 Masakazu Yoshizumi1,2
Masakazu Yoshizumi1,2 Hirokazu Kimura3* Yoshimichi Okayama4* Atsuyoshi Nishina5 Masahiro Noda6 Hiroyuki Tsukagoshi1 Kunihisa Kozawa1
Hirokazu Kimura3* Yoshimichi Okayama4* Atsuyoshi Nishina5 Masahiro Noda6 Hiroyuki Tsukagoshi1 Kunihisa Kozawa1 Masahiko Kurabayashi2
Masahiko Kurabayashi2
- 1 Gunma Prefectural Institute of Public Health and Environmental Sciences, Maebashi-shi, Gunma, Japan
- 2 Department of Medicine and Biological Science, Gunma University Graduate School of Medicine, Maebashi-shi, Gunma, Japan
- 3 Infectious Disease Surveillance Center, National Institute of Infectious Diseases, Musashimurayama-shi, Tokyo, Japan
- 4 Division of Molecular Cell Immunology and Allergology, Advanced Medical Research Center, Nihon University Graduate School of Medical Science, Tokyo, Japan
- 5 Department of Health and Nutrition, Yamagata Prefectural Yonezawa Women’s Junior College, Yonezawa-shi, Yamagata, Japan
- 6 Department of Virology III, National Institute of Infectious Diseases, Musashimurayama-shi, Tokyo, Japan
Respiratory viruses such as parainfluenza virus (PIV) in individuals with certain genetic predispositions in early life are associated with the induction of wheezing, which can progress to the development of asthma. It has been suggested that aberrant production of various cytokines due to viral infection are associated with virus-induced asthma. However, the mechanisms of how respiratory viruses induce and exacerbate asthma have yet to be clarified. To examine cytokine responses to PIV infection, we assessed 27 cytokine levels released from PIV-infected human fetal lung fibroblasts. In addition, we examined relationships between these cytokine responses and signaling pathways (IκB kinase and p38 MAPK) in PIV-infected cells. At 24 h after infection, PIV-infected cells significantly released a number of cytokines, namely, proinflammatory cytokines [interleukins (IL)-1β, IL-6, and tumor necrosis factor-α], anti-inflammatory cytokine (IL-1ra), Th1 cytokines (interferon-γ, and IL-2), Th2 cytokines (IL-4, IL-5, and IL-10), granulopoiesis-inducing cytokines (granulocyte colony-stimulating factor and granulocyte–macrophage colony-stimulating factor), neutrophil recruitment-inducing cytokines (IL-8 and interferon-inducible protein-10), and eosinophil recruitment-inducing cytokines (eotaxin and regulated on activation normal T-cell expressed and secreted). PIV infection enhanced phosphorylation of both IκB and p38 MAPK, but not Akt, in the cells. Signaling pathway inhibitors, BMS-345541 (a specific IκB kinase inhibitor) and SB203580 (a specific p38 MAPK inhibitor), significantly suppressed release of these cytokines from PIV-infected cells. The results indicate that PIV infection induces aberrant production and release of various cytokines through IκB kinase and p38 MAPK pathways in human lung fibroblasts. Overproduction and imbalance of these cytokines may be partially associated with the pathophysiology of virus-induced asthma.
Introduction
Respiratory viruses are responsible for most acute respiratory illnesses in humans, including the common cold, bronchiolitis, and pneumonia (Johnston, 2004). These major pathogens include human parainfluenza viruses (PIV), rhinoviruses (RV), respiratory syncytial virus (RSV), and influenza viruses (Monto, 2004). Respiratory infections resulting from PIV, RV, and RSV have also been implicated in the induction and exacerbation of asthma (Folkerts et al., 1998; Stein et al., 1999; Azevedo et al., 2003; Kotaniemi-Syrjänen et al., 2003; Martinez, 2003; Matsuse et al., 2005; Hershenson and Johnston, 2006; Khetsuriani et al., 2007; Kusel et al., 2007). Most children are infected at least once with PIV early in life, but reinfections occur throughout life. Serologic surveys indicate that at least 60% of children have been infected with PIV3 by 2 years of age while approximately 80% have been infected by 4 years of age. PIV are associated with 0–12% of acute lower respiratory tract infections in adults. PIV infections may be associated with not only respiratory infections but also the induction and exacerbation of asthma (virus-induced asthma) (Azevedo et al., 2003; Matsuse et al., 2005). However, many children who wheeze with viral infections during infancy will not go on to develop asthma, suggesting a role of virus- and/or host-specific factors in the pathogenesis of the disease.
Most types of cytokines may be associated with the pathophysiology of virus-induced asthma or excessive virus-induced inflammation (Mallia and Johnston, 2006; Proud and Chow, 2006). In extreme cases, an overproduction of cytokines due to viral infection (e.g., influenza virus infection) may induce a cytokine storm resulting in systemic inflammatory response syndrome (SIRS) (Watanabe et al., 2003). Thus, viral infection-induced overproduction of cytokines in vivo might be closely associated with the pathophysiology of various diseases including asthma (Message and Johnston, 2004; Mallia and Johnston, 2006; Proud and Chow, 2006). However, cytokine profiles of viral infections including PIV infection remain largely unknown.
IκB kinase, p38 MAPK, and Akt are major signaling molecules thought to play a pivotal role in most types of cytokine production in various cells (Hiscott et al., 2001; Adcock et al., 2006). Single strand RNA such as viral RNA binds and activates helicase, which further activates IκB kinase pathways, resulting in the production of certain types of cytokine (Lee and Lau, 2007). In addition, it is well known that phosphorylation of p38 MAPK is associated with the production of cytokines in many types of cells (Suzuki et al., 1999; Wong et al., 2005). Thus, these signaling molecules may be associated with the production of cytokines in virus-infected cells, although the details are not as yet known.
Human lung fibroblasts are reported to be susceptible to various respiratory viruses such as PIV and RV (Bousse et al., 2006; Jang et al., 2009). In addition, lung fibroblasts may be associated with remodeling involved in fibrosis in the small airways (Puxeddu et al., 2006). In asthmatics, viral infections are more likely to result in lower respiratory tract infection (Gern, 2009), and moreover, these infections are not restricted to the respiratory epithelium, but can spread to the structural cells such as smooth muscle cells and fibroblasts (Holgate et al., 2004b). From these background, in the present study we chose human fetal lung fibroblasts to examine the cytokine milieu of the asthmatic airway under chronic viral infection. We hypothesized that PIV infection might induce virus-induced asthma-related cytokine production in fetal lung fibroblasts. To explore this possibility, we profiled cytokines released from PIV-infected fetal lung fibroblasts and examined the role of the signaling pathways (IκB kinase, p38 MAPK, and Akt).
Materials and Methods
Cells and Cell Culture
Human fetal lung fibroblasts (MRC-5 cells) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were grown in 75-cm2 tissue culture flasks and maintained in Eagle’s minimum essential medium (MEM; Invitrogen Life Technologies, CA, USA) containing 10% fetal bovine serum (FBS), l-glutamine (0.6 mg/ml), and 0.35% NaHCO3 at 36°C in a humidified atmosphere containing 5% CO2 (de Oña et al., 1995).
Viral Propagation and Measurement of Viral Titers
Human PIV type 3 (C 243 strain) were obtained from American Tissue Culture Corrections (Rockville, MD, USA) and propagated in Vero E6 cells. When the PIV-infected Vero E6 cells showed cytopathic effects (CPE), they were centrifuged at 4200 × g for 30 min at 4°C to remove debris. To purify the viruses, the supernatants were layered over 50% sucrose in PBS, then centrifuged at 65,000 × g for 2 h at 4°C as previously described (Ueba, 1978). Aliquots of the viruses were stored at −80°C until required.
Measurement of Cytokine Concentrations
We inoculated 0 (no virus) or 1.0 MOI of each virus suspension (100 μl) in Opti-MEM I to the human fetal lung fibroblasts grown in 96-well microplates. Concentrations of cytokines, chemokines, and growth factors in the culture supernatants from each experimental system (virus-infected, UV-inactivated virus, inhibitor-added, and no virus) were obtained at 24 h. To remove the effect of intracellular cytokines/chemokines, we centrifuged the microplates at 1660 × g for 10 min before carefully harvesting the supernatants.
We measured the following 27 cytokines using an available kit (Cytokine assay 27-Plex Panel kit, Bio-Rad, Hercules, CA, USA): interleukins (IL)-1β, 1ra, 2, 4, 5, 6, 7, 8, 9, 10, 12, 13, 15, and 17, interferon (IFN)-γ, granulocyte colony-stimulating factor (G-CSF), granulocyte–macrophage colony-stimulating factor (GM-CSF), tumor necrosis factor (TNF)-α, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, 1β, interferon-inducible protein (IP)-10, eotaxin, fibroblast growth factor (FGF) basic, platelet-derived growth factor (PDGF)-bb, regulated on activation normal T-cell expressed and secreted (RANTES), and vascular endothelial growth factor (VEGF). Assays were performed using the Bio-Plex suspension array system according to the manufacturer’s instructions (Bio-Rad). Data were automatically processed and analyzed using Bio-Plex Manager Software 5.0 with the standard curve produced from the recombinant cytokine standard. The sensitivity limits for each cytokine were set at 0.1–3 pg/ml (Okazaki et al., 2008; Yoshizumi et al., 2008).
Measurement of Phosphorylation Inhibition of IκB Kinase, p38 MAPK, and Akt
The inhibitors BMS-345541 (4(2′-aminoethyl)amino-1,8-dimethylimidazo(1,2-a)quinoxaline), a specific inhibitor of IκB kinase), SB203580 (4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole, a specific inhibitor of p38 MAPK), and Akt inhibitor X (10-(4-(N-diethylamino)butyl)-2-chlorophenoxazine, HCl) were purchased from Calbiochem (KGaA, Darmstadt, Germany). These compounds were dissolved in DMSO (10 mM) and stored at −80°C until required. The optimal concentration of each of these specific inhibitors of signaling protein phosphorylation was preliminary examined, and 10 μM of each inhibitor was found to be suitable for significant inhibition of the phosphorylation of the kinases, results which are compatible with previous reports (MacMaster et al., 2003; Thimmaiah et al., 2005; Zaheer et al., 2009). Phosphorylation of signal molecules induced by PIV was also assessed using a Bio-Plex suspension array system.
Statistical Analysis
Data are expressed as means ± SE. Statistical analyses of cytokine/chemokine concentrations were performed using the Kruskal–Wallis and Mann–Whitney U methods. Subsequent post hoc analysis was conducted using the Bonferroni-adjusted α method. All statistical analyses were performed using SPSS version 12.0 (SPSS Inc., Chicago, IL, USA). Values of p < 0.05 were considered significant.
Results
Experimental Conditions
To confirm the infection of human fetal lung fibroblasts with PIV, morphological changes in the cells were examined by light microscopy after 6, 24, and 48 h. No changes were observed 6 h after PIV infection, whereas the cells clearly showed CPE after 48 h of PIV infection (data not shown). We confirmed that there were no significant differences in the amounts of cytokines released between the UV-treated virus treatment, medium with 0.1% DMSO, and medium alone (without the addition of viruses) (data not shown). Of the 27 cytokines examined, significantly higher amounts of 18 cytokines were released after addition of 1.0 MOI of PIV compared with no addition of viruses (data not shown). Cytokine release by viruses was MOI-dependent (data not shown).
Categorization of Cytokines Released by PIV-Infected Human Fetal Lung Fibroblasts
Next, we compared the cytokine levels induced between control and PIV-infected fibroblasts. The amounts of 18 cytokines induced by PIV infection were significantly higher, at least at one time point, than those of control cells. The 18 cytokines were categorized into nine groups (Barnes, 2008): proinflammatory cytokines (Figures 1A–C), anti-inflammatory cytokines, (Figure 1D), Th1 cytokines (Figures 1E,F), Th2 cytokines (Figures 1G–I), granulopoiesis-inducing cytokines (Figures 1J,K), neutrophil recruitment-inducing chemokines (Figures 1L,M), eosinophil recruitment-inducing chemokines (Figures 1N,O), and tissue remodeling-related cytokines (Figures 1P,Q).
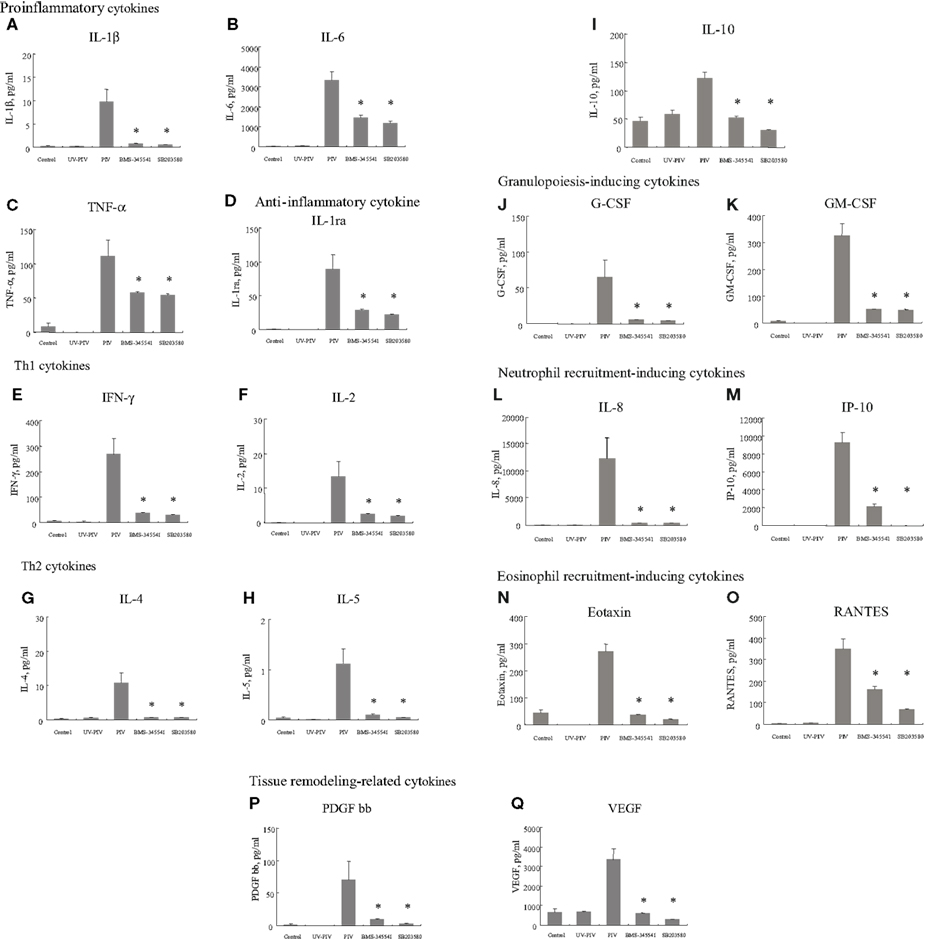
Figure 1. Proinflammatory cytokine, Th1 cytokine, Th2 cytokine, growth factor, chemokine, and tissue remodeling-related cytokine levels in culture supernatants from PIV-infected human fetal lung fibroblasts. All assays were performed in duplicate in five independent experiments. “Control” indicates no addition of virus to the fibroblasts. BMS-345541 and SB203580 indicate addition of each inhibitor within 60 min of PIV infection. Vertical bars represent mean ± SE. *p < 0.05 when compared with the cytokine levels induced by infection with PIV.
Comparison of Proinflammatory and Anti-Inflammatory Cytokines Released by PIV-Infected Human Fetal Lung Fibroblasts
As shown in Figures 1A–C, proinflammatory cytokine (IL-1β, IL-6 and TNF-α levels at 24 h after infection with PIV were significantly higher than levels of control cells. Similarly, the level of IL-1ra (anti-inflammatory cytokine) was significantly higher from PIV-infected cells than from the control cells (Figure 1D).
Comparison of Th1, Th17, and Th2 Cytokines Released by PIV-Infected Human Fetal Lung Fibroblasts
To clarify the differences in Th1, Th17, and Th2 cytokine profiles between PIV-infected human fetal lung fibroblasts and control cells, we compared levels of IFN-γ, IL-2, IL-12, IL-17, IL-4, IL-5, IL-10, and IL-13. We found that small but significant amounts of IFN-γ and IL-2 were produced by PIV-infected fibroblasts compared with control fibroblasts (Figures 1E,F), but there was no significant difference in IL-12 levels (data not shown). In addition, PIV infection did not result in any significant difference in amounts of IL-17 from fibroblasts compared with the control (data not shown). PIV infection did result in significantly higher production of IL-4, IL-5, and IL-10 compared with the control (Figures 1G–I), but not IL-13 (data not shown).
Comparison of Growth Factors and Chemokines Released by PIV-Infected Human Fetal Lung Fibroblasts
G-CSF and GM-CSF levels induced by PIV infection were significantly higher than those induced by the control (Figures 1J,K). Similarly, IL-8, IP-10, eotaxin, and RANTES levels were significantly higher in supernatants from PIV-infected fibroblasts than control fibroblasts (Figures 1L–O). Regarding cytokines related to tissue remodeling, the same tendency was observed; PDGF-bb and VEGF levels were significantly higher in supernatants from PIV-infected fibroblasts compared with control fibroblasts (Figures 1P,Q).
Phosphorylation of IκB Kinase, p38 MAPK, and Akt
To examine possible signal molecules, we investigated the phosphorylation of IκB kinase, p38 MAPK, and Akt. PIV infection dramatically induced the phosphorylation of IκB kinase and p38 MAPK in MRC-5 cells compared with the control (Figures 2A,B). However, no enhancement of Akt phosphorylation was found in the virus-infected cells (Figure 2C). Thus, to define the role of the IκB kinase and p38 MAPK pathways in viral induction of cytokines and chemokines, we used their specific inhibitors. As a result, in MRC-5 cells, inhibition of the NF-κB pathway with BMS-345541 (10 μM) significantly inhibited PIV-induced cytokines, namely, IL-1β, 1ra, 2, 4, 5, 6, 8, 10, IFN-γ, TNF-α, G-CSF, GM-CSF, IP-10, eotaxin, RANTES, PDGF-bb, and VEGF (Figure 1). Similarly, treatment of the p38 MAPK pathway with SB203580 (10 μM) also significantly inhibited the release of these cytokines (Figure 1). The results suggested that both IκB kinase and p38 MAPK phosphorylation was associated with the production and release of various cytokines in the virus-infected cells.
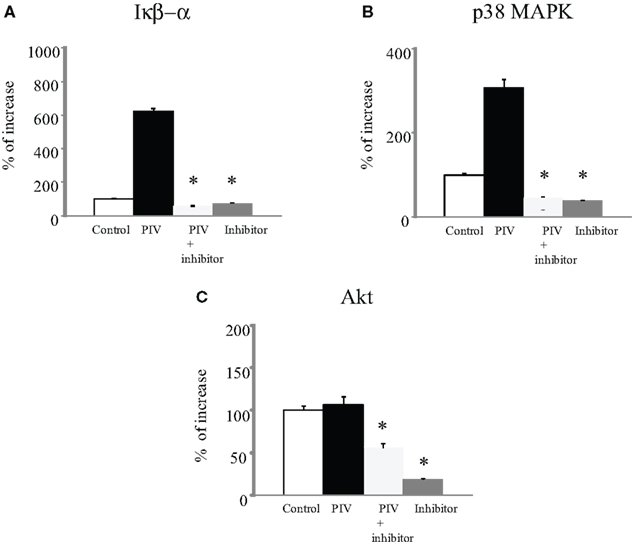
Figure 2. Phosphorylation of IκB kinase, p38 MAPK, and Akt levels in cell lysate from PIV-infected human fetal lung fibroblasts. All assays were performed in duplicate in five independent experiments. “Control” indicates no addition of virus to the fibroblasts. Vertical bars represent mean ± SE. *p < 0.05 when compared with the IκB kinase (A), p38 MAPK (B), and Akt (C) levels induced by infection with PIV.
Discussion
We profiled a number of cytokines from PIV-infected fetal lung fibroblasts. Compared with control (no virus) cells, a significant amount of various cytokines were shown to be released, namely, IL-1β, IL-6, TNF-α, IL-1ra, IFN-γ, IL-2, IL-4, IL-5, IL-10, G-CSF, GM-CSF, IL-8, IP-10, eotaxin, RANTES, PDGF-bb, and VEGF. In addition, we found that most aberrantly released cytokines were associated with phosphorylation of IκB kinase and p38 MAPK. Infection by PIV may induce large-scale production of various cytokines in the airway. Although PIV failed to induce IL-12 and IL-17 production, these results indicate that PIV induces a Th2 cytokine milieu as well as eosinophil and neutrophil recruitment to fibroblasts. As a result, Th2 immunity may be augmented, which is likely to be an important mechanism in the pathophysiology of PIV-induced asthma (virus-induced asthma).
It is suggested that maximal MAP kinase phosphorylation and activity is seen within 60 min after stimulation. In this study, the phosphorylation of IκB kinase and p38 MAP kinase was examined at 24 h after PIV infection. Thus, it is possible that cytokines produced by PIV-infected MRC-5 cells might be responsible for the induction of MAP kinase phosphorylation, since various cytokines produced after PIV infection have been shown to induce IκB kinase and MAP kinase phosphorylation.
We speculate that the effects of PIV on normal fibroblasts can be extrapolated to corresponding cells in asthmatic airways. Previous studies have shown that increased viral replication and impaired IFN response to viral infection occurs in primary epithelial cells and peripheral blood cells from asthmatics compared with normal subjects (Wark et al., 2005; Contoli et al., 2006; Gehlhar et al., 2006). The increase in virus infection-induced inflammation is a probable explanation for these observations. Asthmatics may also be more susceptible to infection (Holgate, 2007). Furthermore, it has also been suggested that infants who are born with poor antiviral responses and/or airway hyper-responsiveness are prone to repetitive severe illnesses (Gehlhar et al., 2006), and these same abnormalities may increase the risk of recurrent wheezing and possibly asthma. Therefore, fibroblasts from those who develop asthma in earlier childhood might possess a defective IFN response to these respiratory viruses. Such partially defective innate immune responses may result in augmented Th2 immunity in asthmatic patients as well as increased and prolonged airway inflammation (Barrett and Austen, 2009).
A direct causative role for these respiratory viruses in the development of asthma has not been proven yet. Lemanske et al. (2005) suggested at least two potential mechanisms for the development of recurrent wheezing in early childhood. First, healthy infants who undergo repetitive severe viral respiratory infections could develop recurrent wheezing because of lung damage and/or airway remodeling. Second, as mentioned above, infants who are born with weak antiviral responses and/or airway hyper-responsiveness are prone to repetitive severe illnesses, which may increase the risk of recurrent wheezing and possibly asthma. A recent study suggests that RSV-induced wheezing is an indicator of genetic predisposition to asthma, but is not associated with allergic sensitization, irrespective of the family history of asthma (Singh et al., 2007). In the present study, PIV-infected human lung fibroblasts produced and released tissue remodeling-related cytokines (PDGF and VEGF) and proinflammatory cytokines (IL-1β, IL-6, and TNF-α) compared with the control. Using animal models, it was also recently suggested that endotracheal inoculation of PIV induces biological and histological changes reminiscent of asthma (Pelaia et al., 2006). In particular, PIV infection elicits an airway influx of inflammatory cells, bronchial hyper-responsiveness, epithelial damage, and bronchiolar fibrosis (Pelaia et al., 2006). These findings suggest that the recurrence of PIV infections in patients with asthma may contribute to prolonged airway inflammation and airway structural changes typical of the disease (Pelaia et al., 2006). Moreover, our results suggest that PIV-induced tissue remodeling-related cytokines and proinflammatory cytokines from human fibroblasts may be associated with respiratory remodeling and prolonged airway inflammation after infection. Human asthma subjects have exhibited significant increases in IFN-γ, in the supernatants of cultured bronchoalveolar cells (Krug et al., 1996). Recently, Magnan et al. (2000) demonstrated increased IFN-γ-producing CD8+ T cells in asthmatic airways, and the levels of IFN-γ correlated with asthma severity, bronchial hyper-responsiveness, and blood eosinophilia (Ishihara et al., 1997). IFN-γ has been reported to be associated with severe and chronic asthma. In the present study, PIV-infected human lung fibroblasts produced and released IFN-γ, which might be associated with severity of asthma.
Cytokine production-related signaling pathways related to viral infections have been reported (Santoro et al., 2003; Kohlmeier et al., 2009; Zaheer et al., 2009). For example, Lee et al. suggested that single strand viral RNA of influenza virus binds to the host cellular helicase resulting in activation of signaling pathways including IκB kinase (Lee and Lau, 2007). In addition, viral single strand or double strand RNA/MKK3/6 complex can also activate p38 MAPK (Stephan, 2007). Finally, activation of these pathways may lead to the production and release of cytokines in virus-infected cells as an antiviral response (Stephan, 2007). In the present study, at 24 h after PIV infection, phosphorylation of IκB kinase and p38 MAPK and the release of cytokines were significantly increased in lung fibroblasts. Furthermore, inhibition of phosphorylation of these kinases by specific inhibitors significantly eliminated release of cytokines (Figure 2). At 24 h after infection, PIV genomes (also single strand RNA) may be significantly propagated (De et al., 1990). A possible reason for this is that the increase in phosphorylation of IκB kinase and p38 MAPK after PIV infection might be associated with propagated PIV genomes in the cells, although we did not observe an increase in the PIV genome/helicase or PIV genome MKK3/6 complex.
Fibroblasts have the potential to play a critical role in remodeling of and changes in the airway matrix (Holgate et al., 2004a). In this study, we found that PIV infection-induced significantly higher production of Th1 and Th2 cytokines from fibroblasts compared to control cells. PIV might promote release of not only inflammatory cytokines but also allergy-related cytokines in virus-infected MRC-5 cells. From the present results and previous our results suggest that aberrant release/production of various cytokines (i.e., cytokine storm) due to PIV or RSV infection is also associated with severe respiratory infection such as pneumonia (Ishioka et al., 2010). In addition, we showed that this cytokine production may be closely linked to phosphorylation of signal molecules such as IκB kinase and p38 MAPK. Recurrent RSV infection may damage the lower respiratory airways and induce remodeling. Finally, our findings suggest that PIV infection-induced aberrant phosphorylation of IκB kinase and p38 MAPK in lung fibroblasts might mediate hypersensitive Th1- and Th2-type responses in subjects with asthma, which is likely an important mechanism in virus-induced asthma.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Mika Saito, Dr. Masahiro Fujita, and Mr. Hayato Takada for their skilful assistance and helpful discussions. This study was supported in part by Research on Emerging and Re-emerging Infectious Diseases, Labour, and Welfare Programs from the Ministry of Health, Labour, and Welfare, Japan and Research on Health and Welfare Programs from the Research Fund for Promoting Science and Technology of Gunma prefecture, Japan.
References
Adcock, I. M., Chung, K. F., Caramori, G., and Ito, K. (2006). Kinase inhibitors and airway inflammation. Eur. J. Pharmacol. 533, 118–132.
Azevedo, A. M., Durigon, E. L., Okasima, V., Queiroz, D. A., de Moraes-Vasconcelos, D., Duarte, A. J., and Grumach, A. S. (2003). Detection of influenza, parainfluenza, adenovirus and respiratory syncytial virus during asthma attacks in children older than 2 years old. Allergol. Immunopathol. (Madr) 31, 311–317.
Barnes, P. J. (2008). The cytokine network in asthma and chronic obstructive pulmonary disease. J. Clin. Invest. 118, 3546–3556.
Barrett, N. A., and Austen, K. F. (2009). Innate cells and T helper 2 cell immunity in airway inflammation. Immunity 31, 425–437.
Bousse, T., Chambers, R. L., Scroggs, R. A., Portner, A., and Takimoto, T. (2006). Human parainfluenza virus type 1 but not Sendai virus replicates in human respiratory cells despite IFN treatment. Virus Res. 121, 23–32.
Contoli, M., Message, S. D., Laza-Stanca, V., Edwards, M. R., Wark, P. A., Bartlett, N. W., Kebadze, T., Mallia, P., Stanciu, L. A., Parker, H. L., Slater, L., Lewis-Antes, A., Kon, O. M., Holgate, S. T., Davies, D. E., Kotenko, S. V., Papi, A., and Johnston, S. L. (2006). Role of deficient type III interferon-lambda production in asthma exacerbations. Nat. Med. 12, 1023–1026.
De, B. P., Galinski, M. S., and Banerjee, A. K. (1990). Characterization of an in vitro system for the synthesis of mRNA from human parainfluenza virus type 3. J. Virol. 64, 1135–1142.
de Oña, M., Melón, S., de la Iglesia, P., Hidalgo, F., and Verdugo, A. F. (1995). Isolation of influenza virus in human lung embryonated fibroblast cells (MRC-5) from clinical samples. J. Clin. Microbiol. 33, 1948–1949.
Folkerts, G., Busse, W. W., Nijkamp, F. P., Sorkness, R., and Gern, J. E. (1998). Virus-induced airway hyperresponsiveness and asthma. Am. J. Respir. Crit. Care Med. 157(Pt 1), 1708–1720.
Gehlhar, K., Bilitewski, C., Reinitz-Rademacher, K., Rohde, G., and Bufe, A. (2006). Impaired virus-induced interferon-alpha2 release in adult asthmatic patients. Clin. Exp. Allergy 36, 331–337.
Gern, J. E. (2009). Rhinovirus and the initiation of asthma. Curr. Opin. Allergy Clin. Immunol. 9, 73–78.
Hershenson, M. B., and Johnston, S. L. (2006). Rhinovirus infections: more than a common cold. Am. J. Respir. Crit. Care Med. 174, 1284–1285.
Hiscott, J., Kwon, H., and Génin, P. (2001). Hostile takeovers: viral appropriation of the NF-kappaB pathway. J. Clin. Invest. 107, 143–151.
Holgate, S. T., Davies, D. E., Rorke, S., Cakebread, J., Murphy, G., Powell, R. M., and Holloway, J. W. (2004a). ADAM 33 and its association with airway remodeling and hyperresponsiveness in asthma. Clin. Rev. Allergy Immunol. 27, 23–34.
Holgate, S. T., Holloway, J., Wilson, S., Bucchieri, F., Puddicombe, S., and Davies, D. E. (2004b). Epithelial–mesenchymal communication in the pathogenesis of chronic asthma. Proc. Am. Thorac. Soc. 1, 93–98.
Ishihara, C., Ochiai, K., Kagami, M., Takashahi, H., Matsuyama, G., Yoshida, S., Tomioka, H., and Koya, N. (1997). Human peripheral eosinophils express functional interferon-gamma receptors (IFN-gammaR). Clin. Exp. Immunol. 110, 524–529.
Ishioka, T., Kimura, H., Kita, H., Obuchi, M., Hoshino, H., Noda, M., Nishina, A., Kozawa, K., and Kato, M. (2010). Effects of respiratory syncytial virus infection and major basic protein derived from eosinophils in pulmonary alveolar epithelial cells (A549). Cell Biol. Int. (in press).
Jang, Y. J., Wang, J. H., Kim, J. S., Kwon, H. J., Yeo, N. K., and Lee, B. J. (2009). Levocetirizine inhibits rhinovirus-induced ICAM-1 and cytokine expression and viral replication in airway epithelial cells. Antiviral Res. 81, 226–233.
Johnston, S. L. (2004). Overview of virus-induced airway disease. Proc. Am. Thorac. Soc. 2, 150–156.
Khetsuriani, N., Kazerouni, N. N., Erdman, D. D., Lu, X., Redd, S. C., Anderson, L. J., and Teague, W. G. (2007). Prevalence of viral respiratory tract infections in children with asthma. J. Allergy Clin. Immunol. 119, 314–321.
Kohlmeier, J. E., Cookenham, T., Miller, S. C., Roberts, A. D., Christensen, J. P., Thomsen, A. R., and Woodland, D. L. (2009). CXCR3 directs antigen-specific effector CD4+ T cell migration to the lung during parainfluenza virus infection. J. Immunol. 183, 4378–4384.
Kotaniemi-Syrjänen, A., Vainionpää, R., Reijonen, T. M., Waris, M., Korhonen, K., and Korppi, M. (2003). Rhinovirus-induced wheezing in infancy--the first sign of childhood asthma? J. Allergy Clin. Immunol. 111, 66–71.
Krug, N., Madden, J., Redington, A. E., Lackie, P., Djukanovic, R., Schauer, U., Holgate, S. T., Frew, A. J., and Howarth, P. H. (1996). T-cell cytokine profile evaluated at the single cell level in BAL and blood in allergic asthma. Am. J. Respir. Cell Mol. Biol. 14, 319–326.
Kusel, M. M., de Klerk, N. H., Kebadze, T., Vohma, V., Holt, P. G., Johnston, S. L., and Sly, P. D. (2007). Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J. Allergy Clin. Immunol. 119, 1105–1110.
Lee, D. C. W., and Lau, A. S. Y. (2007). Avian influenza virus signaling: implications for the disease severity of H5N1 infection. Signal Transduct. 7, 64–80.
Lemanske, R. F. Jr., Jackson, D. J., Gangnon, R. E., Evans, M. D., Li, Z., Shult, P. A., Kirk, C. J., Reisdorf, E., Roberg, K. A., Anderson, E. L., Carlson-Dakes, K. T., Adler, K. J., Gilbertson-White, S., Pappas, T. E., Dasilva, D. F., Tisler, C. J., and Gern, J. E. (2005). Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J. Allergy Clin. Immunol. 116, 571–577.
MacMaster, J. F., Dambach, D. M., Lee, D. B., Berry, K. K., Qiu, Y., Zusi, F. C., and Burke, J. R. (2003). An inhibitor of IkappaB kinase, BMS-345541, blocks endothelial cell adhesion molecule expression and reduces the severity of dextran sulfate sodium-induced colitis in mice. Inflamm. Res. 52, 508–511.
Magnan, A. O., Mély, L. G., Camilla, C. A., Badier, M. M., Montero-Julian, F. A., Guillot, C. M., Casano, B. B., Prato, S. J., Fert, V., Bongrand, P., and Vervloet, D. (2000). Assessment of the Th1/Th2 paradigm in whole blood in atopy and asthma. Increased IFN-gamma-producing CD8(+) T cells in asthma. Am. J. Respir. Crit. Care Med. 161, 1790–1796.
Mallia, P., and Johnston, S. L. (2006). How viral infections cause exacerbation of airway diseases. Chest 130, 1203–1210.
Martinez, F. D. (2003). Respiratory syncytial virus bronchiolitis and the pathogenesis of childhood asthma. Pediatr. Infect. Dis. J. 22, S76–S82.
Matsuse, H., Kondo, Y., Saeki, S., Nakata, H., Fukushima, C., Mizuta, Y., and Kohno, S. (2005). Naturally occurring parainfluenza virus 3 infection in adults induces mild exacerbation of asthma associated with increased sputum concentrations of cysteinyl leukotrienes. Int. Arch. Allergy Immunol. 138, 267–272.
Message, S. D., and Johnston, S. L. (2004). Host defense function of the airway epithelium in health and disease: clinical background. J. Leukoc. Biol. 75, 5–17.
Monto, A. S. (2004). Occurrence of respiratory virus: time, place and person. Pediatr. Infect. Dis. J. 23(Suppl.), S58–S64.
Okazaki, K., Kondo, M., Kato, M., Kakinuma, R., Nishida, A., Noda, M., Taniguchi, K., and Kimura, H. (2008). Serum cytokine and chemokine profiles in neonates with meconium aspiration syndrome. Pediatrics 121, e748–e753.
Pelaia, G., Vatrella, A., Gallelli, L., Renda, T., Cazzola, M., Maselli, R., and Marsico, S. A. (2006). Respiratory infections and asthma. Respir. Med. 100, 775–784.
Proud, D., and Chow, C. W. (2006). Role of viral infections in asthma and chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 35, 513–518.
Puxeddu, I., Bader, R., Piliponsky, A. M., Reich, R., Levi-Schaffer, F., and Berkman, N. (2006). The CC chemokine eotaxin/CCL11 has a selective profibrogenic effect on human lung fibroblasts. J. Allergy Clin. Immunol. 117, 103–110.
Santoro, M. G., Rossi, A., and Amici, C. (2003). NF-kappaB and virus infection: who controls whom. EMBO J. 22, 2552–2560.
Singh, A. M., Moore, P. E., Gern, J. E., Lemanske, R. F. Jr., and Hartert, T. V. (2007). Bronchiolitis to asthma: a review and call for studies of gene-virus interactions in asthma causation. Am. J. Respir. Crit. Care Med. 175, 108–119.
Stein, R. T., Sherrill, D., Morgan, W. J., Holberg, C. J., Halonen, M., Taussig, L. M., Wright, A. L., and Martinez, F. D. (1999). Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet 354, 541–545.
Stephan, L. (2007). Influenza viruses and MAP kinase cascades - Novel targets for an antiviral intervention? Signal Transduct. 7, 81–88.
Suzuki, K., Hino, M., Hato, F., Tatsumi, N., and Kitagawa, S. (1999). Cytokine-specific activation of distinct mitogen-activated protein kinase subtype cascades in human neutrophils stimulated by granulocyte colony-stimulating factor, granulocyte–macrophage colony-stimulating factor, and tumor necrosis factor-alpha. Blood 93, 341–349.
Thimmaiah, K. N., Easton, J. B., Germain, G. S., Morton, C. L., Kamath, S., Buolamwini, J. K., and Houghton, P. J. (2005). Identification of N10-substituted phenoxazines as potent and specific inhibitors of Akt signaling. J. Biol. Chem. 280, 31924–31935.
Ueba, O. (1978). Respiratory syncytial virus. I. Concentration and purification of the infectious virus. Acta Med. Okayama 32, 265–272.
Wark, P. A., Johnston, S. L., Bucchieri, F., Powell, R., Puddicombe, S., Laza-Stanca, V., Holgate, S. T., and Davies, D. E. (2005). Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 201, 937–947.
Watanabe, T., Yoshikawa, H., Abe, Y., Yamazaki, S., Uehara, Y., and Abe, T. (2003). Renal involvement in children with influenza A virus infection. Pediatr. Nephrol. 18, 541–544.
Wong, C. K., Wang, C. B., Ip, W. K., Tian, Y. P., and Lam, C. W. (2005). Role of p38 MAPK and NF-κB for chemokine release in coculture of human eosinophils and bronchial epithelial cells. Clin. Exp. Immunol. 139, 90–100.
Yoshizumi, M., Nakamura, T., Kato, M., Ishioka, T., Kozawa, K., Wakamatsu, K., and Kimura, H. (2008). Release of cytokines/chemokines and cell death in UVB-irradiated human keratinocytes, HaCaT. Cell Biol. Int. 32, 1405–1411.
Keywords: cytokines/chemokines, parainfluenza virus, human fetal lung fibroblast, inflammation, signaling pathway
Citation: Yoshizumi M, Kimura H, Okayama Y, Nishina A, Noda M, Tsukagoshi H, Kozawa K and Kurabayashi M (2010) Relationships between cytokine profiles and signaling pathways (IκB kinase and p38 MAPK) in parainfluenza virus-infected lung fibroblasts. Front. Microbio. 1:124. doi: 10.3389/fmicb.2010.00124
Received: 15 September 2010;
Accepted: 22 October 2010;
Published online: 15 November 2010.
Edited by:
Akihide Ryo, Yokohama City University, JapanReviewed by:
Isao Kosugi, Hamamatsu University School of Medicine, JapanMasatoshi Nakazawa, Yokohama City University, Japan
Copyright: © 2010 Yoshizumi, Kimura, Okayama, Nishina, Noda, Tsukagoshi, Kozawa and Kurabayashi. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Hirokazu Kimura, National Institute of Infectious Diseases, Infectious Disease Surveillance Center, 4-7-1 Gakuen, Musashimurayama-shi, Tokyo 208-0011, Japan. e-mail:a2ltaGlyb0BuaWguZ28uanA=; Yoshimichi Okayama, Division of Molecular Cell Immunology and Allergology, Advanced Medical Research Center, Nihon University Graduate School of Medical Science, 30-1 Oyaguchi-Kamimachi, Itabashi-ku, Tokyo 173-8610, Japan. e-mail:eW9rYXlhbWFAbWVkLm5paG9uLXUuYWMuanA=