



- Department of Pathobiological Sciences, University of Wisconsin-Madison, Madison, WI, USA
Infection with Mycobacterium tuberculosis, the causative agent of tuberculosis, is considered one of the biggest infectious disease killers worldwide. A significant amount of attention has been directed toward revealing genes involved in the virulence and pathogenesis of this air-born pathogen. With the advances in technologies for transcriptional profiling, several groups, including ours, took advantage of DNA microarrays to identify transcriptional units differentially regulated by M. tuberculosis within a host. The main idea behind this approach is that pathogens tend to regulate their gene expression levels depending on the host microenvironment, and preferentially express those needed for survival. Identifying this class of genes will improve our understanding of pathogenesis. In our case, we identified an in vivo expressed genomic island that was preferentially active in murine lungs during early infection, as well as groups of genes active during chronic tuberculosis. Other studies have identified additional gene groups that are active during macrophage infection and even in human lungs. Despite all of these findings, one of the lingering questions remaining was whether in vivo expressed transcripts are relevant to the virulence, pathogenesis, and persistence of the organism. The work of our group and others addressed this question by examining the contribution of in vivo expressed genes using a strategy based on gene deletions followed by animal infections. Overall, the analysis of most of the in vivo expressed genes supported a role of these genes in M. tuberculosis pathogenesis. Further, these data suggest that in vivo transcriptional profiling is a valid approach to identify genes required for bacterial pathogenesis.
Introduction
While we are still uncovering the survival strategies used by M. tuberculosis within a host, it is clear that these strategies are very effective: approximately one-third of the world’s population is infected with tuberculosis (Corbett et al., 2003). Infection progresses to active tuberculosis, characterized by coughing and weakness, in approximately 5–10% of cases. Without proper, long-term treatment, tuberculosis can be fatal. Currently, the death rate from tuberculosis is approximately 1.7 million deaths per year, the highest for any single bacterial pathogen (Glaziou et al., 2009). Infected individuals who do not progress to active tuberculosis are said to develop latent tuberculosis, and represent a potential reservoir for future infection. Bacteria within latently infected individuals are often localized to granulomas, which are dense areas of both live and dead immune cells where the bacteria survive at a low but persistent level. Since the initial sequencing of M. tuberculosis in 1998 (Cole et al., 1998), many expression studies have been published in an attempt to better understand the survival mechanisms of this deadly pathogen (Kendall et al., 2004; Waddell et al., 2008; Haller et al., 2010). In a post-genomic era, we now need to learn how to utilize these expression data to better understand the ever-growing problem of tuberculosis.
Mycobacterium tuberculosis is an intracellular pathogen that infects via aerosolization from an infected host. After inhalation by a naïve host, the bacteria localize to the alveoli of the lungs, where they are phagocytosed by alveolar macrophages. M. tuberculosis is able to reside and even replicate within the normally toxic phagosomal compartment of human macrophages by using a variety of strategies, including the prevention of phagosome:lysosome fusion (Chua et al., 2004), prevention of phagosome acidification (Sturgill-Koszycki et al., 1994), and detoxification of the stresses it encounters in this environment (Hingley-Wilson et al., 2003; Hestvik et al., 2005; Warner and Mizrahi, 2007). One detriment to tuberculosis research is the difficulty of working with the pathogenic M. tuberculosis bacteria. The severity of the disease combined with the extremely low infectious dose (1–10 bacteria) means that research must be conducted within specialized facilities. Further, the lengthy doubling time (approximately 1 day) makes work progress slowly while the unique, fatty-aid rich cell wall makes the extraction of well-preserved RNA difficult. As such, most of the expression studies conducted with M. tuberculosis have taken place in vitro and not within a living host. Until recently, in vivo analysis on a genome-wide level was not even possible.
Fortunately, many in vitro models have been developed that attempt to replicate aspects of the in vivo environment using large-scale analysis approaches, including low oxygen levels (Rustad et al., 2008), low nutrients (Betts et al., 2002), and the addition of exogenous stresses (Stewart et al., 2002; Deb et al., 2009). While these models have been very useful in identifying and characterizing genes within M. tuberculosis, they are limited in their ability to identify survival strategies specific to the M. tuberculosis in vivo lifestyle. Our review will briefly discuss those in vitro models that improved our understanding of tuberculosis pathogenesis. However, this review focuses specifically on large-scale expression profiling experiments that have been conducted in vivo within host systems of tuberculosis. In particular, we emphasize instances where in vivo expression profiling has led to the discovery of genes that have been confirmed experimentally to be required for full virulence and/or survival of M. tuberculosis. These discoveries suggest that in vivo expression profiling is a valid strategy to identify transcripts directly relevant to the development of tuberculosis.
Major Approaches for Transcriptional Profiling of M. tuberculosis
Transcriptional profiling can be performed in several different ways. One of the simplest and most commonly used methods to determine transcript levels within bacteria such as M. tuberculosis is RT-PCR. Because gene-specific primers are required for the amplification and quantification of transcripts, this technique is used for studies of specific genes that are of particular interest to the researcher. Because it measures transcript levels of only a small number of specific, pre-chosen genes, RT-PCR is not generally useful at identifying novel pathways or new targets for research. However, RT-PCR remains a popular and pragmatic technique for measuring gene expression, particularly during in vivo infection (Mariani et al., 2000; Timm et al., 2003; Shi et al., 2005; Srivastava et al., 2008; Kesavan et al., 2009). It is lower-cost than other profiling techniques, and it provides quantization levels that are generally considered to be more accurate than those provided by microarray analyses. It requires less bacterial RNA than other techniques, making it desirable in situations where only small amounts of RNA can be recovered, as is frequently the case in M. tuberculosis in vivo infections. Because it is more accurate than whole-genome techniques, RT-PCR is used as a confirmatory step in almost all microarray studies.
On the other hand, whole-genome microarray analyses provide transcriptional profiling without bias toward previously known genes. In this way, microarrays are able to identify interesting transcriptional changes in genes that may not have been previously characterized or studied, as well as large-scale trends occurring on a genome-wide level. Microarrays have frequently been used for the study of in vitro models of tuberculosis infection, but have been used less frequently to study in vivo models due to the difficulties in obtaining sufficient levels of mycobacterial RNA. Amplification techniques, although not yet widely utilized outside of human clinical samples, can alleviate this limitation in some situations (Waddell et al., 2008). Additionally, mycobacterial genome-directed primers (GDPs) have been developed to ensure that full-genome priming occurs in the presence of contaminating host transcripts (Talaat et al., 2000).
Recently, a novel sequencing-based approach for all RNA transcripts, RNA-Seq, has emerged as an alternative to microarrays for whole-genome transcriptional profiling (Wang et al., 2009). Although RNA-Seq is significantly more expensive than microarray technology, and requires large amounts of high-quality extracted RNA, it also presents many advantages relative to older techniques. The quantization provided by RNA-Seq is significantly more accurate than microarray analysis (Fu et al., 2009). Additionally, RNA-Seq provides the actual transcript sequences simultaneously with quantization data, rather than relying on predicted open reading frames in a given genome. This allows for the identification of other types of RNA (e.g., sRNAs or non-coding RNAs). RNA-Seq may be able to provide particularly interesting data for host-pathogen infection models, as it is theoretically capable of profiling both the host and the pathogen simultaneously. Although RNA-Seq has yet to be used to study M. tuberculosis, other bacterial pathogens have been successfully profiled using this new technology (Cossart and Archambaud, 2009; Albrecht et al., 2010; Sharma et al., 2010). Applying RNA-Seq to tuberculosis is an area of active research in our laboratory and others. In an attempt to establish a protocol for RNA-Seq in our hands, we have used RNA-Seq to analyze the transcriptomes of M. tuberculosis cultures grown in vitro to mid-log phase. We found that using our designed GDPs (Talaat et al., 2000) yielded a better representation of the transcriptomes than using the standard random primers (see Table 1 for detailed results). Clearly, RNA-Seq provided a higher-resolution analysis of M. tuberculosis transcription that cannot be matched by our earlier analysis using DNA microarrays (Talaat et al., 2002).
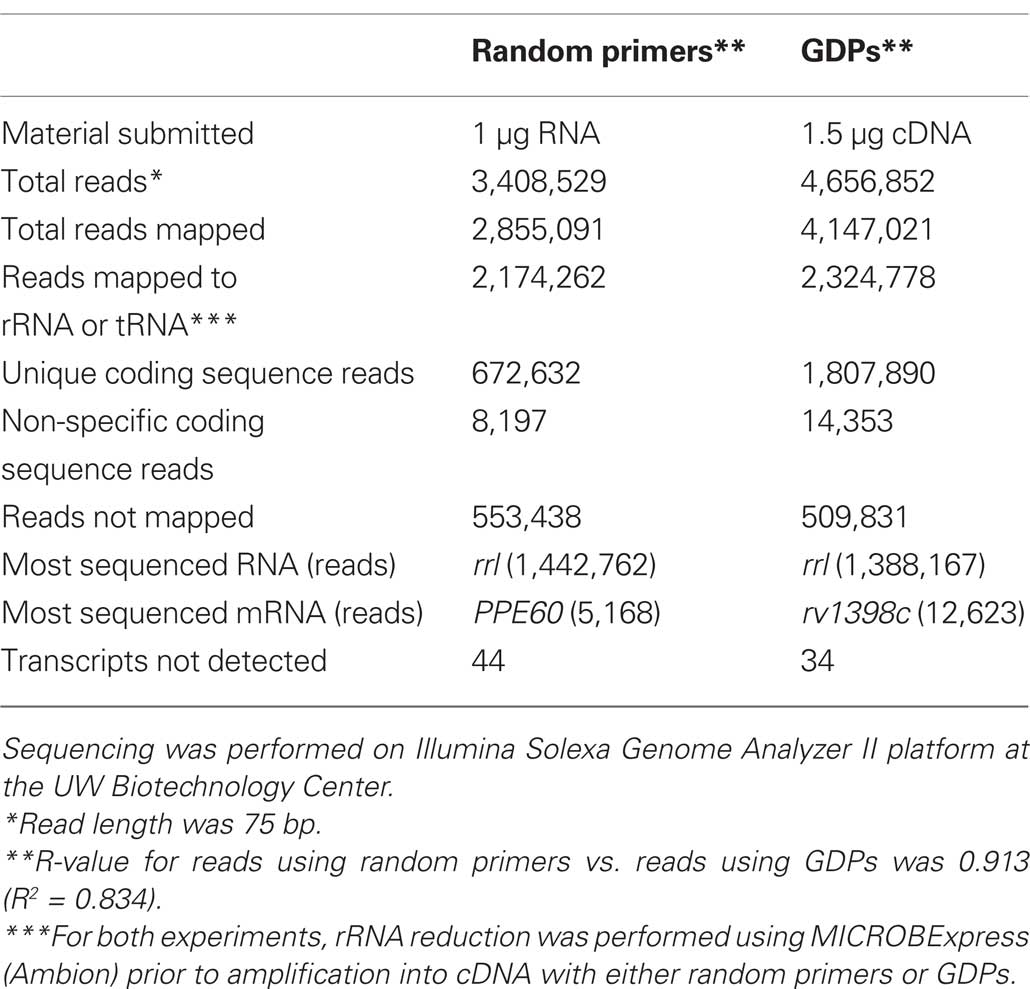
Table 1. Results from RNA-Seq of M. tuberculosis grown in vitro using random primers vs. genome-directed primers (GDPs).
Tuberculosis Models of Infection
To gain insights into the molecular pathogenesis of M. tuberculosis, many transcriptional profiling studies have been conducted within various models of tuberculosis infection. We will highlight the major tuberculosis models of infection suitable for transcriptional profiling techniques before delving into what we have learned from the generated M. tuberculosis transcriptomes. To begin, in vitro models have been developed that incorporate exposure of M. tuberculosis growing in culture to conditions thought to be similar to those experienced within the host microenvironment. For example, studies have been conducted under exposure to acid, oxidative stress, and nutrient starvation (Betts et al., 2002; Fisher et al., 2002; Schnappinger et al., 2003). One of the most well-established in vitro models for M. tuberculosis is the Wayne model, which uses oxygen deprivation to mimic the conditions found in a tuberculosis granuloma (Wayne and Hayes, 1996; Wayne and Sohaskey, 2001). In another recently developed in vitro multiple stress model, M. tuberculosis bacilli are subjected to multiple stresses including low oxygen, high CO2, low nutrient availability, and acidic pH (Deb et al., 2009). These models have been useful in identifying and characterizing the genes within the M. tuberculosis genome.
A prime example of in vitro microarray studies includes those that identified the DosR regulon, a group of genes responsible for transitioning M. tuberculosis from an aerobic state to an anaerobic, persistent state (Park et al., 2003; Converse et al., 2009). However, although a knockout mutant of dosR caused attenuation within the mouse model, it was still able to establish a persistent infection (Converse et al., 2009). In addition to the inability of in vitro models to fully mimic the fluctuation and feedback found in an in vivo system, creating accurate in vitro models is also a difficult task because the conditions found in a mycobacterial phagosome are still not entirely known. Therefore, despite the utility of in vitro host models, particularly those that incorporate multiple stresses, they are unlikely to accurately represent the normal surroundings of intracellular pathogens such as M. tuberculosis. We are convinced that using in vivo models of infection will more closely represent the complex environment found in human lungs and provide a wealth of information related to M. tuberculosis pathogenesis.
Because M. tuberculosis is an intracellular pathogen, ex vivo macrophage infections can provide a model similar to its natural environment. Cell culture infections have been used to study host response to infection (Danelishvili et al., 2003; Xu et al., 2003; McGarvey et al., 2004), and several large-scale studies profiling the mycobacterial transcriptome have been done. Infected THP-1 cells, which are cultured human monocytes, have been used to stimulate a host environment (Fontán et al., 2008). Additionally, primary cultures of murine macrophages as well as human macrophages have been used for microarray analysis (Schnappinger et al., 2003; Cappelli et al., 2006; Rachman et al., 2006a; Tailleux et al., 2008). Although macrophages are more similar to the natural host environment than an in vitro system, the absence of interacting immune cells such as T cell, natural killer cells, and dendritic cells, and the lack of a granulomatous structure, are disadvantages to cell culture systems.
While humans represent the only natural reservoir for M. tuberculosis, many standard laboratory animals are capable of being infected with human strains via aerosolization, which most closely mimics natural infection. Mice represent the most commonly used animal model for tuberculosis, and the murine model provides a relatively tractable system in which to study M. tuberculosis pathogenesis. However, difficulty in isolating sufficient amounts of mycobacterial transcripts from host tissue mean that only a few large-scale transcriptional profiles within this model have been completed (Talaat et al., 2004, 2007). Additionally, the murine model is not able to completely replicate the biology of human tuberculosis. Although mice develop granulomas, they are not as defined and organized as the granulomas observed in humans, or even other animal models such as guinea pigs. Also, mice carry a high bacterial load and display progressively deteriorating lung pathology during chronic infection, whereas the latent stage in humans is asymptomatic and characterized by low bacterial numbers (Flynn, 2006). Other less-common animal models include the use of guinea pigs, rabbits, and non-human primates for infections (Gupta and Katoch, 2005; Flynn, 2006). Financial restrictions and host-specific technical challenges mandates that large-scale transcriptional profiling has yet to be done within these models, although qRT-PCR has been used to profile select gene expression within infected rabbit lung tissue (Kesavan et al., 2009) as well as primate tissue (tbdb.org).
Finally, an additional option to obtain in vivo profiling data of pathogens is to isolate bacterial RNA directly from infected patients. In the case of M. tuberculosis, this requires access to sections of infected lung tissue, a limitation that has made profiling within human tissue rare. However, a few instances of transcriptional profiling within human lungs have been reported (Timm et al., 2003; Rachman et al., 2006b). These studies can be considered the most accurate representation of genes expressed by M. tuberculosis during human infection. However, in addition to the difficulty in obtaining samples, other downsides to profiling within humans include the potential effect of chemotherapy treatment on bacterial transcription, the potential introduction of artifacts during sample transport and storage, and the inability to reproduce experiments due to diverse genetic backgrounds. In the next sections, we will focus our attention on specific examples where large-scale analyses were useful in delineating novel aspects of tuberculosis pathogenesis.
Genes Expressed Inside Macrophages
The Murine Macrophage Model
A few whole-genome transcriptional profiles of M. tuberculosis growing within murine primary cell culture have been conducted using murine bone-marrow derived macrophages. Rachman et al. (2006a) used microarrays to profile the transcriptomes of M. tuberculosis within both active and resting macrophages relative to in vitro culture. Approximately 190 genes were identified as upregulated in response to the resting macrophage vs. liquid bacterial culture in vitro, or the activated macrophage vs. resting macrophage state. The upregulation of siderophore-encoding genes mbtJ and mbtI indicated that the bacteria were responding to iron limitation. Genes associated with amino acid synthesis and lipid metabolism were also upregulated, suggesting nutrient deprivation and the switch to lipids as a carbon source. Genes encoding cell wall components, stress response mechanisms, and regulatory proteins were also identified as induced within murine macrophages.
Similarly, Schnappinger et al. (2003) identified over 400 genes that were induced after phagocytosis by either resting or active murine macrophages relative to bacteria grown in culture. Despite the use of the same ex vivo model, only 27 genes were identified in both the Rachman and Schnappinger study (Figure 1A), possibly reflecting the differences in experimental conditions such as time points and bacterial load. Interestingly, despite the low number of overlap in genes identified in both studies, the functional categories identified were very similar. As with the Rachman study, the Schnappinger study found groups of genes associated with iron scavenging, the cell wall, and lipid metabolism. In addition, the Schnappinger study identified genes associated with anaerobic respiration and dormancy, such as narX and ndh, indicating that a switch to anaerobic respiration may occur in cell culture models, and particularly in macrophages that have been activated with IFN-γ. Curiously, the largest functional category within the overlap between the two studies was a high number of predicted transposases (6/27). Although the role for transposases within host infection is unknown, their upregulation is associated with DNA damage (Boshoff et al., 2003).
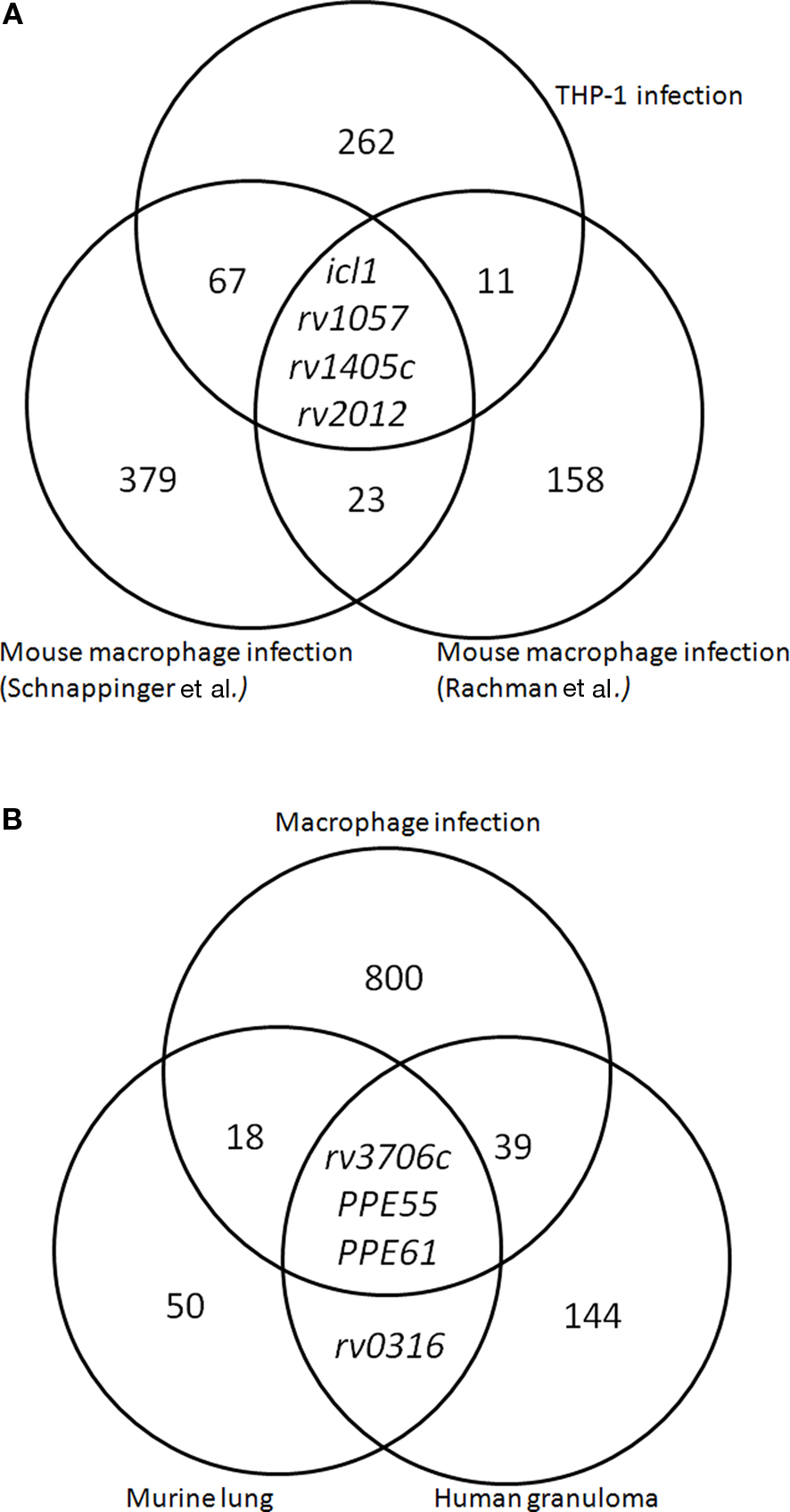
Figure 1. Meta-analysis of M. tuberculosis transcriptomes using variable models of infection. (A) Macrophage expression data following infection with M. tuberculosis reveals overlap of upregulated genes identified in three macrophage models of M. tuberculosis infection (Schnappinger et al., 2003; Rachman et al., 2006a; Fontán et al., 2008). (B) Expression data in three models of tuberculosis infection, including ex vivo macrophage infection, mouse infection, and profiling of infected human lung tissue (Schnappinger et al., 2003; Talaat et al., 2004; Rachman et al., 2006a,b; Fontán et al., 2008).
The Human Macrophage Model
Microarray analysis performed by Fontán et al. (2008) examined macrophage infection using THP-1 cells, a human-derived cultured cell line. Approximately 380 genes were identified as upregulated intracellularly relative to M. tuberculosis grown in broth culture. Unlike the primary mouse macrophages, M. tuberculosis living within cultured human macrophages did not reveal upregulation of iron uptake genes. In fact, upregulation of bfrA, a gene associated with high levels of iron availability, occurred within this model. However, the THP-1 model identified other groups similar to those found in the murine macrophage studies, such as signs of lipid metabolism and cell envelope stress. This study also revealed the upregulation of many transcriptional regulators, including whiB3, ideR, mprA, and dosR. The THP-1 study had only 15 genes in common with the Rachman study, most of which encode uncharacterized hypothetical proteins. On the other hand, it had a 71-gene overlap with the Schnappinger study, including many transcriptional regulators, membrane proteins, and lipid synthesis enzymes.
Additional microarray studies within human macrophages have used primary cultured macrophages isolated from healthy humans (Cappelli et al., 2006; Tailleux et al., 2008). Cappelli et al. (2006) discovered that genes associated with the cell wall, oxidative damage repair, and regulatory functions were upregulated, while iron-associated genes were not. The identification of the upregulated sigma factor sigG in this study was unique to the primary culture human model, and SigG was confirmed to be required for full survival within macrophages (Lee et al., 2008). Tailleux et al. (2008) studied the transcriptional response of M. tuberculosis within human-derived macrophages as well as dendritic cells. The induction of genes associated with anaerobic respiration, including narX, was observed, as well as the induction of genes associated with lipid metabolism. Interestingly, this study concluded that dendritic cells represent a more nutrient-limited environment for M. tuberculosis than macrophages, as evidenced by the increased upregulation of genes associated with amino acid synthesis and cholesterol metabolism.
Overlap in Macrophage-Induced Genes
Comparing the transcriptional data from the two murine cell infections with the human THP-1 infection revealed only four genes that were identified as significantly upregulated in all three analyses, most likely due to differences in experimental setup (Schnappinger et al., 2003; Rachman et al., 2006a; Fontán et al., 2008). Two of these genes (rv1057, rv2012) are completely uncharacterized, and one is a putative methyltransferase (rv1405c) of unknown biological function that was identified as necessary for infection within a mouse model (Sassetti and Rubin, 2003). The only functionally characterized gene identified in all three studies was isocitrate lyase (icl1), which is known to be involved in the utilization of fatty acids within the host (Höner Zu Bentrup et al., 1999). When icl1 was knocked out and the mutant tested in a murine model, studies revealed attenuation in both persistence and virulence (McKinney et al., 2000; Muñoz-Elías and McKinney, 2005). Overall, the macrophage studies have sketched the macrophage as a location where M. tuberculosis responds to oxidative stress, utilizes lipids as a carbon source, and modulates cell wall components. Other areas, such as iron availability, seem to provide different pictures depending on the cell culture model used. The high number of uncharacterized genes that are identified consistently among all macrophage studies emphasizes the need for continued basic research on characterizing the genome of M. tuberculosis.
Genes Expressed During Murine Infection
In 2004, our group published an in vivo microarray analysis study of immune-competent and immune-deficient mice infected with M. tuberculosis (Talaat et al., 2004). Whole-genome expression levels were measured weekly over the course of the first 4 weeks of infection. One of the major findings of this study was a region of 32 consecutive genes that was highly expressed in the mouse lung tissue relative to its expression levels when grown outside the host, in liquid culture media. This region of the genome was termed the iVEGI, for in vivo expressed genomic island (Figure 2A). Since that publication, further characterization of all of the operons within the iVEGI has been an area of active research both in our laboratory and others. We have come to understand more about the functions performed by the gene products encoded in the iVEGI, some of which were previously associated with in vivo survival, such as lipid synthesis, while others represented new insights into environmental stress, such as copper toxicity and novel protease/chaperone activity. Importantly, we have also shown that most of the operons within the iVEGI are required for full virulence within animal host models.
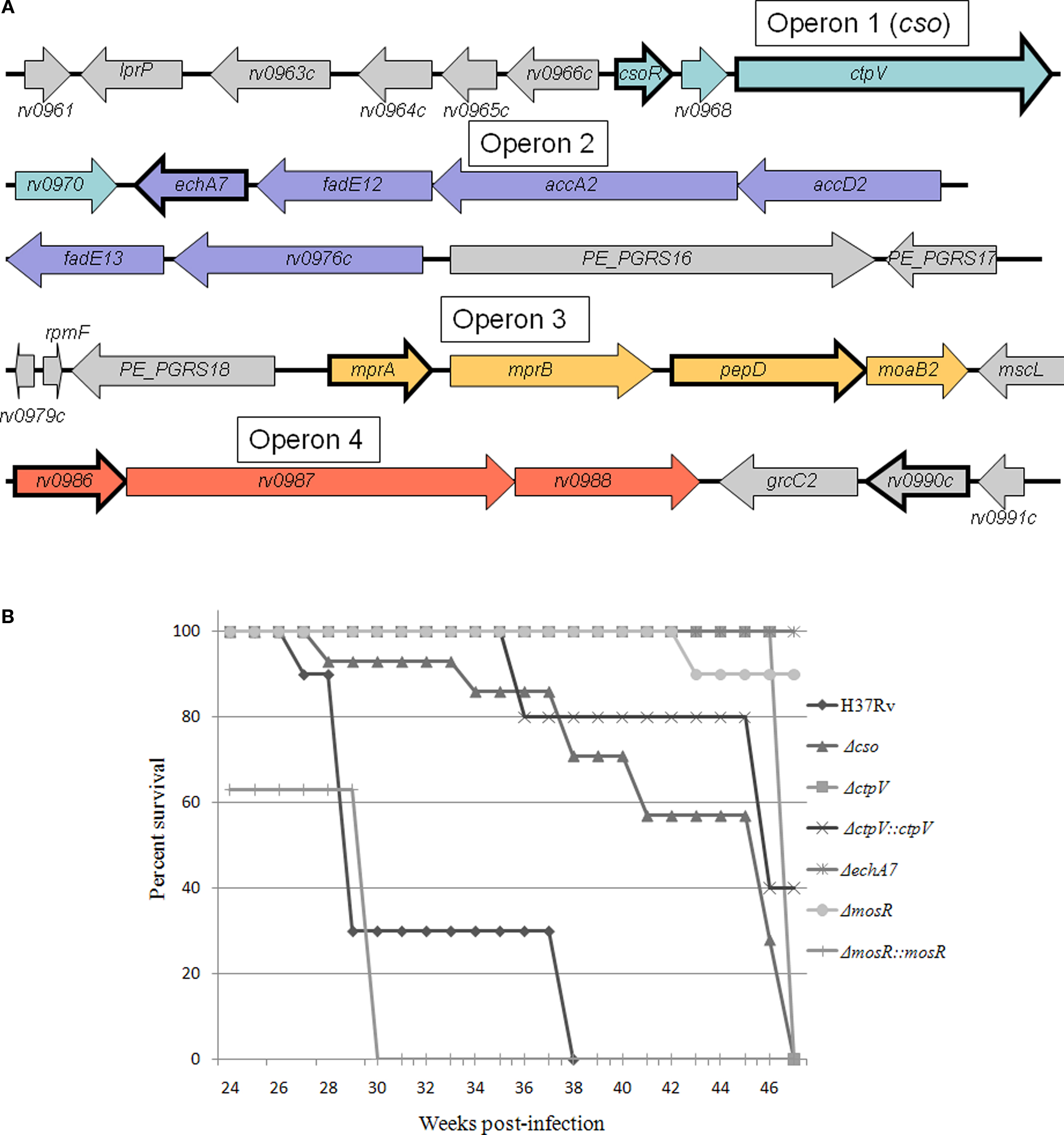
Figure 2. Analysis of key M. tuberculosis expressed genes in mice. (A) Schematic of the genetic organization of the in vivo expressed genomic island (iVEGI). Genes that have been targeted for deletion and tested for virulence within animal model(s) are bolded. (B) Murine survival curves after infection with the wild-type strain (H37Rv), or with targeted deletion strains created based on in vivo transcriptional profiling. Mouse groups were infected using aerosol infection (Glas-col inhalation system) using 100–300 CFU/animal (Abomoelak et al., 2009). The survival of infected mice was monitored over the course of a 1-year infection.
The first operon in the iVEGI, termed cso (copper sensitive operon), encodes four genes: csoR, rv0968, ctpV, and rv0970. The first gene, csoR, has been shown to encode a transcriptional regulator that binds or releases DNA in response to the intracellular level of copper (Liu et al., 2007). At least one of the genes controlled by CsoR, ctpV, is directly involved in reducing the effects of copper toxicity. CtpV is proposed to function as a copper exporter based on sequence analysis and experimental data (Ward et al., 2010). Recent research suggests that copper toxicity may represent a source of host-mediated in vivo stress that successful pathogens must overcome through carefully regulated copper response mechanisms, such as those represented by CsoR and CtpV (Percival, 1998; Wagner et al., 2005; Ward et al., 2010).
A knockout mutant of the entire cso operon was created, Δcso, and was tested in a mouse model. Results showed an approximately 1.5-log reduction in the colonization levels of Δcso relative to H37Rv, the virulent wild-type strain, at 38 weeks post-infection (Abomoelak et al., 2009). Additionally, a single-gene knockout of CtpV was created, ΔctpV. This knockout mutant was tested in both a mouse model and a guinea pig model of infection (Ward et al., 2010). The same phenotype was seen in both host models: although survival of ΔctpV was similar to the wild-type strain H37Rv along the course of infection, damage to host tissue was lessened by the deletion of the ctpV gene, and mice infected with ΔctpV survived longer relative to those infected with H37Rv (Figure 2B). This reduction in pathology and mortality shows that ctpV is required for full virulence of M. tuberculosis, despite not being required for survival within a host.
Operon 2 of the iVEGI encodes six genes predicted to be involved in lipid metabolism. The genes encode a putative enoyl-CoA hydratase thought to be involved in the oxidation of fatty acids (echA7), two predicted acyl-coA dehydrogenases (fadE12, fadE13), two genes involved in the synthesis of mycolic acids (accA2, accD2), as well as one uncharacterized gene (rv0976c). Overall, the operon is likely involved in fatty acid metabolism and anabolism, processes key to M. tuberculosis pathogenesis, including formation of the unique cell wall as well the utilization of fatty acids as a carbon source. The genes involved in synthesizing and breaking down fatty acids are extremely prevalent within the M. tuberculosis genome, making it likely that some or all of the genes in Operon 2 are redundant (Kinsella et al., 2003). A non-polar knockout of the first gene in Operon 2, echA7, was created in our laboratory using a specialized transduction system (Bardarov et al., 2002) and used to infect mice (Figure 2B). Despite its probable functional redundancy, ΔechA7 showed attenuation in an aerosol mouse model of infection, with the ΔechA7 mutant colonizing with approximately one log fewer bacteria relative to H37Rv over the course of infection at both short-term (4 weeks) and long-term (up to 38 weeks) time points (data not shown). The ΔechA7 mutant also displayed extremely reduced virulence, with the infected mice surviving for the entire duration of the mouse survival experiment (1 year) compared to the average time-to-death of mice infected with H37Rv (31 weeks) (Figure 2B).
The third operon of the iVEGI encodes a two-component regulatory system (MprAB; He and Zahrt, 2005), as well as a protease (PepD) (Mohamedmohaideen et al., 2008; White et al., 2010) and an uncharacterized gene thought to be involved in molybdopterin synthesis (moaB2). The MprAB system consists of a response regulator (MprA) and a sensor kinase (MprB). This system has been shown to regulate many genes, including the genes encoding sigma factors SigB and SigE, in response to cellular stresses and particularly membrane stress (He et al., 2006). An ΔmprAB deletion mutant failed to establish a persistent infection in mice, suggesting that it could be required for the entrance into chronic tuberculosis (Zahrt and Deretic, 2001). PepD is proposed to function as a protease and chaperone involved in mycobacterial stress response and is under the control of MprAB (Skeiky et al., 1999; Mohamedmohaideen et al., 2008; White et al., 2010). Similar to the ΔctpV mutant, a ΔpepD mutant did not have a colonization defect within a mouse model, but animals infected with the mutant displayed less lung tissue damage relative to those infected with H37Rv, and mutant-infected mice lived longer than those infected with H37Rv (Mohamedmohaideen et al., 2008).
Operon 4 of the iVEGI encodes three genes: rv0986, rv0987, and rv0988, which together encode components of an ABC transporter (Braibant et al., 2000). Functional characterization of an Δrv0986 knockout mutant revealed that this transporter is required for invasion of host cells as well as the blocking of phagosome–lysosome fusion within the host macrophage cell (Pethe et al., 2004; Rosas-Magallanes et al., 2006). Testing of a knockout mutant within this operon occurred within a murine model for central nervous system tuberculosis, with a knockout mutant of rv0986 less able to invade brain tissue (Be et al., 2008). Operon 4 is thus far the only operon shown to be horizontally transferred within the iVEGI. The operon as a whole is specific to the M. tuberculosis complex and has an unusually low GC content as well as distinct codon usage from the rest of the genome (Rosas-Magallanes et al., 2006). Overall, analysis conducted so far indicates the involvement of the iVEGI in M. tuberculosis pathogenesis and virulence.
In a second microarray study using the murine model that focused on the chronic stage of tuberculosis, mycobacterial RNA were successfully extracted from infected mouse lungs at 28, 45, and 60 days post-infection (Talaat et al., 2007). Surprisingly, these microarrays revealed that M. tuberculosis bacilli remain metabolically active during chronic tuberculosis, even though bacterial counts remain the same. Pathways for carbohydrate metabolism, lipid metabolism, and energy metabolism were significantly upregulated. These analyses identified several clustered sets of genes, including a group of 12 in vivo expressed genes upregulated at all three time points relative to in vitro cultures. Included in this group was mprA of the iVEGI island as well as a previously uncharacterized gene, mosR, which was the most highly expressed gene of the cluster, at 200-fold upregulated. Further investigation of the role of MosR revealed that a ΔmosR strain is attenuated in a murine model of tuberculosis, and affects the expression of operons involved in mycobacterial survival at late stages of infection, likely through transcriptional regulation (Abomoelak et al., 2009).
Profiling of Human Infection
A disadvantage of the in vivo models reviewed thus far is that, while host models can provide a closer insight into human pathogenesis than in vitro models, they are still unable to wholly replicate the natural, human environment of M. tuberculosis. A study of M. tuberculosis transcription within resected human lung tissue was conducted using qRT-PCR, with results compared to expression levels found in the murine model (Timm et al., 2003). Interestingly, this study found that genes associated with iron limitation were not as induced in human lung as they were in mouse lung, which together with the cell culture data presented earlier, suggests that metal availability within the phagosome could be species-specific. Expression levels of icl1, found to be highly expressed in the macrophage model and necessary for pathogenesis in the murine model, showed that icl1 levels vary depending on the lung specimen used, possibly due to differences in oxygen availability between different types of granulomatous tissue.
A second study used microarray analysis to profile the expression of M. tuberculosis in resected lung tissue removed from patients with severe tuberculosis, often caused by multiple drug resistant (MDR) strains (Rachman et al., 2006b). The profiling study revealed upregulation of genes involved in the modification of the cell wall, including fatty and mycolic acids. Included in this group of cell wall-associated genes were many PE and PPE family genes, a family of surface proteins specific to mycobacteria whose function is not yet completely understood (Bottai and Brosch, 2009). Genes for protein chaperones and detoxification activities were also identified, as was seen in the murine model, as well as genes associated with transposition and insertion elements, as had been identified in the macrophage models. Finally, genes involved in both aerobic and anaerobic respiration were seen, and the transcriptional profiles of these genes differed depending on the site of the tissue being studied (e.g., granuloma vs. distant lung tissue), supporting the idea that both aerobic and anaerobic models may be used to represent different sites of human infection.
Although the overall categories of genes identified in the human study were similar to those seen within mouse models of infection, the specific genes identified had little overlap with those found in the model. For example, a comparison of genes upregulated in the human granuloma vs. genes upregulated in chronically infected mice revealed only four genes in common between the two data sets: PPE61, PPE55, rv0316, predicted to be involved in cholesterol utilization, and rv3706c, of unknown function. Between all genes upregulated in the murine and THP-1 macrophage models (860 genes total) and the genes upregulated in the human granuloma (187 genes), only 42 genes overlapped (Table 2). This 42 gene overlap includes PPE61, PPE55, and rv3706 also found in the mouse model (Figure 1B; Table 2). Notably, PPE55 has also been identified as a potential vaccine target based on its antigenicity (Singh et al., 2005; Zvi et al., 2008). However, as has been seen in other models, the largest category of genes identified in the human study was uncharacterized, hypothetical proteins.
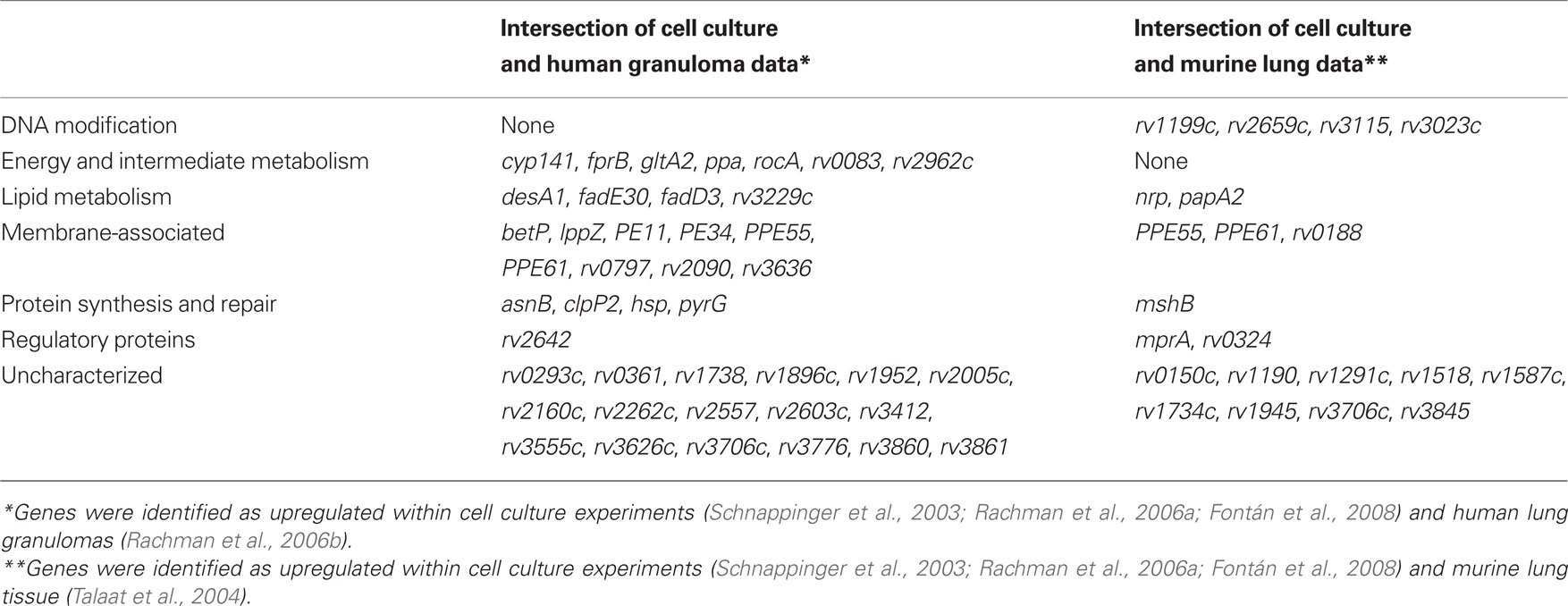
Table 2.The overlap in genes identified in multiple in vivo models, as represented in Figure 1B.
Concluding Remarks
In vivo studies have been very useful as a tool to better understand the host environment that M. tuberculosis experiences during human infection. Studies of macrophages, animal models, and even human lungs have all helped researchers make inferences about the host environment based on the reaction of M. tuberculosis to its surroundings. However, as tuberculosis continues to pose a looming threat to global health, it is important to mine through this expression data to infer clues not only about the host status but also about the factors enabling the virulence and pathogenesis of the bacteria. Studies conducted so far of strains containing mutations in genes identified through in vivo profiling suggest that this could be an effective way to identify gene products important to the bacteria. These genes could serve as interesting targets for applications such as vaccine creation and anti-mycobacterial drug targeting. Serving to complicate this process, the environment of M. tuberculosis is difficult to duplicate. Even within human host tissue itself, gene expression differs based on the biological variation seen not only from patient to patient, but even within different locations of the granuloma itself.
Despite these challenges, comparisons of the available data allows for the discovery of genes that are expressed highly in multiple models and across different locations and time points, which can help distinguish biological phenomena from experimental artifacts. Because no one model can accurately represent the full course of human disease, this type of meta-analysis may be the best way to obtain meaningful data from in vivo models. Therefore, it is important to keep in mind that if whole-genome data is to be utilized efficiently by the research community, it must be fully disseminated using standardized terminology, and in fact, the online database tbdb.org has recently been created for this reason (Galagan et al., 2010). It is equally imperative to prepare ourselves for the types of data that will emerge from new techniques, including advances in proteomics (Rao and Li, 2009) and metabolomics, which will allow future studies to account for post-transcriptional disease regulation, as well as next-generation sequencing, which represents an important new direction for transcriptional profiling studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Tuberculosis research in the Talaat lab has been supported by several NIH grants.
References
Abomoelak, B., Hoye, E., Chi, J., Marcus, S., Laval, F., Bannantine, J., Ward, S., Daffé, M., Liu, H., and Talaat, A. (2009). mosR, a novel transcriptional regulator of hypoxia and virulence in Mycobacterium tuberculosis. J. Bacteriol. 191, 5941–5952.
Albrecht, M., Sharma, C., Reinhardt, R., Vogel, J., and Rudel, T. (2010). Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 38, 868–877.
Bardarov, S., Bardarov, S. J. Jr., Pavelka, M. J. Jr., Sambandamurthy, V., Larsen, M., Tufariello, J., Chan, J., Hatfull, G., and Jacobs Jr, W. (2002). Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148, 3007–3017.
Be, N., Lamichhane, G., Grosset, J., Tyagi, S., Cheng, Q., Kim, K., Bishai, W., and Jain, S. (2008). Murine model to study the invasion and survival of Mycobacterium tuberculosis in the central nervous system. J. Infect. Dis. 198, 1520–1528.
Betts, J., Lukey, P., Robb, L., McAdam, R., and Duncan, K. (2002). Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731.
Boshoff, H. I., Reed, M. B, Barry, C. E. III, and Mizrahi, V. (2003). DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 113, 183–193.
Bottai, D., and Brosch, R. (2009). Mycobacterial PE, PPE and ESX clusters: novel insights into the secretion of these most unusual protein families. Mol. Microbiol. 73, 325–328.
Braibant, M., Gilot, P., and Content, J. (2000). The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 24, 449–467.
Cappelli, G., Volpe, E., Grassi, M., Liseo, B., Colizzi, V., and Mariani, F. (2006). Profiling of Mycobacterium tuberculosis gene expression during human macrophage infection: upregulation of the alternative sigma factor G, a group of transcriptional regulators, and proteins with unknown function. Res. Microbiol. 157, 445–455.
Chua, J., Vergne, I., Master, S., and Deretic, V. (2004). A tale of two lipids: Mycobacterium tuberculosis phagosome maturation arrest. Curr. Opin. Microbiol. 7, 71–77.
Cole, S., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S., Eiglmeier, K., Gas, S., Barry III, C., Tekaia, F., Badcock, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R., Devlin, K., Feltwell, T., Gentles, S., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Krogh, A., McLean, J. Moule, S., Murphy, L., Oliver, K., Osborne, J., Quail, M., Rajandream, M., Rogers, J., Rutter, S., Seeger, K., Skelton, J. Squares, R., Squares, S., Sulston, J., Taylor, K., Whitehead, S., and Barrell, B. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544.
Converse, P., Karakousis, P., Klinkenberg, L., Kesavan, A., Ly, L., Allen, S., Grosset, J., Jain, S., Lamichhane, G., Manabe, Y., McMurray, D., Nuermberger, E., and Bishai, W. (2009). Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect. Immun. 77, 1230–1237.
Corbett, E., Watt, C., Walker, N., Maher, D., Williams, B., Raviglione, M., and Dye, C. (2003). The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch. Intern. Med. 163, 1009–1021.
Cossart, P., and Archambaud, C. (2009). The bacterial pathogen Listeria monocytogenes: an emerging model in prokaryotic transcriptomics. J. Biol. 8, 107.
Danelishvili, L., McGarvey, J., Li, Y., and Bermudez, L. (2003). Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell. Microbiol. 5, 649–660.
Deb, C., Lee, C. M., Dubey, V. S., Daniel, J., Abomoelak, B., Sirakova, T. D., Pawar, S., Rogers, L., and Kolattukudy, P. (2009). A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS ONE 4, e6077. doi: 10.1371/journal.pone.0006077.
Fisher, M., Plikaytis, B., and Shinnick, T. (2002). Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J. Bacteriol. 184, 4025–4032.
Flynn, J. (2006). Lessons from experimental Mycobacterium tuberculosis infections. Microbes Infect. 8, 1179–1188.
Fontán, P., Aris, V., Ghanny, S., Soteropoulos, P., and Smith, I. (2008). Global transcriptional profile of Mycobacterium tuberculosis during THP-1 human macrophage infection. Infect. Immun. 76, 717–725.
Fu, X., Fu, N., Guo, S., Yan, Z., Xu, Y., Hu, H., Menzel, C., Chen, W., Li, Y., Zeng, R., and Khaitovich, P. (2009). Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genomics 10, 161.
Galagan, J., Sisk, P., Stolte, C., Weiner, B., Koehrsen, M., Wymore, F., Reddy, T., Zucker, J., Engels, R., Gellesch, M., Hubble, J., Jin, H., Larson, L., Mao, M., Nitzberg, M., White, J., Zachariah, Z., Sherlock, G., Ball, C., and Schoolnik, G. (2010). TB database 2010: overview and update. Tuberculosis (Edinb.) 90, 225–235.
Glaziou, P., Floyd, K., and Raviglione, M. (2009). Global burden and epidemiology of tuberculosis. Clin. Chest Med. 30, 621–636, vii.
Haller, R., Kennedy, M., Arnold, N., and Rutherford, R. (2010). The transcriptome of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 86, 1–9.
He, H., Hovey, R., Kane, J., Singh, V., and Zahrt, T. (2006). MprAB is a stress-responsive two-component system that directly regulates expression of sigma factors SigB and SigE in Mycobacterium tuberculosis. J. Bacteriol. 188, 2134–2143.
He, H., and Zahrt, T. (2005). Identification and characterization of a regulatory sequence recognized by Mycobacterium tuberculosis persistence regulator MprA. J. Bacteriol. 187, 202–212.
Hestvik, A., Hmama, Z., and Av-Gay, Y. (2005). Mycobacterial manipulation of the host cell. FEMS Microbiol. Rev. 29, 1041–1050.
Hingley-Wilson, S., Sambandamurthy, V., and Jacobs, W. J. (2003). Survival perspectives from the world’s most successful pathogen, Mycobacterium tuberculosis. Nat. Immunol. 4, 949–955.
Höner Zu Bentrup, K., Miczak, A., Swenson, D., and Russell, D. (1999). Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J. Bacteriol. 181, 7161–7167.
Kendall, S., Rison, S., Movahedzadeh, F., Frita, R., and Stoker, N. (2004). What do microarrays really tell us about M. tuberculosis? Trends Microbiol. 12, 537–544.
Kesavan, A., Brooks, M., Tufariello, J., Chan, J., and Manabe, Y. (2009). Tuberculosis genes expressed during persistence and reactivation in the resistant rabbit model. Tuberculosis (Edinb.) 89, 17–21.
Kinsella, R., Fitzpatrick, D., Creevey, C., and McInerney, J. (2003). Fatty acid biosynthesis in Mycobacterium tuberculosis: lateral gene transfer, adaptive evolution, and gene duplication. Proc. Natl. Acad. Sci. U.S.A. 100, 10320–10325.
Lee, J., Geiman, D., and Bishai, W. (2008). Role of stress response sigma factor SigG in Mycobacterium tuberculosis. J. Bacteriol. 190, 1128–1133.
Liu, T., Ramesh, A., Ma, Z., Ward, S., Zhang, L., George, G., Talaat, A., Sacchettini, J., and Giedroc, D. (2007). CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat. Chem. Biol. 3, 60–68.
Mariani, F., Cappelli, G., Riccardi, G., and Colizzi, V. (2000). Mycobacterium tuberculosis H37Rv comparative gene-expression analysis in synthetic medium and human macrophage. Gene 253, 281–291.
McGarvey, J., Wagner, D., and Bermudez, L. (2004). Differential gene expression in mononuclear phagocytes infected with pathogenic and non-pathogenic mycobacteria. Clin. Exp. Immunol. 136, 490–500.
McKinney, J., Höner zu Bentrup, K., Muñoz-Elías, E., Miczak, A., Chen, B., Chan, W., Swenson, D., Sacchettini, J., Jacobs Jr, W., and Russell, D. (2000). Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406, 735–738.
Mohamedmohaideen, N., Palaninathan, S., Morin, P., Williams, B., Braunstein, M., Tichy, S., Locker, J., Russell, D., Jacobs Jr., W., and Sacchettini, J. (2008). Structure and function of the virulence-associated high-temperature requirement A of Mycobacterium tuberculosis. Biochemistry 47, 6092–6102.
Muñoz-Elías, E., and McKinney, J. (2005). Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 11, 638–644.
Park, H., Guinn, K., Harrell, M., Liao, R., Voskuil, M., Tompa, M., Schoolnik, G., and Sherman, D. (2003). Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 48, 833–843.
Pethe, K., Swenson, D., Alonso, S., Anderson, J., Wang, C., and Russell, D. (2004). Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc. Natl. Acad. Sci. U.S.A. 101, 13642–13647.
Rachman, H., Strong, M., Schaible, U., Schuchhardt, J., Hagens, K., Mollenkopf, H., Kosmiadi, G. Eisenberg, D., and Kaufmann, S. (2006a). Mycobacterium tuberculosis gene expression profiling within the context of protein networks. Microbes Infect. 8, 747–757.
Rachman, H., Strong, M., Ulrichs, T., Grode, L., Schuchhardt, J., Mollenkopf, H., Kosmiadi, G. Eisenberg, D., and Kaufmann, S. (2006b). Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 74, 1233–1242.
Rosas-Magallanes, V., Deschavanne, P., Quintana-Murci, L., Brosch, R., Gicquel, B., and Neyrolles, O. (2006). Horizontal transfer of a virulence operon to the ancestor of Mycobacterium tuberculosis. Mol. Biol. Evol. 23, 1129–1135.
Rustad, T., Harrell, M., Liao, R., and Sherman, D. (2008). The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 3, e1502. doi: 10.1371/journal.pone.0001502.
Sassetti, C., and Rubin, E. (2003). Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994.
Schnappinger, D., Ehrt, S., Voskuil, M., Liu, Y., Mangan, J., Monahan, I., Dolganov, G., Efron, B., Butcher, P., Nathan, C., and Schoolnik, G. (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704.
Sharma, C., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiss, S., Sittka, A., Chabas, S., Reiche, K., Hackermüller, J., Reinhardt, R., Stadler, P., and Vogel, J. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255.
Shi, L., Sohaskey, C., Kana, B., Dawes, S., North, R., Mizrahi, V., and Gennaro, M. (2005). Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. U.S.A. 102, 15629–15634.
Singh, K., Dong, Y., Patibandla, S., McMurray, D., Arora, V., and Laal, S. (2005). Immunogenicity of the Mycobacterium tuberculosis PPE55 (Rv3347c) protein during incipient and clinical tuberculosis. Infect. Immun. 73, 5004–5014.
Skeiky, Y., Lodes, M., Guderian, J., Mohamath, R., Bement, T., Alderson, M., and Reed, S. (1999). Cloning, expression, and immunological evaluation of two putative secreted serine protease antigens of Mycobacterium tuberculosis. Infect. Immun. 67, 3998–4007.
Srivastava, V., Jain, A., Srivastava, B., and Srivastava, R. (2008). Selection of genes of Mycobacterium tuberculosis upregulated during residence in lungs of infected mice. Tuberculosis (Edinb.) 88, 171–177.
Stewart, G., Wernisch, L., Stabler, R., Mangan, J., Hinds, J., Laing, K., Young, D., and Butcher, P. (2002). Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology 148, 3129–3138.
Sturgill-Koszycki, S., Schlesinger, P., Chakraborty, P., Haddix, P., Collins, H., Fok, A., Allen, R., Gluck, S., Heuser, J., and Russell, D. (1994). Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263, 678–681.
Tailleux, L., Waddell, S., Pelizzola, M., Mortellaro, A., Withers, M., Tanne, A., Castagnoli, P., Gicquel, B., Stoker, N., Butcher, P., Foti, M., and Neyrolles, O. (2008). Probing host pathogen cross-talk by transcriptional profiling of both Mycobacterium tuberculosis and infected human dendritic cells and macrophages. PLoS ONE 3, e1403. doi: 10.1371/journal.pone.0001403.
Talaat, A., Howard, S., Hale, W. IV, Lyons, R., Garner, H., and Johnston, S. (2002). Genomic DNA standards for gene expression profiling in Mycobacterium tuberculosis. Nucleic Acids Res. 30, e104.
Talaat, A., Hunter, P., and Johnston, S. (2000). Genome-directed primers for selective labeling of bacterial transcripts for DNA microarray analysis. Nat. Biotechnol. 18, 679–682.
Talaat, A., Lyons, R., Howard, S., and Johnston, S. (2004). The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc. Natl. Acad. Sci. U.S.A. 101, 4602–4607.
Talaat, A., Ward, S., Wu, C., Rondon, E., Tavano, C., Bannantine, J., Lyons, R., and Johnston, S. (2007). Mycobacterial bacilli are metabolically active during chronic tuberculosis in murine lungs: insights from genome-wide transcriptional profiling. J. Bacteriol. 189, 4265–4274.
Timm, J., Post, F., Bekker, L., Walther, G., Wainwright, H., Manganelli, R., Chan, W., Tsenova, L., Gold, B., Smith, I., Kaplan, G., and McKinney, J. (2003). Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc. Natl. Acad. Sci. U.S.A. 100, 14321–14326.
Waddell, S., Laing, K., Senner, C., and Butcher, P. (2008). Microarray analysis of defined Mycobacterium tuberculosis populations using RNA amplification strategies. BMC Genomics 9, 94.
Wagner, D., Maser, J., Lai, B., Cai, Z., Barry, C. E. III, Höner Zu Bentrup, K., Russell, D. G., and Bermudez, L. E. (2005). Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J. Immunol. 174, 1491–1500.
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63.
Ward, S., Abomoelak, B., Hoye, E., Steinberg, H., and Talaat, A. (2010). CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol. Microbiol. 77, 1096–1110.
Warner, D., and Mizrahi, V. (2007). The survival kit of Mycobacterium tuberculosis. Nat. Med. 13, 282–284.
Wayne, L., and Hayes, L. (1996). An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64, 2062–2069.
Wayne, L., and Sohaskey, C. (2001). Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55, 139–163.
White, M., He, H., Penoske, R., Twining, S., and Zahrt, T. (2010). PepD participates in the mycobacterial stress response mediated through MprAB and SigE. J. Bacteriol. 192, 1498–1510.
Xu, Y., Xie, J., Li, Y., Yue, J., Chen, J., Chunyu, L., and Wang, H. (2003). Using a cDNA microarray to study cellular gene expression altered by Mycobacterium tuberculosis. Chin. Med. J. 116, 1070–1073.
Zahrt, T., and Deretic, V. (2001). Mycobacterium tuberculosis signal transduction system required for persistent infections. Proc. Natl. Acad. Sci. U.S.A. 98, 12706–12711.
Keywords: tuberculosis, pathogenesis, infection, transcriptional profiling
Citation: Ward SK, Abomoelak B, Marcus SA and Talaat AM (2010) Transcriptional profiling of Mycobacterium tuberculosis during infection: lessons learned Front. Microbio. 1:121. doi: 10.3389/fmicb.2010.00121
Received: 27 August 2010;
Accepted: 12 October 2010;
Published online: 18 November 2010.
Edited by:
Paul De Figueiredo, Texas A&M University, USAReviewed by:
Libo Shan, Texas A&M University, USAMartin Dickman, Texas A&M University, USA
David O’callaghan, Institut National de la Santé et de la Recherche Médicale, France
Yongqun “Oliver” He, University of Michigan chool of Medicine, USA
Yi Xu, Texas A&M Health Science Center, USA
Mark Estes, University of Texas Medical Branch, USA
Copyright: © 2010 Ward, Abomoelak, Marcus and Talaat. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Adel M. Talaat, Laboratory of Bacterial Genomics, Department of Pathobiological Sciences, University of Wisconsin-Madison, 1656 Linden Drive, Madison, WI 53706-1581, USA. e-mail:YXRhbGFhdEB3aXNjLmVkdQ==