



 and
and - 1 Department of Life Sciences, Faculty of Natural Sciences, Imperial College London, London, UK
- 2 Section of Biomolecular Medicine, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, London, UK
- 3 Centre for the Understanding and Prevention of Infection in Children/Division of Infectious and Immunological Diseases, Department of Pediatrics, University of British Columbia, Vancouver, BC, Canada
Cystic fibrosis (CF) is caused by a defect in the CF transmembrane regulator that leads to depletion and dehydration of the airway surface liquid (ASL) of the lung epithelium, providing an environment that can be infected by bacteria leading to increased morbidity and mortality. Pseudomonas aeruginosa chronically infects more than 80% of CF patients and one hallmark of infection is the emergence of a mucoid phenotype associated with a worsening prognosis and more rapid decline in lung function. Hypertonic saline (HS) is a clinically proven treatment that improves mucociliary clearance through partial rehydration of the ASL of the lung. Strikingly, while HS therapy does not alter the prevalence of P. aeruginosa in the CF lung it does decrease the frequency of episodes of acute, severe illness known as infective exacerbations among CF patients. In this article, we propose a hypothesis whereby the positive clinical effects of HS treatment are explained by the osmotic sensitivity of the mucoid sub-population of P. aeruginosa in the CF lung leading to selection against this group in favor of the osmotically resistant non-mucoid variants.
Introduction
In this paper we will discuss the background to the use of hypertonic saline (HS) in the treatment of cystic fibrosis (CF) and propose a hypothesis whereby the positive effects of this approach can be explained by population shifts of Pseudomonas aeruginosa, the major chronic infecting bacterium of the CF lung.
Cystic Fibrosis and Chronic Pseudomonas Aeruginosa Infection
Cystic fibrosis is an inherited condition that results from mutations in the gene encoding the CF transmembrane regulator (CFTR), a cAMP-regulated chloride channel in epithelial tissues. These mutations lead to disruption of normal ion and water transport across the epithelium and so to thickened, dehydrated airway secretions resulting in impaired mucociliary clearance (Collins, 1992; Ratjen and Doring, 2003; Campodonico et al., 2008). There is strong evidence to support an airway surface liquid (ASL) depletion/dehydration theory and a model for ASL volume regulation and hence mucociliary clearance has been described for normal and CF epithelia (see Figure 1). This defect in the mucociliary escalator means that CF lungs are prone to chronic infection, which is the main cause of mortality and morbidity in CF patients (Dodge et al., 2007). While experimental data supports the polymicrobial nature of CF airway infections there is little evidence for serious lung damage occurring in CF in the absence of P. aeruginosa infection (Sibley et al., 2008; Tunney et al., 2008). P. aeruginosa chronically infects more than 80% of CF patients by adulthood leading to significantly poorer lung function and increased risk of mortality than non-infected peers (Govan and Deretic, 1996; Govan et al., 2007; Murray et al., 2007). The high incidence of specific P. aeruginosa infection in CF is probably not explained solely by a defect in mucociliary clearance, as P. aeruginosa infection is less frequently encountered and usually occurs later in life in patients with primary ciliary dyskinesia (Noone et al., 2004; Barbato et al., 2009). Without aggressive treatment of initial P. aeruginosa infection most CF patients will develop chronic lung infection (Gibson et al., 2003).
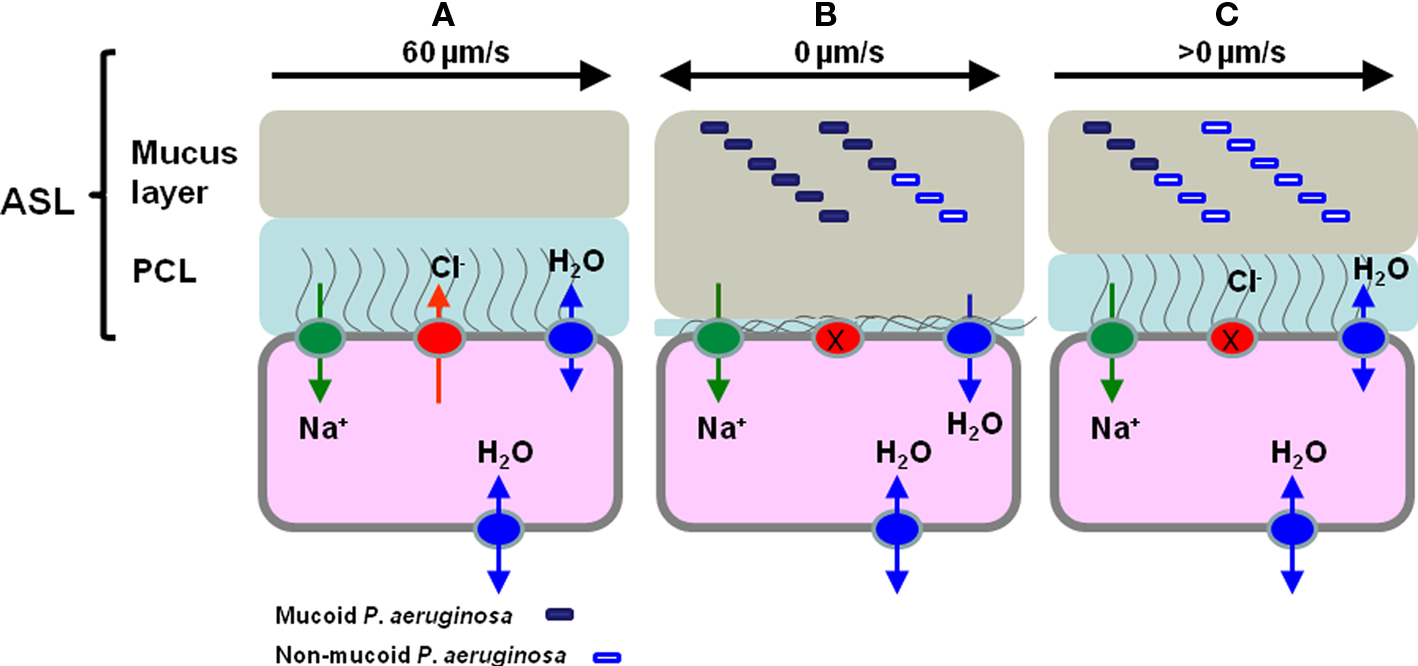
Figure 1. The regulation of airway surface liquid (ASL) volume and Pseudomonas aeruginosa phenotype in normal, CF epithelia and CF epithelia following hypertonic saline treatment. (A) In normal epithelia CFTR (red sphere) plays a vital role in regulating the concentration of NaCl on airways surfaces, ensuring correct hydration of the ASL, which is comprised of the mucus and periciliary layers (PCL). (B) In the epithelia of the CF lung the PCL is depleted because of defective Cl− secretion due to CFTR mutation and through increased adsorption of Na+ ions through ENaC (green sphere), as a consequence of ENaC being locked in the Na+ adsorptive mode. This results in continued absorption of water from the airway surface and resulting depletion of ASL, collapse of the PCL and sticky, dehydrated mucus in contact with epithelial cells (Matsui et al., 1998; Boucher, 2007; Livraghi and Randell, 2007). The lack of PCL lubrication means that cilia cannot beat to move mucus out of the lung and this results in chronic bacterial infection indicated here by non-mucoid (open rectangle) and mucoid (filled rectangle) P. aeruginosa. (C) Following HS treatment it is proposed that ASL volume increases as it encourages transmembrane water movement and that this leads to a partial rehydration of the PCL and less sticky, partially rehydrated mucus and the possibility of some cilial beating-mediated mucus clearance. Furthermore, we propose that the increased salinity of the ASL leads to a shift in the P. aeruginosa population as consequence of the osmotic sensitivity of the mucoid P. aeruginosa and that this population shift impacts on the frequency of infective exacerbations. Note that in this cartoon we do not attempt to distinguish planktonic and biofilm bacteria.
The Mucoid Switch
One hallmark of chronic P. aeruginosa CF infection is the emergence of an alginate over-producing, mucoid phenotype, which is associated with worsening of prognosis and an accelerated loss of lung function (Lyczak et al., 2002; Ramsey and Wozniak, 2005). The algD operon (PA3540–PA3551) encodes the alginate biosynthetic genes and is under the regulatory control of the alternative sigma factor AlgU (AlgT) (Martin et al., 1993; Hershberger et al., 1995). The algU gene is in an operon with the mucABCD genes, which play a role in regulating AlgU activity. In particular, the MucA protein acts as an anti-sigma factor specific for AlgU. MucA, in combination with MucB sequesters AlgU at the membrane surface, preventing it forming a RNA polymerase holoenzyme complex and so directing transcription of its target genes (Schurr et al., 1996; Boucher et al., 1997; Rowen and Deretic, 2000). The most common mechanism of mucoid switching by P. aeruginosa in the CF lung is through mutation of the mucA gene (the mucA22 or muc-22 mutation being most commonly encountered) that prevents the anti-sigma factor from sequestering AlgU, making it available to direct transcription of its target genes including the algD alginate biosynthetic operon. This mutation is a single base pair deletion that results in a truncated, dysfunctional MucA protein. AlgU-responsive promoter sequences are found for at least 35 genes and included many stress-induced genes (Firoved et al., 2002; Firoved and Deretic, 2003). The mutation also affects the production of a number of virulence factors (Whitchurch et al., 1996; Carterson et al., 2004; Wu et al., 2004). The selective advantage of mutation-driven alginate production in the CF lung is unclear, but it has been proposed to protect against phagocytosis (Schwarzmann and Boring, 1971) and reactive oxygen species (Mathee et al., 1999). Mucoidy is not an essential requirement for biofilm formation in vitro, as alginate is not a major component of non-mucoid P. aeruginosa biofilms (Ramsey and Wozniak, 2005), but it is likely that mucoid P. aeruginosa are part of the persistent biofilm communities in the CF lung (Hassett et al., 2009; Hoiby et al., 2010). Indeed, exposure to antibiotics triggers transient alginate production in vitro (Wood et al., 2006; Hoiby et al., 2010) and alginate-overproduction increases resistance to tobramycin in biofilm cultures (Hentzer et al., 2001). As more than 80% of adult CF patients have mucoid P. aeruginosa, mucoid biofilms may act as a reservoir for the infection of previously healthy parts of the lung through biofilm dispersal mechanisms that lead to the release of motile, non-mucoid bacteria (VanDevanter and Van Dalfsen, 2005; Kirov et al., 2007; Bjarnsholt et al., 2010), although this proposal has been challenged (Reid et al., 2006) and mucoid variants have been shown to be seeding dispersal proficient (Kirov et al., 2007). Treatments that would destroy or weaken biofilm structures may be of therapeutic benefit.
Treatment of P. Aeruginosa Infections
The severity of P. aeruginosa infection can be limited by early detection and aggressive antibiotic treatment before the bacteria convert to a mucoid phenotype, but it is extremely difficult to eradicate once established (Doring et al., 2000; Ratjen, 2001, 2006; Davies, 2006; Goss and Burns, 2007; Bendiak and Ratjen, 2009). When chronically infected, the patient will have repeated episodes of acute pulmonary exacerbations characterized by increased breathlessness, sputum production and feeling unwell and a reduction in lung function. Exacerbations are usually treated with a combination of systemic antibiotics (Davies, 2006; Bendiak and Ratjen, 2009; Foweraker, 2009); additionally, antibiotic therapy is supplemented by treatments designed to improve sputum clearance, such as chest physiotherapy and administration of nebulized recombinant deoxyribonuclease (rhDNase), although the later approach is relatively expensive leading to restricted use in many countries (Jones and Wallis, 2003).
Hypertonic Saline Treatment
An alternative or supplementary therapy to improve mucociliary clearance either in the context of a long term maintenance therapy or during infective exacerbations is the use of hypertonic saline hypertonic saline (HS). It is an inexpensive, straightforward treatment during which nebulized HS (3–7% NaCl), is inhaled as a fine mist through a mask or a mouthpiece; typically, 10 ml of saline is inhaled twice a day (Wark and McDonald, 2009).
In vitro studies first indicated that HS might be a useful as a potential mucotropic approach as it was found to reduce the viscoelasticity and increase the predicted cough clearability of sputum (King et al., 1997). A number of clinical studies followed that supported the efficacy of nebulized HS in improving lung function in CF (Robinson et al., 1996, 1997, 1999). Two more recent clinical studies have shown clearly the short and long term benefits of nebulized HS. Elkins et al. (2006) reported a double blind, placebo-controlled, parallel group trial over 48 weeks of HS treatment in which 4 ml of HS (7%) was nebulized twice daily to CF patients with the control group receiving 0.9% saline. The rate of change in lung function was recorded as the forced vital capacity (FVC), forced expiratory volume in 1 s (FEV1) and forced expiratory flow at 25–75% of FVC (FEV25–75). While the individual measures did not show significant differences during the treatment period, a composite end point measurement (averaged FEV1 and FVC across all post-randomization visits) was found to be significantly different between the two groups.
More strikingly, the HS treated group had significantly fewer pulmonary exacerbations and a significantly higher percentage of patients without exacerbations. This is particularly important given that frequency of exacerbations is strong predictor of morbidity and mortality in CF and so a treatment that reduces exacerbations is of major clinical relevance. Interestingly, however, HS treatment did not significantly alter the concentrations or prevalence of either P. aeruginosa or common co-infector Staphylococcus aureus in sputum between treated and non-treated groups.
In a shorter but more intensive 2-week trial in which 5 ml of HS (7% NaCl) was administered four times daily a larger increase in pulmonary function was observed (Donaldson et al., 2006). No microbiological studies were done as part of this study.
A recent independent review of all controlled trials (12 in total) assessing HS compared to placebo or other mucolytic treatments concluded that HS does improve quality of life and reduces pulmonary exacerbations and that there was sufficient evidence to recommend using HS on CF patients (Wark and McDonald, 2009). There are side effects of HS therapy that include more frequent coughing, sore throat and chest tightness in some patients, as HS can irritate the airways, and these can lead to patient compliance problems (Elkins et al., 2006). Inhaled mannitol is an alternative non-ionic osmotic agent to HS to promote airway rehydration and to increase mucociliary clearance. It has shown some promising effects on lung function, but its effects on pulmonary exacerbations and lung microbiology has not been tested (Robinson et al., 1999; Jaques et al., 2008; Minasian et al., 2008; Daviskas et al., 2010).
How does Hypertonic Saline Treatment Work?
The exact mechanism by which HS treatment works is unclear, but several ways as to how it could improve mucus clearance have been proposed: by inducing osmotic flow of water into the mucus layer so improving mucus rheology, through its action directly on the mucus, breaking the ionic bonds leading to lower viscosity and elasticity; and/or by increasing the ion concentration in the mucus and a subsequent reduction of intra and inter-molecular mucus repulsion leading to more compact mucus that could be more effectively cleared (Robinson et al., 1997; Wark and McDonald, 2009).
Donaldson et al. (2006) proposed that HS produced a sustained increase in airway surface hydration and elevation in ASL volume that improves mucociliary clearance from the airways, so protecting relatively non-obstructed airways from exogenous insults that would slow mucus clearance and promote intrapulmonary spread of bacterial infection. They postulated the involvement of airway aquaporin water channels (AQPs) in the modulation of ASL volume. However, experimental evidence has subsequently been reported against this explanation of the data, indicating the need to identify alternate mechanisms (Levin et al., 2006).
An alternative is that HS treatment acts directly on the infecting bacteria in the CF lung, among which P. aeruginosa is usually the most prominent. P. aeruginosa motility was shown to be sensitive to NaCl exposure (Havasi et al., 2008) and the study suggests that the clinical benefits attributed to HS treatment might be in part due to these effects on bacterial motility. Although not specifically discussed in the paper, it is reasonable to think that a reduction in flagellin [which serves as a pathogen associated molecular pattern (PAMP) and activates the innate immune response; Morris et al., 2009; Raoust et al., 2009; Campodonico et al., 2010] could lead to a reduction in inflammatory responses in the CF lung. The importance of motility during chronic infection of the CF lung remains uncertain, as P. aeruginosa is thought to populate the lung in biofilm-like communities (Moreau-Marquis et al., 2008; Davies and Bilton, 2009; Hassett et al., 2010) and following biofilm establishment the bacteria lose their flagella. However, motility might be involved in mobilization of P. aeruginosa to infect new sites in the lung using flagella-based swimming and swarming motility (VanDevanter and Van Dalfsen, 2005; Kirov et al., 2007).
An Alternative Hypothesis to Explain the Effect of Hypertonic Saline Treatment on Infective Exacerbations
Based on recently published data we propose an alternative hypothesis whereby long term HS treatment leads to a shift in P. aeruginosa sub-populations in the CF lung, away from mucoid variants, that are so strongly associated with increased morbidity, to non-mucoid strains.
We recently applied a nuclear magnetic resonance (NMR)-based extracellular metabolomics approach called time resolved metabolic footprinting (TReF; Behrends et al., 2009) to investigate metabolic changes in mucoid P. aeruginosa mucA22 mutants to determine the overall metabolic and physiological changes that are caused by the mucoid switch (Behrends et al., 2010). We found changes in the levels of various metabolites associated with osmotic tolerance, including glycine-betaine, trehalose, and glutamate in the mucoid mucA22 mutant. Quantitative comparison of the endometabolome of wildtype and isogenic mucA22 mutant cells showed differences in the accumulation of osmo-protectants, suggesting a switch in osmo-protectant preference from glycine-betaine to trehalose in the mucA22 mutant. Subsequent physiological experiments showed that the mucA22 mutant was significantly more sensitive to osmotic stress than the isogenic parental P. aeruginosa PA01 strain. The effect was tested over a concentration range of 0.2–0.8 M NaCl with mucoid P. aeruginosa showing increased sensitivity over the complete range (Behrends et al., 2010). The mucA22 mutant was also more sensitive to the alternative osmotic stressor sucrose, showing it was not just an effect of NaCl.
Ambient NaCl concentrations in the CF lung are in the region of 0.15 M (Smith et al., 1996), so while the concentration that NaCl reaches in ASL or sputum following HS treatment is not known, it is likely that P. aeruginosa will encounter osmotic stress when NaCl at a concentration of between 0.5 and 1.2 M is added during HS treatment. Strikingly, the mucA22 mutation affected osmotic tolerance only in the stationary phase of growth in laboratory culture, where it had a growth inhibitory effect on the mucA22 mutant independent of the presence of osmo-protectants in the medium (Behrends et al., 2010). This is in contrast to previously described osmotic stress responsive systems that have an effect on growth in exponential phase and can be rescued by addition of osmo-protectants like glycine-betaine (Aspedon et al., 2006). This is an intriguing observation as it links the effects of the switch to mucoidy to stationary phase physiology. Following initial colonization P. aeruginosa is likely to find itself mostly in a biofilm like structure in which a significant fraction of the cells will spend much of their time in a non-growing state, dividing infrequently and so physiologically resemble the stationary phase of the planktonic growth (Davies and Bilton, 2009; Bjarnsholt et al., 2010; Hassett et al., 2010).
While the mucoid shift in P. aeruginosa that occurs in most adult patients is well documented, it is rarely a complete takeover and the most common situation is the co-existence of mucoid, non-mucoid and other morphotypes in the CF lung population. We hypothesize that the increased osmotic sensitivity of mucoid P. aeruginosa could explain part of the beneficial effects of HS treatment on CF patients. We argue that prolonged HS treatment will select against osmotically sensitive mucoid strains and in favor of the greater osmotically resistant non-mucoid strains (Figure 1).
Given that >80% of adult patients have mucoid P. aeruginosa and the switch to mucoidy is associated with increased prevalence of pulmonary exacerbations, we hypothesize that improvement in exacerbation rates following prolonged treatment with HS solution result from a population shift from mucoid to non-mucoid P. aeruginosa driven by selection against osmotically sensitive mucA strain and a selective pressure for secondary suppressor mutations to reverse the effects of mucoidy, something that occurs easily in the laboratory (Figure 1). If this process was to occur within mucoid biofilms it would weaken their structure, potentially decrease resistance to antibiotics and/or delay the previously hypothesized seeding dispersal of P. aeruginosa to new sites of infection within the lung (VanDevanter and Van Dalfsen, 2005; Kirov et al., 2007; Bjarnsholt et al., 2009). So we suggest that the effect of HS might be two-fold, with high salinity affecting both P. aeruginosa motility (Havasi et al., 2008) and the viability of the mucoid sub-population in biofilms (Behrends et al., 2010).
To test this hypothesis we recommend that in future clinical trials of the effectiveness of HS-treatment a quantitative determination of mucoid and non-mucoid P. aeruginosa populations is undertaken, with the prediction being that the fraction of mucoid P. aeruginosa will fall significantly in the HS treated group. This is an important consideration as if P. aeruginosa phenotypic/genotypic population shifts can result in better patient prognosis then interventions can be tailored to the weaknesses of a specific sub-group of the bacterial population.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aspedon, A., Palmer, K., and Whiteley, M. (2006). Microarray analysis of the osmotic stress response in Pseudomonas aeruginosa. J. Bacteriol. 188, 2721–2725.
Barbato, A., Frischer, T., Kuehni, C. E., Snijders, D., Azevedo, I., Baktai, G., Bartoloni, L., Eber, E., Escribano, A., Haarman, E., Hesselmar, B., Hogg, C., Jorissen, M., Lucas, J., Nielsen, K. G., O'Callaghan, C., Omran, H., Pohunek, P., Strippoli, M. P., and Bush, A. (2009). Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur. Respir. J. 34, 1264–1276.
Behrends, V., Ebbels, T. M., Williams, H. D., and Bundy, J. G. (2009). Time-resolved metabolic footprinting for nonlinear modeling of bacterial substrate utilization. Appl. Environ. Microbiol. 75, 2453–2463.
Behrends, V., Ryall, B., Wang, X., Bundy, J. G., and Williams, H. D. (2010). Metabolic profiling of Pseudomonas aeruginosa demonstrates that the anti-sigma factor MucA modulates osmotic stress tolerance. Mol. Biosyst. 6, 562–569.
Bendiak, G. N., and Ratjen, F. (2009). The approach to Pseudomonas aeruginosa in cystic fibrosis. Semin. Respir. Crit. Care Med. 30, 587–595.
Bjarnsholt, T., Jensen, P. O., Fiandaca, M. J., Pedersen, J., Hansen, C. R., Andersen, C. B., Pressler, T., Givskov, M., and Hoiby, N. (2009). Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 44, 547–558.
Bjarnsholt, T., Tolker-Nielsen, T., Hoiby, N., and Givskov, M. (2010). Interference of Pseudomonas aeruginosa signalling and biofilm formation for infection control. Expert Rev. Mol. Med. 12, e11.
Boucher, J. C., Yu, H., Mudd, M. H., and Deretic, V. (1997). Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect. Immun. 65, 3838–3846.
Boucher, R. C. (2007). Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol. Med. 13, 231–240.
Campodonico, V. L., Gadjeva, M., Paradis-Bleau, C., Uluer, A., and Pier, G. B. (2008). Airway epithelial control of Pseudomonas aeruginosa infection in cystic fibrosis. Trends Mol. Med. 14, 120–133.
Campodonico, V. L., Llosa, N. J., Grout, M., Doring, G., Maira-Litran, T., and Pier, G. B. (2010). Evaluation of flagella and flagellin of Pseudomonas aeruginosa as vaccines. Infect. Immun. 78, 746–755.
Carterson, A. J., Morici, L. A., Jackson, D. W., Frisk, A., Lizewski, S. E., Jupiter, R., Simpson, K., Kunz, D. A., Davis, S. H., Schurr, J. R., Hassett, D. J., and Schurr, M. J. (2004). The transcriptional regulator AlgR controls cyanide production in Pseudomonas aeruginosa. J. Bacteriol. 186, 6837–6844.
Collins, F. S. (1992). Cystic fibrosis: molecular biology and therapeutic implications. Science 256, 774–779.
Davies, J. C., and Bilton, D. (2009). Bugs, biofilms, and resistance in cystic fibrosis. Respir. Care 54, 628–640.
Daviskas, E., Anderson, S. D., Jaques, A., and Charlton, B. (2010). Inhaled mannitol improves the hydration and surface properties of sputum in patients with cystic fibrosis. Chest 137, 861–868.
Dodge, J. A., Lewis, P. A., Stanton, M., and Wilsher, J. (2007). Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur. Respir. J. 29, 522–526.
Donaldson, S. H., Bennett, W. D., Zeman, K. L., Knowles, M. R., Tarran, R., and Boucher, R. C. (2006). Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N. Engl. J. Med. 354, 241–250.
Doring, G., Conway, S. P., Heijerman, H. G., Hodson, M. E., Hoiby, N., Smyth, A., and Touw, D. J. (2000). Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. Eur. Respir. J. 16, 749–767.
Elkins, M. R., Robinson, M., Rose, B. R., Harbour, C., Moriarty, C. P., Marks, G. B., Belousova, E. G., Xuan, W., and Bye, P. T. (2006). A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N. Engl. J. Med. 354, 229–240.
Firoved, A. M., Boucher, J. C., and Deretic, V. (2002). Global genomic analysis of AlgU (sigma(E))-dependent promoters (sigmulon) in Pseudomonas aeruginosa and implications for inflammatory processes in cystic fibrosis. J. Bacteriol. 184, 1057–1064.
Firoved, A. M., and Deretic, V. (2003). Microarray analysis of global gene expression in mucoid Pseudomonas aeruginosa. J. Bacteriol. 185, 1071–1081.
Foweraker, J. (2009). Recent advances in the microbiology of respiratory tract infection in cystic fibrosis. Br. Med. Bull. 89, 93–110.
Gibson, R. L., Burns, J. L., and Ramsey, B. W. (2003). Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 168, 918–951.
Goss, C. H., and Burns, J. L. (2007). Exacerbations in cystic fibrosis. 1: epidemiology and pathogenesis. Thorax 62, 360–367.
Govan, J. R., Brown, A. R., and Jones, A. M. (2007). Evolving epidemiology of Pseudomonas aeruginosa and the Burkholderia cepacia complex in cystic fibrosis lung infection. Future Microbiol. 2, 153–164.
Govan, J. R., and Deretic, V. (1996). Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60, 539–574.
Hassett, D. J., Korfhagen, T. R., Irvin, R. T., Schurr, M. J., Sauer, K., Lau, G. W., Sutton, M. D., Yu, H., and Hoiby, N. (2010). Pseudomonas aeruginosa biofilm infections in cystic fibrosis: insights into pathogenic processes and treatment strategies. Expert Opin. Ther. Targets 14, 117–130.
Hassett, D. J., Sutton, M. D., Schurr, M. J., Herr, A. B., Caldwell, C. C., and Matu, J. O. (2009). Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol. 17, 130–138.
Havasi, V., Hurst, C. O., Briles, T. C., Yang, F., Bains, D. G., Hassett, D. J., and Sorscher, E. (2008). Inhibitory effects of hypertonic saline on P. aeruginosa motility. J. Cyst. Fibros. 7, 267–269.
Hentzer, M., Teitzel, G. M., Balzer, G. J., Heydorn, A., Molin, S., Givskov, M., and Parsek, M. R. (2001). Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J. Bacteriol. 183, 5395–5401.
Hershberger, C. D., Ye, R. W., Parsek, M. R., Xie, Z. D., and Chakrabarty, A. M. (1995). The algT (algU) gene of Pseudomonas aeruginosa, a key regulator involved in alginate biosynthesis, encodes an alternative sigma factor (sigma E). Proc. Natl. Acad. Sci. U.S.A. 92, 7941–7945.
Hoiby, N., Bjarnsholt, T., Givskov, M., Molin, S., and Ciofu, O. (2010). Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 35, 322–332.
Jaques, A., Daviskas, E., Turton, J. A., McKay, K., Cooper, P., Stirling, R. G., Robertson, C. F., Bye, P. T., Lesouef, P. N., Shadbolt, B., Anderson, S. D., and Charlton, B. (2008). Inhaled mannitol improves lung function in cystic fibrosis. Chest 133, 1388–1396.
Jones, A. P., and Wallis, C. (2003). Dornase alfa for cystic fibrosis. Cochrane Database Syst. Rev. CD001127.
King, M., Dasgupta, B., Tomkiewicz, R. P., and Brown, N. E. (1997). Rheology of cystic fibrosis sputum after in vitro treatment with hypertonic saline alone and in combination with recombinant human deoxyribonuclease I. Am. J. Respir. Crit. Care Med. 156, 173–177.
Kirov, S. M., Webb, J. S., O'May, C. Y., Reid, D. W., Woo, J. K., Rice, S. A., and Kjelleberg, S. (2007). Biofilm differentiation and dispersal in mucoid Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 153(Pt 10), 3264–3274.
Levin, M. H., Sullivan, S., Nielson, D., Yang, B., Finkbeiner, W. E., and Verkman, A. S. (2006). Hypertonic saline therapy in cystic fibrosis: evidence against the proposed mechanism involving aquaporins. J. Biol. Chem. 281, 25803–25812.
Livraghi, A., and Randell, S. H. (2007). Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicol. Pathol. 35, 116–129.
Lyczak, J. B., Cannon, C. L., and Pier, G. B. (2002). Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15, 194–222.
Martin, D. W., Schurr, M. J., Mudd, M. H., Govan, J. R., Holloway, B. W., and Deretic, V. (1993). Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 90, 8377–8381.
Mathee, K., Ciofu, O., Sternberg, C., Lindum, P. W., Campbell, J. I., Jensen, P., Johnsen, A. H., Givskov, M., Ohman, D. E., Molin, S., Høiby, N., and Kharazmi, A. (1999). Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145, 1349–1357.
Matsui, H., Grubb, B. R., Tarran, R., Randell, S. H., Gatzy, J. T., Davis, C. W., and Boucher, R. C. (1998). Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95, 1005–1015.
Minasian, C., Wallis, C., Metcalfe, C., and Bush, A. (2008). Bronchial provocation testing with dry powder mannitol in children with cystic fibrosis. Pediatr. Pulmonol. 43, 1078–1084.
Moreau-Marquis, S., Stanton, B. A., and O'Toole, G. A. (2008). Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm. Pharmacol. Ther. 21, 595–599.
Morris, A. E., Liggitt, H. D., Hawn, T. R., and Skerrett, S. J. (2009). Role of Toll-like receptor 5 in the innate immune response to acute P. aeruginosa pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L1112–L1119.
Murray, T. S., Egan, M., and Kazmierczak, B. I. (2007). Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr. Opin. Pediatr. 19, 83–88.
Noone, P. G., Leigh, M. W., Sannuti, A., Minnix, S. L., Carson, J. L., Hazucha, M., Zariwala, M. A., and Knowles, M. R. (2004). Primary ciliary dyskinesia: diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 169, 459–467.
Ramsey, D. M., and Wozniak, D. J. (2005). Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol. Microbiol. 56, 309–322.
Raoust, E., Balloy, V., Garcia-Verdugo, I., Touqui, L., Ramphal, R., and Chignard, M. (2009). Pseudomonas aeruginosa LPS or flagellin are sufficient to activate TLR-dependent signaling in murine alveolar macrophages and airway epithelial cells. PLoS ONE 4, e7259.
Ratjen, F. (2001). Changes in strategies for optimal antibacterial therapy in cystic fibrosis. Int. J. Antimicrob. Agents 17, 93–96.
Ratjen, F. (2006). Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis. Curr. Opin. Pulm. Med. 12, 428–432.
Reid, D. W., Kirov, S. M., Webb, J. S., Carroll, V., Kjelleberg, S., Champion, A., and Sanderson, K. (2006). Biofilm dispersal and exacerbations of cystic fibrosis lung disease. Pediatr. Pulmonol. 41, 1254; author reply 1255.
Robinson, M., Daviskas, E., Eberl, S., Baker, J., Chan, H. K., Anderson, S. D., and Bye, P. T. (1999). The effect of inhaled mannitol on bronchial mucus clearance in cystic fibrosis patients: a pilot study. Eur. Respir. J. 14, 678–685.
Robinson, M., Hemming, A. L., Regnis, J. A., Wong, A. G., Bailey, D. L., Bautovich, G. J., King, M., and Bye, P. T. (1997). Effect of increasing doses of hypertonic saline on mucociliary clearance in patients with cystic fibrosis. Thorax 52, 900–903.
Robinson, M., Regnis, J. A., Bailey, D. L., King, M., Bautovich, G. J., and Bye, P. T. (1996). Effect of hypertonic saline, amiloride, and cough on mucociliary clearance in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 153, 1503–1509.
Rowen, D. W., and Deretic, V. (2000). Membrane-to-cytosol redistribution of ECF sigma factor AlgU and conversion to mucoidy in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Mol. Microbiol. 36, 314–327.
Schurr, M. J., Yu, H., Martinez-Salazar, J. M., Boucher, J. C., and Deretic, V. (1996). Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J. Bacteriol. 178, 4997–5004.
Schwarzmann, S., and Boring, J. R. (1971). Antiphagocytic effect of slime from a mucoid strain of Pseudomonas aeruginosa. Infect. Immun. 3, 762–767.
Sibley, C. D., Parkins, M. D., Rabin, H. R., Duan, K., Norgaard, J. C., and Surette, M. G. (2008). A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 105, 15070–15075.
Smith, J. J., Travis, S. M., Greenberg, E. P., and Welsh, M. J. (1996). Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85, 229–236.
Tunney, M. M., Field, T. R., Moriarty, T. F., Patrick, S., Doering, G., Muhlebach, M. S., Wolfgang, M. C., Boucher, R., Gilpin, D. F., McDowell, A., and Elborn, J. S. (2008). Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 177, 995–1001.
VanDevanter, D. R., and Van Dalfsen, J. M. (2005). How much do Pseudomonas biofilms contribute to symptoms of pulmonary exacerbation in cystic fibrosis? Pediatr. Pulmonol. 39, 504–506.
Wark, P., and McDonald, V. M. (2009). Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst. Rev. CD001506.
Whitchurch, C. B., Alm, R. A., and Mattick, J. S. (1996). The alginate regulator AlgR and an associated sensor FimS are required for twitching motility in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 93, 9839–9843.
Wood, L. F., Leech, A. J., and Ohman, D. E. (2006). Cell wall-inhibitory antibiotics activate the alginate biosynthesis operon in Pseudomonas aeruginosa: roles of sigma (AlgT) and the AlgW and Prc proteases. Mol. Microbiol. 62, 412–426.
Keywords: mucoid, mucA, alginate, infective exacerbation, Pseudomonas aeruginosa, metabolomics
Citation: Williams HD, Behrends V, Bundy JG, Ryall B and Zlosnik JEA (2010) Hypertonic saline therapy in cystic fibrosis: do population shifts caused by the osmotic sensitivity of infecting bacteria explain the effectiveness of this treatment?. Front. Microbio. 1:120. doi:10.3389/fmicb.2010.00120
Received: 20 August 2010;
Accepted: 11 October 2010;
Published online: 11 November 2010.
Edited by:
Michael J. Schurr, University of Colorado, USAReviewed by:
Michael J. Schurr, University of Colorado, USA;Michael L. Vasil, University of Colorado, USA;
Gerald Pier, Harvard Medical School, USA
Copyright: © 2010 Williams, Behrends, Bundy, Ryall and Zlosnik. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Huw D. Williams, Department of Life Sciences, Imperial College London, South Kensington Campus, Sir Alexander Fleming Building, London SW7 2AZ, UK. e-mail:aC5kLndpbGxpYW1zQGltcGVyaWFsLmFjLnVr