Munn, 2005). Culture-independent studies have also reported that Vibrio is a vital component of healthy coral tissues (Harris et al., 2001; Cooney et al., 2002; Frias-Lopez et al., 2002; Rohwer et al., 2002). When the balance of the bacterial consortium becomes disrupted under stressed conditions, Vibrio species may become dominant; in some cases, they activate pathogenic traits (Mao-Jones et al., 2010; Littman et al., 2011; Kimes et al., 2012). Therefore, the higher abundance of Vibrio in the tissue samples of the HV site could be attributed to stressors such as reduced pH. However, none of the coral samples collected from these sites showed any symptoms of infectious diseases possibly because some Vibrio strains are permanent members of diazotrophic assemblages found in tissues of some corals (Olson et al., 2009; Lema et al., 2014; Li et al., 2014; Zhang et al., 2016).
The bacterial composition results revealed that the tissue and mucus samples of T. aurea from the NPP and CZ sites and the tissue samples from the HV site had a higher abundance of Synechococcus (Figure 3A). We assumed that such a higher abundance might have been influenced by high seawater salinity, and the lower abundance of this genus at the CM site might be due to low salinity, which is 25 psu (Table 1). As a support to our assumption, Xia et al. (2017) reported that the abundance and diversity of Synechococcus in freshwater-dominated low-saline waters are low. The marine clade of Synechococcus likely grows abundantly in high-saline seawater with low turbidity rather than in low-saline regions with high turbidity (Li et al., 2021). This condition could possibly explain the higher abundance of Synechococcus in the tissue and mucus samples at the NPP and CZ sites and the tissue samples at the HV sites, where salinity is higher and turbidity is lower than those at the CM site (Table 1). Cyanobacteria are responsible for N fixation in the tissues of hermatypic corals (Shashar et al., 1994). Jeong et al. (2012) reported that Synechococcus is an important source of nitrogen for the associated Symbiodinium in zooxanthellate corals. However, studies have yet to describe the putative functional role of Synechococcus in ahermatypic corals. We hypothesized that T. aurea may trap Synechococcus from the surrounding water column (Naumann et al., 2009; Hoadley et al., 2021), which then contributes to nitrogen cycling in the coral.
Ruegeria contributed to more than 2% of the bacterial communities in HV, NPP, and CZ (Figure 3A). It is one of the abundant bacterial genera associated with several coral hosts, including healthy and diseased ones (Ziegler et al., 2016; Keller-Costa et al., 2017; Huggett and Apprill, 2019). This genus has been recognized as robust among most bacterial lineages and found in a wide range of marine environments; it also has various metabolic capabilities and regulatory mechanisms (Luo and Moran, 2014). Ruegeria species show horizontal gene transfer that may help hosts and their associated microbes quickly adapt to changing environmental conditions (McDaniel et al., 2010; McDaniel et al., 2012). These species also require NaCl or sea salt as an important growth factor (Lee et al., 2007). In our study, the analyzed hydrographic parameters indicated that the CM site had the lowest salinity, which was approximately 30% less than the standard seawater salinity. We assumed that salinity could be the driving factor of the reduced abundance of Ruegeria at the CM site.
Coral mucus samples from the CM and HV sites had a higher abundance of Methylobacterium (Figures 3A, F). Its participation in the nitrogen cycle is well established in deep-sea corals, several other animal and plant hosts, and marine environments (Kellogg, 2019). Additionally, some studies have reported the antimicrobial activities of Methylobacterium isolates against pathogenic microorganisms (Balachandran et al., 2012; Photolo et al., 2020). Methylobacterium species also possess a Type VI secretion system (T6SS), which helps them compete with other competitive bacterial groups (Kempnich and Sison-Mangus, 2020). However, the role of Methylobacterium in the coral holobiont is poorly understood. Previous studies observed that the abundance of Methylobacterium is positively correlated with the presence of Si in seawater (Kempnich and Sison-Mangus, 2020). Its siderophore has high concentrations of Fe (III) ions (Simionato et al., 2006). Consistent with these findings, our results revealed a higher abundance of Methylobacterium in coral mucus from the CM and HV sites (Figure 3), which are characterized by high concentrations of Si and Fe in ambient water (Table 1).
4.2 Bacterial functional profiles vary across habitats
Bacterial functional group analysis demonstrated the heterogeneity in the functional groups of the bacterial genera associated with the coral tissue and mucus samples and with the environmental samples (Supplementary Figure S5A). Bacterial functions in coral host microbiota have been explored. Although some of the bacteria appear to have commensal or anti-infectious functions (Nissimov et al., 2009; Olson et al., 2009), other bacteria are dynamic in nature and change with varying environmental conditions (Bruno et al., 2007). Our samples contained abundant chemoheterotrophic bacteria, which derive their energy from various processes, including the reduction and respiration of nitrate and nitrogen; fermentation; oxidation of sulfur compounds, thiosulfate, and hydrogen; and degradation of aromatic compounds. The cycling of nutrients such as nitrogen, sulfur, phosphorous, and carbon in a holobiont is crucial for the health and development of host organisms in oligotrophic environments (Zhang et al., 2015; Babbin et al., 2021).
Our bacterial function results revealed a higher abundance of nitrate-reducing and nitrogen/nitrate-respiring bacteria in the tissue and mucus samples from the HV site (Supplementary Figure S5B). Yang et al. (2013) reported that the bacterial symbionts of the coral T. coccinea have potential denitrifiers and ammonia-oxidizing bacteria, which consist of the nitrite reductase gene nirK and ammonia monooxygenase subunit A (amoA) genes, respectively. Bacterial groups that contribute to nitrogen cycling are important in maintaining homeostasis (Rädecker et al., 2015), and they may be critical for the resilience of hermatypic corals during ocean acidification to keep up with the higher photosynthetic rate expected under elevated CO2 conditions (Santos et al., 2014; Rädecker et al., 2015; Marcelino et al., 2017). However, the role of nitrogen-cycling bacteria in azooxanthellate Scleractinia corals remains understudied. Biagi et al. (2020) first reported an increase in the abundance of nitrogen-fixing bacteria in the scleractinian coral Asteroides calycularis, which has no symbionts and lives under acidified conditions near CO2 vents on Ischia Island, Gulf of Naples, Italy. Our study revealed a higher abundance of bacteria involved in the reduction and respiration of nitrate and nitrogen from the HV site, where the ambient seawater had pH 5.7 (Table 1; Supplementary Figure S5B).
Chemoautotrophic bacteria with sulfur-oxidizing potential are the common primary producers in vent ecosystems, while phototrophic prokaryotes and eukaryotes are far less abundant (Wang et al., 2015). Wang et al. (2015) reported that the mesophilic chemoautotrophs Sulfurovum and Sulfurimonas are dominant in the vent sediment on Kueishan Island. These Epsilonproteobacteria have the potential to oxidize sulfur compounds (Yamamoto et al., 2010; Han and Perner, 2016). This ability might explain the significant dominance of dark sulfur- and sulfur-compound-oxidizing prokaryotes in our HV site samples (Figure 4). The HV site had a higher abundance of Sulfurovum and Sulfurimonas, while the water sample was dominated by sulfur-oxidizing Gammaproteobacteria, namely, Thiomicrorhabdus (Kojima and Fukui, 2019; Liu et al., 2020; Figure 3).
Photoautotrophic prokaryotic functional groups were significantly abundant in the coral tissue samples of the NPP and CZ sites (Figures 4A, C). Endolithic algal and cyanobacterial photosynthates serve as a nutrient source for the coral host during extreme events (Fine and Loya, 2002; Yang et al., 2019). Yang et al. (2013) reported the presence of cyanobacteria in the tissue samples of T. coccinea. However, knowledge about the interaction between phototrophic prokaryotes and azooxanthellate Scleractinia is limited. These functional groups may fulfil the nutritional requirements and support the skeletal growth of the host T. aurea. However, a comprehensive study should be performed to enhance our understanding of the complex relationship between the coral host and its symbiotic phototrophic functional groups.
4.3 Coral-related bacteria in response to heavy metal pollution
Some bacterial species can be used to predict certain environmental conditions (Burger, 2006). In the present study, we observed the dominance of certain coral-related bacteria, which might serve as bioindicators of high concentrations of Cd, Zn, or Pb in their respective aquatic environments (Figure 6B). These genera include Vallitalea, Thermostilla, Methyloceanibacter, Kryptousia, Hoeflea, Denitrobaculum, Bythopirellula, Aliterella, and Actinomarinicola. Additionally, Marispirochaeta and Kistimonas are highly correlated with Fe pollution in specific water bodies (Figure 6B). However, we could not find any published reports supporting the use of these bacteria as indicators for monitoring heavy metal pollution. Given the adaptability of T. aurea to thrive in reef and non-reef habitats, as well as in extreme environments such as HV and CM sites, we propose that the microbial communities associated with this coral, as revealed by nanopore sequencing technology, could be used to monitor environments with high sulfur and copper contents in the future.
5 Conclusions
Corals associated microbial communities are influenced by various factors reflecting their respective ambient environments. We analyzed the structural and functional dynamics of the bacterial communities in T. aurea by applying the third-generation sequencing technology of ONT to reveal their response to the influence of extreme environmental conditions. Our study highlighted the remarkable adaptability of coral-associated microbial communities in T. aurea in response to extreme environmental stressors, such as acidification and pollution. Despite these challenges, beneficial bacterial genera dominated the microbial composition, which might have enhanced the resilience of the coral. Variations in structural and functional profiles among these communities indicated their role for natural selection mechanisms in promoting bacteria that facilitate the success of T. aurea in extreme habitats. Changes in the expression of Endozoicomonas bacterial species across tissue samples from distinct sites might be attributed to coral-mediated tolerance mechanisms and the existence of transposable elements within bacterial genomes. In addition to the inherent physiological strategies of the coral, this microbial-mediated resilience helped T. aurea adapt to dispersion, range expansion, and potential invasiveness in increasingly severe fluctuating scenarios worldwide. Therefore, these findings emphasized the critical need for comprehensive research to elucidate the mechanisms by which microbial communities aid T. aurea. This study also provided insights into the coral’s ability to outperform traditional reef-building corals in present and future ocean scenarios.
Data availability statement
The raw data are available at the following Bioproject accession number PRJNA937790 (SRA accession numbers: for tissue samples SAMN33419890 - SAMN33419901, for mucus samples SAMN33419914 - SAMN33419925, for water samples SAMN33419902 - SAMN33419913, and for sediment samples SAMN33419926 - SAMN33419937).
Ethics statement
The animal study was approved by Institutional animal care and use committee, College of Life Sciences, National Taiwan Ocean University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
GGK and J-SH conceived the study, GGK and L-CT collected samples, GGK, Y-LC, and P-JM did the lab work, GGK and Y-NH analyzed the data, and GGK wrote the draft manuscript. GGK, L-CT, J-SH and Y-NH finalized the manuscript. Y-NH and J-SH provided conceptualization, methodology, formal analysis, writing, review and editing. All authors contributed to the article and approved the submitted version.
Funding
Financial support was provided by the Ministry of Science and Technology (MOST), now the National Science and Technology Council (NSTC) of Taiwan, through grant no. MOST 109-2313-B- 019 -003-MY2, MOST 110-2621-M-019-003 and MOST 111-2628-M-019-001-MY3 to Y-NH as well as grant no. MOST 109-2621-M-019-002, MOST 110-2621-M-019-001 and MOST 111- 2621-M-019-001, and Center of Excellence for Ocean Engineering (Grant No. 109J 13801-51, 110J13801-51, 111J13801-51) to J-SH, and grant no. MOST 110-2811-M-019-504 and MOST 111-2811-M-019-003 to L-CT, and also the International Panel on Climate Change (IPCC) and the Prince Albert II Monaco Foundation to GG.
Acknowledgments
GG acknowledges the Prince Albert II Monaco Foundation and IPCC for awarding the IPCC scholarship. We are equally thankful to lab members of J-SH’s lab for assisting with field sampling.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1234137/full#supplementary-material
References
Ainsworth T. D., Wasmund K., Ukani L., Seneca F., Yellowlees D., Miller D., et al. (2011). Defining the tipping point. A complex cellular life/death balance in corals in response to stress. Sci. Rep. 1, 160. doi: 10.1038/srep00160
Babbin A. R., Tamasi T., Dumit D., Weber L., Rodríguez M. V. I., Schwartz S. L., et al. (2021). Discovery and quantification of anaerobic nitrogen metabolisms among oxygenated tropical Cuban stony corals. ISME J. 15, 1222–1235. doi: 10.1038/s41396-020-00845-2
Balachandran C., Duraipandiyan V., Ignacimuthu S. (2012). Cytotoxic (A549) and antimicrobial effects of Methylobacterium sp. isolate (ERI-135) from Nilgiris forest soil, India. Asian Pac. J. Trop. BioMed. 2, 712–716. doi: 10.1016/s2221-1691(12)60215-9
Ben-Haim Y., Thompson F. L., Thompson C. C., Cnockaert M. C., Hoste B., Swings J., et al. (2003). Vibrio coralliilyticus sp. nov., a temperature-dependent pathogen of the coral Pocillopora damicornis. Int. J. Syst. Evol. Microbiol. 53, 309–315. doi: 10.1099/ijs.0.02402-0
Biagi E., Caroselli E., Barone M., Pezzimenti M., Teixido N., Soverini M., et al. (2020). Patterns in microbiome composition differ with ocean acidification in anatomic compartments of the Mediterranean coral Astroides calycularis living at CO2 vents. Sci. Total Environ. 274, 138048. doi: 10.1016/j.scitotenv.2020.138048
Bourne D. G., Morrow K. M., Webster N. S. (2016). Insights into the coral microbiome: Underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70, 317–340. doi: 10.1146/annurev-micro-102215-095440
Bourne D. G., Munn C. B. (2005). Diversity of bacteria associated with the coral Pocillopora damicornis from the Great Barrier Reef. Environ. Microbiol. 7, 1162–1174. doi: 10.1111/j.1462-2920.2005.00793.x
Breitwieser F. P., Salzberg S. L. (2020). Pavian: Interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 36, 1303–1304. doi: 10.1093/bioinformatics/btz715
Bruno J. F., Selig E. R., Casey K. S., Page C. A., Willis B. L., Harvell C. D., et al. (2007). Thermal stress and coral cover as drivers of coral disease outbreaks. PloS Biol. 5, e124. doi: 10.1371/journal.pbio.0050124
Burger J. (2006). Bioindicators: Types, development, and use in ecological assessment and research. Environ. Bioindic. 1, 22–39. doi: 10.1080/15555270590966483
Carlos C., Torres T. T., Ottoboni L. M. M. (2013). Bacterial communities and species-specific associations with the mucus of Brazilian coral species. Sci. Rep. 3, 1624. doi: 10.1038/srep01624
Carradec Q., Poulain J., Boissin E., Hume B. C. C., Voolstra C. R., Ziegler M., et al. (2020). A framework for in situ molecular characterization of coral holobionts using nanopore sequencing. Sci. Rep. 10, 1–10. doi: 10.1038/s41598-020-72589-0
Chan B. K. K., Wang T.-W., Chen P.-C., Lin C.-W., Chan T.-Y., Tsang L. M. (2016). Community structure of macrobiota and environmental parameters in shallow water hydrothermal vents off Kueishan Island, Taiwan. PloS One 11, e0148675. doi: 10.1371/journal.pone.0148675
Chan I., Hung J.-J., Peng S.-H., Tseng L.-C., Ho T.-Y., Hwang J.-S. (2014). Comparison of metal accumulation in the azooxanthellate scleractinian coral (Tubastraea coccinea) from different polluted environments. Mar. pollut. Bull. 85, 648–658. doi: 10.1016/j.marpolbul.2013.11.015
Chen C., Chan T.-Y., Chan B. K. K. (2018). Molluscan diversity in shallow water hydrothermal vents off Kueishan Island, Taiwan. Mar. Biodivers. 48, 709–714. doi: 10.1007/s12526-017-0804-2
Chen C.-T. A., Wang B. J., Huang J., Lou J.-Y. (2005a). Investigation into extremely acidic hydrothermal fluids off Kueishan Tao, Taiwan, China. Acta Oceanologica Sin. -English Edition. 24 (1), 125–133.
Chen C.-T. A., Zeng Z., Kuo F. W., Yang T. F., Wang B. J., Tu Y. Y. (2005b). Tide-influenced acidic hydrothermal system offshore NE Taiwan. Chem. Geol. 224, 69–81. doi: 10.1016/j.chemgeo.2005.07.022
Cooney R. P., Pantos O., Le Tissier M. D. A., Barer M. R., O’Donnell A. G., Bythell J. C. (2002). Characterization of the bacterial consortium associated with black band disease in coral using molecular microbiological techniques. Environ. Microbiol. 4, 401–413. doi: 10.1046/j.1462-2920.2002.00308.x
Creed J. C., Fenner D., Sammarco P., Cairns S., Capel K., Junqueira A. O., et al. (2017). The invasion of the azooxanthellate coral Tubastraea (Scleractinia: Dendrophylliidae) throughout the world: history, pathways and vectors. Biol. Invasions 19, 283–305. doi: 10.1007/s10530-016-1279-y
Dahms H. U., Schizas N. V., James R. A., Wang L., Hwang J.-S. (2018). Marine hydrothermal vents as templates for global change scenarios. Hydrobiologia 818, 1–10. doi: 10.1007/s10750-018-3598-8
De Coster W., D’Hert S., Schultz D. T., Cruts M., Van Broeckhoven C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
De Siqueira G. M. V., Pereira-dos-Santos F. M., Silva-Rocha R., Guazzaroni M. E. (2021). Nanopore sequencing provides rapid and reliable insights into microbial profiles of intensive care units. Front. Public Heal. 9. doi: 10.3389/fpubh.2021.710985
Ding J. Y., Shiu J. H., Chen W. M., Chiang Y. R., Tang S. L. (2016). Genomic insight into the host-endosymbiont relationship of Endozoicomonas montiporae CL-33T with its coral host. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00251
Engelen A. H., Aires T., Vermeij M. J. A., Herndl G. J., Serrão E. A., Frade P. R. (2018). Host differentiation and compartmentalization of microbial communities in the azooxanthellate cupcorals Tubastrea coccinea and Rhizopsammia goesi in the Caribbean. Front. Mar. Sci. 5. doi: 10.3389/fmars.2018.00391
Epstein H. E., Smith H. A., Torda G., van Oppen M. J. H. (2019). Microbiome engineering: Enhancing climate resilience in corals. Front. Ecol. Environ. 17, 100–108. doi: 10.1002/fee.2001
Fang J.-N., Yu B.-S., Chen Y.-L., Song S.-R., Lo H.-J., Lin I.-C., et al. (2003). Chemical composition and the origin of suspension particles in Liang-Dong Bay (Yin-Yang Sea), Northern Taiwan. J. Chin. Chem. Soc 50, 465–469. doi: 10.1002/jccs.200300073
Fine M., Loya Y. (2002). Endolithic algae: An alternative source of photoassimilates during coral bleaching. Proc. R. Soc London. Ser. B Biol. Sci. 269, 1205–1210. doi: 10.1098/rspb.2002.1983
Frias-Lopez J., Zerkle A. L., Bonheyo G. T., Fouke B. W. (2002). Partitioning of bacterial communities between seawater and healthy, black band diseased, and dead coral surfaces. Appl. Environ. Microbiol. 68, 2214–2228. doi: 10.1128/aem.68.5.2214-2228.2002
Geffen Y., Ron E. Z., Rosenberg E. (2009). Regulation of release of antibacterials from stressed scleractinian corals. FEMS Microbiol. Lett. 295, 103–109. doi: 10.1111/j.1574-6968.2009.01590.x
Giovannelli D., d’Errico G., Manini E., Yakimov M., Vetriani C. (2013). Diversity and phylogenetic analyses of bacteria from a shallow-water hydrothermal vent in Milos Island (Greece). Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00184
Han Y., Perner M. (2016). Sulfide consumption in Sulfurimonas denitrificans and heterologous expression of its three sulfide-quinone reductase homologs. J. Bacteriol. 198, 1260–1267. doi: 10.1128/jb.01021-15
Harris J. B., Grubb B. D., Maltin C. A., Dixon R. (2001). Diversity of bacteria associated with the Caribbean coral Montastraea franksi. Coral Reefs 20, 85–91. doi: 10.1007/s003380100138
Ho Y.-N., Chen Y.-L., Liu D.-Y. (2021). Portable and rapid sequencing device with microbial community-guided culture strategies for precious field and environmental samples. mSystems 6 (4), e0074821. doi: 10.1128/msystems.00748-21
Hoadley K. D., Hamilton M., Poirier C. L., Choi C. J., Yung C. M., Worden A. Z. (2021). Selective uptake of pelagic microbial community members by Caribbean reef corals. Appl. Environ. Microbiol. 87, 1–16. doi: 10.1128/aem.03175-20
Hoegh-Guldberg O. R., Cai E. S., Poloczanska P. G., Brewer S., Sundby K., Hilmi V. J., et al. (2014). The ocean. In: Climate Change 2014: Impacts, Adaptation, and Vulnerability. Part B: Regional Aspects. Contribution of Working Group II to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Barros V. R., Field C. B., Dokken D. J., Mastrandrea M. D., Mach K. J., Bilir T. E., et al. eds. (Cambridge, United Kingdom and New York, NY, USA: Cambridge University Press), 1655–1731. doi: 10.1128/aem.03175-20
Huang Y. C. A., Huang S. C., Meng P. J., Hsieh H. J., Chen C. A. (2012). Influence of strong monsoon winds on the water quality around a marine cage-culture zone in a shallow and semi-enclosed bay in Taiwan. Mar. pollut. Bull. 64, 851–860. doi: 10.1016/j.marpolbul.2012.01.012
Huggett M. J., Apprill A. (2019). Coral microbiome database: Integration of sequences reveals high diversity and relatedness of coral-associated microbes. Environ. Microbiol. Rep. 11, 372–385. doi: 10.1111/1758-2229.12686
Hügler M., Wirsen C. O., Fuchs G., Taylor C. D., Sievert S. M. (2005). Evidence for autotrophic CO2 fixation via the reductive tricarboxylic acid cycle by members of the ϵ subdivision of proteobacteria. J. Bacteriol. 187, 3020–3027. doi: 10.1128/JB.187.9.3020-3027.2005
Inagaki F., Takai K., Nealson K. H., Horikoshi K. (2004). Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the E-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 54, 1477–1482. doi: 10.1099/ijs.0.03042-0
IUCN Brief Issues. (2021). Invasive alien species and climate change. Available at: https://www.iucn.org/resources/issues-brief/invasive-alien-species-and-climate-change (Accessed on 12/May/2023).
Jeong H. J., Yoo Y., Kang N. S., Lim A. S., Seong K. A., Lee S. Y., et al. (2012). Heterotrophic feeding as a newly identified survival strategy of the dinoflagellate Symbiodinium. Proc. Natl. Acad. Sci. U.S.A. 109, 12604–12609. doi: 10.1073/pnas.1204302109
Keller-Costa T., Eriksson D., Goncalves J. M. S., Gomes N. C. M., Lago-Leston A., Costa R. (2017). The gorgonian coral Eunicella labiata hosts a distinct prokaryotic consortium amenable to cultivation. FEMS Microbiol. Ecol. 93, 143. doi: 10.1093/femsec/fix143
Kellogg C. A. (2019). Microbiomes of stony and soft deep-sea corals share rare core bacteria. Microbiome 7, 1–13. doi: 10.1186/s40168-019-0697-3
Kempnich M. W., Sison-Mangus M. P. (2020). Presence and abundance of bacteria with the Type VI secretion system in a coastal environment and in the global oceans. PloS One 15, e0244217. doi: 10.1371/journal.pone.0244217
Kim D., Song L., Breitwieser F. P., Salzberg S. L. (2016). Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729. doi: 10.1101/gr.210641.116
Kimes N. E., Grim C. J., Johnson W. R., Hasan N. A., Tall B. D., Kothary M. H., et al. (2012). Temperature regulation of virulence factors in the pathogen Vibrio coralliilyticus. ISME J. 6, 835–846. doi: 10.1038/ismej.2011.154
Kitahara M. V., Cairns S. D., Stolarski J., Blair D., Miller D. J. (2010). A comprehensive phylogenetic analysis of the scleractinia (cnidaria, anthozoa) based on mitochondrial CO1 sequence data. PloS One 5, e11490. doi: 10.1371/journal.pone.0011490
Kojima H., Fukui M. (2019). Thiomicrorhabdus aquaedulcis sp. nov., a sulfur-oxidizing bacterium isolated from lake water. Int. J. Syst. Evol. Microbiol. 69, 2849–2853. doi: 10.1099/ijsem.0.003567
Kuo J., Shibuno T., Kanamoto Z., Noro T. (2001). Halophila ovalis (R. Br.) Hook. f. from a submarine hot spring in southern Japan. Aquat. Bot. 70, 329–335. doi: 10.1016/s0304-3770(01)00162-0
Lee K., Choo Y. J., Giovannoni S. J., Cho J. C. (2007). Ruegeria pelagia sp. nov., isolated from the Sargasso Sea, Atlantic Ocean. Int. J. Syst. Evol. Microbiol. 57, 1815–1818. doi: 10.1099/ijs.0.65032-0
Lee P. W., Tseng L. C., Hwang J. S. (2018). Comparison of mesozooplankton mortality impacted by the cooling systems of two nuclear power plants at the northern Taiwan coast, southern East China Sea. Mar. pollut. Bull. 136, 114–124. doi: 10.1016/j.marpolbul.2018.09.003
Lema K. A., Willis B. L., Bourne D. G. (2014). Amplicon pyrosequencing reveals spatial and temporal consistency in diazotroph assemblages of the Acropora millepora microbiome. Environ. Microbiol. 16, 3345–3359. doi: 10.1111/1462-2920.12366
Li J., Chen Q., Long L. J., Dong J. D., Yang J., Zhang S. (2014). Bacterial dynamics within the mucus, tissue and skeleton of the coral Porites lutea during different seasons. Sci. Rep. 4, 1–8. doi: 10.1038/srep07320
Li G., Song Q., Zheng P., Zhang X., Zou S., Li Y., et al. (2021). Dynamics and distribution of marine Synechococcus abundance and genotypes during seasonal hypoxia in a coastal marine ranch. J. Mar. Sci. Eng. 9, 549. doi: 10.3390/jmse9050549
Littman R., Willis B. L., Bourne D. G. (2011). Metagenomic analysis of the coral holobiont during a natural bleaching event on the Great Barrier Reef. Environ. Microbiol. Rep. 3, 651–660. doi: 10.1111/j.1758-2229.2010.00234.x
Liu X., Jiang L., Hu Q., Lyu J., Shao Z. (2020). Thiomicrorhabdus indica sp. nov., an obligately chemolithoautotrophic, sulfur-oxidizing bacterium isolated from a deep-sea hydrothermal vent environment. Int. J. Syst. Evol. Microbiol. 70, 234–239. doi: 10.1099/ijsem.0.003744
Louca S., Parfrey L. W., Doebeli M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/S13059-014-0550-8
Luo H., Moran M. A. (2014). Evolutionary ecology of the marine Roseobacter clade. Microbiol. Mol. Biol. Rev. 78, 573–587. doi: 10.1128/mmbr.00020-14
Magnuson E., Mykytczuk N. C. S., Pellerin A., Goordial J., Twine S. M., Wing B., et al. (2020). Thiomicrorhabdus streamers and sulfur cycling in perennial hypersaline cold springs in the Canadian high Arctic. Environ. Microbiol. 23 (7), 3384–3400. doi: 10.1111/1462-2920.14916
Mao-Jones J., Ritchie K. B., Jones L. E., Ellner S. P. (2010). How microbial community composition regulates coral disease development. PloS Biol. 8, 1000345. doi: 10.1371/journal.pbio.1000345
Marcelino V. R., Morrow K. M., van Oppen M. J. H., Bourne D. G., Verbruggen H. (2017). Diversity and stability of coral endolithic microbial communities at a naturally high pCO2 reef. Mol. Ecol. 26, 5344–5357. doi: 10.1111/mec.14268
Matsuo Y., Komiya S., Yasumizu Y., Yasuoka Y., Mizushima K., Takagi T., et al. (2021). Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution. BMC Microbiol. 21, 1–13. doi: 10.1186/s12866-021-02094-5
McDaniel L. D., Young E., Delaney J., Ruhnau F., Ritchie K. B., Paul J. H. (2010). High frequency of horizontal gene transfer in the oceans. Science 330 (6000), 50. doi: 10.1126/science.1192243
McDaniel L. D., Young E. C., Ritchie K. B., Paul J. H. (2012). Environmental factors influencing gene transfer agent (GTA) mediated transduction in the subtropical ocean. PloS One 7, 43506. doi: 10.1371/journal.pone.0043506
Meng P. J., Lee H. J., Wang J. T., Chen C. C., Lin H. J., Tew K. S., et al. (2008). A long-term survey on anthropogenic impacts to the water quality of coral reefs, southern Taiwan. Environ. pollut. 156, 67–75. doi: 10.1016/j.envpol.2007.12.039
Meron D., Atias E., Iasur Kruh L., Elifantz H., Minz D., Fine M., et al. (2011). The impact of reduced pH on the microbial community of the coral Acropora eurystoma. ISME J. 5, 51–60. doi: 10.1038/ismej.2010.102
Metsalu T., Vilo J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43 (W1), W566–W570. doi: 10.1093/nar/gkv468
Meyer J. L., Paul V. J., Teplitski M. (2014). Community shifts in the surface microbiomes of the coral Porites astreoides with unusual lesions. PloS One 9, e100316. doi: 10.1371/journal.pone.0100316
Miranda R. J., Tagliafico A., Kelaher B. P., Mariano-Neto E., Barros F. (2018). Impact of invasive corals Tubastrea spp. on native coral recruitment. Mar. Ecol. Prog. Ser. 605, 125–133. doi: 10.3354/meps12731
Morrow K. M., Bourne D. G., Humphrey C., Botté E. S., Laffy P., Zaneveld J., et al. (2015). Natural volcanic CO2 seeps reveal future trajectories for host-microbial associations in corals and sponges. ISME J. 9, 894–908. doi: 10.1038/ismej.2014.188
Naumann M. S., Richter C., El-Zibdah M., Wild C. (2009). Coral mucus as an efficient trap for picoplanktonic cyanobacteria: implications for pelagic–benthic coupling in the reef ecosystem. Mar. Ecol. Prog. Ser. 385, 65–76. doi: 10.3354/meps08073
Neave M. J., Apprill A., Ferrier-Pagès C., Voolstra C. R. (2016a). Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl. Microbiol. Biotechnol. 100, 8315–8324. doi: 10.1007/s00253-016-7777-0
Neave M. J., Michell C. T., Apprill A., Voolstra C. R. (2017). Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci. Rep. 7, 1–12. doi: 10.1038/srep40579
Neave M. J., Rachmawati R., Xun L., Michell C. T., Bourne D. G., Apprill A., et al. (2016b). Differential specificity between closely related corals andabundant Endozoicomonas endosymbionts across global scales. ISME J. 11, 186–200. doi: 10.1038/ismej.2016.95
Nissimov J., Rosenberg E., Munn C. B. (2009). Antimicrobial properties of resident coral mucus bacteria of Oculina patagonica. FEMS Microbiol. Lett. 292, 210–215. doi: 10.1111/j.1574-6968.2009.01490.x
Olson N. D., Ainsworth T. D., Gates R. D., Takabayashi M. (2009). Diazotrophic bacteria associated with Hawaiian Montipora corals: Diversity and abundance in correlation with symbiotic dinoflagellates. J. Exp. Mar. Bio. Ecol. 371, 140–146. doi: 10.1016/j.jembe.2009.01.012
Pai S. C. (1988). Pre-concentration efficiency of chelex-100 resin for heavy metals in seawater Part 2. Distribution of heavy metals on a Chelex-100 column and optimization of the column efficiency by a plate simulation method. Anal. Chim. Acta 211, 271–280.
Pai S.-C., Tsau Y.-J., Yang T.-I. (2001). pH and buffering capacity problems involved in the determination of ammonia in saline water using the indophenol blue spectrophotometric method. Anal. Chim. Acta 434, 209–216. doi: 10.1016/S0003-2670(01)00851-0
Peixoto R. S., Rosado P. M., Leite D. C., de A., Rosado A. S., Bourne D. G. (2017). Beneficial microorganisms for corals (BMC): Proposed mechanisms for coral health and resilience. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00341
Photolo M. M., Mavumengwana V., Sitole L., Tlou M. G. (2020). Antimicrobial and antioxidant properties of a bacterial endophyte, Methylobacterium radiotolerans MAMP 4754, isolated from Combretum erythrophyllum seeds. Int. J. Microbiol. 2020, 9483670. doi: 10.1155/2020/9483670
Precht W. F., Hickerson E. L., Schmahl G. P., Aronson R. B. (2014). The invasive coral Tubastraea coccinea (Lesson 1829): implications for natural habitats in the Gulf of Mexico and the Florida Keys. Gulf Mexico Sci. 32 (1), 5. doi: 10.18785/goms.3201.05
Rädecker N., Pogoreutz C., Voolstra C. R., Wiedenmann J., Wild C. (2015). Nitrogen cycling in corals: the key to understanding holobiont functioning? Trends Microbiol. 23, 490–497. doi: 10.1016/j.tim.2015.03.008
Reigel A. M., Paz-García D. A., Hellberg M. E. (2021). Microbiome of a reef-building coral displays signs of acclimation to a stressful shallow hydrothermal vent habitat. Front. Mar. Sci. 8, 652633. doi: 10.3389/fmars.2021.652633
Riul P., Targino C. H., Júnior L. A., Creed J. C., Horta P. A., Costa G. C. (2013). Invasive potential of the coral Tubastraea coccinea in the southwest Atlantic. Mar. Ecol. Prog. Ser. 480, 73–81. doi: 10.3354/meps10200
Rohwer F., Seguritan V., Azam F., Knowlton N. (2002). Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243, 1–10. doi: 10.3354/meps243001
Rosenberg E., Falkovitz L. (2004). The Vibrio shiloi/ Oculina patagonica model system of coral bleaching. Annu. Rev. Microbiol. 58, 143–159. doi: 10.1146/annurev.micro.58.030603.123610
Rypien K. L., Ward J. R., Azam F. (2010). Antagonistic interactions among coral-associated bacteria. Environ. Microbiol. 12, 28–39. doi: 10.1111/j.1462-2920.2009.02027.x
Santos H. F., Carmo F. L., Duarte G., Dini-Andreote F., Castro C. B., Rosado A. S., et al. (2014). Climate change affects key nitrogen-fixing bacterial populations on coral reefs. ISME J. 8, 2272–2279. doi: 10.1038/ismej.2014.70
Shashar N., Cohen Y., Loya Y., Sar N. (1994). Nitrogen fixation (acetylene reduction) in stony corals - Evidence for coral-bacteria interactions. Mar. Ecol. Prog. Ser. 111, 259–264. doi: 10.3354/meps111259
Shore A., Day R. D., Stewart J. A., Burge C. A. (2021). Dichotomy between regulation of coral bacterial communities and calcification physiology under ocean acidification conditions. Invertebr. Microbiol. 87 (6), e02189–e02120. doi: 10.1128/aem.02189-20
Simionato A. V. C., Simó C., Cifuentes A., Lacava P. T., Araújo W. L., Azevedo J. L., et al. (2006). Capillary electrophoresis-mass spectrometry of citrus endophytic bacteria siderophores. Electrophoresis 27, 2567–2574. doi: 10.1002/elps.200500933
Tait K., Hutchison Z., Thompson F. L., Munn C. B. (2010). Quorum sensing signal production and inhibition by coral-associated vibrios. Environ. Microbiol. Rep. 2, 145–150. doi: 10.1111/j.1758-2229.2009.00122.x
Tandon K., Chiou Y.-J., Yu S.-P., Hsieh H.-J., Lu C.-Y., Hsu M.-T., et al. (2022). Microbiome restructuring: dominant coral bacterium Endozoicomonas species respond differentially to environmental changes. mSystems 7, e00359–e00322. doi: 10.1128/msystems.00359-22
Tandon K., Lu C. Y., Chiang P. W., Wada N., Yang S. H., Chan Y. F., et al. (2020). Comparative genomics: dominant coral-bacterium Endozoicomonas acroporae metabolizes dimethylsulfoniopropionate (DMSP). ISME J. 14, 1290–1303. doi: 10.1038/s41396-020-0610-x
Tang K., Liu K., Jiao N., Zhang Y., Chen C. T. A. (2013). Functional metagenomic investigations of microbial communities in a shallow-sea hydrothermal system. PloS One 8, e72958. doi: 10.1371/journal.pone.0072958
Thompson J. R., Rivera H. E., Closek C. J., Medina M. (2014). Microbes in the coral holobiont: Partners through evolution, development, and ecological interactions. Front. Cell. Infect. Microbiol. 4. doi: 10.3389/fcimb.2014.00176
Thurber R. V., Willner-Hall D., Rodriguez-Mueller B., Desnues C., Edwards R. A., Angly F., et al. (2009). Metagenomic analysis of stressed coral holobionts. Environ. Microbiol. 11, 2148–2163. doi: 10.1111/j.1462-2920.2009.01935.x
Torondel B., Ensink J. H. J., Gundogdu O., Ijaz U. Z., Parkhill J., Abdelahi F., et al. (2016). Assessment of the influence of intrinsic environmental and geographical factors on the bacterial ecology of pit latrines. Microb. Biotechnol. 9, 209–223. doi: 10.1111/1751-7915.12334
Urban L., Holzer A., Baronas J. J., Hall M. B., Braeuninger-Weimer P., Scherm, et al. (2021). Freshwater monitoring by nanopore sequencing. Elife 10, 1–27. doi: 10.7554/eLife.61504
Van Oppen M. J., Gates R. D., Blackall L. L., Cantin N., Chakravarti L. J., Chan W. Y., et al. (2017). Shifting paradigms in restoration of the world's coral reefs. Glob Chang Biol. 23 (9), 3437–3448. doi: 10.1111/gcb.13647
Vezzulli L., Pezzati E., Huete-Stauffer C., Pruzzo C., Cerrano C. (2013). 16SrDNA pyrosequencing of the mediterranean gorgonian Paramuricea clavata reveals a link among alterations in bacterial holobiont members, anthropogenic influence and disease outbreaks. PloS One 8, e67745. doi: 10.1371/journal.pone.0067745
Wang L., Cheung M. K., Kwan H. S., Hwang J.-S., Wong C. K. (2015). Microbial diversity in shallow-water hydrothermal sediments of Kueishan Island, Taiwan as revealed by pyrosequencing. J. Basic Microbiol. 55, 1308–1318. doi: 10.1002/jobm.201400811
Wang L., Cheung M. K., Liu R., Wong C. K., Kwan H. S., Hwang J.-S. (2017). Diversity of total bacterial communities and chemoautotrophic populations in sulfur-rich sediments of shallow-water hydrothermal vents off Kueishan Island, Taiwan. Environ. Microbiol. 73, 571–582. doi: 10.1007/s00248-016-0898-2
Weiss S., Xu Z. Z., Peddada S., Amir A., Bittinger K., Gonzalez, et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5, 27. doi: 10.1186/s40168-017-0237-y
Wickham H. (2009). Ggplot2: Elegant graphics for data analysis. 2nd Edition (New York: Springer). doi: 10.1007/978-0-387-98141-3
Xia X., Guo W., Tan S., Liu H. (2017). Synechococcus assemblages across the salinity gradient in a salt wedge estuary. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01254
Yamamoto M., Nakagawa S., Shimamura S., Takai K., Horikoshi K. (2010). Molecular characterization of inorganic sulfur-compound metabolism in the deep-sea epsilonproteobacterium Sulfurovum sp. NBC37-1. Environ. Microbiol. 12, 1144–1153. doi: 10.1111/j.1462-2920.2010.02155.x
Yang S., Sun W., Zhang F., Li Z. (2013). Phylogenetically diverse denitrifying and ammonia-oxidizing bacteria in corals Alcyonium gracillimum and Tubastraea coccinea. Mar. Biotechnol 15, 540–551. doi: 10.1007/s10126-013-9503-6
Yang S. H., Tandon K., Lu C. Y., Wada N., Shih C. J., Hsiao S. S. Y., et al. (2019). Metagenomic, phylogenetic, and functional characterization of predominant endolithic green sulfur bacteria in the coral Isopora palifera. Microbiome 7, 1–13. doi: 10.1186/s40168-018-0616-z
Zaneveld J. R., Burkepile D. E., Shantz A. A., Pritchard C. E., McMinds R., Payet, et al. (2016). Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat. Commun. 7, 11833. doi: 10.1038/ncomms11833
Zhang Y., Ling J., Yang Q., Wen C., Yan Q., Sun H., et al. (2015). The functional gene composition and metabolic potential of coral-associated microbial communities. Sci. Rep. 5, 1–11. doi: 10.1038/srep16191
Zhang Y., Yang Q., Ling J., Van Nostrand J. D., Shi Z., Zhou J., et al. (2016). The shifts of diazotrophic communities in spring and summer associated with coral Galaxea astreata, Pavona decussata, and Porites lutea. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.01870
Ziegler M., Grupstra C. G. B., Barreto M. M., Eaton M., BaOmar J., Zubier K., et al. (2019). Coral bacterial community structure responds to environmental change in a host-specific manner. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-019-10969-5
Ziegler M., Roik A., Porter A., Zubier K., Mudarris M. S., Ormond R., et al. (2016). Coral microbial community dynamics in response to anthropogenic impacts near a major city in the central Red Sea. Mar. pollut. Bull. 105, 629–640. doi: 10.1016/j.marpolbul.2015.12.045
Keywords: copper pollution, hydrothermal vent, sulfur, acidification, functional group, Oxford Nanopore Technologies
Citation: Girija GK, Tseng L-C, Chen Y-L, Meng P-J, Hwang J-S and Ho Y-N (2023) Microbiome variability in invasive coral (Tubastraea aurea) in response to diverse environmental stressors. Front. Mar. Sci. 10:1234137. doi: 10.3389/fmars.2023.1234137
Received: 03 June 2023; Accepted: 29 September 2023;
Published: 12 October 2023.
Edited by:
Christian Robert Voolstra, University of Konstanz, GermanyReviewed by:
Robert W. Thacker, Stony Brook University, United StatesFluvio Modolon da Silva, Federal University of Rio de Janeiro, Brazil
Copyright © 2023 Girija, Tseng, Chen, Meng, Hwang and Ho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying-Ning Ho, eW5ob0BtYWlsLm50b3UuZWR1LnR3
†ORCID: Gowri Krishna Girija, orcid.org/0000-0002-5227-2935