

- Centre for Molecular Microbiology and Infection, Division of Cell and Molecular Biology, Imperial College London, London, UK
An inhibitor of host cell lysophospholipid acyltransferase, an enzyme involved in lipid metabolism blocked growth of the obligate intracellular pathogen Chlamydia through its action on the transport of transferrin (Tf) via the slow pathway of recycling. A detailed characterization of this inhibition revealed that Tf accumulated in vesicles positive for Rab11, with a concomitant reduction in the level of Tf found within the transport intermediate Rab4/11 hybrid vesicles. The net result was the failure to be recycled to the plasma membrane. In chlamydiae-infected cells, the Tf-containing Rab11-positive vesicles were typically found intimately associated with the inclusion, and treatment with the inhibitor caused their accumulation, suggesting that the timely progression and completion of Tf recycling was necessary for proper chlamydial growth. Growth inhibition by the compound could be negated by the simple removal of the Tf-containing fraction of the serum, a further indication that accumulation of Tf around the chlamydial inclusion was deleterious to the pathogen. Thus, it appears that manipulating the slow recycling pathway can have biological consequences for Chlamydia and implies the need to regulate carefully the interaction of the inclusion with this host trafficking pathway.
Introduction
Chlamydia is a developmentally regulated bacterium that resides within its target host cell in a pathogen-defined protective niche called an inclusion (Hackstadt, 2000). Chlamydiae do not acquire markers of the endocytic pathway but rather appear to intercept exocytic traffic as demonstrated by the acquisition of sphingomyelin (SM) en route from the trans-Golgi network to the plasma membrane (Hackstadt et al., 1996). Furthermore, the chlamydial inclusion displays selectivity in its interactions with the exocytic pathway by excluding vesicles containing glycoprotein cargo (Scidmore et al., 1996a). The significance of SM transport to the inclusion is unclear although a recent report by Heuer et al. (2009) demonstrated the role of infection-induced Golgi fragmentation in chlamydial growth, which correlated the fragmentation of the Golgi apparatus with enhanced SM transport to the inclusion. However, inhibiting this process had only modest effects on chlamydial growth.
Recent reports from a number of laboratories have begun to challenge the limited fusogenicity of the inclusion with vesicular transport pathways. Several Rab GTPases, including Rab4 and Rab11, have been localized to the inclusion membrane, and chlamydial proteins that interact with them have been identified (Rzomp et al., 2003, 2006; Cortes et al., 2007). In addition, lipid droplets have been found inside chlamydial inclusions (Cocchiaro et al., 2008), thus the repertoire of organelles and vesicles with which Chlamydia interacts is likely to grow.
The observation that Tf-containing vesicles localized around the inclusion throughout the chlamydial developmental cycle has been repeatedly confirmed (Scidmore et al., 1996b, 2003; van Ooij et al., 1997; Al-Younes et al., 1999; Rzomp et al., 2003). However, the significance to Chlamydia of this association has not been elucidated. Expression of dominant-negative mutants and knockdown by RNA interference to interfere with Rab4 function failed to reveal any role in chlamydial growth. Recently, the Rab11 GTPase, which functions in the slow pathway of Tf recycling, was implicated in chlamydial growth but at the level of the Golgi-inclusion interaction (Rejman Lipinski et al., 2009). Without any data that correlates inhibition of the transferrin (Tf) recycling pathway with abrogation of chlamydial growth, the significance of the localization of these vesicles relative to the inclusion remains uncertain.
Transferrin recycling could be divided into two pathways based on the kinetics of recycling to the plasma membrane. Approximately 80–90% of internalized Tf are recycled through the fast/bulk pathway within 30 min after internalization; and the recycling Tf transits through vesicular compartments positive for Rab5 and/or Rab4 GTPases. The remaining population of Tf is retained much longer in Rab4 and/or Rab11 positive vesicles within the endocytic recycling compartment, and has a longer duration (60–90 min) of recycling (Sönnichsen et al., 2000). Given this added complexity of Tf recycling, we sought to examine the involvement of both pathways in chlamydial growth.
Here, we describe the characterization of a potent inhibitor of Tf recycling with regards to its effect on chlamydial growth. A screen of a small molecule library identified this compound as a potent inhibitor of chlamydial growth. The inhibitor (FR179254) targeted both the enzyme acyl-CoA:cholesterol acyltransferase (ACAT) and lysophospholipid acyltransferase (LPAT). The demonstrated lack of anti-chlamydial activity of an ACAT-specific inhibitor (Sandoz 58-035) or the normal growth of chlamydia in ACAT-deficient cell lines indicated that the relevant activity was indeed LPAT. This was confirmed by the use of the CI-976 compound, which has been characterized to specifically inhibit LPAT (Chambers and Brown, 2004). FR179254 targeted the transport of Tf through the Rab4/Rab11 double-positive vesicle, which is an intermediate transport compartment of the slow recycling pathway. The net result was the accumulation of Tf in Rab11-positive vesicles. This inhibitor-induced Tf accumulation was observed in both infected and uninfected cells. Interestingly, we found that the removal of Tf-containing fractions from the serum in an attempt to prevent accumulation rendered FR179254 ineffective in inhibiting chlamydial growth thus linking abnormal Tf recycling with chlamydia development. Furthermore, both Tf accumulation and inhibition of chlamydial growth could be mimicked partially by overexpressing the dominant-negative mutants of Rab4 and Rab11, confirming a role for these Rab GTPases in this process. The observed biological link between Tf-containing vesicles and the chlamydial inclusion reveals a different picture of the inclusion – a pathogen-defined organelle that is highly interactive with host vesicular traffic.
Materials and Methods
Organisms and Cell Culture
Chlamydia trachomatis L2 EBs were harvested from infected HEp-2 or HeLa cell cultures at 37°C with 5% CO2, purified by discontinuous density gradient centrifugation in Renografin (Bracco Diagnostics, Princeton, NJ, USA), and titered for infectivity as measured by inclusion forming units (IFU). Escherichia coli MG1655 and the temperature-sensitive ΔplsC (Coli Genetic Stock Center, Yale University, New Haven, CT, USA) bacteria were cultured on LB plates or broth using standard bacterial culture protocols with the mutant being routinely cultured at 30°C. HEp-2, HeLa, COS7, CHO, or AC29 (a kind gift of Dr. Chang, Dartmouth University) cells were routinely cultivated at 37°C with 5% CO2 in IMDM supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 10 μg/mL gentamicin (all from Invitrogen, Carlsbad, CA, USA). For some experiments, IMDM lacking serum (iIMDM) was used in the presence or absence of FR179254 (Calbiochem, La Jolla, CA, USA). Serum fractionations were performed sequentially using 50 or 100 kDa cut-off filters (Millipore) to obtain three fractions representing less than 50 kDa, 50–100 kDa, and greater than 100 kDa. Fractions were analyzed by standard SDS-PAGE techniques and Western blot using a rabbit polyclonal anti-Tf antibody (Abcam, Cambridge, UK), and compared to total serum and purified holo-Tf (Sigma, St. Louis, MO, USA). Each fraction was normalized for volume and subsequently added to iIMDM to a final concentration of 10%.
Quantification of IFUs from Infected Cell Cultures
HEp-2 cells were plated in 6-well culture plates at a density of 106 cells per well. In a subset of wells, cells were plated onto glass cover slips for immunofluorescence microscopy. Approximately 18 h later, cells were infected at an MOI of 1 by direct addition to each well and left untreated or treated with 20 μM FR179254, 20 μM U18666a (Calbiochem, La Jolla, CA, USA), 5 μM Triacsin C, 50 μM CI-976, or 5 μg/mL Sandoz 58-035 (all from Sigma, St. Louis, MO, USA) and infected cells were incubated at 37°C with 5% CO2. For oleic acid experiments, cells were treated with 300 μM oleic acid (Sigma) at the time of plating or during reactivation as indicated. For recovery experiments, cells were cultured in serum-free IMDM or IMDM supplemented with serum fractions in the presence and absence of FR179254. For all samples, addition of inoculum to wells marks the time of infection (t = 0 h). Medium was aspirated from the treatment samples of infected cells at 24 h p.i., and 1 mL of SPG was added to each well. Cells were scraped and collected from each well into a 1.5-mL microfuge tube with three glass beads. Samples were vortexed for 45 s and frozen at −80°C. Samples were titered for infectivity on fresh cell layers as described elsewhere to quantify the number of IFU per well (Ouellette et al., 2006). At 24 h p.i., cells plated on cover slips were washed with PBS, fixed with methanol for 10 min, and carefully washed three times with PBS plus 0.025% sodium azide. Infected cultures were stained with primary human polyclonal antibody (Ab) against chlamydiae and secondary Ab goat anti-human Alexa488 (Invitrogen). Immunofluorescence was viewed on an Olympus Fluoview 500 Laser Scanning Microscope.
Fluorescent Microscopy
For all of these assays, HEp-2 cells were plated at 105 cells per well on 10 mm cover slips in 24-well tissue culture plates. Cells were processed as described after 24 h infection and/or treatment with drugs and visualized by confocal microscopy except where indicated. For visualization of lipid droplets, live cells were incubated in the presence of 1 μg/mL Bodipy 493/503 (Invitrogen) under normal culture conditions for approximately 10 min, rinsed 3× with HBSS, and viewed live on a Zeiss Axio Imager.M1 deconvolution microscope. For fluid-phase uptake, live cells were incubated in 50 μg/mL dextran-647 (10000 MW, fixable; Invitrogen) for 2 h, rinsed 3× with HBSS, fixed in paraformaldehyde, and imaged. All images were acquired using the same exposure settings on the microscope. CD63-GFP cells were a kind gift of Dr. Frank Dorsey (Scripps FL). Cells were infected and treated as indicated and viewed live 24 h p.i. The GFP signal was verified to co-localize with a CD63 Ab (data not shown). For SM labeling, control or pre-treated cells were pulse-labeled with 5 μM BodipyFL-ceramide complexed with defatted BSA (dfBSA) (Invitrogen) diluted in IMDM without supplements for 30 min at 4°C, washed 3× with HBSS, and chased for 1 h at 37°C in back-extraction medium (IMDM + 1% dfBSA) with or without drugs as indicated. Cells were then imaged live to visualize inclusions and Golgi. For retrograde trafficking, control cells or cells pre-treated with drugs were labeled with 10 μg/mL of Alexa488 labeled cholera toxin subunit B (CTxB-488; Invitrogen) diluted in IMDM medium without any supplements. CTxB-488 was added to cells for 1 h at 4°C to pulse-label cell membranes, washed 3× with HBSS, and chased by incubating for 1 h at 37°C in medium with or without drugs as indicated. Separate samples were fixed in paraformaldehyde after the pulse and chase and imaged.
Transfections
PCR-amplified LAMP-1 sequences were inserted into pEYFP-N1 (CLONTECH Laboratories, Mountain View, CA, USA) to create YFP–LAMP-1 (from Dr. Joel Swanson: Henry et al., 2004). Human cDNA to CD4 cloned into pCMV6-XL5 was obtained from Origene (Rockville, MD, USA). Rab4-GFP (wild-type and dominant-negative) was a kind gift of Dr. Marci Scidmore (Cornell University: Rzomp et al., 2006). The 2x-FYVE-GFP construct was a kind gift of Dr. Jean Celli (Rocky Mountain Labs, NIAID). Rab11 and Rab5 constructs were obtained from Addgene. One microgram of plasmid was transfected into cells using Fugene 6 (Roche, Indianapolis, IN, USA) according to the manufacturer’s directions. Briefly, control or pre-treated cells were incubated in Opti-MEM (Invitrogen). One microgram of plasmid DNA was mixed with Fugene 6 reagent diluted in Opti-MEM for 30 min and then added to cells for 4 h. Medium with or without drugs was added to cells, and cultures were incubated overnight. The following day, cells were fixed in paraformaldehyde, in some cases after a pulse with fluorescently tagged Tf and chased as described. In most cases, cells were visualized directly. For CD4, non-permeabilized cells were incubated with primary Ab mouse anti-human CD4 (R&D Systems, Minneapolis, MN, USA) followed by secondary goat anti-mouse Alexa488 (Invitrogen). The antibody stained cells were then visualized. For the dominant-negative studies, inclusion sizes within infected cells were quantified using ImageJ software (NIH, Bethesda, MD, USA).
Transferrin Recycling
Transferrin conjugated to Alexa488 or Alexa647 (Tf488 or Tf647, respectively; Invitrogen) was diluted to 50 μg/mL in IMDM without supplements. Control or pre-treated cells were pulse-labeled with 250 μL of Tf488/Tf647 medium for 1 h at 4°C, washed 3× with HBSS, and chased by incubating at 37°C in medium with or without drugs. At the indicated times, cells were fixed with 4% paraformaldehyde overnight at 4°C and viewed by confocal microscopy. Images were taken from multiple fields of view across the cover slip using the same exposure settings on the microscope and analyzed for total fluorescence intensity within the field of view using ImageJ software.
Assay for Inhibition of plsC
The plsC mutant phenotype has been characterized by Coleman as showing a growth defect at 42°C (Coleman, 1990). Wild-type E. coli MG1655 or the ΔplsC strain from a single colony were grown overnight at 37°C or 30°C in 5 mL LB broth, respectively. The following day, bacteria were quantified by OD600 and diluted in 5 mL LB broth to normalize bacterial numbers. Cultures were grown for 4 h to ensure mid-log phase growth and then sub-cultured again (after normalizing bacterial numbers) in 5 mL broth in the presence of 100 μM FR179254 or CI-976, or 50 μg/mL 58-035, or 34 μg/mL chloramphenicol, or 0.5% DMSO and grown for up to 8 h at either 37°C/30°C or 42°C. OD600 readings were taken to quantify bacterial growth.
Results
FR179254 Blocks Chlamydial Growth
We screened a library of low-molecular weight compounds for their ability to abolish chlamydial growth. One of the compounds identified from the screen was a known inhibitor of the host ACAT, FR179254 (Tanaka et al., 1998). A more detailed characterization of its anti-chlamydial activity showed FR179254 at 20 μM to dramatically inhibit growth of the rapidly growing C. trachomatis serovar L2, resulting smaller and less numerous inclusions and lower yields of IFU assay (Figure 1A). In contrast, the unrelated compound U18666a, which affects a number of transport pathways due to its effects on cellular cholesterol homeostasis (Higgins et al., 1999), did not have observable effects on the inclusion phenotype and only minimal effects on IFU yield. This indicated that the inhibition of chlamydial growth may be specific to FR179254. The activity of FR179254 was verified biochemically by monitoring the levels of unesterified cholesterol that would result from inhibition of the ACAT enzyme by the compound. U18666A did not show inhibitory activity toward cholesterol ester synthesis. In testing other ACAT inhibitors, we found the structurally unrelated CI-976, which has been reported to inhibit the host cell LPAT (Chambers and Brown, 2004), to also block chlamydial growth to a similar extent as FR179254 (Figure 1A). We concluded, therefore, that FR179254, like CI-976, is blocking chlamydial growth through its inhibitory action on LPAT.
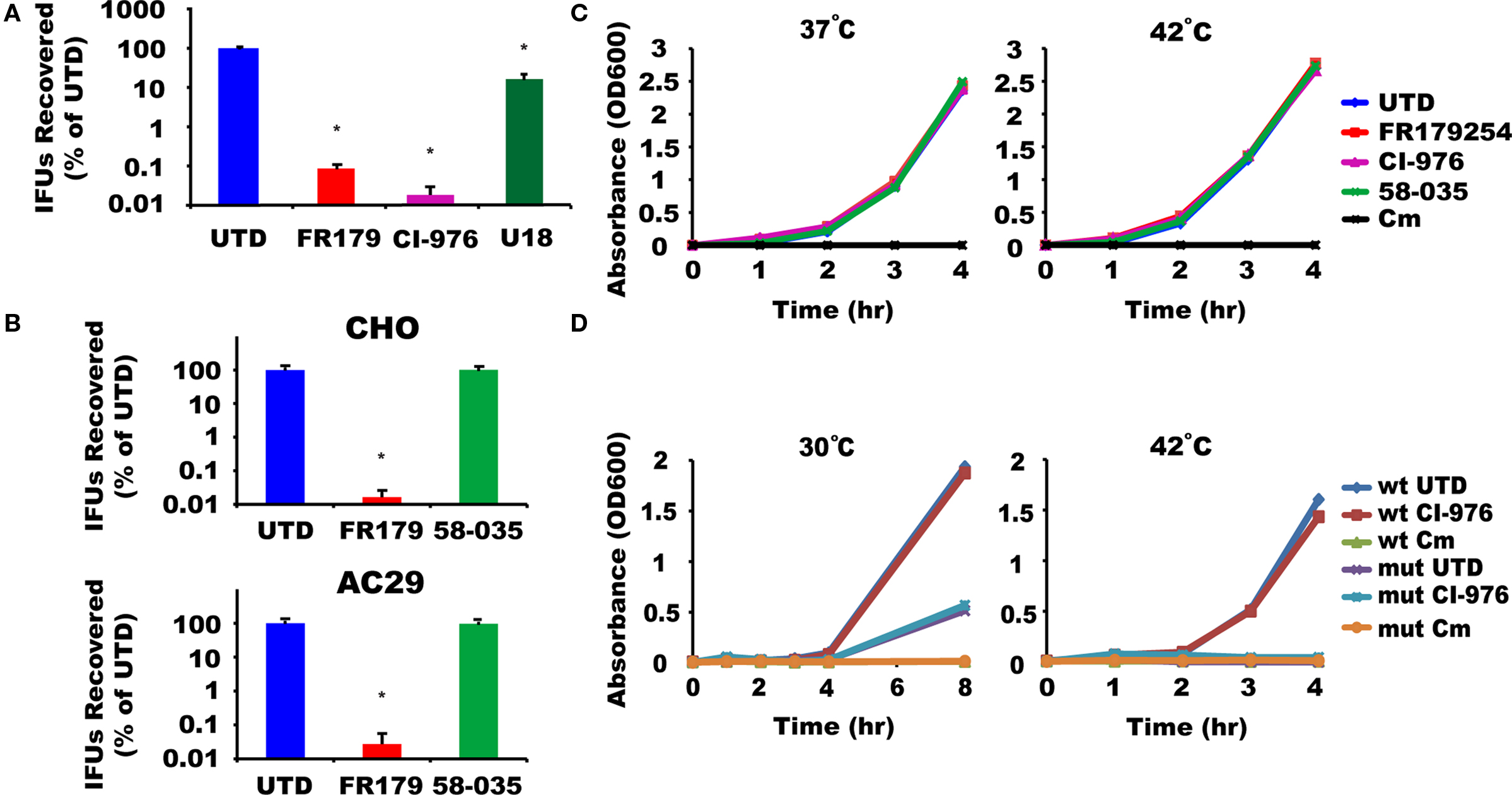
Figure 1. FR179254 blocks chlamydial growth by targeting host cell LPAT and not ACAT or bacterial enzymes. (A) FR179254 (FR179) and CI-976 block chlamydial growth. Effect of indicated treatments on recovery of inclusion forming units (IFUs), a measure for infectious EBs. (B) Chlamydiae grow in the absence of ACAT activity. CHO or AC29 cells, a CHO cell mutant which lacks ACAT activity, were infected with Chlamydia trachomatis serovar L2 and treated with or without FR179254 or the ACAT inhibitor 58-035 and recovery of infectious EBs was monitored after 24 h infection. (C) Assay for LPAT inhibition by compounds in Escherichia coli. Wild-type E. coli MG1655 were grown in broth for 4 h in the presence or absence of LPAT and ACAT inhibitors or chloramphenicol (Cm) as indicated, and growth was monitored by OD600 at 37°C or 42°C. (D) Assay for effect of compounds on ΔplsC mutant. Wild-type or mutant bacteria were grown in broth for up to 8 h in the presence or absence of the LPAT inhibitor CI-976 or chloramphenicol and growth monitored by OD600 at 30°C or 42°C.
FR179254 Abrogates Chlamydial Growth Independently of ACAT Activity
We wanted to further exclude host cell ACAT activity in chlamydial growth. To examine this, we used two approaches – comparing different compounds with established specificity and the use of a mutant Chinese hamster ovary (CHO) cell derivative that lacks significant ACAT activity (AC29) (Cadigan et al., 1988). Firstly, using wild-type CHO cells, we monitored chlamydial growth in cells treated with a specific ACAT inhibitor, Sandoz 58-035. This compound had no effect on chlamydial growth in contrast to FR179254 (Figure 1B). Secondly, the CHO mutant AC29 cells remained competent in supporting chlamydial growth. As with CHO cells, 58-035 had no effect on chlamydial growth in AC29 cells whereas FR179254 severely blocked growth in an ACAT-deficient background (Figure 1B). These data give further support to LPAT, and not ACAT, as the target of FR179254.
Prokaryotic LPAT-Like Enzymes rae not a Target for FR179254
We wanted to disprove that chlamydiae were the target of the LPAT inhibitors. This is particularly important because chlamydiae have a predicted LPAT-like activity. In E. coli the homologous gene, which encodes the LPAT-like activity, is plsC (Zhang and Rock, 2008), and mutants of plsC show a growth defect at 42°C (Coleman, 1990). Because of the genetic intractability of chlamydiae, we took advantage of the E. coli surrogate experimental system, and assayed the effects of our various inhibitors during growth at both 37°C and 42°C, with the latter mimicking the temperature-sensitive phenotype of the ΔplsC mutant. We reasoned that if any of our inhibitors were targeting PlsC, then growth should be abrogated at 42°C. We cultured E. coli in the presence of 100 μM inhibitor (5× the concentration used to inhibit Chlamydia in vitro) or chloramphenicol as a negative control. As a reference, we also monitored the growth of the reference ΔplsC mutant at both temperatures. None of the inhibitors we tested, aside from chloramphenicol, was effective at blocking E. coli growth at either temperature as judged by OD readings and colony forming units (Figure 1C; data not shown). The ΔplsC mutant strain showed the expected growth inhibitory phenotype at the non-permissive temperature (Figure 1D). Consequently, it is unlikely that CI-976 or FR179254 are targeting the prokaryotic LPAT homolog.
FR179254 has no Discernible Effect on Lipid Droplets, Endocytic, Exocytic, or Retrograde Trafficking in Treated Cells
The chlamydial inclusion is a pathogen-designed organelle with limited interaction with host vesicular trafficking pathways. However, this image of limited fusogenicity is being challenged by a number of reports that describes inclusion interactions with select Rab GTPases (Cortes et al., 2007; Rzomp et al., 2003, 2006), which are important for vesicular traffic. Displaying a broader range of vesicular interactions, especially transport compartments that are nutrient-laden, would maximize the potential for robust growth. To determine how FR179254 inhibited chlamydial growth, we screened a number of intracellular trafficking pathways using fluorescent microscopic techniques. In particular we sought a phenotype unique to FR179254 by comparing it to untreated and U18666a-treated cells. To initiate these studies, we assessed lipid droplets in untreated and treated cells (Figure 2A). FR179254 treatment, in spite of its inhibitory effects on chlamydial growth did not result in a change in lipid droplet contents. Furthermore, inclusions were present in cells with no detectable lipid droplets. Thus, growth inhibition could not be attributed to effects of the compound on lipid droplets. A converse experiment was performed in which lipid droplets were induced in the presence of FR179254, in an effort to rescue chlamydial growth. We observed no such rescue when compared to the untreated group (Figure 3).
We continued our studies by examining endocytosis by monitoring fluid-phase uptake as measured by Alexa488-dextran labeling of endocytic compartments. In untreated cells, strong labeling could be seen whereas fluorescence was slightly reduced in both the FR179254- and U18666a-treated cells (Figure 2B). The similar effects on dextran labeling in the two samples did not correlate with the different effects of the two compounds on chlamydial growth, and thus, fluid-phase uptake could be excluded as the targeted process.
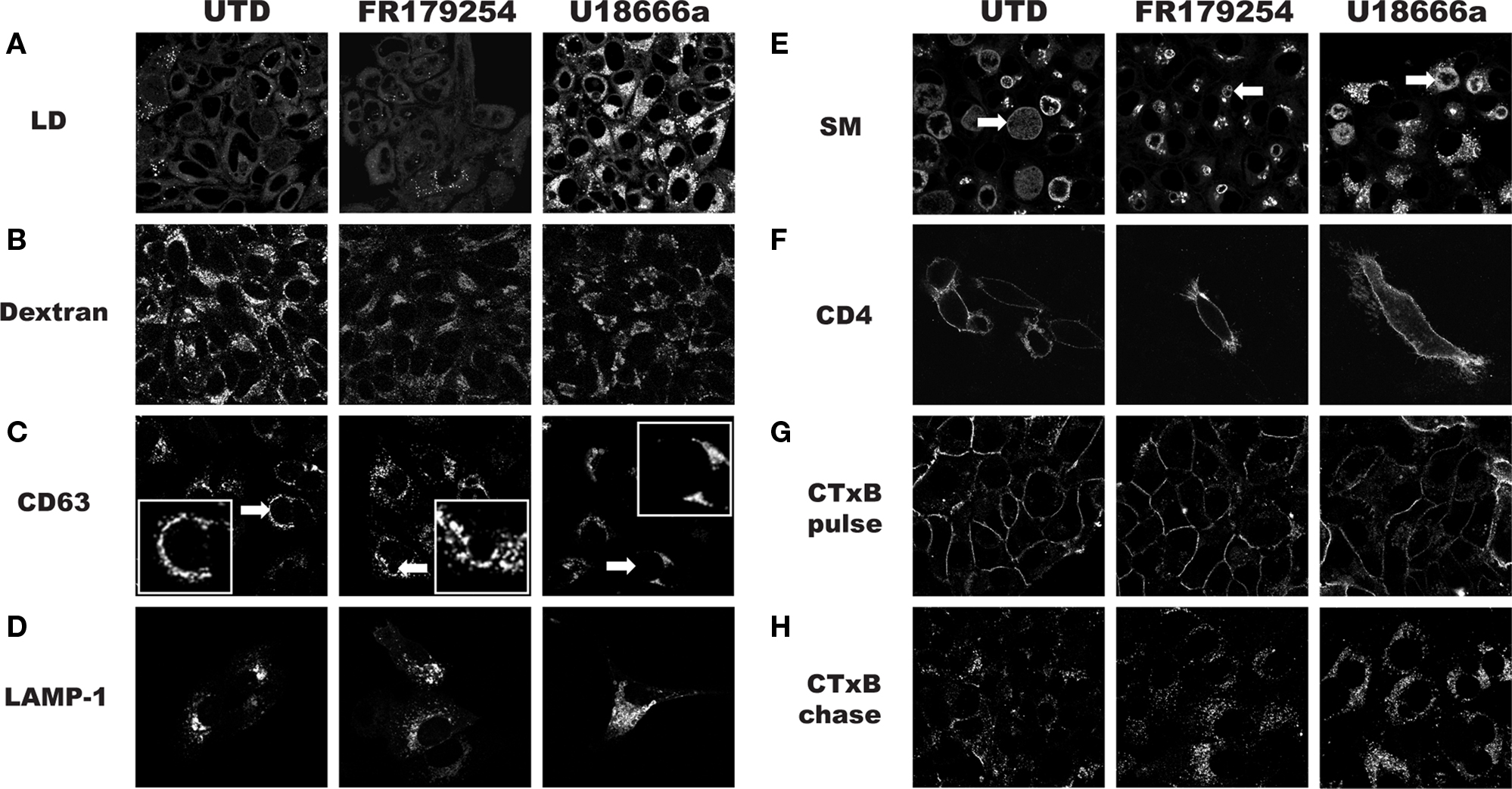
Figure 2. Analysis of host cell trafficking pathways during treatment with FR179254. HEp-2 cells were treated as indicated, and trafficking pathways were analyzed as described in Section “Materials and Methods.” (A) Lipid droplet (LD) visualization with the neutral lipid stain Bodipy 493. (B) Fluid-phase uptake as measured by fluorescent dextran. (C) Live cell visualization of multivesicular bodies/late endosomes using CD63-GFP expressing HEp-2 cells. (D) Analysis of late endosomes/lysosomes in LAMP1-YFP transfected cells. (E) Exocytic trafficking as measured by incorporation of fluorescent sphingomyelin (SM) in Golgi and chlamydiae. (F) Exocytic trafficking of glycoproteins as measured by localization of CD4 on the surface of transfected cells. (G,H) Analysis of retrograde trafficking using fluorescently tagged cholera toxin subunit B (CTxB) after pulse-chase labeling. Arrows within (C) and (E) indicate inclusions. Note the lack of significant staining in (C) (inset) and the presence of staining in (E), which is contrast to a published study (Beatty, 2006).
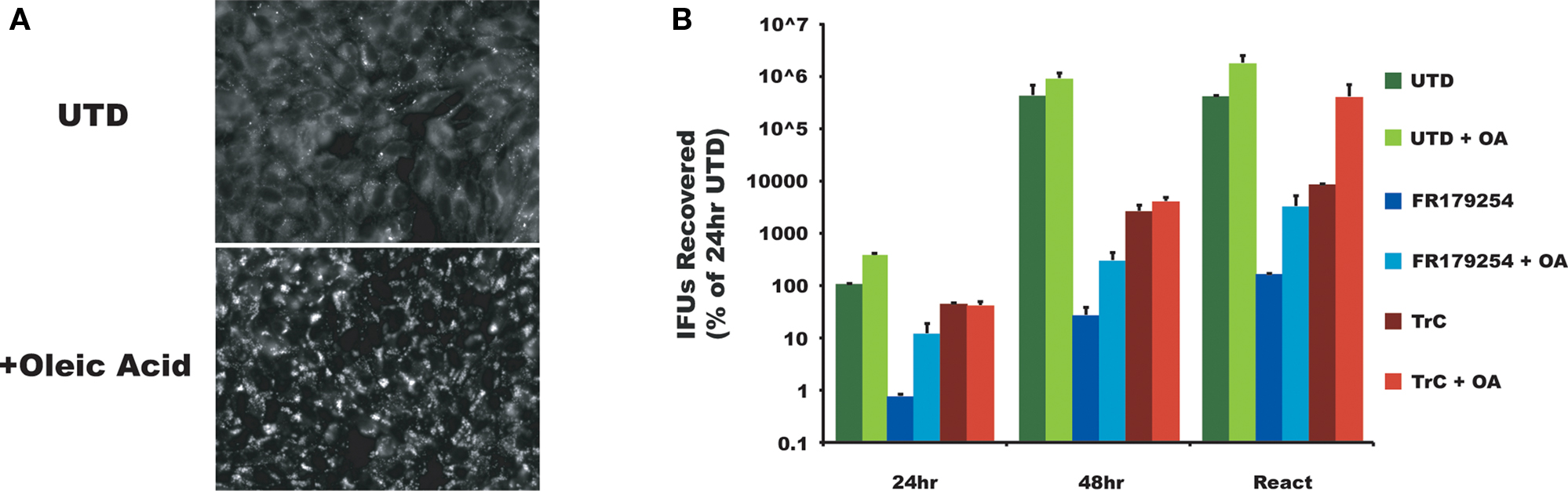
Figure 3. Excess oleic acid fails to reverse the effect of FR179254. Lipid droplets are not the targeted process of FR179254. Cells were cultured in normal medium or medium containing excess oleic acid (OA) to induce lipid droplets (A) and then infected and treated as indicated with either FR179254 or Triacsin C (TrC), a fatty acid synthase and lipid droplet inhibitor. (B) Chlamydial growth was assessed at 24 and 48 h post-infection. One set of samples were reactivated (React) by treating with indicated drugs for 24 h, washing three times, and then culturing in the presence or absence of excess oleic acid in fresh medium lacking drugs. Reactivated samples were collected after 24 h (48 h total culture time). All data were normalized to chlamydial growth in untreated cells at 24 h post-infection (arbitrarily 100%).
We next examined CD63-GFP expressing cells, as a marker for late endosome/multivesicular bodies, and LAMP1-transfected cells as a marker for the lysosomal compartment. We saw no obvious differences in the localization of these markers in FR179254-treated or untreated cells, but U18666a induced the accumulation of these markers (Figures 2C,D). Contrary to a published study (Beatty, 2006), we did not see significant CD63 localization in the chlamydial inclusion under any condition examined unless we fixed and permeabilized the cells following the methods of Beatty (2006; data not shown) for immunofluorescence confocal microscopy: our CD63-GFP cells were visualized live. We concluded from these experiments that the Chlamydia-relevant transport pathway targeted by FR179254 was unlikely to be the endocytic pathway.
We next assayed the exocytic pathway by monitoring the transport of sphingolipid and glycoprotein cargos to the inclusion and plasma membrane, respectively. Many studies have confirmed the observation that SM-containing vesicles are trafficked to the inclusion via a novel route that excludes glycoproteins (Scidmore et al., 1996a). To better aid visualization, we delayed treatment with drugs until 8 h post-infection to allow for inclusion development. When we monitored SM trafficking to the inclusion at 24 h post-infection in untreated and treated cells by live cell microscopy, we observed no defect in labeling of the chlamydiae (Figure 2E). This is in contrast to a report by Beatty (2006) that suggested U18666a blocked the trafficking of SM to the inclusion during observation of fixed cells. The reason for this discrepancy is not known and warrants further investigation, but may be explained by the difference in methodology vis-à-vis live cell imaging versus observation of fixed cells. To verify that trafficking of proteins to the cell membrane was not affected by treatment, we monitored CD4 or vesicular stomatitis virus G-glycoprotein (VSV-G) transfected, but uninfected cells (Figure 2F; data not shown). Transfected cells were pre-treated with drugs where indicated and maintained in the presence of drugs after transfection. CD4 and VSV-G was visualized on the cell surface without permeabilization of the cells. Here too, we saw no defects in labeling and concluded that FR179254 is not affecting the exocytic pathway in a way that impacts chlamydial growth.
Another prominent intracellular trafficking pathway is the retrograde transport, which allows for certain endocytosed molecules to be diverted to the exocytic pathway where they transit back to the ER. We examined the retrograde trafficking of fluorescently tagged cholera toxin subunit B (CTxB) in untreated and treated cells. After an initial pulse, cells showed good labeling of the membrane, regardless of the conditions used (Figure 2G). Cells were then chased in medium for 2 h, fixed, and viewed by confocal microscopy. We did not observe any noticeable differences between untreated and FR179254-treated cells (Figure 2H). We concluded that retrograde transport was unlikely to be the sought-after target of FR179254. It appears that FR179254 treatment did not result in any qualitative differences in the staining profile and morphology of the compartments within the endolysosomal, exocytic, and retrograde transport pathways.
FR179254 Induces a Delay in the Slow Transferrin Recycling Pathway
Because Tf has been localized to the chlamydial vacuole (Scidmore et al., 2003; van Ooij et al., 1997), we examined Tf recycling in treated and untreated cells. Cells were pulse-labeled with fluorescent Tf for 1 h at 4°C to prevent uptake, washed, and shifted to 37°C to allow uptake and recycling. Labeling of cells was unaffected by treatment. Surprisingly, FR179254 induced a marked retention of Tf compared to untreated or U18666a-treated cells that was both quantifiable and easily seen in fluorescent images (Figure 4A). We also found that Sandoz 58-035 and lipid droplet inhibitors had no effect on Tf recycling (data not shown). Thus, it seems that the growth inhibitory effects of FR179254 on chlamydiae are due to its ability to delay Tf recycling.
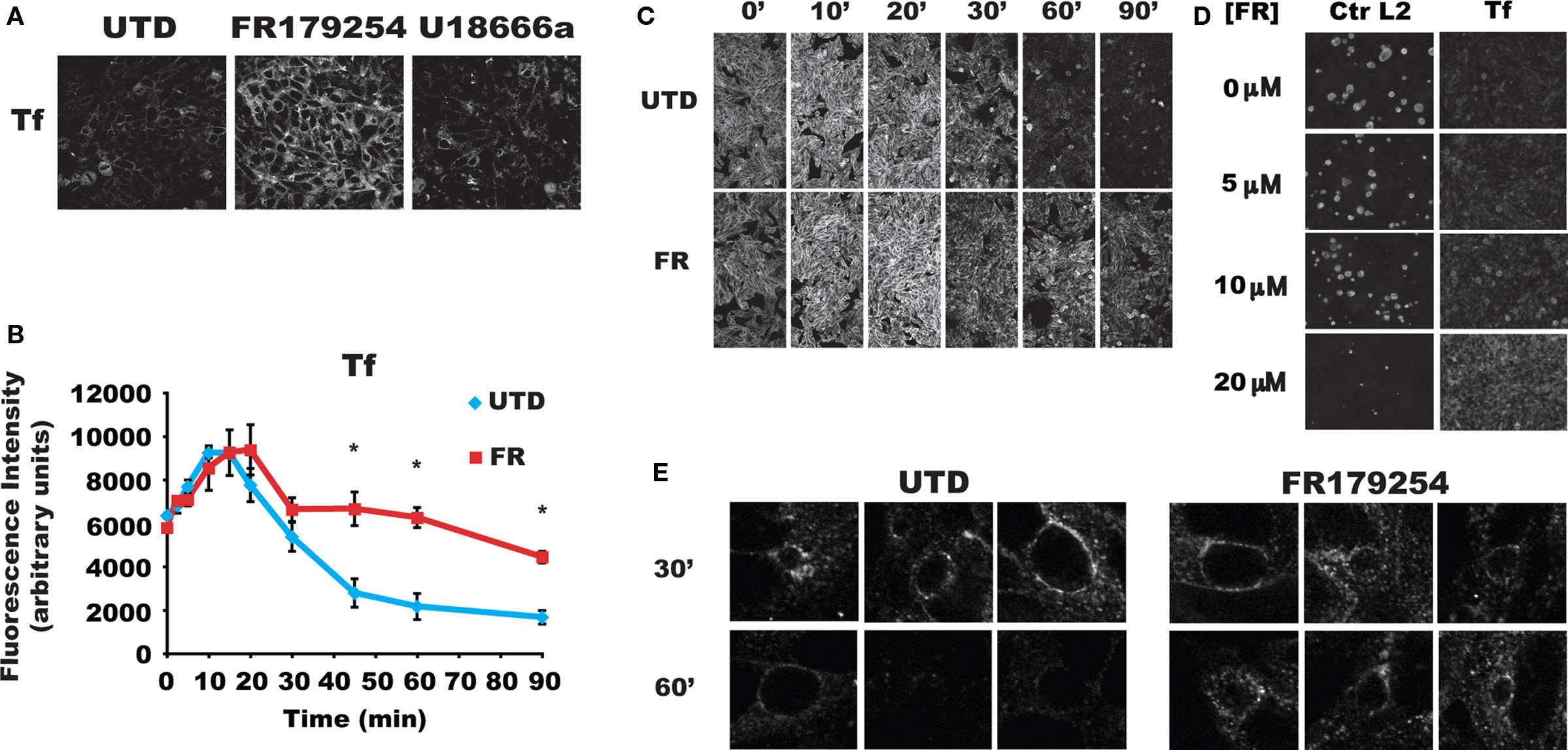
Figure 4. Slow transferrin recycling is delayed in FR179254 treated cells. HEp-2 cells were treated as indicated, pulsed with fluorescent transferrin, and chased for the times shown. (A) Fluorescent micrographs of transferrin pulse-chased cells from different conditions at 600× magnification acquired by laser scanning confocal microscopy using identical settings. (B) Quantification of transferrin fluorescence in treated cells measured over a time course during chase. (C) Representative images used for quantification. FR179254 shows no effect on transferrin recycling during the early (up to 30′) part of the chase but significantly delays recycling during the latter phases. “*” Indicates p < 0.005 compared to UTD calculated by Student’s t-test. (D) HEp-2 cells were infected and treated with different concentrations of FR179254 as indicated and processed for immunofluorescence of chlamydial inclusions after 24 h infection. Separate samples were treated as indicated and pulse-chased with fluorescent transferrin as described previously and imaged at 60′ chase time. Chlamydial inclusions were imaged at 400× magnification whereas transferrin staining was imaged at 100× magnification using the same settings on the microscope. Images were acquired on an epifluorescent microscope. (E) Transferrin is recruited to the inclusion periphery and remains there for an extended time during FR179254 treatment. Cells were infected and treated with or without FR179254 at 12 h post-infection to allow inclusions to develop for easy visualization. Cells were then pulse-chased with fluorescent transferrin as described at 24 h post-infection.
To further characterize the effects of FR179254 on Tf recycling, we monitored retention of fluorescent Tf within treated and untreated cells over a time course during the chase period. Based on the distinct differences in Tf retention of the untreated and FR179254-treated samples (Figure 4A), the use of fluorescent Tf rather than radioactively labeled ones was deemed sufficient and informative. Indeed, we observed that the bulk (approximately 80%) of endocytosed Tf is rapidly recycled back to the plasma membrane within 30 min whereas the remainder is recycled more slowly. Up to 30 min chase, there were no significant differences between retained fluorescence (Figures 4B,C), marking the initial uptake and bulk recycling of Tf through the fast recycling pathway. However, after 30 min, we noticed obvious, significant retention of labeled Tf within treated cells, which corresponds with the slow pathway of Tf recycling (Figures 4B,C). We concluded from these data that FR179254 specifically affects the transit of Tf through the slow recycling pathway while having no effect on the fast recycling pathway.
To correlate the delay in Tf recycling with chlamydial growth, we monitored chlamydial inclusion development and Tf recycling over a dose curve of FR179254. Chlamydial inclusion formation was significantly reduced at a concentration of 20 μM, accompanied by the marked retention of fluorescent Tf in parallel samples (Figure 4D). Lower concentrations failed to show an obvious microscopic effect on either chlamydial inclusions development or Tf retention, although 10 μM FR179254 inhibited chlamydial recovery by approximately 0.5 log (data not shown). An excellent concordance was seen between the effect of FR179254 on Tf recycling and chlamydial inclusion formation, supporting the notion that interference with the Tf recycling pathway was linked to chlamydial growth.
We next examined the effects of FR179254 treatment on the subpopulation of Tf that localize around the chlamydial inclusion. Would they be refractory to the inhibitor, and thus belong to the fast/bulk recycling, or would they be retained similarly as the Tf in the slow recycling pathway? We analyzed confocal images of treated and untreated infected cells. As others have reported (Al-Younes et al., 1999; Rzomp et al., 2006; Scidmore et al., 1996b, 2003; van Ooij et al., 1997), we could detect Tf around inclusions at 30 min chase time (Figure 4E). By 60 min, most of this staining was absent, indicating that the Tf had been recycled away from the inclusion, apparently with a kinetics consistent with that of slow recycling. Treatment with FR179254 resulted in the retention of Tf around the chlamydial inclusion at 30 and 60 min chase times (Figure 4E). Based on the susceptibility of this peri-inclusion Tf to FR179254 treatment, it is very likely that this Tf subpopulation belong to the slow pathway of recycling.
Multiple Influxes of Transferrin are Delayed by FR179254 Treatment
If a pulse of labeled Tf was found to be retained for a longer duration, it would be logical to assume that under steady state labeling or even growth in Tf-rich serum would result in accumulation. To investigate further the apparent retention of Tf, we performed a double-pulse-chase experiments using Tf conjugated to different fluorophores. Untreated and treated cells were initially pulsed with Alexa488-Tf, and after 30 min chase time, were pulsed again with Alexa647-Tf and chased for a further 60 min. We found that each fluorescence label was retained in FR179254-treated cells with similar kinetics (see Figure 4) whereas, in untreated or Sandoz 58-035-treated cells, the fluorescence associated with both Tf pulses was lost quite rapidly (Figure 5). These results are consistent with a chronic delay of Tf recycling through the slow pathway such that each wave of Tf endocytosed by the cell is similarly affected.
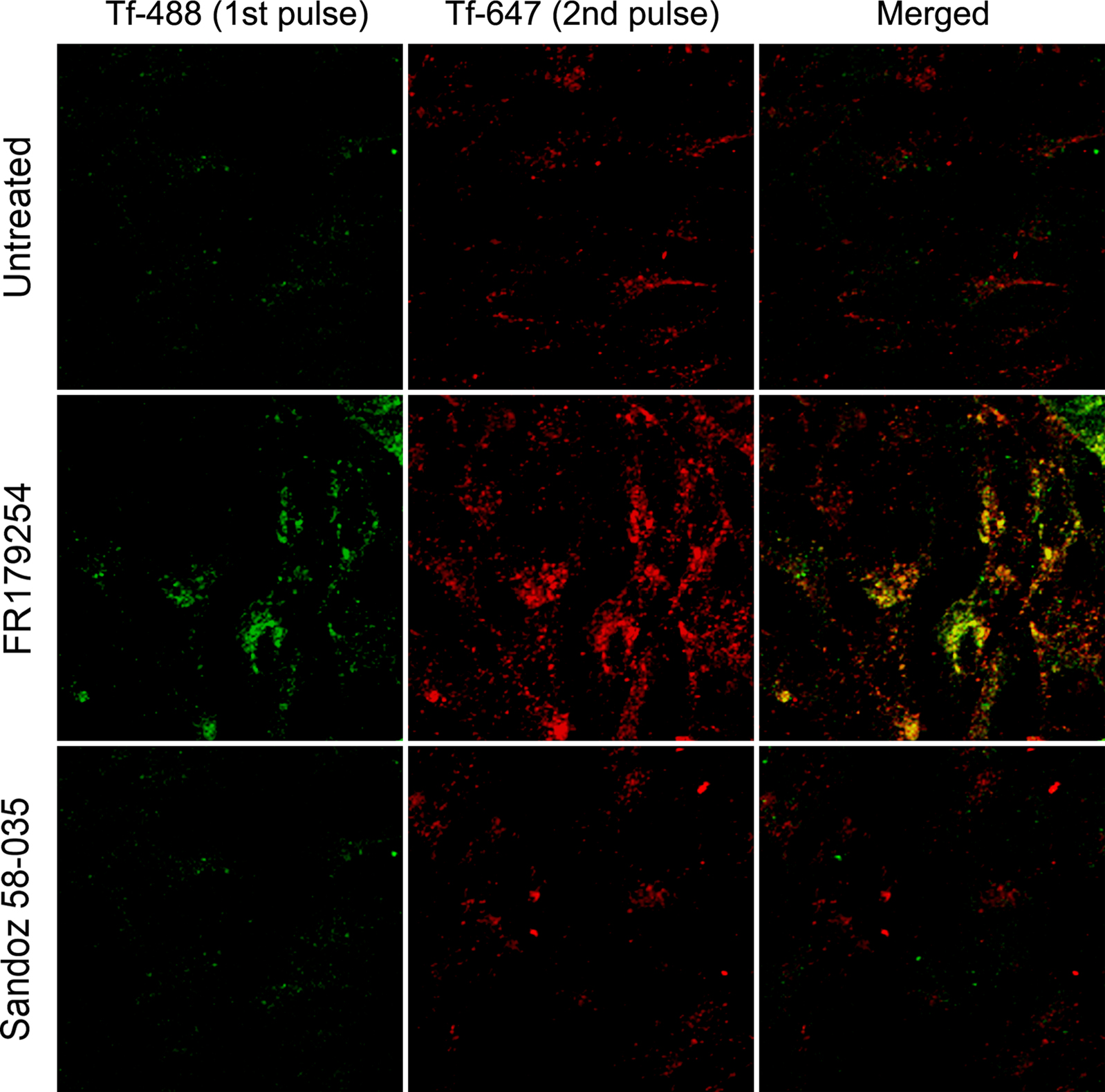
Figure 5. Successive waves of internalized transferrin are delayed by FR179254 treatment. Cells were treated as indicated and pulse-chased with Tf-488. At 30′ chase, cells were subsequently pulsed with Tf-647. After a total of 90′ chase time (60′ for Tf-647), cells were fixed and analyzed by confocal microscopy. Noticeable accumulation of both fluorescent labels is present in FR179254-treated but not Sandoz 58-035-treated or untreated cells.
FR179254 Treatment Inhibits the Transport of Transferrin to the Rab4/11 Hybrid Recycling Vesicle
The previously reported effects of CI-976 include delaying the recycling of Tf, and causing the accumulation of Tf in Rab11-positive compartments (Chambers et al., 2005). Therefore, we sought to determine if the same is true for FR179254. Also, we extended this observation by including in the analysis the Rab4/5 and Rab4/11 hybrid vesicle intermediates that may function in sorting the Tf/Tf receptor from the endosomal compartments for recycling to the plasma membrane via the fast and slow recycling pathways, respectively. Cells were transfected with one of three markers either individually or in combination with Rab4 (a marker for recycling vesicles): Rab5 or 2x-FYVE domain (early endosome markers) or Rab11 (late endosome/endocytic recycling compartment). Cells were infected and then pulse-chased with Tf and processed for confocal microscopy. The number of vesicles co-localizing with Tf were quantified.
In untreated cells, Tf-containing vesicles co-localizing with the singly transfected markers increase between the end of the Tf pulse and 30 min chase (Figure 6A) and decline to “starting” levels by 60 min chase (Figure 6B). A similar pattern was seen for Tf co-localizing with the FYVE domain in FR179254-treated cells. In contrast, Tf-positive Rab4 vesicles did not decrease after 30 min, and Tf-positive Rab11 vesicles accumulated over time (Figures 6A,B), consistent with published effects of LPAT inhibition (Chambers et al., 2005). To further characterize the compartment in which Tf was delayed, we monitored doubly transfected cells for Tf localization. There were no differences in Tf localization in Rab5/Rab4 doubly positive compartments between untreated or FR179254-treated cells (Figures 6C,D). Curiously, there was a marked decrease in the levels of Tf in FR179254-treated Rab4/Rab11 doubly positive compartments (Figures 6C,D). However, assessing singly transfected Rab11 positive cells within the same cell population revealed that this deficit in Tf/Rab4/Rab11 co-localization was compensated by an increase in the levels of Tf/Rab11 co-localization compared to untreated cells (Figure 6). These data indicated that there was a defect in the formation of the Tf-containing Rab4/11 hybrid vesicles. These same observations applied to infected cells, but enumeration of vesicles was difficult due to the vesicles around the inclusions being less distinct or sometimes appearing fused (data not shown). Our observation of Tf transiting through the Rab4/11 hybrid vesicle on its way to the plasma membrane concurs with the model of Tf recycling proposed (Sönnichsen et al., 2000).
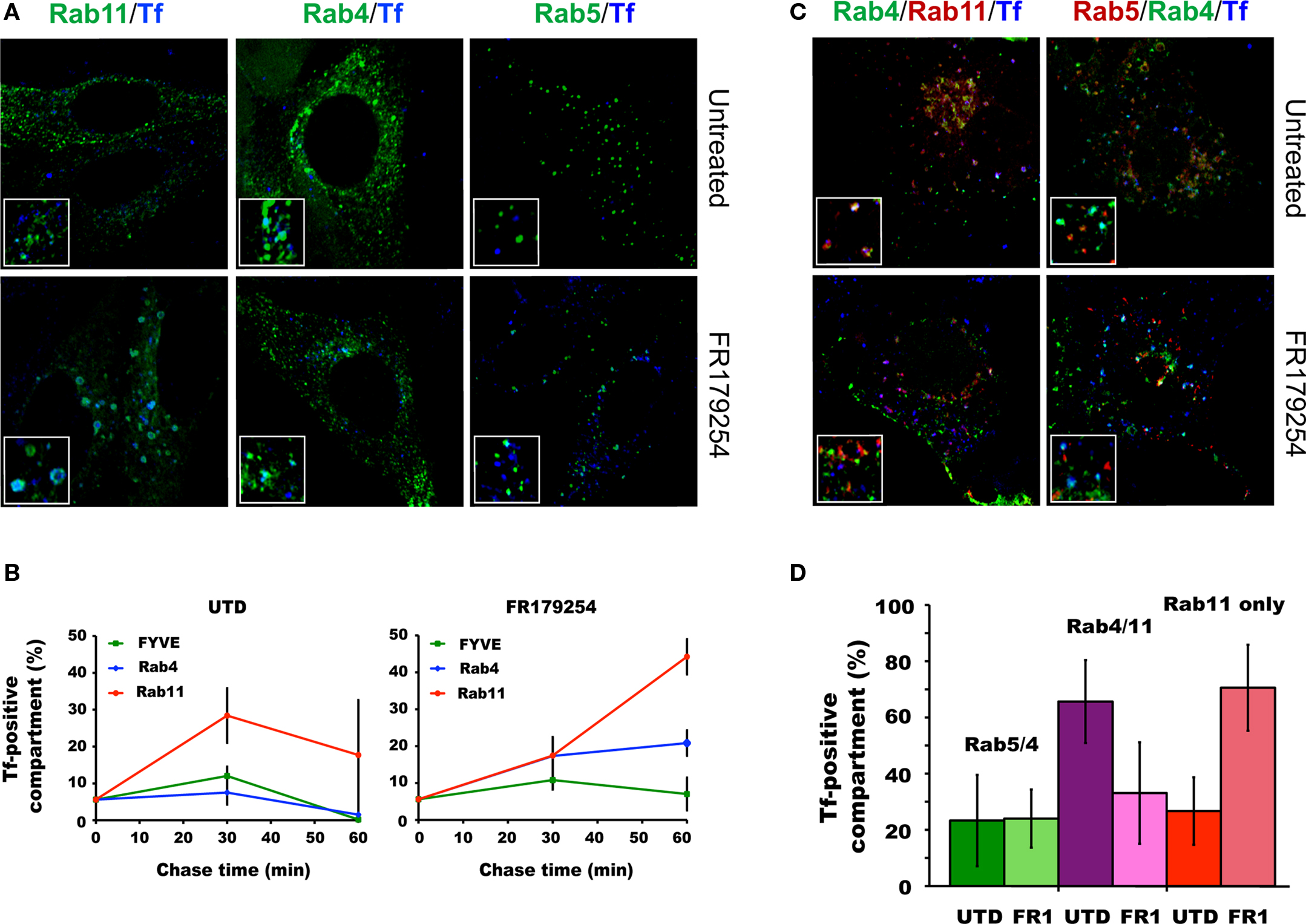
Figure 6. FR179254 causes a delay in the trafficking of transferrin from Rab11 compartments to Rab4/11 recycling compartments. Transfected cells were pulse-chased with fluorescent transferrin (Tf647) as described previously and monitored for co-localization with vesicles by confocal microscopy. Representative confocal images (A) and quantification (B) of co-localization of transferrin with individual markers in singly transfected cells in untreated or FR179254-treated conditions. Representative confocal images (C) and quantification (D) of co-localization of transferrin with indicated markers in doubly transfected cells.
Co-Expression of Dominant-Negative Constructs for RAB4 and RAB11 Causes Retention of Transferrin and Delays Inclusion Development
Previous reports investigating the ability of dominant-negative Rabs to inhibit chlamydial growth failed to show any biologically significant effects (Rzomp et al., 2006; Rejman Lipinski et al., 2009). Possible explanations for this include the compensatory functions provided by the wild-type, endogenous proteins and/or the ability of chlamydial proteins to selectively interact with them (Rzomp et al., 2006). Given the apparent involvement of both Rab4 and Rab11 in the slow recycling pathway, we speculated that co-expression of both dominant-negative Rab4 and Rab11 may lead to Tf retention and chlamydial growth inhibition. Cells were transfected with both constructs, infected, and fixed after 24 h. Tf retention and inclusion sizes were respectively monitored and measured in untransfected, singly transfected, and doubly transfected cells within the same population. For the Tf experiment, chlamydiae-infected cells were incubated with Alexa647-Tf for a 1-h pulse, and chased for 90 min. Figure 7A displays a representative visual field with cells harboring chlamydial inclusions (white arrowhead). Only cells that expressed both the dominant-negative forms of Rab4 and Rab11 specifically retained Tf that circumscribed the inclusions (p < 0.002; chi-square test) whereas expression of Rab4 or Rab11 DN only led to a mild retention of Tf fluorescence and untransfected cells showed very little Tf retained around the inclusion. Thus, much like FR179254, the co-expression of the dominant-negative mutants of Rab4 and Rab11 led to relatively higher levels of retained Tf when compared to untransfected cells.
As shown previously, singly transfected cells expressing either Rab4 or Rab11 dominant-negative GTPases failed to significantly reduce chlamydial inclusion size, a correlate for IFU output. Similarly, the dominant-negative constructs failed to circumferentially stain the inclusion. However, inclusion sizes in cells expressing both dominant-negative GTPases were significantly reduced albeit not to the same extent as for FR179254-treated cells (Figure 7B). In contrast to co-expression, the lone expression of each dominant-negative mutant did not result in a statistically significant decrease in inclusion size, suggestive of a synergistic effect of co-expression (Figure 7C).
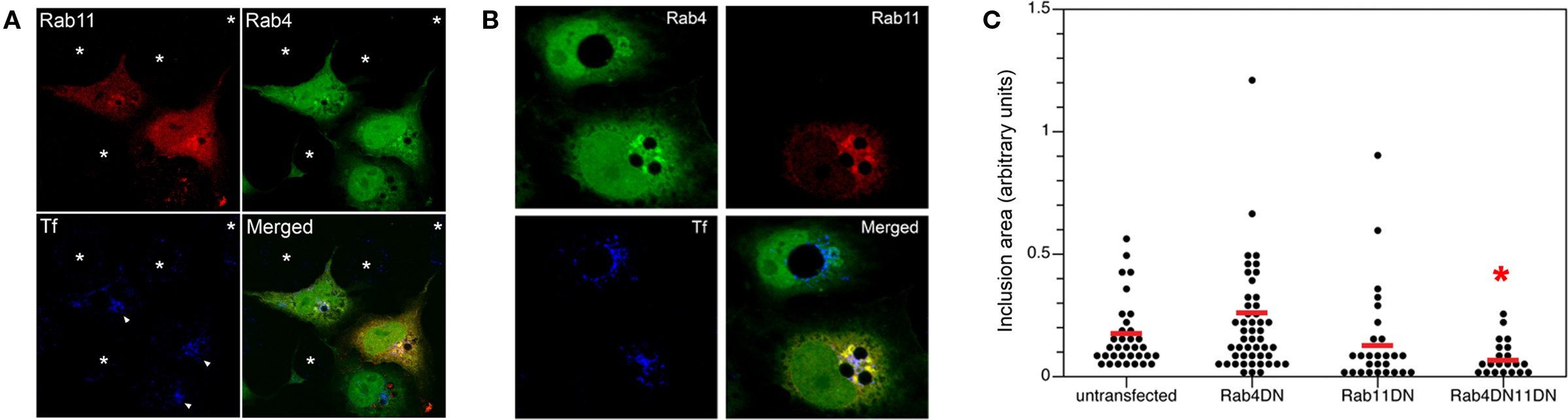
Figure 7. Co-expression of dominant-negative constructs of Rab4 and Rab11 GTPases causes retention of Tf and reduces chlamydial inclusion growth. Cells were transfected with dominant-negative (DN) constructs of Rab4 or Rab11 either individually or in conjunction. (A) Transfected cells were pulse-chased with fluorescent Tf to monitor Tf retention in these cell types. (B,C) Quantification of inclusion size in cells transfected with indicated construct(s) with accompanying representative confocal image. * indicates p < 0.001 compared to UTD by Kruskal–Wallis test.
FR179254 Inhibition of Growth of Chlamydia is Reversed by Omitting Serum from the Medium
We have shown above that FR19254 treatment resulted in the significant retention of Tf (Figure 5), and that the Tf retention correlated with decreased inclusion development (Figure 4D). This correlation was also observed in dominant-negative transfection experiments (Figure 7). If Tf accumulation was the primary cause of growth inhibition of Chlamydia by FR179254, we reasoned that omission of Tf from the growth media would relieve this inhibition. We monitored chlamydial growth in media lacking whole serum or supplemented only with the Tf-containing fraction of the serum in FR179254-treated and untreated cells. An SDS-PAGE and Western blot analysis of the serum fractions (less than 50 kDa, 50–100 kDa, and greater than 100 kDa) compared to purified Tf or complete serum demonstrated that the fraction with the greatest amount of Tf was the >100 kDa fraction (Figure 8), owing to the tendency of native Tf proteins to aggregate during the concentration process. In the absence of serum, FR179254 had no effect on chlamydial growth, as measured by IFU recovery and visualized by fluorescence microscopy of inclusions (Figure 8 and data not shown). Consequently, the supplementation of the Tf-containing fraction of the serum to the growth media restored the inhibitory effects of the compound (red bars in Figure 8 indicate no significant difference compared to FR). No such restoration of inhibition could be observed when other fractions of the serum were used individually (purple bars represent a significant difference in growth compared to FR treatment; Figure 7). Therefore, we concluded that the inhibitor activity of FR179254 only manifested in the presence of Tf in the growth media. An equally important result obtained from this experiment is the normal growth of chlamydia in media lacking serum, but containing the inhibitor, which supports our data above (Figures 1 and 4) that the target of the compound is not the bacteria.
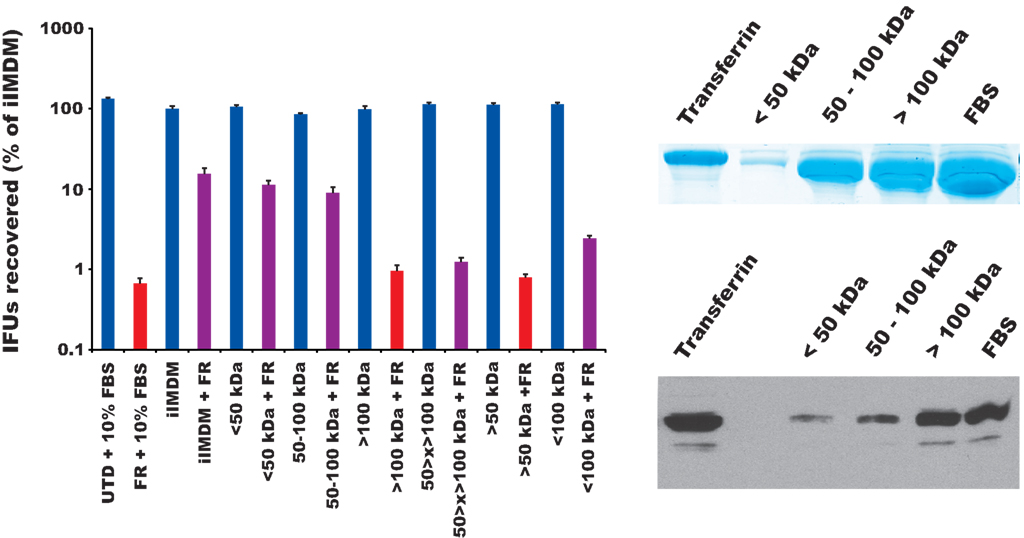
Figure 8. The effect of FR179254 can be reversed by omission of the transferrin-containing fraction of serum from the medium. Infected cells were treated with or without FR179254 (FR) and cultured in the presence or absence of serum or serum fractions as described in Section “Materials and Methods.” IFUs were collected and quantified. Red bars indicate no statistically significant difference compared to FR treatment, hence restoration of inhibition; whereas purple bars indicate a significant difference (p < 0.05 by Student’s t-test). SDS-PAGE analysis and Western blot of serum fractions for Tf content compared to complete serum (FBS) and purified, commercially available Tf.
Discussion
Chlamydia is a medically important pathogen that has proven difficult to genetically manipulate, thus alternative methods must be employed to investigate its biology at a mechanistic level. We used a small molecule inhibitor approach to identify chlamydial and host processes required for survival of the pathogen, and here, we report the identification of an inhibitor of LPAT, which interrupted the proper recycling of Tf – specifically via the slow recycling pathway. In the context of Chlamydia-infected cells, interference of this pathway using the small molecule inhibitor suppressed chlamydial growth, and thus providing additional insight into the significance of the oft-reported localization of Tf around the inclusion.
The chlamydial inclusion lacks markers of the endolysosomal pathway and is distinct from the phagosomes of other intracellular bacteria, which, aside from Coxiella, appear to be arrested within the endolysosomal transport pathway (Scott et al., 2003). This unusual characteristic of the inclusion was reinforced by the reported interaction with exocytic Golgi-derived vesicles (Hackstadt et al., 1996; Carabeo et al., 2003), and the localization to the inclusion of Rab GTPases associated with various intracellular trafficking pathways challenged this notion of restricted fusogenicity (Rzomp et al., 2003). In this report we demonstrated the requirement for the proper recycling of Tf through the slow recycling pathway in inclusion development and the completion of the developmental cycle of Chlamydia. A model is proposed below demonstrating the potential interaction of the chlamydial inclusion with the recycling pathway (Figure 9). The preferential interaction with the slow recycling pathway is supported by our FR179254 experiments.
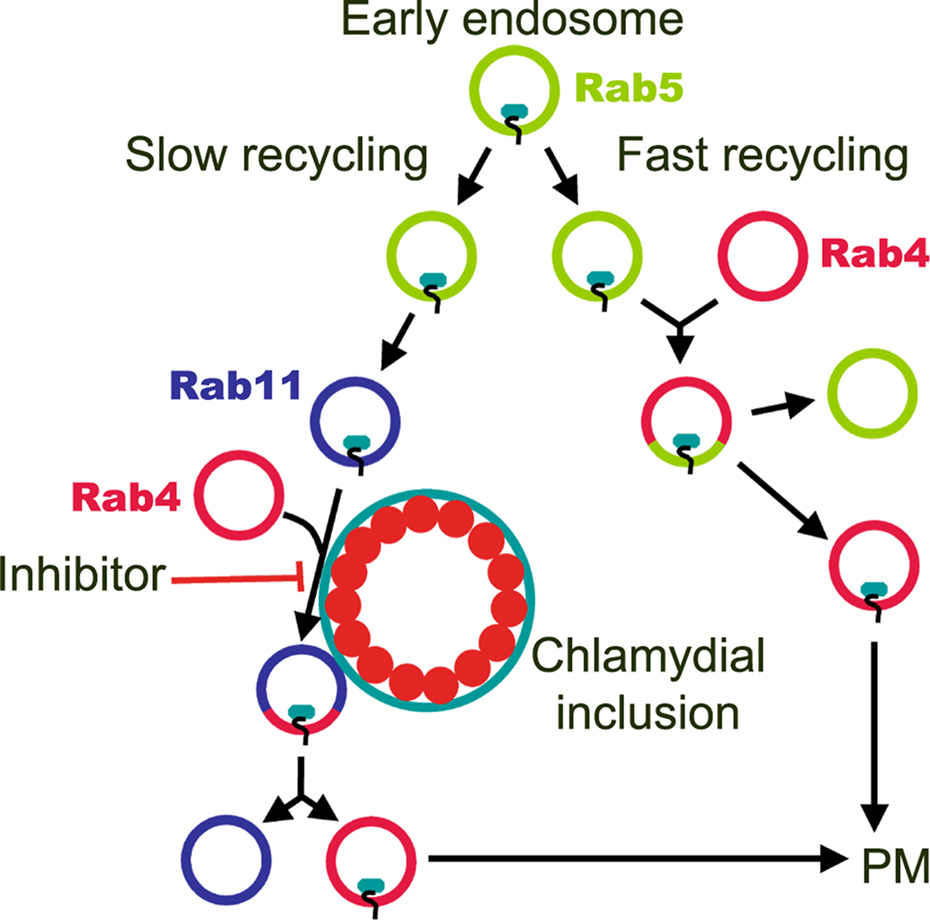
Figure 9. A model for the action of FR179254 against chlamydiae. Transferrin is recycled through two main pathways. For fast recycling, the majority of transferrin (∼80%) is endocytosed into a Rab5 positive early endosome, which subsequently fuses with Rab4 recycling endosomes for transport back to the plasma membrane (PM) in Rab4 vesicles. For slow recycling, transferrin (∼20%) is endocytosed into Rab5 early endosomes, which mature to Rab11 compartments that then fuse with Rab4 for recycling back to the PM. FR179254 delays the fusion of transferrin-containing Rab11 vesicles with Rab4 recycling vesicles, possibly at the inclusion membrane, leading to chlamydial growth abrogation.
The negative effects of LPAT inhibition on the slow recycling pathway has been reported previously (Chambers et al., 2005), and here, we extend it to Chlamydia-infected cells. We observed that the previously reported accumulation of Tf in Rab11 vesicles was at the expense of Tf localizing to the Rab4/11 hybrid transport intermediate, which Sönnichsen et al. (2000) have proposed to be involved in the recycling of Tf to the plasma membrane. This defect in the slow recycling pathway could also be observed in infected cells, with Tf accumulating around the inclusion. We suspect that the Tf accumulation is directly linked to inhibition of chlamydial growth because the removal of the Tf-containing fraction from the serum alleviated the growth inhibitory effects of FR179254. It is possible that chlamydia may have requirements for Tf-independent recycling pathway. However, the near-complete restoration of the inhibition of chlamydial growth of FR179254 treatment by the simple addition of Tf-containing fractions indicate that Tf accumulation contributes significantly to inhibition of chlamydial growth. This observation is also important in demonstrating that the target of the anti-chlamydial activity of this compound is of host origin; otherwise, alleviation of growth inhibition would not have been observed in the absence of Tf, while in the continued presence of the inhibitor. We could not observe a similar growth inhibition using commercially available purified holo-Tf (unpublished observation) either due to the difficulty in reaching the Tf concentration level found in the serum or the possibly insufficient level of iron loading of the commercial preparations of holo-Tf. Further evidence for a host target is our demonstration that the co-expression of the dominant-negative mutants of Rab4 and Rab11 partially mimicked LPAT growth inhibition of chlamydial inclusions, and that the prokaryotic LPAT homolog, 1-acylglycerol-3-phosphate acyltransferase encoded by plsC in E. coli was refractory to the inhibition of FR179254. Prokaryotes have an enzyme with predicted LPAT-like activity (Zhang and Rock, 2008). This enzyme participates in the synthesis of phospholipids from phosphatidic acid. Given the conservation of this pathway among prokaryotes and the relatively high levels of homology of the enzymes within this biosynthetic pathway, it is likely the inhibitor would be similarly ineffective toward Chlamydia. Furthermore, the PlsC pathway may be dispensable in chlamydia, as work by Su et al. (2004) demonstrated that chlamydia uses host cell precursors to generate phospholipids, thus circumventing any requirement for the Pls system.
How the inhibition of the host cell LPAT links to defects in the slow recycling pathway is unknown. There are at least eight LPAT isoforms that display differential subcellular localization (Shindou and Shimizu, 2009), and Brown and colleagues have reported (Chambers and Brown, 2004) that the relevant LPAT targeted by CI-976, which we also showed to have similar effects with regards to chlamydial infection and Tf transport as FR179254, is the Golgi-associated isoform. Because the Golgi apparatus is involved in the biogenesis of vesicles in the endosomal compartments, it is possible that interference with the Golgi-associated LPAT may affect the Tf-containing Rab11 compartment. However, we did not observed defects in the transport to the plasma membrane of protein (CD4 and VSV-G) cargos, or the delivery of SM to the chlamydial inclusion. It is tempting to speculate that a drastic change induced by LPAT inhibition in the general lipid composition of intracellular compartments may contribute to the delayed recycling of Tf via the slow pathway. However, this possibility seems unlikely as transport of other cargos via the endocytic, retrograde, and anterograde pathways seemed unaffected in cells treated with the LPAT inhibitor. The apparent specific inhibition of Tf recycling by the LPAT inhibitors may be due to unique properties of the slow recycling compartment, possibly vesicle tubulation, which is critical for recycling, or the localization of critical proteins to the slow recycling vesicles. The molecular link between LPAT and Tf transport requires further investigation.
How might the slow Tf recycling pathway contribute to chlamydial growth? The lack of obvious siderophore homologs necessitates the capture of iron prior to release to the host cytosol, where the metal is immediately sequestered by host ferritin. Such a model would imply the presence of iron in the slow recycling pathway. Indeed, the divalent metal transporter 1 (Dmt1) has been localized to the endocytic recycling compartments (Tabuchi et al., 2000), suggesting that iron may be present in these vesicles. The close association of the inclusion with these Rab11-positive vesicles would allow Chlamydia access to a possible iron source, perhaps by transient “kiss-and-run” interaction as has been suggested for phospholipid acquisition (Hatch and McClarty, 1998). In the presence of LPAT inhibitors, the resulting retention and subsequent accumulation of iron-laden compartments around the chlamydial inclusion would lead to iron overload and toxicity. Preliminary results indicate that chlamydial genes induced under limiting iron availability were, in contrast, repressed under conditions of iron overload. Thus, we are now investigating the possibility of the slow recycling pathway as the source of iron Chlamydia and that the dysregulation of its recycling, such as that induced by the LPAT inhibitors results in the uncontrolled transfer of potently oxidative Fe2+. Because of the deleterious effects of a severely delayed transport of Tf through the slow recycling pathway, any interaction the inclusion may have with this trafficking compartment would need to be regulated by Chlamydia to ensure minimal interference with the recycling of Tf to the plasmamembrane.
As with most living organisms, chlamydia absolutely requires iron. Iron limitation under in vitro conditions induces aberrant growth, growth arrest, and a transcriptional response in chlamydia (Timms et al., 2009; Wehrl et al., 2004; Wyrick, 2010). In vivo, interferon-γ, being a potent anti-chlamydial cytokine also modulates iron acquisition of and metabolism in the host to starve intracellular pathogens of this essential metal (Beekhuizen and van de Gevel, 2007; Sow et al., 2007; Nairz et al., 2008). Hence, iron limitation induced by this cytokine could act in concert with other anti-bacterial effectors to effectively eliminate infection. It is tempting to speculate that the slow recycling compartment could be the source of iron that chlamydia utilizes. By interacting directly with this pathway, chlamydia bypasses the need to compete with host cytosolic ferritins. This mechanism of iron acquisition would be especially important for a non-siderophore synthesizing intracellular pathogen.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Drs. Joel Swanson (University of Michigan), Marci Scidmore (Cornell University), and Frank Dorsey (Scripps Florida) for their generous gifts of reagents. The authors also acknowledge Charla Mutchler and Serkan Belkaya for the screen of the small molecule library, Denise Malcolm for her technical assistance, and members of the Carabeo lab for their helpful discussions. This work was supported by the NIH (AI065545) and MRC (G0900213). Author contributions: Scot P. Ouellette and Rey A. Carabeo designed, performed, and analyzed the experiments and prepared the manuscript.
References
Al-Younes, H. M., Rudel, T., and Meyer, T. F. (1999). Characterization and intracellular trafficking patterns of vacuoles containing Chlamydia pneumoniae in human epithelial cells. Cell. Microbiol. 1, 237–247.
Beatty, W. L. (2006). Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J. Cell Sci. 119, 350–359.
Beekhuizen, H., and van de Gevel, J. S. (2007). Gamma interferon confers resistance to infection with Staphylococcus aureus in human vascular endothelial cells by cooperative proinflammatory and enhanced intrinsic antibacterial activities. Infect. Immun. 75, 5615–5626.
Cadigan, K. M., Heider, J. G., and Chang, T. Y. (1988). Isolation and characterization of Chinese hamster ovary cell mutants deficient in acyl-coenzyme A:cholesterol acyltransferase activity. J. Biol. Chem. 263, 274–282.
Carabeo, R. A., Mead, D. J., and Hackstadt, T. (2003). Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc. Natl. Acad. Sci. U.S.A. 100, 6771–6776.
Chambers, K., and Brown, W. J. (2004). Characterization of a novel CI-976-sensitive lysophospholipid acyltransferase that is associated with the Golgi complex. Biochem. Biophys. Res. Commun. 313, 681–686.
Chambers, K., Judson, B., and Brown, W. J. (2005). A unique lysophospholipid acyltransferase (LPAT) antagonist, CI-976, affects secretory and endocytic membrane trafficking pathways. J. Cell Sci. 118, 3061–3071.
Cocchiaro, J. L., Kumar, Y., Fischer, E. R., Hackstadt, T., and Valdivia, R. H. (2008). Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. U.S.A. 195, 9379–9384.
Coleman, J. (1990). Characterization of Escherichia coli cells deficient in 1-acyl-sn-glycerol-3-phosphate acyltransferase activity. J. Biol. Chem. 265, 17215–17211.
Cortes, C., Rzomp, K. A., Tvinnereim, A., Scidmore, M. A., and Wizel, B. (2007). Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infect. Immun. 75, 5586–5596.
Hackstadt, T. (2000). Redirection of host vesicle trafficking pathways by intracellular parasites. Traffic 1, 93–99.
Hackstadt, T., Rockey, D. D., Heinzen, R. A., and Scidmore, M. A. (1996). Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 15, 964–977.
Hatch, G. M., and McClarty, G. (1998). Phospholipid composition of purified Chlamydia trachomatis mimics that of the eucaryotic host cell. Infect. Immun. 66, 3725–3735.
Henry, R. M., Hoppe, A. D., Joshi, N., and Swanson, J. A. (2004). The uniformity of phagosome maturation in macrophages. J. Cell Biol. 164, 185–194.
Heuer, D., Lipinski, A. R., Machuy, N., Karlas, A., Wehrens, A., Siedler, F., Brinkmann, V., and Meyer, T. F. (2009). Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature 457, 731–735.
Higgins, M. E., Davies, J. P., Chen, F. W., and Ioannou, Y. A. (1999). Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol. Genet. Metab. 68, 1–13.
Nairz, M., Fritsche, G., Brunner, P., Talasz, H., Hantke, K., and Weiss, G. (2008). Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur. J. Immunol. 38, 1923–1936.
Ouellette, S. P., Hatch, T. P., Abdel-Rahman, Y. M., Rose, L. A., Belland, R. J., and Byrne, G. I. (2006). Global transcriptional upregulation in the absence of increased translation in Chlamydia during IFN gamma-mediated host cell tryptophan starvation. Mol. Microbiol. 62, 1387–1401.
Rejman Lipinski, A., Heymann, J., Meissner, C., Karlas, A., Brinkmann, V., Meyer, T. F., and Heuer, D. (2009). Rab6 and Rab11 regulate Chlamydia trachomatis development and golgin-84-dependent Golgi fragmentation. PLoS Pathog. 5, e1000615. doi: 10.1371/journal.ppat.1000615.
Rzomp, K. A., Moorhead, A. R., and Scidmore, M. A. (2006). The GTPase Rab4 interacts with Chlamydia trachomatis inclusion membrane protein CT229. Infect. Immun. 74, 5362–5373.
Rzomp, K. A., Scholtes, L. D., Briggs, B. J., Whittaker, G. R., and Scidmore, M. A. (2003). Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect. Immun. 71, 5855–5870.
Scidmore, M. A., Fischer, E. R., and Hackstadt, T. (1996a). Sphingolipids and glycoproteins are differentially trafficked to the Chlamydia trachomatis inclusion. J. Cell Biol. 134, 363–374.
Scidmore, M. A., Rockey, D. D., Fischer, E. R., Heinzen, R. A., and Hackstadt, T. (1996b). Vesicular interactions of the Chlamydia trachomatis inclusion are determined by chlamydial early protein synthesis rather than route of entry. Infect. Immun. 64, 5366–5372.
Scidmore, M. A., Fischer, E. R., and Hackstadt, T. (2003). Restricted fusion of Chlamydia trachomatis vesicles with endocytic compartments during the initial stages of infection. Infect. Immun. 71, 973–984.
Scott, C. C., Botelho, R. J., and Grinstein, S. (2003). Phagosome maturation: a few bugs in the system. J. Membr. Biol. 193, 137–152.
Shindou, H., and Shimizu, T. (2009). Acyl-CoA:lysophospholipid acyltransferases. J. Biol. Chem. 284, 1–5.
Sönnichsen, B., De Renzis, S., Nielsen, E., Rietdorf, J., and Zerial, M. (2000). Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149, 901–914.
Sow, F. B., Florence, W. C., Satoskar, A. R., Schlesinger, L. S., Zwilling, B. S., and Lafuse, W. P. (2007). Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J. Leukoc. Biol. 82, 934–945.
Su, H., McClarty, G., Dong, F., Hatch, G. M., Pan, Z. K., and Zhong, G. (2004). Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J. Biol. Chem. 279, 9409–9416.
Tabuchi, M., Yoshimori, T., Yamaguchi, K., Yoshida, T., and Kishi, F. (2000). Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J. Biol. Chem. 275, 22220–22228.
Tanaka, A., Terasawa, T., Hagihara, H., Sakuma, Y., Ishibe, N., Sawada, M., Takasugi, H., and Tanaka, H. (1998). Inhibitors of acyl-CoA:cholesterol O-acyltransferase (ACAT). Part 1: identification and structure-activity relationships of a novel series of substituted N-alkyl-N-biphenylylmethyl-N′-arylureas. Bioorg. Med. Chem. 6, 15–30.
Timms, P., Good, D., Wan, C., Theodoropoulos, C., Mukhopadhyay, S., Summersgill, J., and Mathews, S. (2009). Differential transcriptional responses between the interferon-gamma-induction and iron-limitation models of persistence for Chlamydia pneumoniae. J. Microbiol. Immunol. Infect. 42, 27–37.
van Ooij, C., Apodaca, G., and Engel, J. (1997). Characterization of the Chlamydia trachomatis vacuole and its interaction with the host endocytic pathway in HeLa cells. Infect. Immun. 65, 758–766.
Wehrl, W., Meyer, T. F., Jungblut, P. R., Müller, E. C., and Szczepek, A. J. (2004). Action and reaction: Chlamydophila pneumoniae proteome alteration in a persistent infection induced by iron deficiency. Proteomics 4, 2969–2981.
Wyrick, P. B. (2010). Chlamydia trachomatis persistence in vitro: an overview. J. Infect. Dis. 201(Suppl. 2), S88–S95.
Keywords: Chlamydia, inclusion, slow transferrin recycling pathway, LPAT, iron
Citation: Ouellette SP and Carabeo RA (2010) A functional slow recycling pathway of transferrin is required for growth of Chlamydia. Front. Microbio. 1:112. doi: 10.3389/fmicb.2010.00112
Received: 29 July 2010;
Paper pending published: 14 August 2010;
Accepted: 10 September 2010;
Published online: 08 October 2010.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
Luiz Bermudez, Oregon State University, USADaniel E. Voth, University of Arkansas for Medical Sciences, USA
Copyright: © 2010 Ouellette and Carabeo. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Rey A. Carabeo, Centre for Molecular Microbiology and Infection, Flowers Building, Imperial College London, London SW7 2AZ, UK. e-mail:ci5jYXJhYmVvQGltcGVyaWFsLmFjLnVr