

- Division of Pulmonary, Allergy, Critical Care and Sleep Medicine, Center for Microbial Interface Biology and the Department of Internal Medicine, Ohio State University, Columbus, OH, USA
Legionella pneumophila is the causative agent of Legionnaires’ disease, a serious and often fatal form of pneumonia. The susceptibility to L. pneumophila arises from the ability of this intracellular pathogen to multiply in human alveolar macrophages and monocytes. L. pneumophila also replicates in several professional and non-professional phagocytic human-derived cell lines. With the exception of the A/J mouse strain, most mice strains are restrictive, thus they do not support L. pneumophila replication. Mice lacking the NOD-like receptor Nlrc4 or caspase-1 are also susceptible to L. pneumophila. On the other hand, in the susceptible human hosts, L. pneumophila utilizes several strategies to ensure intracellular replication and protect itself against the host immune system. Most of these strategies converge to prevent the fusion of the L. pneumophila phagosome with the lysosome, inhibiting host cell apoptosis, activating survival pathways, and sequestering essential nutrients for replication and pathogenesis. In this review, we summarize survival mechanisms employed by L. pneumophila to maintain its replication in human cells. In addition, we highlight different human-derived cell lines that support the multiplication of this intracellular bacterium. Therefore, these in vitro models can be applicable and are reproducible when investigating L. pneumophila/phagocyte interactions at the molecular and cellular levels in the human host.
Legionella Pneumophila
Legionella pneumophila is a facultative intracellular lung pathogen (Horwitz and Silverstein, 1980) and the causative agent of Legionnaires’ disease (LD), a serious and often fatal, life-threatening bacterial pneumonia. LD is also a relatively common cause of community-acquired and nosocomial pneumonia in adults. L. pneumophila was recognized in 1976 after an outbreak of pneumonia at an American Legion convention in Philadelphia. Soon after, the etiologic agent was identified as a fastidious Gram-negative bacillus and named L. pneumophila. Although several other species of the genus Legionella were subsequently identified, the great majority of LD is caused by L. pneumophila (Marston et al., 1997; Yu et al., 2002). Most nosocomial infections and hospital outbreaks have been linked to contaminated air or water supplies. L. pneumophila has been recovered from different aquatic habitats including showers, streams, whirlpools, air conditioners, cooling towers, fountains, and spa baths (Fraser et al., 1979; Fliermans et al., 1981; Sethi and Brandis, 1983; Spitalny et al., 1984; Lettinga et al., 2002). Within water systems, L. pneumophila colonizes into biofilm; which are complex bacterial communities attached to a substratum by means of exopolysaccharides (EPS; Rogers et al., 1994). Biofilm develop into mushroom-like structures with water channels that allow access to nutrients and oxygen within these bacterial communities. L. pneumophila is able to obtain nutrients such as amino acids and organic carbon sources from the microbial consortium located in the biofilm (Watnick and Kolter, 2000). Humans are accidental dead-end hosts for L. pneumophila, thus, there is no person-to-person transmission whereas, protozoa are considered the environmental hosts for this intracellular pathogen, and are required for replication of biofilm-associated L. pneumophila (Rowbotham, 1980, 1986; Nash et al., 1984; Kuiper et al., 2004). While providing a niche for L. pneumophila replication, amoebae also protect L. pneumophila from harsh environmental conditions. Replication within different protozoa enhances bacterial multiplication in human alveolar macrophages. Growth within the protozoa also induces resistance to chemical disinfectant, biocides and antibiotics and induces phenotypic changes in L. pneumophila (Barker et al., 1992, 1995; Abu Kwaik et al., 1993; Cirillo et al., 1994).
Risk Factors
The majority of people exposed to L. pneumophila remains asymptomatic or suffer only mild self-limiting infection. Susceptible patients to LD disease are likely to exhibit a defect in cell-mediated immunity rendering them less capable of limiting the early multiplication of L. pneumophila. Cigaret smoking, chronic lung disease, and immunosuppression (especially that caused by corticosteroid therapy) have been consistently implicated as risk factors (Carratala et al., 1994). Surgery is a major predisposing factor in nosocomial infection, with transplant recipients at highest risk. Other factors associated with the development of LD include, old age, cancer, and alcohol intake (Marston et al., 1994; Den Boer et al., 2002).
L. Pneumophila Multiplies in Human Monocytes, Alveolar Macrophages, and Human-Derived Cell Lines
Legionella pneumophila multiplies in human monocytes (Horwitz and Silverstein, 1980; Horwitz, 1983b). The intracellular fate of L. pneumophila in both human and protozoa is similar, where the bacteria evade fusion of the phagosome to lysosome, although the mechanism maybe different in some aspects (Horwitz, 1983a,b; Horwitz and Maxfield, 1984; Bozue and Johnson, 1996). Attachments and binding of L. pneumophila to the complement receptor 1 (CR1), and complement receptor 3 (CR3) on human phagocytic cells is followed by entry into these cells. In some cases, the entry of the pathogen is mediated by “coiling phagocytosis,” in which a single long phagocyte pseudopod coils around the bacterium as it is internalized (Horwitz, 1984; Payne and Horwitz, 1987; Figure 1). Following phagocytosis, L. pneumophila avoids interaction with endosomes, early and late lysosomes, and instead, fuses transiently with mitochondria and intercepts the endoplasmic reticulum (ER) exit vesicles (Horwitz, 1983b; Swanson and Isberg, 1995; Kagan and Roy, 2002; Liang et al., 2006; Isberg et al., 2009). The bacteria maintain interactions with ER-derived vesicles and replicate in a vacuole surrounded by a membrane that resembles rough ER (Horwitz and Silverstein, 1980; Horwitz, 1983b; Figure 1). The L. pneumophila-containing vacuole (LCV) remains studded with ribosomes until hundreds of the bacteria fill the vacuole and the monocyte ruptures (Figure 1; Nash et al., 1984). In contrast, formalin-killed L. pneumophila does not inhibit fusion or induce the formation of specialized phagosome and the dead bacteria are degraded within the phagolysosome (Horwitz and Silverstein, 1980; Horwitz, 1983b).
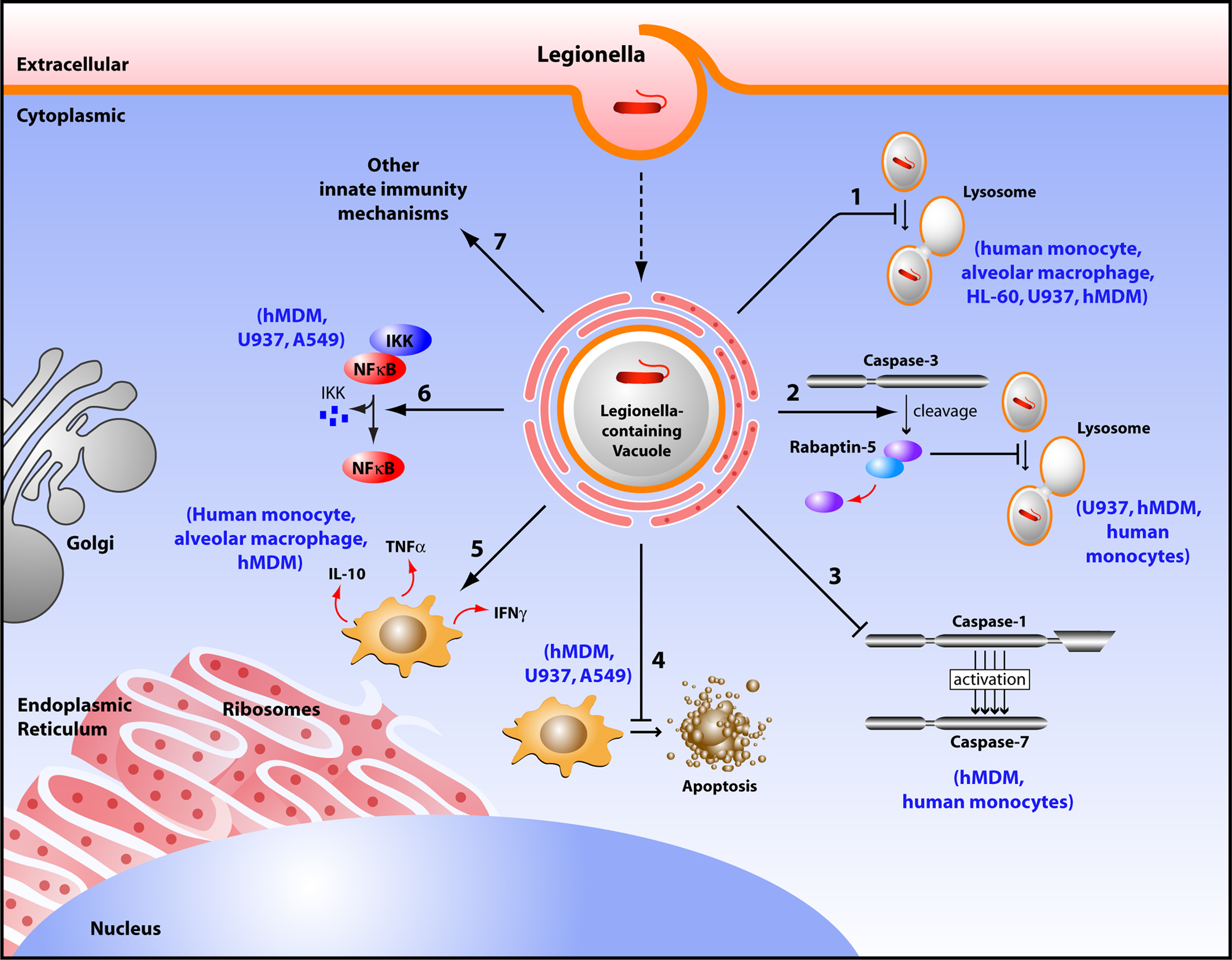
Figure 1. Diagram depicting survival mechanisms employed by L. pneumophila in human phagocytes. Upon entry into a human phagocyte, L. pneumophila becomes contained into a vacuole called Legionella-containing phagosome that avoids the typical fusion with the lysosome, and instead remodels into an ER-like vesicle allowing bacterial replication (1). L. pneumophila promotes the cleavage of Rabaptin-5 by caspase-3, thus preventing the default phagosome–lysosome fusion (2). L. pneumophila does not activate caspase-1 and -7 in human monocytes, consequently aborting the phagosome–lysosome fusion (3). This pathogen inhibits host cell apoptosis by up regulating anti-apoptotic genes (4). L. pneumophila controls the local balance of activating cytokines (INF-γ, TNF-α) that inhibit its replication, and inhibiting cytokines (IL-10) that allow its survival (5). L. pneumophila activates the NF-κB pathway to maintain host cell survival (6). L. pneumophila modulates other innate immune responses to establish a replicative niche (7).
Alveolar macrophages are the most abundant potential effector cells in the lung and play a role in host defense against L. pneumophila (Nash et al., 1984). L. pneumophila that reaches the alveoli encounters and multiplies in human alveolar macrophages at the site of infection, which is crucial to the pathogenesis of LD (Green, 1970; Goldstein et al., 1974). Lung tissue specimens from LD patients frequently contain large numbers of intracellular L. pneumophila (Winn Jr. and Myerowitz, 1981; Nash et al., 1984). Furthermore, alveolar macrophages are a major feature of the alveolar secretions in lung biopsies from patients with LD (Winn Jr. and Myerowitz, 1981; Nash et al., 1984).
Beside human monocytes and alveolar macrophages, investigators have employed a variety of cell lines that not only have similar characteristics to human phagocytes, but are also capable of supporting growth of this intracellular pathogen. For example, L. pneumophila multiplies within and kills HL-60 cells, a macrophage-like cell line that is derived from promyelocyte (Marra et al., 1990). In HL-60 cells, L. pneumophila enters by adhering to the CR3 and is taken by coiling phagocytosis. Similar to human phagocytes, the LCV is surrounded by ER-derived vesicles studded with ribosomes and therefore, HL-60 cells are utilized as a model to study the interaction of human macrophages with the LD bacterium (Marra et al., 1990). Similarly, L. pneumophila grows within U937 cells; a human monocytes lymphoma cell line that displays many monocytic features and can be maintained continuously in culture (Pearlman et al., 1988). These cells can be differentiated to adherent, non-replicative cells with characteristics of tissue macrophages. Additionally, they express surface markers specific for cells of monocyte lineage and are able to differentiate between virulent and avirulent strains of L. pneumophila. Their activation state is controlled experimentally and consequently can be standardized (Rovera et al., 1979). THP-1 is another human monocytic cell line that matures into macrophage-like adherent cells following stimulation with phorbol esters or 1,25-dihydroxyvitamin D3. Cirillo et al. (1994) reported that L. pneumophila could invade and multiply within THP-1 cells in a manner similar to that in human monocytes and macrophages. Mono Mac 6 (MM6) is an additional example of human monocytic cell line. MM6 was generated from peripheral blood of patient with monoblastic leukemia. Phenotypically and functionally, MM6 represents a more mature macrophage-like cell line. Unlike the other monoblastic cell lines U937 and THP-1, MM6 does not require stimulation by phorbol esters or 1,25-dihydroxyvitamin D3 and expresses several markers that are lacking in U937 and THP-1 (Ziegler-Heitbrock et al., 1988; Neumeister et al., 1997). MM6 has been successfully utilized to study the molecular pathogenesis of L. pneumophila and other intracellular pathogens that can replicate within human monocytes (Neumeister, 2004). Furthermore, non-professional phagocytic cell lines have been utilized to study L. pneumophila replication such as A549, a human epithelial cell line derived from a human lung cancer. They are non-professional phagocytes and have characteristics of type II alveolar epithelial cells (Maruta et al., 1998). It has been shown that L. pneumophila is internalized and replicate effectively in A549 cells (Maruta et al., 1998; Vinzing et al., 2008; Bartfeld et al., 2009). Therefore, many human-derived cell lines have been used to characterize the molecular mechanisms associated with L. pneumophila pathogenesis. While some features are unique to the interactions between L. pneumophila and specific host cells, the primary mechanisms of infection appears to be the same.
The Innate Immune Response of Primary Human Cells and Human-Derived Cell Lines to L. Pneumophila
Host cells express a range of receptors that act as microbial sensors. These receptors sense microorganisms and transduce signals that activate immune responses. Pathogen-associated molecular patterns (PAMPs) such as peptidoglycan, bacterial flagellin, and nucleic acid can be recognized by means of cell surface pattern recognition receptors (PRRs) named Toll-like receptors (TLRs) (Janeway Jr. and Medzhitov, 2002; Underhill et al., 1999; Takeda et al., 2003). TLRs recognize PAMPs via an extracellular domain and initiate inflammatory signaling pathways through an intracellular domain (Janeway Jr. and Medzhitov, 2002; Underhill et al., 1999; Takeda et al., 2003). Humans express functional TLRs 1–10 whereas mice express TLRs 1–7 and 9–13 (Table 1; Vinzing et al., 2008).
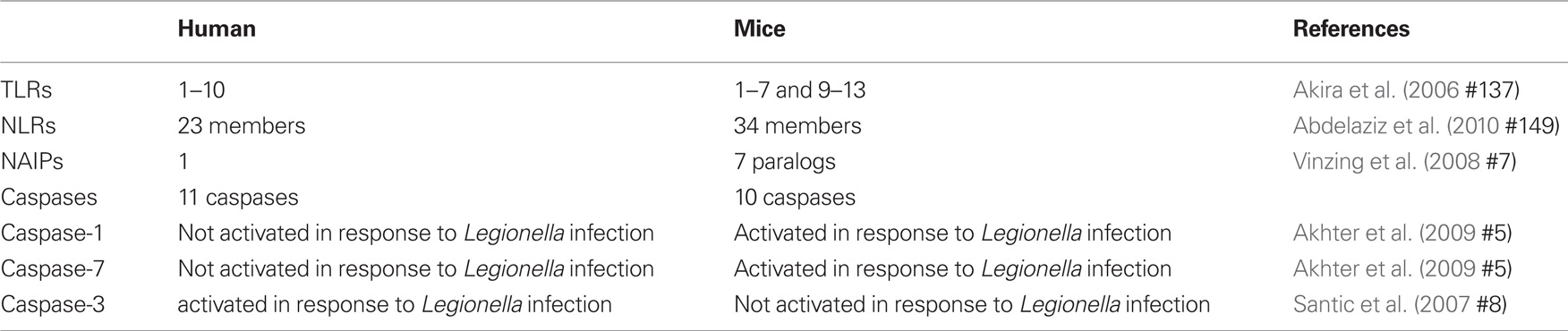
Table 1. Differences between human and mice cells that affects Legionella permissiveness.
Other means of sensing the presence of microbes or their factors in the host cytosol is mediated by NOD-like receptors (NLRs), which in turn, initiate signaling cascades that mediate the production of inflammatory cytokines, recruitment of phagocytic cells and direction of the innate and the acquired immune responses. There are 23 NLRs in human and 34 in mice (Abdelaziz et al., 2010; Amer, 2010). Typically, NLRs are composed of a leucine-rich repeat (LRR) domain, a central nucleotide oligomerization domain (NOD) and N-terminal effector binding domain, which consists of caspase recruitment domain (CARD) and the pyrin domain.
One of the major NLRs contributing to the restriction of L. pneumophila infection in human and in mice is NLRC4 (IPAF) (Damiano et al., 2001; Poyet et al., 2001; Amer et al., 2006; Vinzing et al., 2008; Akhter et al., 2009). Human NLRC4 is highly homologous to the mouse Nlrc4 and is expressed in human macrophages but not in human lung epithelial cells (Vinzing et al., 2008). Like most NLRs, mouse Nlrc4 assembles in a large multiprotein complex called the inflammasome leading to the activation of caspase-1. Caspases are family of cysteine proteases that play a distinct role in apoptosis and inflammation (Fink and Cookson, 2005). Caspases can be divided into two groups; one category of caspases is involved in apoptosis such as caspase-3 and -7 (Martinon and Tschopp, 2004). Another category is involved in inflammation and is required for cytokine processing (Creagh et al., 2003). As an inflammatory caspase, caspase-1 is required for the activation of cytokines such as IL-1β, IL-18, and IL-33 and induces a form of cell death named pyroptosis (Fink and Cookson, 2005). Wild-type mouse macrophages restrict L. pneumophila replication via caspase-1-mediated caspase-7 activation resulting in fusion of the LCV with the lysosome leading to bacterial degradation and growth restriction (Akhter et al., 2009). However, human monocytes do not activate caspase-1 and -7 upon L. pneumophila infection, allowing bacterial growth (Table 1; Santic et al., 2007; Akhter et al., 2009). Although THP-1 cells were not shown to activate caspase-1 in response to L. pneumophila, the depletion of human NLRC4 allows more L. pneumophila replication (Vinzing et al., 2008). This data implies the existence of uncharacterized caspase-1-independent restriction mechanism of L. pneumophila replication in human phagocytes.
A subset of NLRs contains baculovirus inhibitor of apoptosis repeat domains (BIR) instead of CARD or pyrin domains. These proteins are called the neuronal apoptosis-inhibitory proteins (NAIPs). The LRR domain recognizes microbial products, while the BIR domains are essential for their interactions with caspase-3 and -7 (Liston et al., 1996; Takahashi et al., 1998; Diez et al., 2003). The importance of Naip5 during L. pneumophila infection stemmed from studies showing that the A/J mice are permissive to L. pneumophila in contrast to most other inbred mouse strains (Diez et al., 2003; Wright et al., 2003). Gros and his group identified 14 amino acid substitutions in the A/J derived Naip5 when compared to wild-type Naip5 and they proposed that several of these mutations are responsible for permissiveness to L. pneumophila (Beckers et al., 1995; Diez et al., 2003). Furthermore, in contrast to the seven mouse Naips, only one NAIP has been identified in human and exhibits 68% homology to the mouse C57BL/6 and A/J Naip5. Human NAIP is expressed in THP-1, primary macrophages, and A549 epithelial cells. Furthermore, depletion of the human orthologs of Naip5 results in increased replication of L. pneumophila in human THP-1 macrophages and A549 lung epithelial cells (Vinzing et al., 2008). However, the mechanism by which human NAIP restricts L. pneumophila replication is yet to be elucidated.
The Activation of Survival Pathways in L. Pneumophila-Infected Human Cells
Studies with the human macrophage-like U937 and human epithelial cells A549 showed that L. pneumophila induces the activation of the transcriptional regulator nuclear factor κB (NF-κB) upon infection (Losick and Isberg, 2006; Abu-Zant et al., 2007; Schmeck et al., 2007; Bartfeld et al., 2009). L. pneumophila induces biphasic activation of the NF-κB pathway. The early phase is strong, but transient and is flagellin, TLR5 and MYD88-dependent. It is terminated quickly to prevent the autotoxic overproduction of inflammatory mediators. L. pneumophila activates the NF-κB pathway in human epithelial cells leading to secretion of several cytokines and chemokines (Schmeck et al., 2007). This activation is beneficial for the host since it triggers the innate immune response. The second phase of NF-κB activation is characterized by long-term activation for up to 30 h. However, this phase is flagellin, TLR5, MYD88-independent (Bartfeld et al., 2009). The long-term activation of NF-κB is linked to bacterial replication and upregulation of anti-apoptotic genes (Bartfeld et al., 2009). Therefore, this phase seems to be beneficial for the pathogen. The mechanism for this late stage NF-κB activation is still unclear.
The Role of Caspase-3 in L. Pneumophila Infection of Human Cells
The host can employ several mechanisms to overcome intracellular infection. Among these, is the elimination of infected cells by caspase-mediated apoptosis. Arising reports demonstrate new distinct roles for executioner caspases independent of cell death (Li and Yuan, 2008; Walsh et al., 2008; Amer, 2010). L. pneumophila induces caspase-3 activation in human macrophages and U937 (Gao and Au Kwaik, 1999a,b; Zink et al., 2002; Molmeret et al., 2004; Santic et al., 2007). The earlier studies with macrophages and epithelial cells demonstrated that high numbers of L. pneumophila induces apoptosis through activation of caspase-3 (Gao and Abu Kwaik, 1999a; Neumeister et al., 2001). Remarkably, other studies demonstrated that the early caspase-3 activation by L. pneumophila is independent of the intrinsic and extrinsic pathways of apoptosis (Molmeret et al., 2004). Notably, at physiological levels of infection, L. pneumophila utilizes the early mild caspase-3 activation in human cells to evade the phagosome–lysosome fusion. Activation of caspase-3 by low numbers of L. pneumophila is accompanied with cleavage of rabaptin-5, a downstream effector of rab-5 (Molmeret et al., 2004). Rab-5 GTPase is involved in the maturation of the early endosome. Therefore, according to the number of L. pneumophila invading a human cell, caspase-3 activation can lead either to apoptosis or to delayed phagosome maturation.
Modulation of the Ubiquitination Machinery by L. Pneumophila
Several studies showed that in human cells, the LCV is decorated with polyubiquitinated proteins upon L. pneumophila infection (Ivanov and Roy, 2009). Ubiquitination is a reversible post-translational modification that controls the abundance of many critical regulatory proteins (Craig and Tyers, 1999; Lomma et al., 2010). Degradation of intracellular proteins is mediated via means of the ubiquitin system and its specificity is determined at the level of substrate recognition by the E3 ubiquitin ligases, that catalyzes binding of the activated ubiquitin to the target protein for degradation (Craig and Tyers, 1999). Additionally, the L. pneumophila genome encodes for proteins with F-box and U-box domains (Cazalet et al., 2004; Price et al., 2009; Lomma et al., 2010) that are similar to eukaryotic SCF ubiquitin ligases. F-box proteins are known components of E3 ubiquitin ligases named SCF complexes. The F-box motif within the F-box protein interacts with the components of the ubiquitin machinery while the protein–protein interaction domain selectively binds to specific substrate. The U-box is a domain of approximately 70 amino acids that is present in proteins from yeast to humans. Mammalian U-box proteins in conjugation with E1 and E2 mediate protein ubiquitination, and thus have been classified as E3 ligases (Hatakeyama et al., 2001). Three F-box-containing proteins have been shown to be required for intracellular replication of L. pneumophila in THP-1 macrophages and A549 cells, and single, double, or triple mutations in the F-box proteins impair infection within these cells (Lomma et al., 2010). Al-Khodor et al. (2008) showed that the F-box-containing protein AnkB is required for intracellular replication of L. pneumophila within human monocytes-derived macrophages (hMDM). Furthermore, AnkB translocated to the host cytosol via the type IV secretion system mediates L. pneumophila replication via decorating the LCV with polyubiquitinated proteins (Price et al., 2009). The focal adhesion protein ParvB, is the target for AnkB (Lomma et al., 2010), and AnkB interferes with the ubiquitination of the ParvB, likely by competing with the eukaryotic E3 ligase for the protein–protein interaction domain of ParvB (Lomma et al., 2010). Therefore, L. pneumophila hijacks the host cell ubiquitination system to mediate intracellular replication in human cells.
The Role of Cytokines during L. Pneumophila Infection in Human Cells
Other innate immune mechanisms might modulate L. pneumophila infection in human cells, including cytokine production such as Interferon gamma-γ (INF-γ) and Tumor necrosis-α (TNF-α) (Coers et al., 2007). Induction of innate immunity includes macrophage activation by the antimicrobial cytokine IFN-γ, protection by TNF-α, and production of IL-6 and IL-1, as well as chemokines from inflammatory cells (Friedman et al., 2002). In vitro studies with primary human macrophages or cell lines derived from human macrophages that are activated with INF-γ and TNF-α restricted L. pneumophila replication (Bhardwaj et al., 1986; Nash et al., 1988; Matsiota-Bernard et al., 1993). Furthermore, pretreatment of hMDM with INF-γ inhibited intracellular replication of L. pneumophila and restricted the multiplication of the pathogen (Santic et al., 2005). The authors speculate that these processes are distinct between human and mouse macrophages and ultimately determine the permissiveness to L. pneumophila. TNF-α has been detected in the broncho-alveolar lavage fluid during L. pneumophila infection (Blanchard et al., 1987). It is produced readily by human peripheral blood lymphocytes in response to L. pneumophila antigens (Friedman et al., 2002). TNF-α is a potent cytokine that enhances the bactericidal activity of the macrophages. Furthermore, inhibition of TNF-α production is associated with increased bacterial replication (Nash et al., 1984; Matsiota-Bernard et al., 1993).
In contrast to the macrophage activating cytokines INF-γ and TNF-α, Interleukin-10 (IL-10) is an inhibitory cytokine. IL-10 treatment significantly enhances L. pneumophila growth in monocytes, and completely reverses the protective effect of INF-γ on L. pneumophila replication (Park and Skerrett, 1996). IL-10 has similar, but less potent, effects on alveolar macrophages and INF-γ-activated alveolar macrophages are less sensitive to deactivation by IL-10. Park and Skerrett (1996) identified important differences in alveolar macrophages and monocytes response to L. pneumophila infection and to IL-10. They also suggested that the induction of IL-10 during L. pneumophila infection may be a virulence mechanism that promotes intracellular bacterial replication. Therefore, the local balance between the INF-γ and IL-10 production by L. pneumophila might determine if monocytes that are recruited to the site of infection will become effector cells or develop into susceptible targets available for bacterial growth.
Cell-mediated immunity seems to play a critical role in restricting L. pneumophila replication, and the activated human monocytes have the capacity to inhibit the multiplication of virulent L. pneumophila both via decreasing the phagocytosis and the rate of intracellular multiplication (Horwitz and Silverstein, 1981b). Consistent with that, patients with LD produce less INF-γ than non-LD patients, suggesting that impairment in the INF-γ response may increase the susceptibility to the disease (Lettinga et al., 2003). However, patients with acute LD are able to restrict L. pneumophila replication as a result elevated level of the Th1-cytokines such as INF-γ and IL-12 (Takeda et al., 2003). Sera from patients with L. pneumophila contain antibodies to L. pneumophila protein antigens (Friedman et al., 2002). However, humoral immunity seems to play a limited role in host defense against LD. Horwitz and Silverstein (1981a) demonstrated that complement and antibody, which promote entry of L. pneumophila into monocytes, do not inhibit the rate of L. pneumophila multiplication in monocytes.
Other Immune Responses to L. Pneumophila in Human Cells
Another major strategy of innate resistance to intracellular infection is to sequester key nutrients, such as iron, from invading bacteria. Within host cells, iron is predominantly bound to proteins and unavailable in free soluble form. Under iron-limited conditions, L. pneumophila secretes a high affinity iron chelator named siderophore, which sequesters iron from the host and the surrounding environment (Liles et al., 2000; Cianciotto, 2007). L. pneumophila utilizes iron to maintain its replication in human monocytes and macrophages. Consistently, treatment of human monocytes and macrophages with iron chelator aborts L. pneumophila replication (Gebran et al., 1994; Viswanathan et al., 2000). Accordingly, restrictive monocytes seem to exhibit low level of expression of transferrin receptor similar to the low level of expression of transferrin receptor in INF-γ activated monocytes (Byrd and Horwitz, 2000). Consequently, supplementation of iron abolishes the effect of INF-γ-mediated growth restriction of L. pneumophila in hMDM (Santic et al., 2005).
Bacterial infection stimulates the production of reactive nitrogen intermediate by the inducible nitric oxide synthase (iNOS). Transcription of NOS2 is mediated by type I and type II interferons; suggesting that the production of nitric oxide (NO) is not an immediate response for the infection. NO mediates its action on bacteria in the vacuole or the cytosol. However, stimulation with 1,25-dihydroxyvitamin D(3) to enhance NO production or the inhibition of NO production by treatment with N(G)-methyl-L-arginine did not modify the intracellular multiplication of L. pneumophila within MM6. This study suggested that NO radicals do not play a role in restricting different L. pneumophila species in human monocytic cell lines (Neumeister et al., 2001). So far, the role of NO radicals in human macrophages seems to be controversial (Neumeister et al., 2001; Radtke and O’Riordan, 2006).
Genetic Susceptibility to Legionnaires’ Disease
While the genetic factors that predispose individuals for LD are not well known, it has been shown that TLR5 influences human susceptibility to L. pneumophila infection. TLR5 recognizes bacterial flagellin (Hayashi et al., 2001) and is the predominant mediator of Interleukin-8 (IL-8) in lung epithelial cells A549. A common stop codon polymorphism in the ligand-binding domain of TLR5 (TLR5329STOP) occurs in 10% of the population resulting in the inability to mediate flagellin signaling and is associated with increased risk to LD (Hawn et al., 2003). TLR5329STOP has a dominant negative effect and peripheral blood monocytes (PBMCs) from heterozygous individuals stimulated with flagellin had less IL-6 and IL-8 secretion. Likely, TLR5329STOP mutation increases susceptibility to LD by decreasing the cytokine secretion. However, the role of the mouse TLR5 in L. pneumophila infection is controversial (Hawn et al., 2007; Archer et al., 2009). On the other hand, polymorphism in the TLR4 receptor, SNP A896G, is associated with resistance to LD in heterozygous individuals (Hawn et al., 2005).
Another factor associated with resistance to L. pneumophila infection is the mannose-binding lectin (MBL), a PRR of the innate immune system that activates complement via the lectin pathway (Turner, 1996). MBL deficiency compromises opsonization by complement leading to an increased risk of infection. In infection with intracellular pathogens, the role of MBL deficiency is more ambiguous, as more intracellular pathogens use opsonization by MBL to enter their host cell. Herpers et al. (2009) showed that Legionellosis is not associated with MBL deficiency. However, a large number of patients with Legionellosis displayed deficient MBL-mediated complement activation even though they carried MBL-sufficient genotypes. Furthermore, patients with MBL-sufficient genotypes showed functional deficiency at the acute phase of disease, yet the lectin pathway functionality was restored at convalescence. However, the deficiency of lectin pathway seems to be an effect rather than a risk factor for LD development (Herpers et al., 2009).
Conclusions
Legionella pneumophila exploits several strategies to overcome the innate immune defense in human phagocytes (Figure 1). However, most of these converge toward preventing the fusion of its phagosome with the lysosome, inhibiting host cell apoptosis, maintaining host cell survival, modulating the local balance between activating and inhibiting cytokines, sequestering nutrients from the host and overcoming the killing mechanisms available within phagocytes.
Conflict of Interest Statement:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Kyle Caution for editing the manuscript and Dr. Tim Eubank for the figure. Studies in Dr. Amal Amer laboratory are supported by grants R01HL094586 and R21AI083871 from the National Institute of Health (NIH) and GRT00013604 from the American Lung Association (ALA).
References
Abdelaziz, D. H., Amr, K., and Amer, A. O. (2010). Nlrc4/Ipaf/CLAN/CARD12: more than a flagellin sensor. Int. J. Biochem. Cell Biol. 42, 789–791.
Abu Kwaik, Y., Eisenstein, B. I., and Engleberg, N. C. (1993). Phenotypic modulation by Legionella pneumophila upon infection of macrophages. Infect. Immun. 61, 1320–1329.
Abu-Zant, A., Jones, S., Asare, R., Suttles, J., Price, C., Graham, J., and Kwaik, Y. A. (2007). Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell. Microbiol. 9, 246–264.
Akhter, A., Gavrilin, M. A., Frantz, L., Washington, S., Ditty, C., Limoli, D., Day, C., Sarkar, A., Newland, C., Butchar, J., Marsh, C. B., Wewers, M. D., Tridandapani, S., Kanneganti, T.-D., and Amer, A. O. (2009). Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. 5, e1000361. doi: 10.1371/journal.ppat.1000361.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Al-Khodor, S., Price, C. T., Habyarimana, F., Kalia, A., and Abu Kwaik, Y. (2008). A Dot/Icm-translocated ankyrin protein of Legionella pneumophila is required for intracellular proliferation within human macrophages and protozoa. Mol. Microbiol. 70, 908–923.
Amer, A., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Ozören, N., Brady, G., Meshinchi, S., Jagirdar, R., Gewirtz, A., Akira, S., and Núñez, G. (2006). Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223.
Amer, A. O. (2010). Modulation of caspases and their non-apoptotic functions by Legionella pneumophila. Cell. Microbiol. 12, 140–147.
Archer, K. A., Alexopoulou, L., Flavell, R. A., and Roy, C. R. (2009). Multiple MyD88-dependent responses contribute to pulmonary clearance of Legionella pneumophila. Cell. Microbiol. 11, 21–36.
Barker, J., Brown, M. R., Collier, P. J., Farrell, I., and Gilbert, P. (1992). Relationship between Legionella pneumophila and Acanthamoeba polyphaga: physiological status and susceptibility to chemical inactivation. Appl. Environ. Microbiol. 58, 2420–2425.
Barker, J., Scaife, H., and Brown, M. R. (1995). Intraphagocytic growth induces an antibiotic-resistant phenotype of Legionella pneumophila. Antimicrob. Agents Chemother. 39, 2684–2688.
Bartfeld, S., Engels, C., Bauer, B., Aurass, P., Flieger, A., Bruggemann, H., and Meyer, T. F. (2009). Temporal resolution of two-tracked NF-kappaB activation by Legionella pneumophila. Cell. Microbiol. 11, 1638–1651.
Beckers, M. C., Yoshida, S., Morgan, K., Skamene, E., and Gros, P. (1995). Natural resistance to infection with Legionella pneumophila: chromosomal localization of the Lgn1 susceptibility gene. Mamm. Genome 6, 540–545.
Bhardwaj, N., Nash, T. W., and Horwitz, M. A. (1986). Interferon-gamma-activated human monocytes inhibit the intracellular multiplication of Legionella pneumophila. J. Immunol. 137, 2662–2669.
Blanchard, D. K., Djeu, J. Y., Klein, T. W., Friedman, H., and Stewart, W. E. (1987). Induction of tumor necrosis factor by Legionella pneumophila. Infect. Immun. 55, 433–437.
Bozue, J. A., and Johnson, W. (1996). Interaction of Legionella pneumophila with Acanthamoeba castellanii: uptake by coiling phagocytosis and inhibition of phagosome–lysosome fusion. Infect. Immun. 64, 668–673.
Byrd, T. F., and Horwitz, M. A. (2000). Aberrantly low transferrin receptor expression on human monocytes is associated with nonpermissiveness for Legionella pneumophila growth. J. Infect. Dis. 181, 1394–1400.
Carratala, J., Gudiol, F., Pallares, R., Dorca, J., Verdaguer, R., Ariza, J., and Manresa, F. (1994). Risk factors for nosocomial Legionella pneumophila pneumonia. Am. J. Respir. Crit. Care Med. 149(Pt 1), 625–629.
Cazalet, C., Rusniok, C., Brüggemann, H., Zidane, N., Magnier, A., Ma, L., Tichit, M., Jarraud, S., Bouchier, C., Vandenesch, F., Kunst, F., Etienne, J., Glaser, P., and Buchrieser, C. (2004). Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat. Genet. 36, 1165–1173.
Cirillo, J. D., Falkow, S., and Tompkins, L. S. (1994). Growth of Legionella pneumophila in Acanthamoeba castellanii enhances invasion. Infect. Immun. 62, 3254–3261.
Coers, J., Vance, R. E., Fontana, M. F., and Dietrich, W. F. (2007). Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol. 9, 2344–2357.
Craig, K. L., and Tyers, M. (1999). The F-box: a new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction. Prog. Biophys. Mol. Biol. 72, 299–328.
Creagh, E. M., Conroy, H., and Martin, S. J. (2003). Caspase-activation pathways in apoptosis and immunity. Immunol. Rev. 193, 10–21.
Damiano, J. S., Stehlik, C., Pio, F., Godzik, A., and Reed, J. C. (2001). CLAN, a novel human CED-4-like gene. Genomics 75, 77–83.
Den Boer, J. W., Yzerman, E. P., Schellekens, J., Lettinga, K. D., Boshuizen, H. C., Van Steenbergen, J. E., Bosman, A., Van den Hof, S., Van Vliet, H. A., Peeters, M. F., Van Ketel, R. J., Speelman, P., Kool, J. L., and Conyn-Van Spaendonck, M. A. (2002). A large outbreak of Legionnaires’ disease at a flower show, the Netherlands, 1999. Emerging Infect. Dis. 8, 37–43.
Diez, E., Lee, S. H., Gauthier, S., Yaraghi, Z., Tremblay, M., Vidal, S., and Gros, P. (2003). Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat. Genet. 33, 55–60.
Fink, S. L., and Cookson, B. T. (2005). Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916.
Fliermans, C. B., Cherry, W. B., Orrison, L. H., Smith, S. J., Tison, D. L., and Pope, D. H. (1981). Ecological distribution of Legionella pneumophila. Appl. Environ. Microbiol. 41, 9–16.
Fraser, D. W., Deubner, D. C., Hill, D. L., and Gilliam, D. K. (1979). Nonpneumonic, short-incubation-period Legionellosis (Pontiac fever) in men who cleaned a steam turbine condenser. Science 205, 690–691.
Friedman, H., Yamamoto, Y., and Klein, T. W. (2002). Legionella pneumophila pathogenesis and immunity. Semin. Pediatr. Infect. Dis. 13, 273–279.
Gao, L. Y., and Abu Kwaik, Y. (1999a). Activation of caspase 3 during Legionella pneumophila-induced apoptosis. Infect. Immun. 67, 4886–4894.
Gao, L. Y., and Abu Kwaik, Y. (1999b). Apoptosis in macrophages and alveolar epithelial cells during early stages of infection by Legionella pneumophila and its role in cytopathogenicity. Infect. Immun. 67, 862–870.
Gebran, S. J., Newton, C., Yamamoto, Y., Widen, R., Klein, T. W., and Friedman, H. (1994). Macrophage permissiveness for Legionella pneumophila growth modulated by iron. Infect. Immun. 62, 564–568.
Goldstein, E., Lippert, W., and Warshauer, D. (1974). Pulmonary alveolar macrophage. Defender against bacterial infection of the lung. J. Clin. Invest. 54, 519–528.
Green, G. M. (1970). The J. Burns Amberson Lecture – in defense of the lung. Am. Rev. Respir. Dis. 102, 691–703.
Hatakeyama, S., Yada, M., Matsumoto, M., Ishida, N., and Nakayama, K. I. (2001). U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276, 33111–33120.
Hawn, T. R., Berrington, W. R., Smith, I. A., Uematsu, S., Akira, S., Aderem, A., Smith, K. D., and Skerrett, S. J. (2007). Altered inflammatory responses in TLR5-deficient mice infected with Legionella pneumophila. J. Immunol. 179, 6981–6987.
Hawn, T. R., Verbon, A., Lettinga, K. D., Zhao, L. P., Li, S. S., Laws, R. J. Skerrett, S. J., Beutler, B., Schroeder, L., Nachman, A., Ozinsky, A., Smith, K. D., and Aderem, A. (2003). A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires’ disease. J. Exp. Med. 198, 1563–1572.
Hawn, T. R., Verbon, A., Janer, M., Zhao, L. P., Beutler, B., and Aderem, A. (2005). Toll-like receptor 4 polymorphisms are associated with resistance to Legionnaires’ disease. Proc. Natl. Acad. Sci. U.S.A. 102, 2487–2489.
Hayashi, F., Smith, K. D., Ozinsky, A., Hawn, T. R., Yi, E. C., Goodlett, D. R., Eng, J. K., Akira, S., Underhill, D. M., and Aderem, A. (2001). The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103.
Herpers, B. L., Yzerman, E. P., de Jong, B. A., Bruin, J. P., Lettinga, K. D., Kuipers, S., Den Boer, J. W., van Hannen, E. J., Rijkers, G. T., van Velzen-Blad, H., and de Jongh, B. M. (2009). Deficient mannose-binding lectin-mediated complement activation despite mannose-binding lectin-sufficient genotypes in an outbreak of Legionella pneumophila pneumonia. Hum. Immunol. 70, 125–129.
Horwitz, M. A. (1983a). Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158, 1319–1331.
Horwitz, M. A. (1983b). The Legionnaires’ disease bacterium (Legionella pneumophila) inhibits phagosome–lysosome fusion in human monocytes. J. Exp. Med. 158, 2108–2126.
Horwitz, M. A. (1984). Phagocytosis of the Legionnaires’ disease bacterium (Legionella pneumophila) occurs by a novel mechanism: engulfment within a pseudopod coil. Cell 36, 27–33.
Horwitz, M. A., and Maxfield, F. R. (1984). Legionella pneumophila inhibits acidification of its phagosome in human monocytes. J. Cell Biol. 99, 1936–1943.
Horwitz, M. A., and Silverstein, S. C. (1980). Legionnaires’ disease bacterium (Legionella pneumophila) multiples intracellularly in human monocytes. J. Clin. Invest. 66, 441–450.
Horwitz, M. A., and Silverstein, S. C. (1981a). Activated human monocytes inhibit the intracellular multiplication of Legionnaires’ disease bacteria. J. Exp. Med. 154, 1618–1635.
Horwitz, M. A., and Silverstein, S. C. (1981b). Interaction of the Legionnaires’ disease bacterium (Legionella pneumophila) with human phagocytes. I. L. pneumophila resists killing by polymorphonuclear leukocytes, antibody, and complement. J. Exp. Med. 153, 386–397.
Isberg, R. R., O’Connor, T. J., and Heidtman, M. (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24.
Ivanov, S. S., and Roy, C. R. (2009). Modulation of ubiquitin dynamics and suppression of DALIS formation by the Legionella pneumophila Dot/Icm system. Cell. Microbiol. 11, 261–278.
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216.
Kagan, J. C., and Roy, C. R. (2002). Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4, 945–954.
Kuiper, M. W., Wullings, B. A., Akkermans, A. D., Beumer, R. R., and van der Kooij, D. (2004). Intracellular proliferation of Legionella pneumophila in Hartmannella vermiformis in aquatic biofilms grown on plasticized polyvinyl chloride. Appl. Environ. Microbiol. 70, 6826–6833.
Lettinga, K. D., Verbon, A., Weverling, G. J., Schellekens, J. F., Den Boer, J. W., Yzerman, E. P., Prins, J., Boersma, W. G., van Ketel, R. J., Prins, J. M., and Speelman, P. (2002). Legionnaires’ disease at a Dutch flower show: prognostic factors and impact of therapy. Emerging Infect. Dis. 8, 1448–1454.
Lettinga, K. D., Weijer, S., Speelman, P., Prins, J. M., Van Der Poll, T., and Verbon, A. (2003). Reduced interferon-gamma release in patients recovered from Legionnaires’ disease. Thorax 58, 63–67.
Liang, J. L., Dziuban, E. J., Craun, G. F., Hill, V., Moore, M. R., Gelting, R. J., Calderon, R. L., Beach, M. J., Roy, S. L., and Centers for Disease Control and Prevention (CDC). (2006). Surveillance for waterborne disease and outbreaks associated with drinking water and water not intended for drinking – United States, 2003–2004. MMWR Surveill. Summ. 55, 31–65.
Liles, M.R., Scheel, T. A., and Cianciotto, N. P. (2000). Discovery of a nonclassical siderophore, legiobactin, produced by strains of Legionella pneumophila. J. Bacteriol. 182, 749–757.
Liston, P., Roy, N., Tamai, K., Lefebvre, C., Baird, S., Cherton-Horvat, G., Farahani, R., McLean, M., Ikeda, J. E., MacKenzie, A., and Korneluk, R. G. (1996). Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 379, 349–353.
Lomma, M., Dervins-Ravault, D., Rolando, M., Nora, T., Newton, H. J., Samson, F. M., Sahr, T., Gomez-Valero, L., Jules, M., Hartland, E. L., and Buchrieser, C. (2010). The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cell. Microbiol. 12, 1272–1291.
Losick, V. P., and Isberg, R. R. (2006). NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203, 2177–2189.
Marra, A., Horwitz, M. A., and Shuman, H. A. (1990). The HL-60 model for the interaction of human macrophages with the Legionnaires’ disease bacterium. J. Immunol. 144, 2738–2744.
Marston, B. J., Lipman, H. B., and Breiman, R. F. (1994). Surveillance for Legionnaires’ disease. Risk factors for morbidity and mortality. Arch. Intern. Med. 154, 2417–2422.
Marston, B. J., Plouffe, J. F., File, T. M., Jr., Hackman, B. A., Salstrom, S. J., Lipman, H. B., Kolczak, M. S., and Breiman, R. F. (1997). Incidence of community-acquired pneumonia requiring hospitalization. Results of a population-based active surveillance Study in Ohio. The Community-Based Pneumonia Incidence Study Group. Arch. Intern. Med. 157, 1709–1718.
Martinon, F., and Tschopp, J. (2004). Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117, 561–574.
Maruta, K., Miyamoto, H., Hamada, T., Ogawa, M., Taniguchi, H., and Yoshida, S. (1998). Entry and intracellular growth of Legionella dumoffii in alveolar epithelial cells. Am. J. Respir. Crit. Care Med. 157(Pt 1), 1967–1974.
Matsiota-Bernard, P., Lefebre, C., Sedqui, M., Cornillet, P., and Guenounou, M.(1993). Involvement of tumor necrosis factor alpha in intracellular multiplication of Legionella pneumophila in human monocytes. Infect. Immun. 61, 4980–4983.
Molmeret, M., Zink, S. D., Han, L., Abu-Zant, A., Asari, R., Bitar, D. M., and Abu Kwaik, Y. (2004). Activation of caspase-3 by the Dot/Icm virulence system is essential for arrested biogenesis of the Legionella-containing phagosome. Cell. Microbiol. 6, 33–48.
Nash, T. W., Libby, D. M., and Horwitz, M. A. (1984). Interaction between the legionnaires’ disease bacterium (Legionella pneumophila) and human alveolar macrophages. Influence of antibody, lymphokines, and hydrocortisone. J. Clin. Invest. 74, 771–782.
Nash, T. W., Libby, D. M., and Horwitz, M. A. (1988). IFN-gamma-activated human alveolar macrophages inhibit the intracellular multiplication of Legionella pneumophila. J. Immunol. 140, 3978–3981.
Neumeister, B. (2004). Intracellular multiplication of Legionella species and the influence of amoebae on their intracellular growth in human monocytes: mono mac 6 cells and Acanthamoeba castellanii as suitable in vitro models. Methods Mol. Biol. 268, 141–151.
Neumeister, B., Bach, V., Faigle, M., and Northoff, H. (2001). Induction of iNOS in human monocytes infected with different Legionella species. FEMS Microbiol. Lett. 202, 31–38.
Neumeister, B., Schoniger, S., Faigle, M., Eichner, M., and Dietz, K. (1997). Multiplication of different Legionella species in Mono Mac 6 cells and in Acanthamoeba castellanii. Appl. Environ. Microbiol. 63, 1219–1224.
Park, D. R., and Skerrett, S. J. (1996). IL-10 enhances the growth of Legionella pneumophila in human mononuclear phagocytes and reverses the protective effect of IFN-gamma: differential responses of blood monocytes and alveolar macrophages. J. Immunol. 157, 2528–2538.
Payne, N. R., and Horwitz, M. A. (1987). Phagocytosis of Legionella pneumophila is mediated by human monocyte complement receptors. J. Exp. Med. 166, 1377–1389.
Pearlman, E., Jiwa, A. H., Engleberg, N. C., and Eisenstein, B. I. (1988). Growth of Legionella pneumophila in a human macrophage-like (U937) cell line. Microb. Pathog. 5, 87–95.
Poyet, J.L., Srinivasula, S. M., Tnani, M., Razmara, M., Fernandes-Alnemri, T., and Alnemri, E. S. (2001). Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 276, 28309–28313.
Price, C. T., Al-Khodor, S., Al-Quadan, T., Santic, M., Habyarimana, F., Kalia, A., and Kwaik, Y. A. (2009). Molecular mimicry by an F-box effector of Legionella pneumophila hijacks a conserved polyubiquitination machinery within macrophages and protozoa. PLoS Pathog. 5, e1000704. doi: 10.1371/journal.ppat.1000704.
Radtke, A. L., and O’Riordan, M. X. (2006). Intracellular innate resistance to bacterial pathogens. Cell. Microbiol. 8, 1720–1729.
Rogers, J., Dowsett, A. B., Dennis, P. J., Lee, J. V., and Keevil, C. W. (1994). Influence of plumbing materials on biofilm formation and growth of Legionella pneumophila in potable water systems. Appl. Environ. Microbiol. 60, 1842–1851.
Rovera, G., Santoli, D., and Damsky, C. (1979). Human promyelocytic leukemia cells in culture differentiate into macrophage-like cells when treated with a phorbol diester. Proc. Natl. Acad. Sci. U.S.A. 76, 2779–2783.
Rowbotham, T. J. (1980). Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33, 1179–1183.
Rowbotham, T. J. (1986). Current views on the relationships between amoebae, legionellae and man. Isr. J. Med. Sci. 22, 678–689.
Santic, M., Asare, R., Doric, M., and Abu Kwaik, Y. (2007). Host-dependent trigger of caspases and apoptosis by Legionella pneumophila. Infect. Immun. 75, 2903–2913.
Santic, M., Molmeret, M., and Abu Kwaik, Y. (2005). Maturation of the Legionella pneumophila-containing phagosome into a phagolysosome within gamma interferon-activated macrophages. Infect. Immun. 73, 3166–3171.
Schmeck, B., N’Guessan, P. D., Ollomang, M., Lorenz, J., Zahlten, J., Opitz, B., Flieger, A., Suttorp, N., and Hippenstiel, S. (2007). Legionella pneumophila-induced NF-kappaB- and MAPK-dependent cytokine release by lung epithelial cells. Eur. Respir. J. 29, 25–33.
Sethi, K. K., and Brandis, H. (1983). Direct demonstration and isolation of Legionella pneumophila (serogroup 1) from bathroom water specimens in a hotel. Zentralbl. Bakteriol. Mikrobiol. Hyg. B 177, 402–405.
Spitalny, K. C., Vogt, R. L., Orciari, L. A., Witherell, L. E., Etkind, P., and Novick, L. F. (1984). Pontiac fever associated with a whirlpool spa. Am. J. Epidemiol. 120, 809–817.
Swanson, M. S., and Isberg, R. R. (1995). Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63, 3609–3620.
Takahashi, R., Deveraux, Q., Tamm, I., Welsh, K., Assa-Munt, N., Salvesen, G. S., and Reed, J. C. (1998). A single BIR domain of XIAP sufficient for inhibiting caspases. J. Biol. Chem. 273, 7787–7790.
Turner, M. W. (1996). Mannose-binding lectin: the pluripotent molecule of the innate immune system. Immunol. Today 17, 532–540.
Underhill, D. M., Ozinsky, A., Smith, K. D. and Aderem, A. (1999). Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. U.S.A. 96, 14459–14463.
Vinzing, M., Eitel, J., Lippmann, J., Hocke, A. C., Zahlten, J., Slevogt, H., N’guessan, P. D., Günther, S., Schmeck, B., Hippenstiel, S., Flieger, A., Suttorp, N., and Opitz, B. (2008). NAIP and Ipaf control Legionella pneumophila replication in human cells. J. Immunol. 180, 6808–6815.
Viswanathan, V. K., Edelstein, P. H., Pope, C. D., and Cianciotto, N. P. (2000). The Legionella pneumophila iraAB locus is required for iron assimilation, intracellular infection, and virulence. Infect. Immun. 68, 1069–1079.
Walsh, J. G., Cullen, S. P., Sheridan, C., Luthi, A. U., Gerner, C., and Martin, S. J. (2008). Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. U.S.A. 105, 12815–12819.
Winn, W. C. Jr., and Myerowitz, R. L. (1981). The pathology of the Legionella pneumonias. A review of 74 cases and the literature. Hum. Pathol. 12, 401–422.
Wright, E. K., Goodart, S. A., Growney, J. D., Hadinoto, V., Endrizzi, M. G., Long, E. M., Sadigh, K., Abney, A. L., Bernstein-Hanley, I., and Dietrich, W. F. (2003). Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr. Biol. 13, 27–36.
Yu, V. L., Plouffe, J. F., Pastoris, M. C., Stout, J. E., Schousboe, M., Widmer, A., Summersgill, J., File, T., Heath, C. M., Paterson, D. L., and Chereshsky, A. (2002). Distribution of Legionella species and serogroups isolated by culture in patients with sporadic community-acquired legionellosis: an international collaborative survey. J. Infect. Dis. 186, 127–128.
Ziegler-Heitbrock, H. W., Thiel, E., Futterer, A., Herzog, V., Wirtz, A., and Riethmuller, G. (1988). Establishment of a human cell line (Mono Mac 6) with characteristics of mature monocytes. Int. J. Cancer 41, 456–461.
Keywords: NOD-like receptors, Toll-like receptors, pathogen-associated molecular patterns, neuronal apoptosis-inhibitory proteins
Citation: Khweek AA and Amer A (2010) Replication of Legionella pneumophila in human cells: why are we susceptible? Front. Microbio. 1:133. doi: 10.3389/fmicb.2010.00133
Received: 02 August 2010;
Accepted: 16 November 2010;
Published online: 28 December 2010.
Edited by:
Elizabeth L. Hartland, The University of Melbourne, AustraliaReviewed by:
Sang S. Yoon, Yonsei University College of Medicine, South KoreaElizabeth L. Hartland, The University of Melbourne, Australia
Copyright: © 2010 Khweek and Amer. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Amal Amer, Division of Pulmonary, Allergy, Critical Care and Sleep Medicine, Center for Microbial Interface Biology, Department of Internal Medicine, Biological Research Tower, 460W 12th Avenue, Room 1014, Columbus, OH 43210, USA. e-mail:YW1hbC5hbWVyQG9zdW1jLmVkdQ==