
- 1 Department of Immunology, National Institute of Infectious Diseases, Tokyo, Japan
- 2 The Immunology Laboratory, Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA
RNA silencing mediated by microRNAs (miRNAs) is a recently discovered gene regulatory mechanism involved in various aspects of biology, such as development, cell differentiation and proliferation, and innate immunity against viral infections. miRNAs, which are a class of small (21–25 nucleotides) RNAs, target messenger RNA (mRNA) through incomplete base-pairing with their target sequences resulting in mRNA degradation or translational repression. Although studies of miRNAs have led to numerous sensational discoveries in biology, many fundamental questions about their expression and function still remain. In this review, we discuss the dynamics of the mammalian miRNA machinery and the biological function of miRNAs, focusing on RNA viruses and the various therapeutic applications of miRNAs against viral infections.
Introduction
The human genome is about 3,300 Mb in size and comprises 1% exons and 24% introns (the remainder being transposons (45%) and repetitive sequences). It is assumed that less than 40,000 protein-coding genes are present (Lewin, 2004), so why do we have such a large genome containing so many non-coding regions? Historically, the idea that RNA regulates the gene expression profile of each cell was proposed by Britten and Davidson (1969). This hypothesis was realized in the 1990s, especially after the first key finding (in Caenorhabditis elegans) that double-stranded RNA is cleaved by the ribonuclease (RNase), Dicer, into small, ∼22 nucleotide (nt) RNAs, and induces gene silencing (RNA silencing) (Fire et al., 1998). Subsequent extensive studies and computer-based bioinformatics approaches shed light on “the world of small RNAs”, which consist of small RNAs (∼20–30 nt) with a variable origin, including exogenous and endogenous short interfering RNAs (siRNA), Piwi (P-element-induced whimpy testes)-interacting (pi)RNAs and microRNAs (miRNAs) (see review in Zamore and Haley, 2005; Choudhuri, 2009).
The post-transcriptional gene silencing (PTGS) mechanism mediated by these small RNAs plays a fundamental role in development, differentiation, proliferation, and transposon mobility (Bartel, 2004; Zamore and Haley, 2005; Berkhout and Jeang, 2007). Currently, nearly 1,000 mature human miRNAs are registered in the miRNA data base (http://www.mirbase.org/). Although our knowledge of small RNAs is expanding rapidly, the expression profiles and function of many of these miRNAs remains unclear. In this expanding field of science, it seems impossible to cover all the aspects of miRNA research and there are already numerous reviews of miRNA focusing, for example, on the immune system (Baltimore et al., 2008; Lodish et al., 2008; Xiao and Rajewsky, 2009; O’Connell et al., 2010), cancer (Croce, 2009), and virus-encoded miRNAs (Gottwein and Cullen, 2008; Boss et al., 2009; Cullen, 2009). Here, we discuss recent topics regarding miRNAs (mostly human) with a focus on RNA virus infection and the development of novel technologies that take advantage of the particular characteristics of miRNA for therapeutic purposes.
Basic Knowledge of Mammalian miRNA
Structure of miRNA Processing Machinery
At present, siRNAs, which are exogenously induced double-stranded or short hairpin RNAs, are utilized as an essential tool for gene-specific silencing. In contrast, miRNAs play an important role in both post-transcriptional and translational regulation (Bartel, 2004). To obtain insights into the molecular mechanisms underlying RNA silencing pathways, the three-dimensional structures of proteins participating in miRNA biosynthesis have been deduced (see review in Jinek and Doudna, 2009). The protein structure of the human miRNA processing machinery is illustrated in Figure 1.
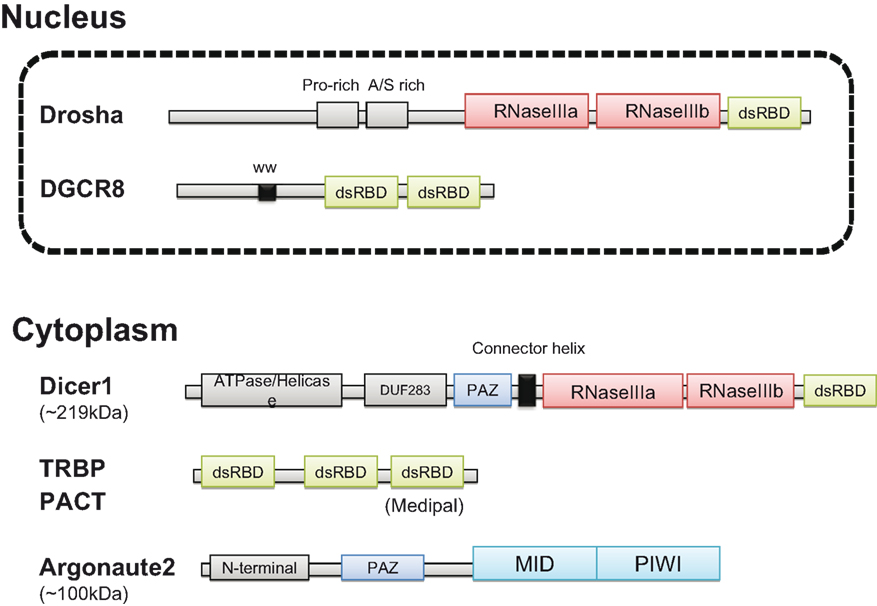
Figure 1. Structure of the human miRNA processing machinery. The predicted structures of Drosha and DGCR8 in the nucleus are shown in the upper panel. Dicer1, TRBP, and PACT interact in the cytoplasm and recruit Argonaute2. TRBP and PACT have a similar structure with the three dsRBDs (green). Two RNase III domains (red) are common in Drosha and Dicer1. PAZ domain (blue) is common in Dicer1 and Argonaute2. In Argonaute2, MID domain provides a RNA binding pocket and PIWI domain has endonuclease activity (light blue) (see the text in detail).
Drosha, a nuclear RNaseIII enzyme, has two RNaseIII catalytic sites with a double strand RNA binding domain (dsRBD) at the C terminus (Lee, 2003) and a proline-rich domain and arginine/serine-rich domains at the N terminus. DGCR8 has two dsRBDs and a WW domain containing two conserved tryptophan (W) residues. DGCR8 binds to the base of the long primary transcript (pri-)miRNA hairpin, positioning Drosha to cleave the pri-miRNA stem at a distance of 11 base pairs from the junction between the dsRNA stem and the flanking ssRNA regions (Han et al., 2006). The core region of human DGCR8 has been crystallized (Sohn et al., 2007).
Dicer, cytoplasmic RNaseIII, has ATPase and helicase domains at the N-terminus, followed by DUF283 (unknown function) and PAZ (PIWI/Argonaute/Zwille) domains, in addition to two RNaseIII domains and one dsRBD (Bernstein et al., 2001; Jinek and Doudna, 2009). The PAZ domain binds specifically to the 3′ end of single-stranded RNA. The crystal structure of Giardia Dicer suggests that it functions as a molecular ruler by anchoring the 3′ dinucleotide of the dsRNA bound to the PAZ domain, cleaving it at a fixed distance from that end, and generating products of defined length (Macrae et al., 2006). A connector helix may be the main determinant of product size.
TAR RNA-binding protein (TRBP) and PACT have similar structures, with three dsRBDs that directly interact with each other and with Dicer (Kok et al., 2007). Interestingly, PACT is an activator of protein kinase R (PKR), whereas TRBP is an inhibitor. One dsRBD known as Medipal in TRBP is a protein–protein interaction domain that binds Merlin, Dicer, and PACT. The TRBP C-terminus of Madipal interacts with Dicer via its ATPase/Helicase domain (Daniels et al., 2009).
Like Dicer, the Argonaute protein also has a PAZ domain, which binds to the 3′ end of guide RNA (Wang et al., 2008). The MID domain provides a binding pocket for the 5′-phosphate of guide RNA and the PIWI domain adopts an RNaseH fold and has endonuclease activity (Boland et al., 2010). In humans, there are four genes encoding Argonaute and only one of these, Ago2, has endonuclease activity. The mechanism by which the Ago protein mediates translational repression remains controversial and may be different in different cell types (for details, see review in Peters and Meister, 2007).
Biogenesis of miRNA
Although there are considerable variations in miRNA biogenesis and regulatory pathways (Winter et al., 2009), a representative pathway is shown in Figure 2.
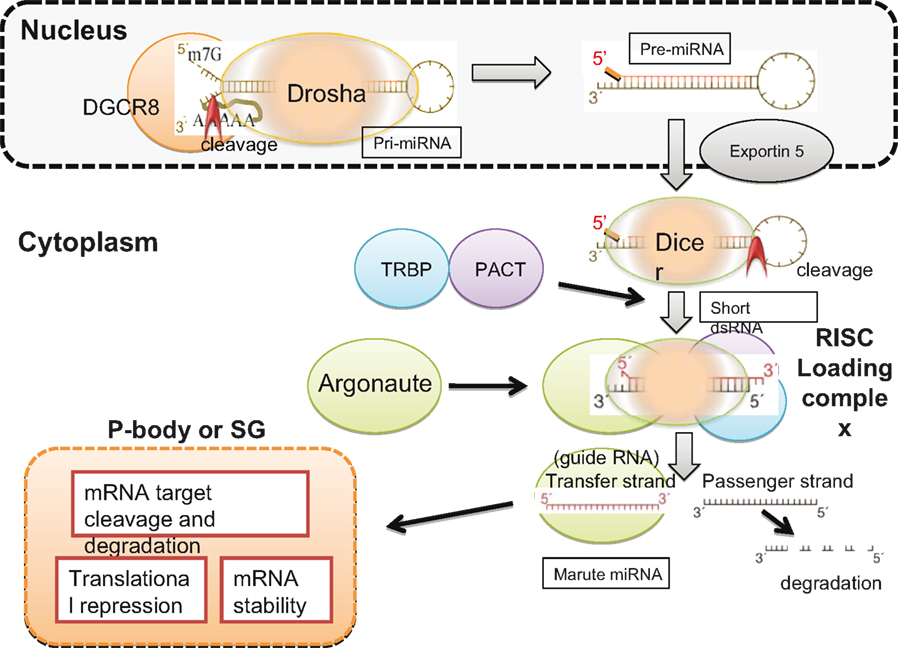
Figure 2. Biogenesis of miRNA. Drosha recognizes the specific hairpin structure of pri-miRNA and, in the presence of the cofactor DGCR8, cleaves the un-stemmed ssRNA region of pri-miRNA to generate pre-mRNA. Pre-miRNA is exported by exportin 5 to the cytoplasm, where Dicer recognizes the hairpin structure of dsRNA and cleaves, and short deRNA is generated. TRBP and PACT bind to Dicer, and Argonaute is recruited to form the RISC loading complex. Mature miRNA, complexed with Argonaute, is known as the transfer strand and is carried to the P-body or stress granules (SG), depending on the condition of the cell. The mRNA is rich in P-bodies, and either degradation or translational repression of target mRNA occurs. The red arrowheads in Pri- and Pre-miRNA indicate cleavage sites.
Human miRNAs are present within the introns of coding genes and the introns and exons of non-coding transcripts (Berkhout and Jeang, 2007). pri-miRNA, which is transcribed by RNA polymerase II, forms a distinctive hairpin structure with a 5′-capped poly(A) tail. This characteristic hairpin structure is recognized by Drosha (Bartel, 2004). Drosha forms a complex with DGCR8 and cleaves pri-miRNA at the unpaired flanking regions, leaving a monophosphate group at the 5′ ends and a two-nt overhang at the 3′ ends (Han et al., 2006). The products of these processed pri-miRNAs are ∼70-nt stem-loop RNA intermediates (pre-miRNA). The pre-miRNA is exported from the nucleus to the cytoplasm by exportin 5 in a Ran guanosine triphosphate-dependent manner (Lund et al., 2004), where it is processed by Dicer.
Human Dicer works together with its dsRNA-binding partner proteins, TRBP and PACT, to cleave pre-miRNA into ∼22-nt long dsRNAs, though TRBP and PACT are not essential for cleavage (Haase et al., 2005; Lee et al., 2006). The complex formed by Dicer, dsRNA, TRBP, and PACT then recruits Argonaute to form the RNA-induced silencing complex (RISC) loading complex (Chendrimada et al., 2005). Finally, Argonaute delivers mature miRNA to the processing bodies (PBs) or stress granules (SGs), where the target messenger RNA (mRNA) is located (Peters and Meister, 2007).
Mature miRNA, carried by Argonaute, can destroy target mRNA with complementary sequences or repress protein synthesis by partially binding to the 3′ untranslated region (UTR) of target mRNA. Because seed sequences of only 7–8 nt are required to loosely base-pair with the target sequence, a single miRNA is estimated, on average, to broadly target ∼200 species of RNA (Lewis et al., 2005; Bartel, 2009).
High Throughput Profiling Analysis
Expression of miRNAs may change temporally, in a spatial and tissue- or cell-type-specific manner. Large scale cloning and expression analyses of mammalian miRNAs were carried out by Landgraf et al. (2007) and the expression profiles of distinct human and mouse mature miRNAs were characterized. Comparison of miRNA expression between the hematopoietic system and all other organ systems indicated that only five miRNAs are highly specific to cells of the hematopoietic lineage (miR-142, miR-144, miR-150, miR-155, and miR-223). It is worth noting that species-specificity is also demonstrated by the data; miR-150 and miR-155 are highly hematopoietic-specific in mice, but not in humans. They also observed that the miRNA expression profiles of different sorted mature T cell types were similar and clustered together, and that no striking differences in the miRNA expression profiles of the different myeloid sub-lineages existed, except in granulocyte/monocyte-specific miR-223 expression. Thus, exclusive expression of miRNA by particular tissues or cell types is rare.
To identify the functionally important targets of miRNA, Selbach et al. (2008) employed a novel isotope pulse-labeling method of proteins (pulsed stable isotype labeling with amino acids in cell culture, pSILAC) in combination with mass-spectrometry-based proteomics. They evaluated changes in production of ∼5000 proteins associated with the overexpression of several miRNAs in HeLa cells, which included tissue-specific miRNAs not expressed in HeLa cells (miR-1 and miR-155) and those expressed ubiquitously (miR-16, miR-30a, and let-7b). This pSILAC technology was quite useful in identifying functionally important targets of miRNAs at the protein level. They demonstrated that a single miRNA can directly downregulate the production of hundreds of proteins, and identified the seed sequence in the 3′ UTR as the primary motif for miRNA-mediated regulation of protein synthesis, probably acting via both mRNA degradation and translational repression. Notably, the repressive effect on individual proteins was relatively small, which may reflect the paucity (less than 3) of seed sites for each miRNA in individual target 3′ UTRs. Importantly, the results obtained from miRNA overexpression systems are equivalent to those obtained from knock down systems, confirming that their conclusions were not experimentally biased.
Baek et al. (2008) demonstrated the proteomic impact of overexpressed miRNAs (miR-1, miR-181, and miR-124) using the same SILAC technology. They also analyzed the relationship between seed sequence matches and protein repression in HeLa cells and found that mRNAs with single 7-mer or 8-mer site matches, but not 6-mers, were significantly downregulated by miRNA. The seed sites within the 3′ UTRs were generally more effective than those in coding regions. Again, they demonstrated that the targeting principles elucidated from overexpressed exogenous miRNAs also apply to endogenous miRNA targeting at the level of protein downregulation, as argued by Selbach et al. (2008). We assume that coordinated post-transcriptional gene regulation by miRNAs contributes to the fine tuning of protein expression by cells in a tissue-, lineage-, differentiation-, proliferation-, or cytokine signal-specific manner. Accumulating knowledge about miRNA expression profiles and their biological functions will be used to build sophisticated bioinformatics programs to clarify various aspects of miRNA biology. Such computer-based tools for predicting the target genes of miRNAs are available at http://www.targetscan.org/, http://www.microrna.org/ and other sites (Bartel, 2009), and will prove helpful for the identification of targets with physiological relevance.
Virus–Host Interactions and miRNA
Recently, miRNAs were broadly implicated in viral infection of mammalian cells, having either positive, or negative effects on virus replication and host immune responses. Herpes-family viruses, which have a large DNA genome, are known to encode viral miRNA (v-miRNA), probably for the regulation of their own replication and latency and to control cellular defense mechanisms. However, the existence of v-miRNA in smaller DNA and RNA viruses remains controversial. It is argued that small viruses have size constraints that mean a greater proportion of their genome is required for coding purposes and is, therefore, less likely to retain non-coding RNAs (Berkhout and Jeang, 2007). In contrast, long-term, latently infected viruses such as herpes viruses derive a greater survival benefit from utilizing the cellular miRNA machinery (Cullen, 2010).
Because DNA virus-encoded v-miRNAs have been reviewed in detail elsewhere (see review in Gottwein and Cullen, 2008; Boss et al., 2009; Cullen, 2009), we will focus on the interaction of cellular miRNAs with RNA viruses as a host defense mechanism against viral infection.
Implications of miRNA Contributions to Antiviral Activity
In plants, worms and flies, RNA silencing suppresses the mobilization of endogenous retroviruses. Therefore, it is expected that the miRNA silencing machinery may be involved in defense against viral infections in vertebrates. Triboulet et al. (2007) demonstrated that, by silencing RNase III, Dicer, or Drosha, HIV-1 replication is inhibited in PBMCs from HIV-infected donors and in latently infected U1 cells in which HIV-1 is not efficiently transcribed through its promoter (the long terminal repeat, LTR) due to a Tat mutation. They analyzed the level of miRNA in Jurkat cells using microarray analysis and found that a polycistronic miRNA cluster, miR-17/92, was markedly decreased upon HIV-1 infection. Although miR-17/92 does not directly target the viral genome, the histone acetylase, PCAF, which is a cofactor for Tat transactivation, has four potential target sequences for miR-17-5p and 20a in its 3′ UTR. Triboulet et al. clearly demonstrated the relationship between HIV-1 replication and the expression of miRNA and PCAF/Tat. Thus, the miRNA biogenesis machinery contributes to host defense against HIV-1. However, additional cellular miRNAs, or other inhibitory mechanisms, may also be involved in virus–host interactions.
Mammals have a single Dicer gene, Dicer1. Inactivation of Dicer1 in mice causes embryonic lethality or severe damage to tissue morphogenesis, cell differentiation and development (Bernstein et al., 2003). Otsuka et al. (2007) managed to produce such Dicer1-deficient mice expressing low levels of Dicer in some tissues and almost none in peritoneal macrophages. Using these mice, they demonstrated for the first time in vivo that miRNAs target the RNA genome of vesicular stomatitis virus (VSV). Interestingly, these Dicer1-deficient mice were susceptible to VSV, but not to other RNA viruses, including encephalomyocarditis virus (EMCV), lymphocytic choriomeningitis virus (LCMV), and Influenza A virus. Computer prediction models allowed them to select 24 candidate miRNAs to potentially target the positive-strand sequence of VSV. They demonstrated that miR-93 and miR-24 suppress VSV replication by targeting the P (polymerase cofactor)- and L (RNA-dependent RNA polymerase)-encoding regions of VSV, respectively.
Cellular miRNAs Directly Target Viral RNA
For the inhibition of virus replication, the seed sequences of miRNA must be complementary to the viral targets for silencing (Gottwein and Cullen, 2008). Although a number of computer-based programs that search for homology between human miRNAs and viral genomes can predict complimentarity to various regions of viral sequences, the effects of RNAi need to be demonstrated in infected cells. The following are examples of successful approaches used to identify cellular miRNAs that target viral RNA.
Lecellier et al. (2005) found no evidence of virus-derived siRNAs within primate foamy virus type 1 (PFV-1), which is a complex retrovirus closely related to HIV and encodes two accessory factors, Bet and Tas. Instead, by fusing viral fragments to the UTR of a GFP-tagged reporter gene, they found that the 3′ end fragment of the PVF-1 genome, which encodes Bet and Env/Bet proteins (and is also within the 3′ UTR), reduced GFP levels and was a target for miR-32. Thus, miR-32 exerts a direct, sequence-specific effect against PFV-1. Moreover, they demonstrated that the viral Tas protein interacts with cellular miRNAs and acts as a broadly effective silencing suppressor. Similar, virally coded, RNA silencing suppressor (RSS) activity was shown for HIV-1 Tat (Bennasser et al., 2005), vaccinia virus E3L, influenza A virus NS1 and Ebola virus VP35 proteins (Haasnoot et al., 2007).
Huang et al. (2007) inserted DNA fragments from the 3′ UTR of HIV-1 RNA into the 3′ UTR of the pEGFP vector and identified the region targeted by a potential miRNA-mediated mechanism in resting primary CD4+ T cells. They found that miRNAs, such as miR-28, miR-125b, miR-150, miR-223, and miR-382, are abundant in resting T cells, but not in activated T cells, and the replication of latent viruses in patients on HAART was prevented by inhibiting these miRNAs. However, there are more than 100 cellular mRNA targets for each miRNA (Lewis et al., 2005) and the consequences of modulating the expression of these miRNAs may differ between T-cell subpopulations at distinct differentiation stages (see below). Furthermore, because transfection of RNA or DNA into resting T cells is a very inefficient procedure, applying miRNA expression in a therapeutic context may be very difficult.
Using microarray technology, Pedersen et al. (2007) analyzed RNA derived from interferon (IFN)-stimulated cells and found that eight miRNAs induced by IFN had seed sequences that were 100% complementary with hepatitis C virus (HCV). Of these putative miRNAs, five (miR-196, miR-296, miR-351, mir-431, and miR-448) were able to attenuate HCV replication in Huh7 cells. miR-196 and miR-448 did not prevent the replication of HCV containing mutant target sequences, indicating that the antiviral effect is not induced by non-specific alterations in cellular gene expression. Furthermore, miR-122, which is essential for HCV replication (Jopling et al., 2005), was downregulated in response to IFN-β. Thus, these results suggest that the modulation of the expression levels of these miRNAs has an important role in the antiviral effects of IFN-β against HCV. However, a recent analysis of liver biopsy samples from chronic hepatitis C patients revealed no correlation between miR-122 expression and viral load (Sarasin-Filipowicz et al., 2009). Moreover, most of the miRNAs that mediate the effects of IFN-β on HCV replication were present only at very low levels, even after IFN treatment, arguing against a protective role for these miRNAs. Nevertheless, the finding that IFN-treatment significantly reduces miR-122 expression in non-responders, as opposed to complete responders, indicates the usefulness of miRNA profiling as a prognostic marker for IFN therapy.
Altered Expression of Cellular miRNA Upon Viral Infection: Pathogenicity
The involvement of RNAi in influenza virus infection was identified by knocking down Dicer in Vero cells, which lack type I IFN genes (Matskevich and Moelling, 2007). Any acute virus infection may alter the expression pattern of both mRNA and miRNA in infected cells. Li et al. (2010) analyzed miRNA expression profiles in the lungs of mice infected with a highly pathogenic 1918 influenza A virus and compared them with those of mice infected with a non-lethal seasonal influenza A virus. By analyzing the inverse correlation between altered miRNA expression and the expression of predicted mRNA targets, they demonstrated that the target mRNAs of one altered miRNA, miR-200a, were associated with the type I IFN signaling pathway, which plays an important role in the pathogenesis in 1918 influenza A virus infection in mice (Kash et al., 2006). They also found that miR-223 indirectly downregulated CREB activity, which is required for the maintenance of cell survival and growth.
Another example can be found in HIV-1 infection. Cells of the monocyte/macrophage lineage are susceptible to HIV-1 infection; however, only macrophages can support massive virus production. This differentiation-dependent restriction mechanism has been attributed to the distinct expression of the APOBEC3 subfamily by monocytes (Peng et al., 2007). Recently, Wang et al. (2009) reported that high expression of miR-28, miR-150, miR-223, and miR-382 was correlated with low susceptibility of monocytes to HIV-1 infection. Although the target mRNAs of these miRNAs were not identified, the results suggest that monocyte differentiation and HIV-1 susceptibility are linked by a common set of miRNAs. In relation to monocyte/macrophage differentiation, the expression of Cyclin T1, required for transactivation by HIV-1 Tat, increases during macrophage differentiation and enhances HIV-1 replication within macrophages (Liou et al., 2002). A recent report indicates that miR-198-mediated repression of Cyclin T1 may contribute to HIV-1 replication during monocyte differentiation into macrophages (Sung and Rice, 2009).
These results indicate that RNA viruses utilize cellular miRNAs to manipulate the expression of cellular genes to enhance their own survival and expansion. The known interactions between cellular miRNA and RNA viruses are summarized in Figure 3.
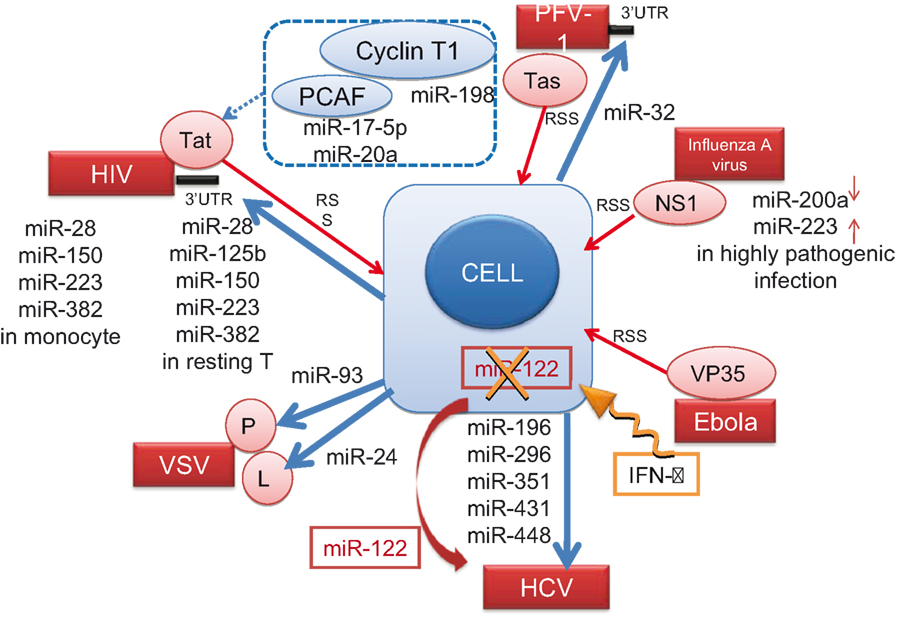
Figure 3. Cellular miRNAs known to interact with RNA viruses. The direct and indirect interactions between cellular miRNA molecules or cellular proteins (blue) with the viral genome (black bar) or viral proteins (red) are depicted. Blue arrows from the cell indicate negative effect of miRNAs on the virus, and a dotted blue line indicates the indirect interaction (see the text in detail). The IFN-β treatment (an orange winding arrow) induces several miRNAs, whereas it downregulates (X) miR-122 essential for HCV replication (a red ribbon arrow). Red thin arrows indicate the RNA silencing suppressor (RSS) activity of viral proteins.
Effects of miRNAs on Immune Cell Differentiation and Viral Infection
The conditional knockdown of Dicer in murine T or B lymphocytes causes abnormal lymphocyte differentiation (Cobb et al., 2005; Muljo et al., 2005). Antigen-induced immune cell differentiation is a key feature of host defense and elicits coordinated immune responses to protect the host from pathogens (Baltimore et al., 2008). For example, profound changes in gene expression occur during antigen-induced CD8+ T cell differentiation in mice (Wu et al., 2007). Wu et al. identified seven miRNAs (miR-16, miR-21, miR-142-3, miR-142-5, miR-150, miR-15b, and let-7f) frequently expressed in all T cells and observed alterations in the miR-21 expression signature as cells differentiated from naïve to effector cells. We have also shown the existence of similar, but not identical, differentiation-associated miRNA expression profiles in human CD8+ T lymphocytes. The function of these miRNAs requires further investigation.
Recent microarray analysis studies show that monocyte differentiation into dendritic cells is regulated and coordinated by miR-34a and miR-21 (Hashimi et al., 2009). JAG1 and WNT1 were identified as targets for these miRNAs using a bioinformatic target ranking system. Considering that monocyte-derived dendritic cells (MDDCs) are poor producers of HIV-1 compared with monocyte-derived macrophages (Tsunetsugu-Yokota, 2008), it is highly likely that miRNA expression associated with cell differentiation from monocytes to either macrophages or MDDCs may also regulate HIV replication in monocytes (Wang et al., 2009). Although such differentiation-associated miRNAs have any impact on virus replication within infected cells remains to be addressed, the results of extensive miRNA profiling analyses of viral infections both in vitro and in vivo will be highly informative for the diagnosis and treatment of viral infections in humans.
Therapeutic Application of miRNAs to Viral Infections
Knowledge regarding the basic biology and function of mammalian miRNAs has increased greatly in recent times. The idea of utilizing RNA interference by miRNA for genetic manipulation is already being realized. Taking advantage of the tissue-specificity of miRNAs, Kelly et al. inserted the target sequences of muscle-specific miRNAs (miR-133a and miR-206) into the 3′ UTR of Coxsackievirus A21 (CVA21), a pathogenic picornavirus that causes lethal myositis and oncolysis in tumor-baring mice (Kelly et al., 2008). The result was tissue-specific attenuation that allowed the virus to replicate well in muscle cells without being pathogenic. Likewise, by incorporating target sites for the neuron-specific miR-124a into the 3′ end of the 5′ UTR and between the structural and non-structural genes within the coding region of the poliovirus, an attenuated poliovirus vaccine lacking any neurovirulence was developed (Barnes et al., 2008). Similar engineering strategies were also applied to the VSV by inserting neurotropic miR-125 target sequences into the 3′ UTR of the viral polymerase gene (Kelly et al., 2010) and to adenoviruses by inserting a hepatocyte-selective miR-122 target site into the 3′ UTR of the E1A transcription cassette (Cawood et al., 2009). The structures of these attenuated viruses are illustrated in Figure 4. In this context, Perez et al. successfully attenuated the influenza A virus by incorporating species-specific, non-avian microRNA (miR-93) targets into nucleoproteins. The result was attenuated viral activity in mice, but not in eggs as expected (Perez et al., 2009). Thus, miRNA-mediated control of viral replication is a promising technology that can be used to develop safe attenuated viruses for cancer therapy and vaccines.
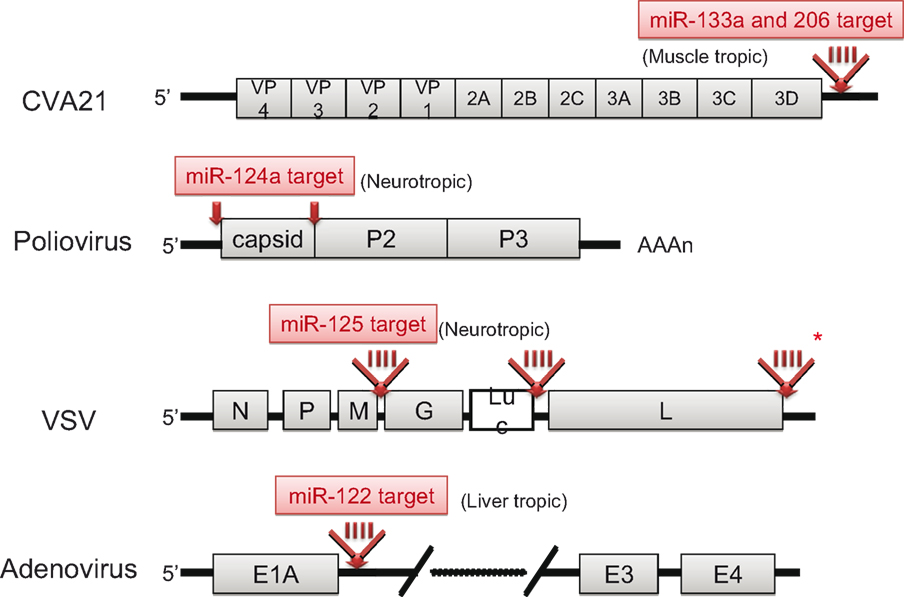
Figure 4. Schematic genome structures of attenuated viruses. The localization of miRNA target insertion site in each virus is depicted (red arrow). In the case of CVA21, VSV and adenovirus, 4-tandem repeat target sequences (red bars) were inserted. Asterisk indicates the most effective site for attenuation of VSV.
The RNAi-based gene delivery systems using lentivirus vectors have been developed for the control of HIV-1 infection, and such lentivirus can be utilized as potential AIDS vaccine candidates (Morris and Rossi, 2006; Rossi et al., 2007; Yamamoto and Tsunetsugu-Yokota, 2008). Likewise, considering the therapeutic applications of miRNA, efficient in vivo miRNA delivery systems by lentivirus vectors are a promising gene-transfer medium. However, because the VSV envelope glycoprotein used to encapsulate the transfer gene is pantropic, transgene expression in all cells could be problematic. To overcome the problem of non-specific gene transfer, Brown et al. (2007) systemically treated a mouse model of hemophilia B with a lentivirus vector expressing clotting factor IX (F.IX) under the control of a hepatocyte-specific promoter. Although F.IX is only expressed in the liver, an anti-F.IX immune response was elicited, probably due to off-target expression by the hepatocyte-specific promoter in hematopoietic cells. By inserting four tandem-repeat target sequences of the hematopoietic-specific miR-142-3p into the 3′ UTR of the transgene expression cassette, they were able to achieve sustained F.IX production in hemophilia B mice. In these mice, miR-142-3p levels were 50-fold higher in the spleen than in the liver. Importantly, the introduction of lentivirus containing miR-142-3p target sequences did not affect the normal in vivo level of miRNA, including that of miR-142-3p. Thus, by combining a highly cell lineage-specific promoter with PTGS using cell state- or lineage-specific miRNAs, the engineered lentivirus delivery system should make it possible to achieve specific expression of transgenes within the desired cells or tissues (Brown et al., 2007).
As described in the previous section (4.2), the abundantly expressed, liver-specific miRNA, miR-122, binds to two closely spaced target sites within the 5′ UTR of the HCV genome and is required to maintain high HCV RNA abundance in liver cells (Jopling et al., 2005, 2008). Interestingly, the same miR-122 binding site placed in the 3′ UTR of a reporter mRNA downregulated mRNA expression, which clearly demonstrates location-dependent gene regulation by miRNA (Jopling et al., 2008). Based on these findings, Lanford et al. (2010) utilized a locked nucleic acid (LNA)-modified phosphorothioate oligonucleotide (SPC3649) complementary to miR-122 to treat chimpanzees chronically infected with HCV. Four animals (two each) received a high or low dose (5 or 1 mg/kg) of SPC3649 intravenously once a week for 12 weeks and were observed over a 17-week treatment-free period. They showed long-standing suppression of HCV viremia without obvious disease, side effects, virus mutation, or virus rebound, which are all consistent with potent and sustained suppression of miR-122 in the liver. Their results clearly show the feasibility and future potential of LNA drug therapy to antagonize specific miRNA functions.
All these studies indicate a promising future for antiviral therapy via the regulation of cellular miRNA expression pathways. By further understanding the biological functions of miRNAs and applying them for diagnostic and therapeutic purposes, we may yet achieve unexpected and exciting discoveries.
Conclusion
In the post-human genome era, our research efforts have shifted more toward the regulation of gene expression and to understanding the function of non-coding regions. Now, accumulating evidence indicates that the coordinated expression of distinct sets of miRNAs regulate many aspects of biological events occurring in cells, such as differentiation, cell-cycle progression, and proliferation, in a cell- or tissue-specific manner. We are just beginning to understand the physiological and pathological roles played by miRNAs in viral infections. We expect that, as we learn more about the targets of miRNAs and their regulatory function in cell physiology, we may be able to develop more sophisticated technologies to treat infectious diseases in humans.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our colleagues in the first laboratory, Department of Immunology, NIID, Japan, for their help and discussion. This paper was supported by grants from the Ministry of Health, Labor and Welfare of Japan and from the Health Science Foundation of Japan.
References
Baek, D., Villen, J., Shin, C., Camargo, F. D., Gygi, S. P., and Bartel, D. P. (2008). The impact of microRNAs on protein output. Nature 455, 64–71.
Baltimore, D., Boldin, M. P., O’Connell, R. M., Rao, D. S., and Taganov, K. D. (2008). MicroRNAs: new regulators of immune cell development and function. Nat. Immunol. 9, 839–845.
Barnes, D., Kunitomi, M., Vignuzzi, M., Saksela, K., and Andino, R. (2008). Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe 4, 239–248.
Bennasser, Y., Le, S. Y., Benkirane, M., and Jeang, K. T. (2005). Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 22, 607–619.
Berkhout, B., and Jeang, K. T. (2007). RISCy business: microRNAs, pathogenesis, and viruses. J. Biol. Chem. 282, 26641–26645.
Bernstein, E., Caudy, A. A., Hammond, S. M., and Hannon, G. J. (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366.
Bernstein, E., Kim, S. Y., Carmell, M. A., Murchison, E. P., Alcorn, H., Li, M. Z., Mills, A. A., Elledge, S. J., Anderson, K. V., and Hannon, G. J. (2003). Dicer is essential for mouse development. Nat. Genet. 35, 215–217.
Boland, A., Tritschler, F., Heimstadt, S., Izaurralde, E., and Weichenrieder, O. (2010). Crystal structure and ligand binding of the MID domain of a eukaryotic Argonaute protein. EMBO Rep. 11, 522–527.
Boss, I. W., Plaisance, K. B., and Renne, R. (2009). Role of virus-encoded microRNAs in herpesvirus biology. Trends Microbiol. 17, 544–553.
Britten, R. J., and Davidson, E. H. (1969). Gene regulation for higher cells: a theory. Science 165, 349–357.
Brown, B. D., Cantore, A., Annoni, A., Sergi, L. S., Lombardo, A., Della Valle, P. D’Angelo, A., and Naldini, L. (2007). A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice. Blood 110, 4144–4152.
Brown, B. D., Gentner, B., Cantore, A., Colleoni, S., Amendola, M., Zingale, A. Baccarini, A., Lazzari, G. Galli, C., and Naldini, L. (2007). Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 25, 1457–1467.
Cawood, R., Chen, H., Carroll, F., Bazan-Peregrino, M., van Rooijen, N., and Seymour, L. W. (2009). Use of tissue-specific microRNA to control pathology of wild-type adenovirus without attenuation of its ability to kill cancer cells. PLoS Pathog. 5, e1000440. doi:10.1371/journal.ppat.1000440.
Chendrimada, T. P., Gregory, R. I., Kumaraswamy, E., Norman, J., Cooch, N., Nishikura, K., and Shiekhattar, R. (2005). TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436, 740–744.
Cobb, B. S., Nesterova, T. B., Thompson, E., Hertweck, A., O’Connor, E., Godwin, J., Wilson, C. B., Brockdorff, N., Fisher, A. G., Smale, S. T., and Merkenschlager, M. (2005). T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J. Exp. Med. 201, 1367–1373.
Croce, C. M. (2009). Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 10, 704–714.
Cullen, B. R. (2009). Viral and cellular messenger RNA targets of viral microRNAs. Nature 457, 421–425.
Cullen, B. R. (2010). Five questions about viruses and microRNAs. PLoS Pathog. 6, e1000787. doi:10.1371/journal.ppat.1000787.
Daniels, S. M., Melendez-Pena, C. E., Scarborough, R. J., Daher, A., Christensen, H. S., El Far, M., Purcell, D. F., Laine, S., and Gatignol, A. (2009). Characterization of the TRBP domain required for dicer interaction and function in RNA interference. BMC Mol. Biol. 10, 38.
Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., and Mello, C. C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811.
Gottwein, E., and Cullen, B. R. (2008). Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 3, 375–387.
Haase, A. D., Jaskiewicz, L., Zhang, H., Laine, S., Sack, R., Gatignol, A., and Filipowicz, W. (2005). TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 6, 961–967.
Haasnoot, J., de Vries, W., Geutjes, E. J., Prins, M., de Haan, P., and Berkhout, B. (2007). The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 3, e86. doi:10.1371/journal.ppat.0030086.
Han, J., Lee, Y., Yeom, K. H., Nam, J. W., Heo, I., Rhee, J. K., Sohn, S. Y., Cho, Y., Zhang, B. T., and Kim, V. N. (2006). Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125, 887–901.
Hashimi, S. T., Fulcher, J. A., Chang, M. H., Gov, L., Wang, S., and Lee, B. (2009). MicroRNA profiling identifies miR-34a and miR-21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood 114, 404–414.
Huang, J., Wang, F., Argyris, E., Chen, K., Liang, Z., Tian, H., Huang, W., Squires, K. Verlinghieri, G., and Zhang, H. (2007). Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 13, 1241–1247.
Jinek, M., and Doudna, J. A. (2009). A three-dimensional view of the molecular machinery of RNA interference. Nature 457, 405–412.
Jopling, C. L., Schutz, S., and Sarnow, P. (2008). Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4, 77–85.
Jopling, C. L., Yi, M., Lancaster, A. M., Lemon, S. M., and Sarnow, P. (2005). Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309, 1577–1581.
Kash, J. C., Tumpey, T. M., Proll, S. C., Carter, V., Perwitasari, O., Thomas, M. J., Basler, C. F., Palese, P., Taubenberger, J. K., Garcia-Sastre, A., Swayne, D. E., and Katze, M. G. (2006). Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443, 578–581.
Kelly, E. J., Hadac, E. M., Greiner, S., and Russell, S. J. (2008). Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 14, 1278–1283.
Kelly, E. J., Nace, R., Barber, G. N., and Russell, S. J. (2010). Attenuation of vesicular stomatitis virus encephalitis through microRNA targeting. J. Virol. 84, 1550–1562.
Kok, K. H., Ng, M. H. Ching, Y. P., and Jin, D. Y. (2007). Human TRBP and PACT directly interact with each other and associate with dicer to facilitate the production of small interfering RNA. J. Biol. Chem. 282, 17649–17657.
Landgraf, P., Rusu, M., Sheridan, R., Sewer, A., Iovino, N., Aravin, A., Pfeffer, S., Rice, A., Kamphorst, A. O., Landthaler, M., Lin, C., Socci, N. D., Hermida, L., Fulci, V., Chiaretti, S., Foa, R., Schliwka, J., Fuchs, U., Novosel, A., Muller, R. U., Schermer, B., Bissels, U., Inman, J., Phan, Q., Chien, M., Weir, D. B., Choksi, R., De Vita, G., Frezzetti, D., Trompeter, H. I., Hornung, V., Teng, G., Hartmann, G., Palkovits, M., Di Lauro, R., Wernet, P., Macino, G., Rogler, C. E., Nagle, J. W., Ju, J., Papavasiliou, F. N., Benzing, T., Lichter, P., Tam, W. Brownstein, M. J. Bosio, A. Borkhardt, A. Russo, J. J. Sander, C. Zavolan, M., and Tuschl, T. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414.
Lanford, R. E., Hildebrandt-Eriksen, E. S., Petri, A., Persson, R., Lindow, M., Munk, M. E., Kauppinen, S., and Orum, H. (2010). Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327, 198–201.
Lecellier, C. H., Dunoyer, P., Arar, K., Lehmann-Che, J., Eyquem, S., Himber, C., Saib, A., and Voinnet, O. (2005). A cellular microRNA mediates antiviral defense in human cells. Science 308, 557–560.
Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., Lee, J., Provost, P., Radmark, O., Kim S., and Kim, V. N. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419.
Lee, Y., Hur, I., Park, S. Y., Kim, Y. K., Suh, M. R., and Kim, V. N. (2006). The role of PACT in the RNA silencing pathway. EMBO J. 25, 522–532.
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20.
Li, Y., Chan, E. Y., Li, J., Ni, C., Peng, X., Rosenzweig, E., Tumpey, T. M., and Katze, M. G. (2010). MicroRNA expression and virulence in pandemic influenza virus-infected mice. J. Virol. 84, 3023–3032.
Liou, L. Y., Herrmann, C. H., and Rice, A. P. (2002). Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J. Virol. 76, 10579–10587.
Lodish, H. F., Zhou, B. Liu, G., and Chen, C. Z. (2008). Micromanagement of the immune system by microRNAs. Nat. Rev. Immunol. 8, 120–130.
Lund, E., Guttinger, S., Calado, A., Dahlberg, J. E., and Kutay, U. (2004). Nuclear export of microRNA precursors. Science 303, 95–98.
Macrae, I. J., Zhou, K. Li, F., Repic, A., Brooks, A. N., Cande, W. Z., Adams, P. D., and Doudna, J. A. (2006). Structural basis for double-stranded RNA processing by Dicer. Science 311, 195–198.
Matskevich, A. A., and Moelling, K. (2007). Dicer is involved in protection against influenza A virus infection. J. Gen. Virol. 88, 2627–2635.
Morris, K. V., and Rossi, J. J. (2006). Lentiviral-mediated delivery of siRNAs for antiviral therapy. Gene Ther. 13, 553–558.
Muljo, S. A., Ansel, K. M., Kanellopoulou, C., Livingston, D. M., Rao, A., and Rajewsky, K. (2005). Aberrant T cell differentiation in the absence of Dicer. J. Exp. Med. 202, 261–269.
O’Connell, R. M., Rao, D. S., Chaudhuri, A. A., and Baltimore, D. (2010). Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 10, 111–122.
Otsuka, M., Jing, Q. Georgel, P., New, L., Chen, J., Mols, J., Kang, Y. J., Jiang, Z., Du, X., Cook, R., Das, S. C. Pattnaik, A. K., Beutler, B., and Han, J. (2007). Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 27, 123–134.
Pedersen, I. M., Cheng, G., Wieland, S., Volinia, S., Croce, C. M., Chisari, F. V., and David, M. (2007). Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449, 919–922.
Peng, G., Greenwell-Wild, T., Nares, S., Jin, W., Lei, K. J., Rangel, Z. G., Munson, P. J., and Wahl, S. M. (2007). Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 110, 393–400.
Perez, J. T., Pham, A. M., Lorini, M. H., Chua, M. A., Steel, J., and tenOever, B. R. (2009). MicroRNA-mediated species-specific attenuation of influenza A virus. Nat. Biotechnol. 27, 572–576.
Peters, L., and Meister, G. (2007). Argonaute proteins: mediators of RNA silencing. Mol. Cell 26, 611–623.
Rossi, J. J., June, C. H., and Kohn, D. B. (2007). Genetic therapies against HIV. Nat. Biotechnol. 25, 1444–1454.
Sarasin-Filipowicz, M., Krol, J., Markiewicz, I., Heim, M. H., and Filipowicz, W. (2009). Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat. Med. 15, 31–33.
Selbach, M., Schwanhausser, B., Thierfelder, N., Fang, Z., Khanin, R., and Rajewsky, N. (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63.
Sohn, S. Y., Bae, W. J., Kim, J. J., Yeom, K. H., Kim, V. N., and Cho, Y. (2007). Crystal structure of human DGCR8 core. Nat. Struct. Mol. Biol. 14, 847–853.
Sung, T. L., and Rice, A. P. (2009). miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 5, e1000263. doi:10.1371/journal.ppat.1000263.
Triboulet, R., Mari, B., Lin, Y. L., Chable-Bessia, C., Bennasser, Y., Lebrigand, K., Cardinaud, B., Maurin, T., Barbry, P., Baillat, V., Reynes, J., Corbeau, P., Jeang, K. T., and Benkirane, M. (2007). Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 315, 1579–1582.
Tsunetsugu-Yokota, Y. (2008). “Transmission of HIV from dendritic cells to CD4+ T cells: a promising target for vaccination and therapeutic intervention,” in AIDS Vaccines, HIV Receptors and AIDS Research, ed. L. B. Kendow (New York: Nova Science Publishers, Inc.), 117–128.
Wang, X., Ye, L., Hou, W., Zhou, Y., Wang, Y. J., Metzger, D. S., and Ho, W. Z. (2009). Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 113, 671–674.
Wang, Y., Sheng, G., Juranek, S., Tuschl, T., and Patel, D. J. (2008). Structure of the guide-strand-containing argonaute silencing complex. Nature 456, 209–213.
Winter, J., Jung, S., Keller, S., Gregory, R. I., and Diederichs, S. (2009). Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 11, 228–234.
Wu, H., Neilson, J. R., Kumar, P., Manocha, M., Shankar, P., Sharp, P. A., and Manjunath, N. (2007). miRNA profiling of naive, effector and memory CD8 T cells. PLoS One 2, e1020. doi:10.1371/journal.pone.0001020.
Xiao, C., and Rajewsky, K. (2009). MicroRNA control in the immune system: basic principles. Cell 136, 26–36.
Yamamoto, T., and Tsunetsugu-Yokota, Y. (2008). Prospects for the therapeutic application of lentivirus-based gene therapy to HIV-1 infection. Curr. Gene Ther. 8, 1–8.
Keywords: mammalian microRNAs, RNA virus infection, host–virus interaction, gene therapy
Citation: Tsunetsugu-Yokota Y and Yamamoto T (2010) Mammalian microRNAs: post-transcriptional gene regulation in RNA virus infection and therapeutic applications. Front. Microbio. 1:108. doi: 10.3389/fmicb.2010.00108
Received: 22 July 2010;
Paper pending published: 01 August 2010;
Accepted: 10 August 2010;
Published online: 13 October 2010.
Edited by:
Hironori Sato, National Institute of Infectious Diseases, JapanReviewed by:
Akio Kanai, Keio University, JapanTakamasa Takeuchi, National Institute of Infectious Diseases, Japan
Copyright: © 2010 Tsunetsugu-Yokota and Yamamoto. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Yasuko Tsunetsugu-Yokota, Department of Immunology, National Institute of Infectious Diseases, 1-23-1 Toyama, Shinjuku, Tokyo 162-8640, Japan. e-mail:eXlva290YUBuaWguZ28uanA=