 S. Kauser Hussain
S. Kauser Hussain
- Department of Microbiology and Immunology, University of Arkansas for Medical Sciences, Little Rock, AR, USA
Coxiella burnetii is the etiologic agent of human Q fever and targets alveolar phagocytic cells in vivo wherein the pathogen generates a phagolysosome-like parasitophorous vacuole (PV) for replication. C. burnetii displays a prolonged growth cycle, making PV maintenance critical for bacterial survival. Previous studies showed that C. burnetii mediates activation of eukaryotic kinases to inhibit cell death, indicating the importance of host signaling during infection. In the current study, we examined the role of eukaryotic kinase signaling in PV establishment. A panel of 113 inhibitors was analyzed for their impact on C. burnetii infection of human THP-1 macrophage-like cells and HeLa cells. Inhibition of 11 kinases or two phosphatases altered PV formation and prevented pathogen growth, with most inhibitor-treated cells harboring organisms in tight-fitting phagosomes, indicating kinase/phosphatase activation is required for PV maturation. Five inhibitors targeted protein kinase C (PKC), suggesting a critical role for this protein during intracellular growth. The PKC-specific substrate MARCKS was phosphorylated at 24 h post-infection and remained phosphorylated through 5 days post-infection, indicating prolonged regulation of the PKC pathway by C. burnetii. Infection also altered the activation status of p38, myosin light chain kinase, and cAMP-dependent protein kinase, suggesting C. burnetii subverts numerous phosphorylation cascades. These results underscore the importance of intracellular host signaling for C. burnetii PV biogenesis.
Introduction
Intracellular bacterial pathogens have evolved sophisticated mechanisms to subvert host cell function and establish a protected replication vacuole. To support intravacuolar growth, these pathogens modulate host processes such as vesicular fusion and trafficking, cytokine production, and cell survival to avoid delivery to degradative lysosomes, recognition by the host immune system, and loss of a viable host cell, respectively (Knodler et al., 2001; Bhavsar et al., 2007). Eukaryotic phosphorylation cascades are efficient regulatory networks that control processes involved in recognition, uptake, and elimination of foreign material such as bacterial pathogens. Therefore, host kinases and phosphatases are often at the forefront of host–pathogen interactions.
Coxiella burnetii is an intracellular bacterial pathogen that causes the zoonosis human Q fever. The pathogen exhibits a global distribution and is primarily spread by contaminated aerosols. Humans are typically exposed to infectious organisms through contact with infected livestock or their products (Maurin and Raoult, 1999). Aside from infrequent abortion in goats, infected animals generally do not display overt signs of disease, but shed high numbers of bacteria into the environment, particularly during parturition. In humans, Q fever typically presents as an acute flu-like illness characterized by prolonged high fever, with some patients developing pneumonia or hepatitis (Raoult et al., 2005). Spread from the site of acute disease can lead to chronic infections, typically in immunocompromised individuals. By mechanisms that are not clearly understood, chronic infections can reactivate months or years following an initial infection and cause serious illness, such as endocarditis, that exhibits a much higher mortality rate than acute disease (Marrie and Raoult, 2002). Although Q fever remains somewhat rare in the United States, a large recent outbreak in the Netherlands (Delsing and Kullberg, 2008; Schimmer et al., 2009) underscores the need to better understand C. burnetii pathogenic mechanisms and develop efficacious treatments. Indeed, since 2007, over 3500 cases of Q fever have been diagnosed in the Netherlands and six deaths reported (Schimmer et al., 2009; Schneeberger et al., 2010).
In vivo, C. burnetii initially infects alveolar phagocytic cells and directs biogenesis of a phagolysosome-like parasitophorous vacuole (PV) in which to replicate (Voth and Heinzen, 2007). C. burnetii enters the host cell by passive phagocytosis and resides in a tight-fitting nascent phagosome during the first 4–6 h post-infection (Howe and Mallavia, 2000). After this phagosomal stall, the vacuole matures along the endolysosomal pathway and culminates in a PV with degradative lysosomal characteristics (Howe et al., 2010). The PV lumen is acidic (pH ∼ 5) and contains active hydrolases and vacuolar conditions are sufficient to degrade other bacterial cells (Howe et al., 2010). The PV acquires membrane via heterotypic fusion with endosomes, autophagosomes, and lysosomes while expanding to occupy most of the host cell cytoplasm (Voth and Heinzen, 2007). In this phagolysosomal PV, C. burnetii replicates to high numbers throughout a lengthy infectious cycle (doubling time ∼ 11 h; Coleman et al., 2004). Formation and maintenance of the PV requires continual C. burnetii protein synthesis as treatment with chloramphenicol causes PV collapse and cessation of bacterial replication (Howe et al., 2003). This requirement for de novo protein synthesis presumably involves production and function of the pathogen’s Dot/Icm type IV secretion system and associated effector proteins (Pan et al., 2008; Voth and Heinzen, 2009a; Voth et al., 2009).
The prolonged duration of C. burnetii infection implies that the pathogen continually modulates host cell processes. We and others recently reported the ability of C. burnetii to potently antagonize apoptotic cell death, presumably as a mechanism to sustain the host cell. C. burnetii actively inhibits activation of caspase-dependent apoptosis in cultured and primary phagocytic cells and non-phagocytic cells. Activation of the intrinsic (mitochondrial-mediated) and extrinsic (death receptor-mediated) pathways is prevented during infection, resulting in potent inhibition of host cell death machinery (Luhrmann and Roy, 2007; Voth et al., 2007). C. burnetii also activates two pro-survival kinases, Akt and Erk1/2, an event needed for full protection from apoptosis (Voth and Heinzen, 2009b). Akt and Erk1/2 phosphorylation is sustained through at least 72 h post-infection, a time at which the PV is filling with replicating organisms. Interestingly, inhibition of Akt or Erk1/2 does not have an obvious deleterious effect on PV formation, only C. burnetii’s ability to antagonize host cell death. Although many intracellular pathogens modulate host kinase cascades, the scope of C. burnetii signaling regulation is poorly understood.
In the current study, we probed the role of host kinases and phosphatases in C. burnetii PV formation and found that multiple signaling proteins regulate infection. Numerous kinases, including protein kinase C (PKC) and cAMP-dependent protein kinase (PKA), promote PV development as cells infected in the presence of distinct inhibitors do not support vacuole formation or bacterial growth. Additionally, p38, myosin light chain kinase (MLCK), PKA, and PKC are activated during intracellular growth, suggesting C. burnetii regulates numerous host pathways during infection. Collectively, the current results underline the importance of signaling pathway modulation by C. burnetii for PV establishment.
Materials and Methods
Mammalian Cell Culture and C. burnetii
THP-1 human monocytes (TIB-202; American Type Culture Collection, Manassas, VA, USA) and HeLa (human epithelioid carcinoma) cells (CCL-2; ATCC) were maintained at 37°C and 5% CO2 in RPMI 1640 medium containing 10% fetal calf serum (HyClone Laboratories, South Logan, UT, USA). Prior to infections, phorbol 12-myristate 13-acetate (PMA; EMD Biosciences, San Diego, CA, USA) was added to medium for 24 h at a final concentration of 200 nM to induce differentiation into a macrophage-like cell as previously described (Voth et al., 2007). Medium containing PMA was then removed and cell cultures replenished with complete medium lacking PMA prior to infections.
Virulent C. burnetii Nine Mile phase I (RSA493) and G (Q212) isolates and avirulent Nine Mile phase II (clone 4; RSA439) organisms (generously provided by Dr. Robert Heinzen, Rocky Mountain Laboratories, Hamilton, MT, USA) were propagated in Vero (African green monkey kidney) cells (CCL-81; ATCC) and purified as previously described (Coleman et al., 2004). For infections, avirulent C. burnetii was used at a multiplicity of infection (MOI) of 10 and virulent isolates at an MOI of 100. The lower in vitro infectivity of virulent C. burnetii necessitates using a higher MOI (Voth and Heinzen, 2007). Handling of virulent C. burnetii isolates was performed in the CDC-approved biosafety level-3 facility at the University of Arkansas for Medical Sciences.
Kinase and Phosphatase Inhibitor Analysis
THP-1 cells or HeLa cells were cultured in 96-well plates for initial assessment of inhibitor treatments. Cells were infected with avirulent C. burnetii in the presence of individual inhibitors at a final concentration of 10 μM and all compounds were added at the time of infection or 4 hpi. Two inhibitor panels were assessed in this study: a kinase inhibitor library containing 80 established pharmacologic compounds and a phosphatase inhibitor library containing 33 compounds (EnzoLife Sciences, Plymouth Meeting, MA, USA) in dimethylsulfoxide (DMSO). Medium containing fresh inhibitors was replenished every 24 h and PV development was assessed at 24 and 48 h post-infection (hpi) by light microscopy. Images were acquired using a Nikon Ti-U microscope (Nikon, Tokyo, Japan), a 40× objective, and a D5-Qi1Mc digital camera. Each inhibitor panel was screened during infection three times independently using THP-1 cells and once using HeLa cells. Mammalian cell morphology and viability was affected by less than 10% of inhibitors as assessed by light microscopy and nuclear staining followed by fluorescence microscopy (data not shown).
Fluorescence Microscopy
HeLa cells were cultured in 24-well plates on 12-mm cover slips and infected with avirulent C. burnetii in the presence or absence of a subset of kinase (GF 109203X, SB-203580, H-89, ML-7, or KN-93) or phosphatase (pentamidine) inhibitors. At 48 hpi, cells were fixed and permeabilized using 100% ice-cold methanol for 3 min, then incubated for 1 h in phosphate–buffered saline (PBS; HyClone Laboratories) containing 0.5% bovine serum albumin (BSA; Cell Signaling, Danvers, MA, USA) and 0.05% sodium azide (ISC BioExpress, Solon, OH, USA). Cells were incubated with PBS–BSA solution containing rabbit anti-C. burnetii and mouse anti-CD63 (LAMP-3; BD Biosciences, San Jose, CA, USA) primary antibodies to detect bacteria and the PV membrane, respectively, for 1 h at room temperature. Primary antibodies were subsequently detected using AlexaFlour-594 anti-rabbit and AlexaFluor-488 anti-mouse secondary antibodies (Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature. Eukaryotic and bacterial DNA was stained with 4′,6-diamidino-2-phenylindole, dilactate (DAPI; Invitrogen). Samples were imaged using a Nikon Ti-U microscope with a 60× oil immersion objective and images captured with a D5-Qi1Mc digital camera (Nikon). Images were analyzed and rendered using NIS-Elements software (Nikon).
Inhibitor Reversibility Assays
HeLa cells were cultured in 24-well plates on 12-mm cover slips and infected with avirulent C. burnetii at an MOI of 10. Infected cells were treated with GF 109203X, H-89, ML-7, KN-93, or pentamidine at 10 μM final concentration at the time of infection. Inhibitor-containing medium was replaced with medium lacking inhibitors at 48 hpi and cells incubated in the absence of inhibitors for an additional 48 h to allow potential PV formation. Cells were then fixed and processed as described above and PV containing replicating bacteria detected using anti-C. burnetii and anti-CD63 antibodies. Inhibitor effects were considered reversible when cells contained PV similar to untreated, infected cells at 48 h post-washout.
Immunoblot Analysis
THP-I cells were infected with virulent or avirulent C. burnetii isolates for 3–120 h, then harvested by lysis in buffer containing 50 mM Tris, 5 mM EDTA, and 1% sodium dodecyl sulfate (SDS) followed by 10 passages through a 21-gage needle. The detergent- compatible DC protein assay (Bio-Rad, Hercules, CA, USA) was used to determine total protein concentration in each lysate. Ten micrograms of total protein was separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to a 0.2 μm PVDF membrane (Bio-Rad). After transfer, membranes were incubated for 1 h at room temperature in Tris–buffered saline (TBS; 150 mM NaCl, 100 mM Tris–HCl, pH 7.6) containing 0.1% Tween-20 and 5% non-fat milk. After blocking, membranes were incubated with rabbit polyclonal antibodies directed against phosphorylated PKA substrates, PKC substrates, MARCKS, p38, or MLC2 (Cell Signaling) in TBS-Tween-20 containing 5% BSA at 4°C overnight. Lysates were probed for β-tubulin as a loading control using mouse monoclonal antibody clone SAP.4G5 (Sigma-Aldrich, St. Louis, MO, USA). Reacting proteins were detected with anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (Cell Signaling) and observed by enhanced chemiluminescence using Femto reagent (Pierce, Rockford, IL, USA) following exposure to film. Immunoblot images were acquired using a Fluorchem FC2 gel documentation system (Alpha Innotech Corp., San Leandro, CA, USA).
PKA and PKC activity was assessed by probing phosphorylation of a panel of downstream substrates. These assays were performed using rabbit polyclonal antibodies (Cell Signaling) that recognize proteins phosphorylated at distinct serine or threonine residues. For PKA, this antibody recognizes proteins with an arginine 3 positions upstream of the phosphorylated serine or threonine residue. The PKC antibody recognizes proteins with phosphorylated serine residues surrounded by arginine and lysine residues.
Results
Host Kinases and Phosphatases are Involved in C. burnetii PV Formation
Because C. burnetii subverts host signaling to control apoptotic cell death, we predicted the pathogen would also regulate distinct pathways to promote generation of its vacuolar niche. To assess host signaling pathways involved in PV biogenesis, we examined vacuole development in the presence of individual mammalian kinase or phosphatase inhibitors. For these studies, we used differentiated THP-1 human macrophage-like cells and HeLa (human epithelial) cells, which are reliable models of C. burnetii–host cell interactions (Voth et al., 2007; Voth and Heinzen, 2009b). THP-1 or HeLa cells were infected with avirulent C. burnetii for 48 h in the presence of individual inhibitors from a panel of 113 well-characterized compounds. C. burnetii directs the synthesis of a large replication compartment easily visible by light microscopy (Voth and Heinzen, 2007). Thus, we examined inhibitor-treated cells by light microscopy for the presence of prototypical PV as compared to untreated, infected cell cultures. As shown in Table 1, inhibitors were categorized as having a negative impact on vacuole formation when less than 10% of cells contained typical PV at 48 hpi. Twenty kinase and two phosphatase inhibitors antagonized PV formation (negative effect), while two inhibitors targeting p38 MAPK induced abnormal early PV expansion (positive effect; Figure 1). Collectively, inhibitors with demonstrable effects on infection targeted 11 kinases and two phosphatases. p38-inhibited cells contained large PV at 24 hpi, a time when PV are not typically apparent by phase contrast microscopy in untreated, infected cell cultures (Figure 1A). In contrast, inhibition of the remaining kinases and phosphatases (Table 1) prevented PV formation (Figure 1B and data not shown). In agreement with previous results from our laboratory (Voth and Heinzen, 2009b), the phosphatidylinositol-3-kinase (PI3K) inhibitors wortmannin and LY-294002 and the MEK1/2 antagonist U0126 had no obvious impact on PV formation (data not shown). Inhibitors that caused rapid cell death at the concentration tested (<10% of those tested) were not further analyzed (data not shown).

Table 1. Inhibitors altering PV formation.
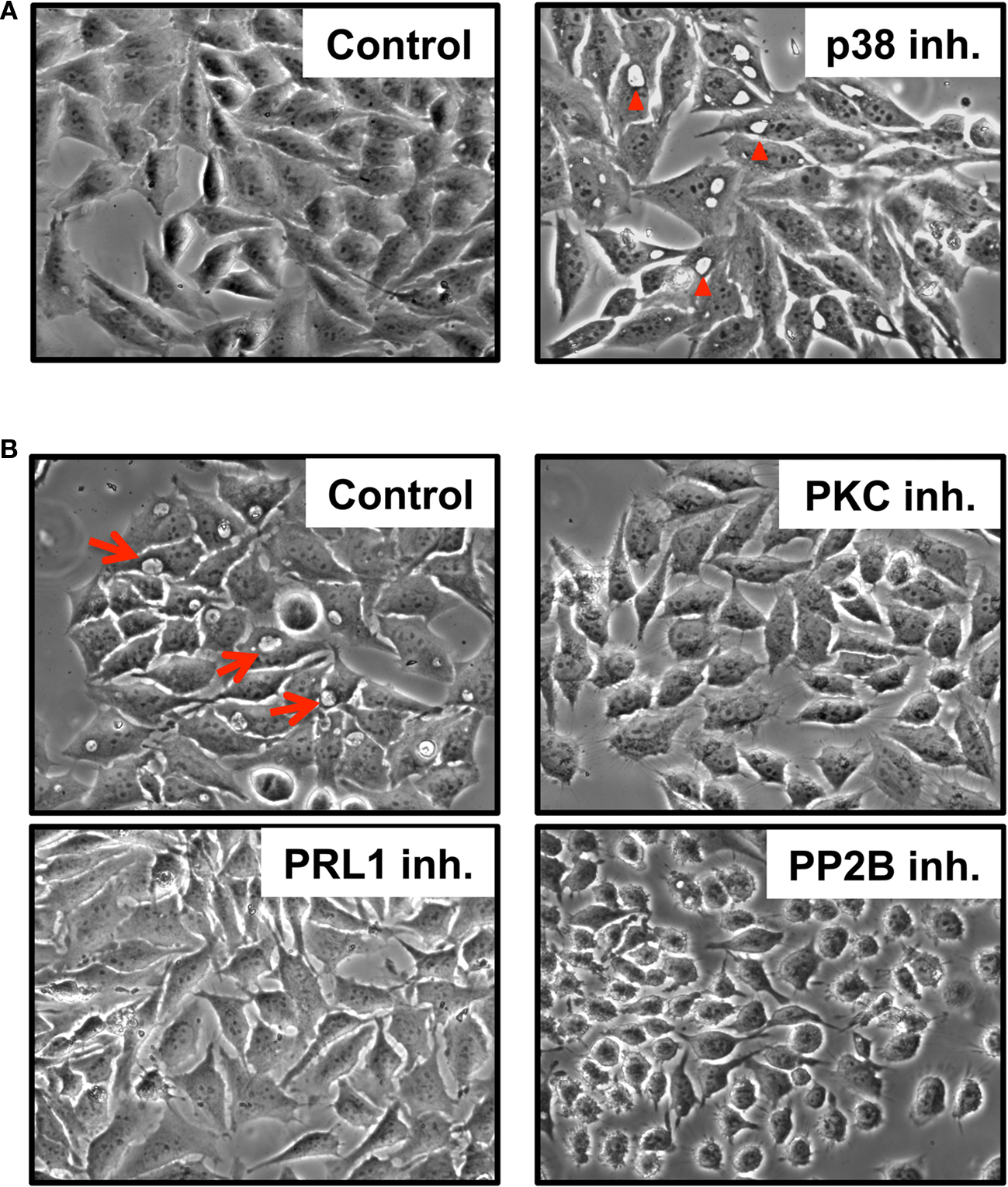
Figure 1. Host kinases and phosphatases are involved in prototypical PV formation. HeLa cells were infected for 24 h (A) or 48 h (B) with C. burnetii in the presence of p38 (SB-203580), PKC (GF 109203X), PRL1 (pentamidine), or PP2B (gossypol) inhibitors and visualized by phase contrast microscopy. Typical phase translucent PV are indicated by red arrows (B). p38 inhibitors dramatically enhanced PV development [(A) right panel, arrowheads] at early times post-infection while PKC, PRL1, and PP2B inhibitors prevented vacuole formation (B). Control = infected, untreated cells.
Inhibitor-Treated Cells Harbor Non-Replicating Organisms in Phagolysosome-Like Compartments
The C. burnetii PV exhibits characteristics strikingly similar to host phagolysosomes (Voth and Heinzen, 2007; Howe et al., 2010). Indeed, the PV is a hydrolytic, degradative compartment, and pathogen metabolism is activated at acidic pH (pH ∼ 5) where replication ensues, making vacuole maturation critical for intracellular growth. Therefore, we examined the localization of C. burnetii in inhibitor-treated cells to determine if the pathogen was precluded from entry into the cell and transit to a phagolysosome-like compartment. HeLa cells were infected in the presence of individual inhibitors for 48 h, then processed for fluorescence microscopy. Aside from p38-inhibited cells, all inhibitor-treated cells contained individual bacteria in tight-fitting, phagolysosome-like compartments (Figure 2 and data not shown) as evidenced by CD63 (LAMP-3) labeling, which is commonly used to identify mature PV (Howe et al., 2010). PRL1-, PKC-, PKA-, and CaMKII-inhibited cells are shown as examples. Hence, in inhibitor-treated cells, C. burnetii trafficked to a vacuole with characteristics that should support growth but was unable to replicate. Conversely, infected cells treated with p38 inhibitors housed replicating C. burnetii in unusually large CD63-positive vacuoles (Figure 2). However, uninfected cells treated with p38 inhibitors did not contain large vacuoles, indicating the effects of these compounds were specific to infected cells (data not shown). Similar results were obtained using THP-1 cells (data not shown).
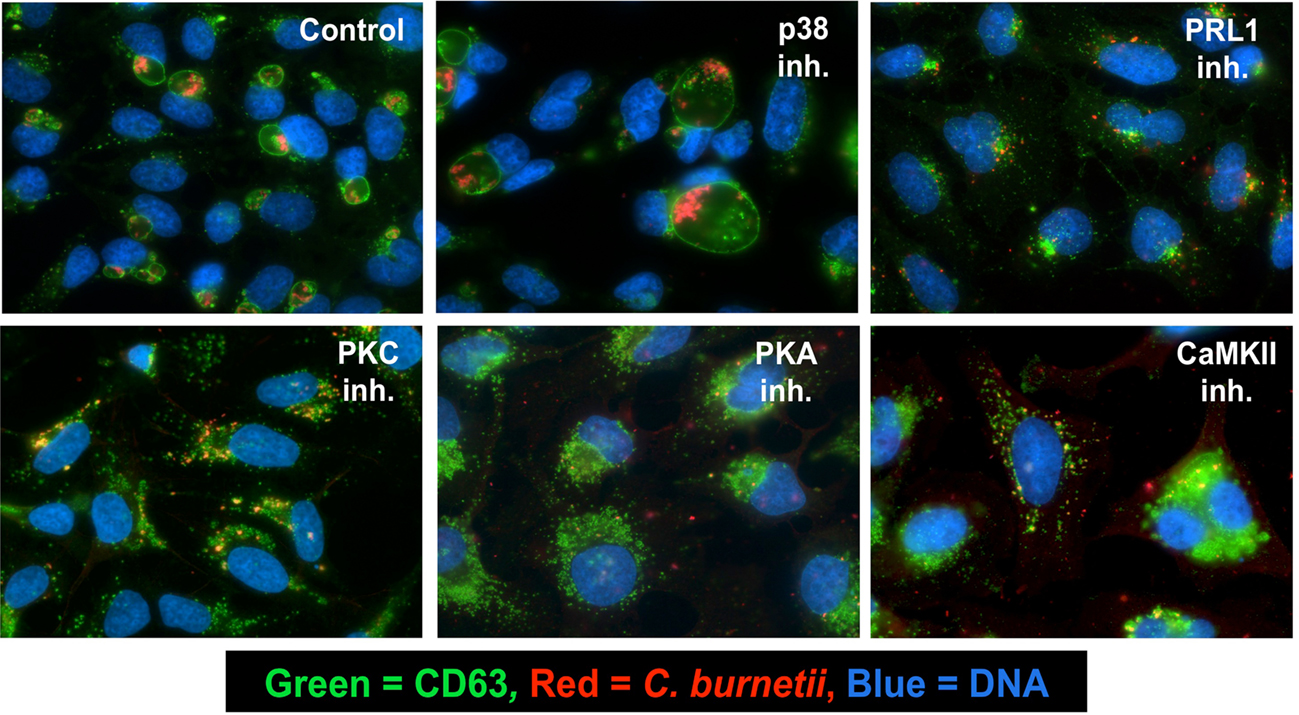
Figure 2. Inhibitor-treated cells harbor C. burnetii in CD63-positive compartments. HeLa cells were infected with C. burnetii for 48 h in the presence of the indicated kinase or phosphatase inhibitors. Cells were then processed for fluorescence microscopy. A primary antibody recognizing CD63 was used to confirm the phagolysosomal nature of the PV, anti-C. burnetii antibody was used to detect bacteria, and DAPI was used to stain DNA. Colors are indicated in the figure. Cells treated with PRL1 (pentamidine), PKC (GF 109203X), PKA (H-89), or CaMKII (KN-93) inhibitors housed organisms in tight-fitting phagolysosome-like compartments, whereas p38-inhibited cells contained replicating organisms in abnormally enlarged PV. Control = infected, untreated cells.
Inhibition of PRL1, PKC, PKA, and MLCK is Reversible
Our inhibitor-based screen suggested that host kinases and phosphatases are intimately involved in prototypical PV maturation. We next analyzed the reversibility of the observed inhibitor effects to determine whether the inhibitors exerted direct cytotoxic effects on C. burnetii. PRL1, PKC, PKA, MLCK, and CaMKII inhibitors were removed from infected HeLa cells at 48 hpi and cells incubated an additional 48 h to allow potential PV formation. As shown in Figure 3, when PRL1, PKC, PKA, and MLCK inhibitors were removed from infected cells, PV maturation ensued, with CD63-positive vacuoles present at 48 h post-washout. Additionally, these newly expanded vacuoles contained increasing numbers of replicating C. burnetii. In contrast, the CaMKII antagonist KN-93 appeared to exert a toxic effect on C. burnetii, as evidenced by a lack of expanded PV at 48 h post-washout (Figure 3). These results further suggest that numerous signaling proteins, including PRL1, PKC, PKA, and MLCK are involved in PV maturation events and do not have bactericidal effects on C. burnetii.
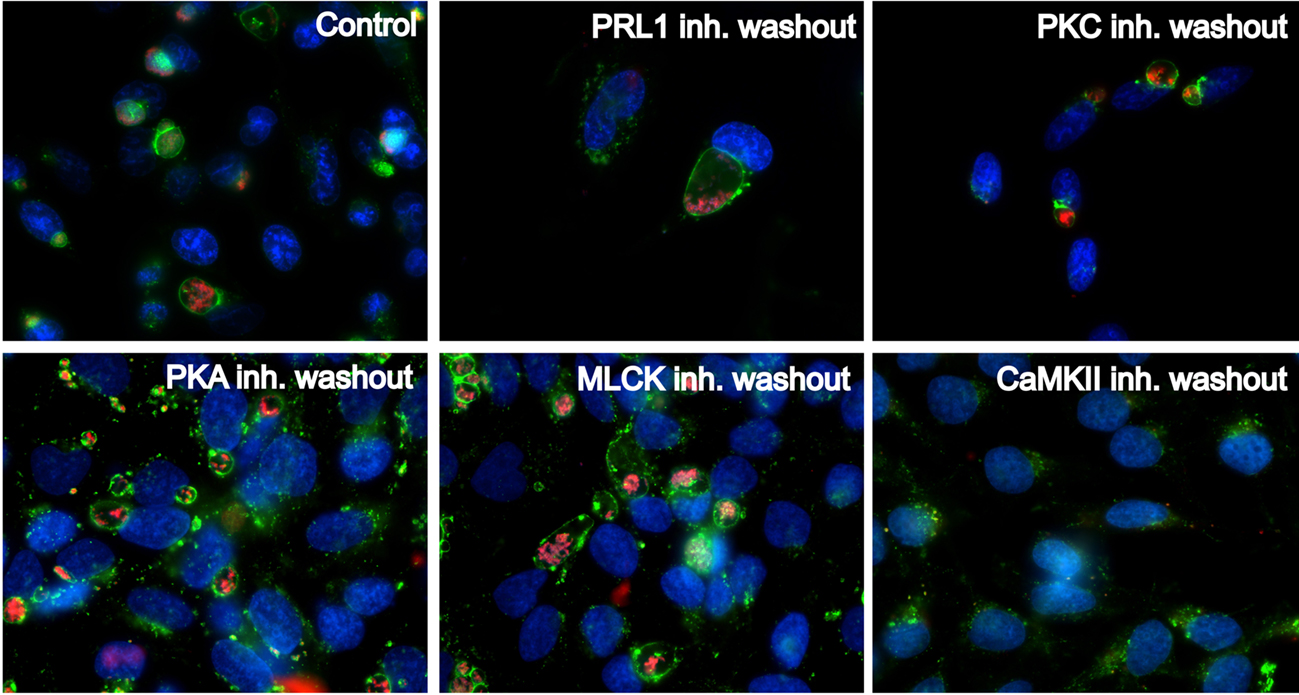
Figure 3. Kinase and phosphatase antagonists reversibly inhibit PV formation and C. burnetii growth. HeLa cells were infected with C. burnetii for 48 h in the presence of the indicated kinase inhibitors, then inhibitors were removed from cells for an additional 48 h. At 48 h post-washout, cells were processed for fluorescence microscopy using anti-CD63 antibody (green) to detect PV and anti-C. burnetii antibody (red) to detect bacteria. DAPI was used to stain host and bacterial DNA (blue). Following washout, CD63-positive PV formed in cells previously treated with PRL1 (pentamidine), PKC (GF 109203X), PKA (H-89), or MLCK (ML-7) inhibitors, indicating these kinases are involved in PV formation and are not directly toxic to C. burnetii. In contrast, washout of KN-93 did not allow PV expansion or bacterial replication, indicating the effects of this inhibitor are not reversible. Control = infected, untreated cells.
C. burnetii Infection Induces Differential Phosphorylation of Eukaryotic Kinases and Downstream Substrates
Due to the negative effects of several kinase inhibitors on PV formation (Table 1), we predicted that C. burnetii might activate a distinct panel of kinases during normal infection. To test this hypothesis, THP-1 cells were infected up to 4 days in the absence of inhibitors and lysates probed for phosphorylated forms of individual kinases at distinct times post-infection. Phosphorylation is a reliable indicator of kinase activation status and is required for regulation of downstream target activity. As shown in Figure 4, C. burnetii infection causes differential phosphorylation of p38 and myosin light chain-2 (MLC-2), which is the downstream target of MLCK. Specifically, levels of phosphorylated p38 increased over those of uninfected cells at 2 hpi (Figure 4A), then declined to near basal level between 48 and 72 hpi similar to previous results (Voth and Heinzen, 2009b). MLC-2 was also phosphorylated by 3 hpi; however, levels continued to increase throughout infection (Figure 4B).
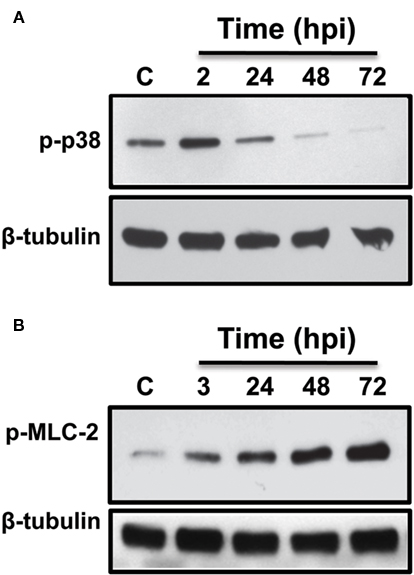
Figure 4. C. burnetii induces differential phosphorylation of p38 and MLC-2. C. burnetii-infected THP-1 cells were harvested at the indicated times and equal amounts of total lysate protein subjected to immunoblot analysis using phosphorylation-specific antibodies to detect the proteins designated in the figure. (A) Increased p38 phosphorylation was observed at 2 hpi, then decreased to basal level by 48–72 hpi. (B) MLC-2 phosphorylation levels increased above those of uninfected cells at 3 hpi and levels continued to increase through 72 hpi, indicating prolonged kinase activation. Tubulin is included as a loading control. C = uninfected cells.
As shown above (Table 1; Figure 2), the PKA inhibitor H-89 completely negated PV formation. Therefore, we predicted that PKA activation is essential for PV biogenesis and C. burnetii growth. To assess kinase activity, we examined the profiles of downstream substrates phosphorylated on PKA-specific serine or threonine residues during a time course of C. burnetii infection (Figure 5). This is a commonly used method for assessing overall PKA activity and identifying novel downstream targets. As shown in Figure 5, phosphorylation levels of three PKA substrates substantially decreased by 48–72 hpi. Conversely, four proteins exhibited increased phosphorylation at later times of infection (24–96 hpi), suggesting C. burnetii engages the PKA pathway at different stages of intracellular growth for controlled regulation of this versatile signaling cascade.
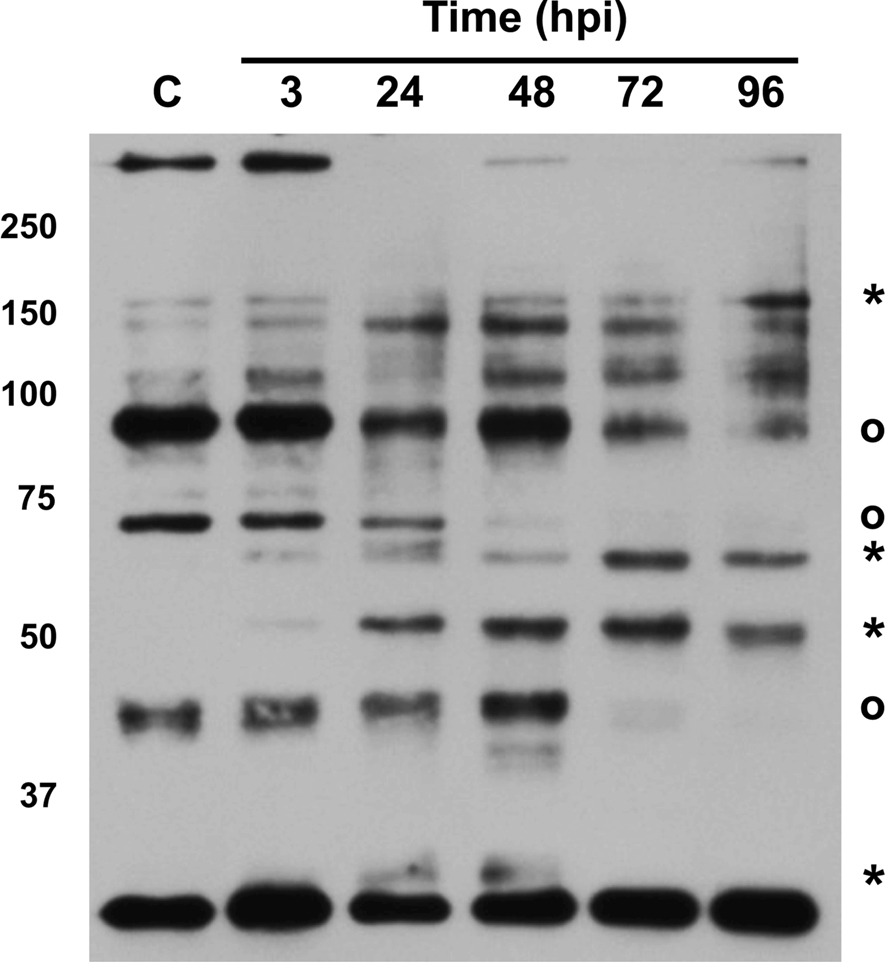
Figure 5. Protein kinase A is differentially activated during C. burnetii infection. Equal amounts of total lysate protein from THP-1 cells infected for the indicated times were analyzed by immunoblotting for PKA substrates phosphorylated at distinct serine or threonine residues, indicative of overall kinase activity. Molecular weight is given on the left of the blot. Asterisks represent proteins with increased phosphorylation levels at 24–96 hpi and circles denote proteins with decreased phosphorylation levels after 48 hpi. At least seven PKA substrates showed altered phosphorylation levels during infection, with increased phosphorylation of three proteins sustained through 96 hpi. C = uninfected cells.
C. burnetii Triggers Sustained PKC Activation
Five inhibitors in our directed screen targeted PKC, with one compound specifically antagonizing PKC-δ, suggesting that C. burnetii requires PKC activity for PV establishment. Twelve PKC isoforms have been described and are regulated via phosphorylation (Reyland, 2009). Due to the complexity of isoform activation, we probed total PKC activity during infection by monitoring phosphorylation of downstream substrates (Figure 6A). THP-1 cells were infected for differing periods of time and total cell lysates subjected to immunoblot analysis using an antibody to detect PKC substrates phosphorylated on serine or threonine residues. By 24 hpi, a substantial overall increase in total PKC substrate phosphorylation was evident, indicative of kinase activation (Figure 6A). Furthermore, phosphorylation of myristoylated PKC substrate (MARCKs) was sustained through 5 days post-infection (Figure 6B), indicating prolonged perturbation of the PKC pathway by C. burnetii during infection.
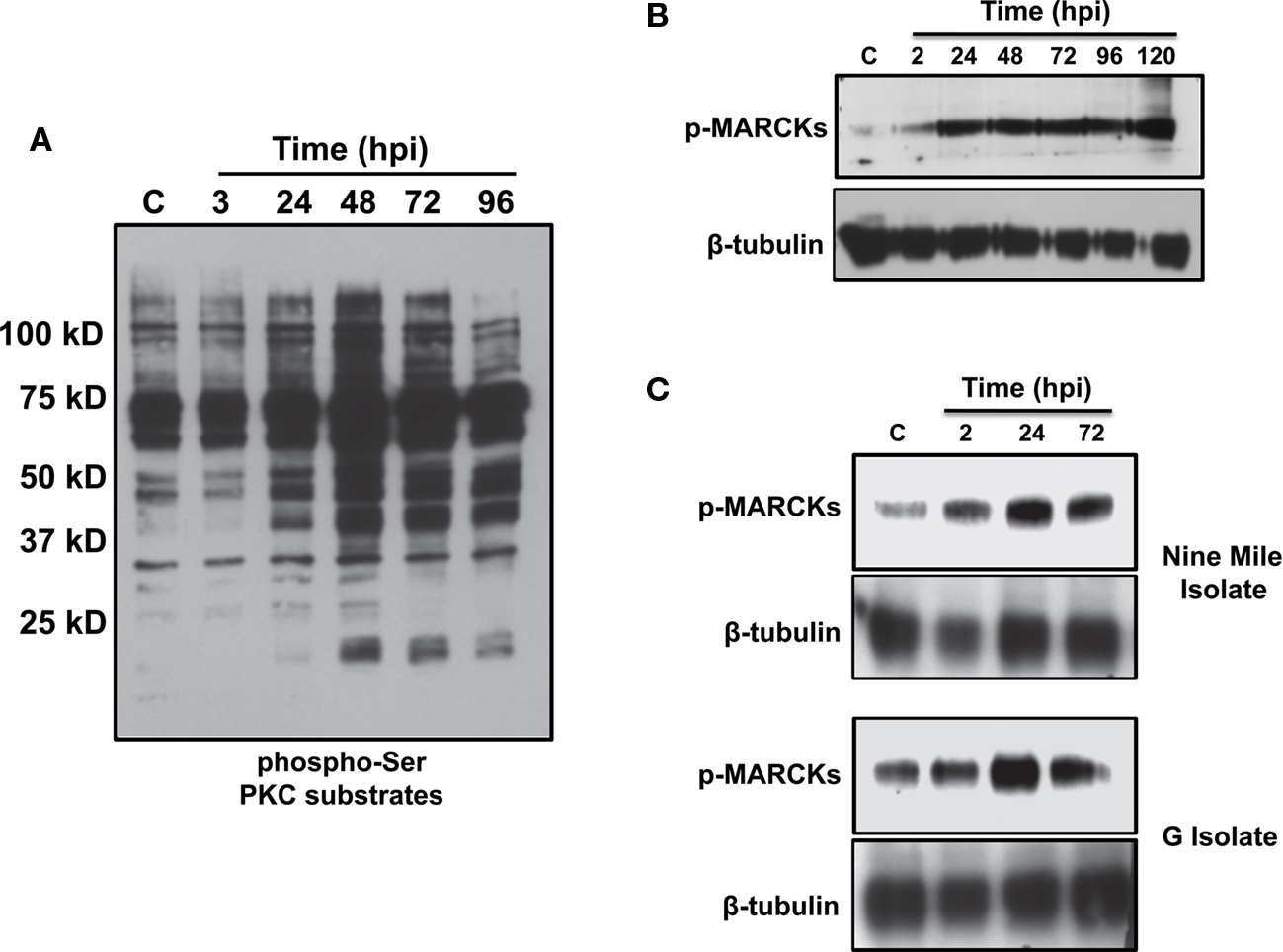
Figure 6. C. burnetii infection elicits prolonged PKC activation. THP-1 cells were infected with C. burnetii for the indicated time periods, harvested, and equal amounts of total lysate protein subjected to immunoblot analysis. Phosphorylation of total PKC substrates and MARCKs were used as readouts of PKC activity. Molecular weight is provided in (A). Increased PKC substrate phosphorylation was observed through 96 hpi (A) and MARCKs phosphorylation was sustained through 120 hpi (B). (C) Virulent C. burnetii isolates activate PKC.THP-1 cells were infected with the C. burnetii Nine Mile or G isolates for the indicated time periods, harvested, and equal amounts of total protein subjected to immunoblot analysis to detect phosphorylated MARCKs. Cells infected with C. burnetii Nine Mile or G contained increased levels of phosphorylated MARCKs, indicating these organisms activate PKC during intracellular growth. Tubulin levels are shown as loading controls. C = uninfected cells.
The experiments described above were performed using avirulent C. burnetii organisms in phase II producing truncated lipopolysaccharide (LPS; Hackstadt, 1986). Accumulating studies demonstrate that this isolate is a reliable model of C. burnetii intracellular activity (Voth et al., 2007; Vazquez and Colombo, 2009; Howe et al., 2010). However, it is also important to assess conditions that alter PV biogenesis in the context of a virulent C. burnetii infection. Therefore, THP-1 cells were infected with one of two virulent C. burnetii pathotypes in phase I producing full-length LPS (Hackstadt, 1986) and lysates probed for MARCKs phosphorylation indicative of PKC activity. Specifically, the Nine Mile (acute disease) and G (chronic endocarditis) isolates were analyzed for potential pathotype-specific responses. As shown in Figure 6C, MARCKs phosphorylation increased at 2–24 hpi with virulent isolates and levels remained elevated above those of uninfected cells through 72 hpi, corresponding to that observed during avirulent C. burnetii infection. These results suggest PKC activation is a conserved event during C. burnetii infection.
Discussion
Here, we demonstrate that 13 eukaryotic phosphorylation-based signaling components participate in biogenesis of the C. burnetii PV. During infection, activity of four kinases substantially increases and inhibition of individual kinases or phosphatases prevents PV formation. Vacuole development and maintenance is essential for replication of many intracellular bacterial pathogens (Brumell and Scidmore, 2007; Deretic, 2008; Isberg et al., 2009).To date, pathogen replication vacuole studies have appropriately focused on the role of fusogenic components such as Rab and SNARE proteins (Brumell and Scidmore, 2007), which are essential for membrane fusion and phagosome maturation in eukaryotic cells. Results from the current study suggest a major role for host kinase-dependent signaling in generation of bacterial growth compartments.
Bacterial pathogens commonly usurp host signaling to influence infection events for the benefit of the pathogen (Bhavsar et al., 2007). One conserved bacterial strategy is subversion of host apoptosis to antagonize cell death and/or promote proliferation, providing a viable niche for replication. For example, Salmonella typhimurium activates the host pro-survival kinase Akt during early stages of epithelial cell infection (Knodler et al., 2005). Additionally, Mycobacterium tuberculosis alters PKC activity during Schwann cell infection, leading to increased proliferation throughout a long infectious cycle (Tapinos and Rambukkana, 2005). Moreover, C. burnetii potently antagonizes macrophage apoptosis in part by triggering sustained activation of Akt and Erk1/2 (Luhrmann and Roy, 2007; Voth et al., 2007; Voth and Heinzen, 2009b).
Apart from apoptosis, intracellular pathogens also utilize host signaling for cellular entry, alteration of cytokine production, and regulation of the host cell cycle (Tse et al., 2002; Buchholz and Stephens, 2007).Conversely, the role of eukaryotic kinases and phosphatases in pathogen vacuole formation has not been elucidated. Pharmacologic inhibitor-based screens provide a directed approach to identify pathogen virulence mechanisms and host proteins involved in infection. For example, a recent inhibitor-based study showed that Listeria monocytogenes engages multiple host pathways to provide a suitable environment for replication (Lieberman and Higgins, 2009). Interestingly, this study implicated a role for PKC in L. monocytogenes infection, similar to our current results for C. burnetii. Inhibitor-based screens have also been used to uncover host processes subverted by Vibrio cholerae (Hung et al., 2005), Toxoplasma gondii (Carey et al., 2004), and Pseudomonas aeruginosa (De La Fuente et al., 2006), highlighting the utility of chemical inhibitor screening in host–pathogen interaction studies. Moreover, recent siRNA-based screens have uncovered roles for host signaling in the intracellular lifestyles of M. tuberculosis (Jayaswal et al., 2010), Brucella melitensis, and Brucella abortus (Qin et al., 2008).
It is important to note that pharmacologic inhibitors can alter the activity of proteins other than their intended target. For example, Rottlerin inhibits PKC-δ, but can also affect PKC-α, -β, and -γ and CaMKII activity (Gschwendt et al., 1994). Thus, we used the inhibitor panel in this study solely as a tool to implicate host pathway involvement in PV biogenesis, not as definitive proof of kinase importance. This panel allowed us to generate a number of hypotheses about the role of host signaling during C. burnetii infection and directed our assessments of p38, MLCK, PKA, and PKC activity, which indicate distinct cascades are manipulated during infection. However, the absolute specificity of each inhibitor during infection has not been confirmed for the compounds in our panel.
Two contrasting effects are observed when C. burnetii-infected cells are incubated with kinase/phosphatase inhibitors. First, cells treated with p38 antagonists harbor bacteria in abnormally expanded CD63-positive vacuoles at 24–48 hpi, suggesting the kinase regulates early PV formation events. Second, inhibition of 10 kinases or 2 phosphatases impedes PV development. Interestingly, inhibitor treatment does not prevent delivery of C. burnetii to its favored phagolysosome-like compartment as single organisms are housed in tight-fitting CD63-positive vacuoles, but fail to replicate. These results suggest host signaling is needed for PV maturation steps including membrane acquisition and expansion and is not simply required for phagocytosis of C. burnetii or phagosome–lysosome fusion. Furthermore, cells treated with distinct inhibitors 4 h after uptake of C. burnetii contain small PV with non-replicating organisms (data not shown). Importantly, these adverse effects are reversible, with PV expanding after inhibitor removal in most cases, indicating kinases are required for PV biogenesis and inhibitors are not cytotoxic to C. burnetii.
Three major classes of kinases are involved in PV generation. First, seven inhibitors target the cell surface receptor kinases EGFRK, PDGFRK, and NGFRK. C. burnetii interactions with these proteins likely occur during entry into a host cell. Thus, we did not study these kinases further as our goal was to identify components required for PV development following phagocytosis. Second, five inhibitors target PKC, a versatile protein with diverse activities in eukaryotic systems. Twelve PKC isoforms have been described, with each regulating distinct events via phosphorylation of downstream targets including MARCKs and GSK-3β (Gonelli et al., 2009). PKC also localizes to phagosomal compartments (Ng Yan Hing et al., 2004; Parent et al., 2009). Thus, it is intriguing to predict that C. burnetii proteins may interact with PKC at the PV membrane. Intracellular pathogens often target their proteins to the vacuolar membrane surface for efficient interaction with host proteins (Ragaz et al., 2008; Ninio et al., 2009). Indeed, C. burnetii produces two secreted effector proteins, AnkN and AnkO, that decorate the PV membrane when ectopically expressed in eukaryotic cells (Voth et al., 2009). However, whether AnkN and/or AnkO interact with PKC or another host protein(s) remains to be determined.
C. burnetii modulates PKC activity during infection as evidenced by downstream substrate phosphorylation profiles. Total PKC substrate phosphorylation increases at 24 hpi, indicating a post-phagocytosis role for the kinase. In agreement with this finding, the PKC-specific substrate MARCKs is phosphorylated throughout infection (up to 5 dpi), indicating prolonged manipulation of this signaling cascade during intracellular growth. These sustained effects are similar to our previous results regarding prolonged activation of the pro-survival kinases Akt and Erk1/2 (Voth and Heinzen, 2009b).
Currently, little is known regarding distinct mechanisms by which virulent C. burnetii causes Q fever and avirulent bacteria are cleared from immunocompetent hosts. However, inside a host cell, virulent and avirulent C. burnetii traffic to similar, if not identical, compartments. A recent study showed that phase I and phase II isolate PV exhibit similar development kinetics and fuse with phagolysosomes (Howe et al., 2010). Both vacuoles are also degradative as evidenced by destruction of Escherichia coli in the PV, further confirming that the avirulent C. burnetii model is representative of a virulent isolate infection. In agreement with these findings, both avirulent and virulent C. burnetii, including acute and chronic disease isolates, trigger sustained activation of PKC. Furthermore, virulent C. burnetii isolates activate p38 with similar kinetics to avirulent phase II organisms (Voth, unpublished results). These results indicate that many C. burnetii signaling activities are conserved among pathotypes.
The third category of kinases involved in PV generation includes proteins that participate in host calcium and cAMP signaling. Inhibition of PKA, CaMKII, or MLCK results in defective vacuole expansion and decreased bacterial replication. PKA is regulated by cAMP levels and controls activation of downstream components including CREB, GSK-3β, and nuclear factor-κB (Pearce et al., 2010). Nothing is known regarding C. burnetii interaction with PKA or second messenger molecules. However, other intracellular pathogens, such as Brucella suis, modulate PKA activity during infection (Gross et al., 2003). CaMKII and MLCK are calmodulin-activated kinases involved in cellular functions such as cytoskeletal regulation (Bayer and Schulman, 2001). CaMKII has been implicated in M. tuberculosis infection whereby the kinase controls delivery of infectious organisms to lysosomes where they are destroyed (Malik et al., 2001). It is unlikely that CaMKII or MLCK are solely needed for delivery of C. burnetii to a phagolysosome, as the pathogen resides in CD63-positive vacuoles following inhibitor treatment. However, the PV decorates with actin and actin-related proteins (Aguilera et al., 2009); thus, CaMKII and MLCK may regulate PV expansion and/or trafficking in the cell.
One inhibitor tested, pentamidine, targets the phosphatase PRL1 and has recently been employed to examine the mechanism of C. burnetii intron activity. Minnick et al. (2010) showed that pentamidine reduces pathogen growth in host cells by ∼73%. Pentamidine is an anti-fungal and anti-protozoal agent used to treat Pneumocystis carinii and trypanosome infections (Soeiro et al., 2008; Cushion and Walzer, 2009) and is predicted to act on two C. burnetii self-splicing group I introns (Minnick et al., 2010). Likewise, our current results show that pentamidine antagonizes PV biogenesis and reduces C. burnetii growth. However, the role of PRL1 in PV formation is unknown. Interestingly, the protein has been implicated in cancer cell tumorigenicity and down-regulation of pro-apoptotic p53 (Min et al., 2009) leading to sustained cell viability. Thus, it is intriguing to predict that C. burnetii requires functional PRL1 to promote host cell survival.
Collectively, our current results underscore the importance of intracellular host signaling in C. burnetii infection. Many kinases in our study have been targeted in clinical trials as disease therapeutics. For example, PKC inhibitors have been used to combat ischemia-related heart conditions and pancreatic cancer (Budas et al., 2007; El-Rayes et al., 2008). Therefore, host kinases may represent targets for therapies designed to combat intracellular parasitic infections. At the cellular level, we have identified novel components needed for C. burnetii host cell parasitism that can be targeted for further dissection of pathogenic mechanisms. It is also important to identify pathogen proteins that modulate host kinase activity. These bacterial proteins are likely delivered to the host cytosol by the Dot/Icm type IV secretion system (Voth and Heinzen, 2009a). Numerous C. burnetii Dot/Icm substrates have been identified (Pan et al., 2008; Voth et al., 2009) and can now be tested for their impact on host signaling pathway activation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Robert Heinzen for critical reading of the manuscript. This research was supported by funding to Daniel E. Voth from the American Heart Association (BGIA3080001), NIH/NIAID (K22AI081753 and R01AI087669), and the Arkansas Biosciences Institute.
References
Aguilera, M., Salinas, R., Rosales, E., Carminati, S., Colombo, M. I., and Beron, W. (2009). Actin dynamics and Rho GTPases regulate the size and formation of parasitophorous vacuoles containing Coxiella burnetii. Infect. Immun. 77, 4609–4620.
Bayer, K. U., and Schulman, H. (2001). Regulation of signal transduction by protein targeting: the case for CaMKII. Biochem. Biophys. Res. Commun. 289, 917–923.
Bhavsar, A. P., Guttman, J. A., and Finlay, B. B. (2007). Manipulation of host-cell pathways by bacterial pathogens. Nature 449, 827–834.
Brumell, J. H., and Scidmore, M. A. (2007). Manipulation of rab GTPase function by intracellular bacterial pathogens. Microbiol. Mol. Biol. Rev. 71, 636–652.
Buchholz, K. R., and Stephens, R. S. (2007). The extracellular signal-regulated kinase/mitogen-activated protein kinase pathway induces the inflammatory factor interleukin-8 following Chlamydia trachomatis infection. Infect. Immun. 75, 5924–5929.
Budas, G. R., Churchill, E. N., and Mochly-Rosen, D. (2007). Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol. Res. 55, 523–536.
Carey, K. L., Westwood, N. J., Mitchison, T. J., and Ward, G. E. (2004). A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 101, 7433–7438.
Coleman, S. A., Fischer, E. R., Howe, D., Mead, D. J., and Heinzen, R. A. (2004). Temporal analysis of Coxiella burnetii morphological differentiation. J. Bacteriol. 186, 7344–7352.
Cushion, M. T., and Walzer, P. D. (2009). Preclinical drug discovery for new anti-pneumocystis compounds. Curr. Med. Chem. 16, 2514–2530.
De La Fuente, R., Sonawane, N. D., Arumainayagam, D., and Verkman, A. S. (2006). Small molecules with antimicrobial activity against E. coli and P. aeruginosa identified by high-throughput screening. Br. J. Pharmacol. 149, 551–559.
Delsing, C. E., and Kullberg, B. J. (2008). Q fever in the Netherlands: a concise overview and implications of the largest ongoing outbreak. Neth. J. Med. 66, 365–367.
Deretic, V. (2008). Autophagy, an immunologic magic bullet: Mycobacterium tuberculosis phagosome maturation block and how to bypass it. Future Microbiol. 3, 517–524.
El-Rayes, B. F., Ali, S., Philip, P. A., and Sarkar, F. H. (2008). Protein kinase C: a target for therapy in pancreatic cancer. Pancreas 36, 346–352.
Gonelli, A., Mischiati, C., Guerrini, R., Voltan, R., Salvadori, S., and Zauli, G. (2009). Perspectives of protein kinase C (PKC) inhibitors as anti-cancer agents. Mini Rev. Med. Chem. 9, 498–509.
Gross, A., Bouaboula, M., Casellas, P., Liautard, J. P., and Dornand, J. (2003). Subversion and utilization of the host cell cyclic adenosine 5′-monophosphate/protein kinase A pathway by Brucella during macrophage infection. J. Immunol. 170, 5607–5614.
Gschwendt, M., Muller, H. J., Kielbassa, K., Zang, R., Kittstein, W., Rincke, G., and Marks, F. (1994). Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun. 199, 93–98.
Hackstadt, T. (1986). Antigenic variation in the phase I lipopolysaccharide of Coxiella burnetii isolates. Infect. Immun. 52, 337–340.
Howe, D., and Mallavia, L. P. (2000). Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A.1 cells. Infect. Immun. 68, 3815–3821.
Howe, D., Melnicakova, J., Barak, I., and Heinzen, R. A. (2003). Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cell. Microbiol. 5, 469–480.
Howe, D., Shannon, J. G., Winfree, S., Dorward, D. W., and Heinzen, R. A. (2010). Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect. Immun. 78, 3465–3474.
Hung, D. T., Shakhnovich, E. A., Pierson, E., and Mekalanos, J. J. (2005). Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310, 670–674.
Isberg, R. R., O’Connor, T. J., and Heidtman, M. (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24.
Jayaswal, S., Kamal, M. A., Dua, R., Gupta, S., Majumdar, T., Das, G., Kumar, D., and Rao, K. V. (2010). Identification of host-dependent survival factors for intracellular Mycobacterium tuberculosis through an siRNA screen. PLoS Pathog. 6, e1000839. doi: 10.1371/journal.ppat.1000839.
Knodler, L. A., Celli, J., and Finlay, B. B. (2001). Pathogenic trickery: deception of host cell processes. Nat. Rev. Mol. Cell Biol. 2, 578–588.
Knodler, L. A., Finlay, B. B., and Steele-Mortimer, O. (2005). The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J. Biol. Chem. 280, 9058–9064.
Lieberman, L. A., and Higgins, D. E. (2009). A small-molecule screen identifies the antipsychotic drug pimozide as an inhibitor of Listeria monocytogenes infection. Antimicrob. Agents Chemother. 53, 756–764.
Luhrmann, A., and Roy, C. R. (2007). Coxiella burnetii inhibits activation of host cell apoptosis through a mechanism that involves preventing cytochrome c release from mitochondria. Infect. Immun. 75, 5282–5289.
Malik, Z. A., Iyer, S. S., and Kusner, D. J. (2001). Mycobacterium tuberculosis phagosomes exhibit altered calmodulin-dependent signal transduction: contribution to inhibition of phagosome–lysosome fusion and intracellular survival in human macrophages. J. Immunol. 166, 3392–3401.
Marrie, T. J., and Raoult, D. (2002). Update on Q fever, including Q fever endocarditis. Curr. Clin. Top. Infect. Dis. 22, 97–124.
Min, S. H., Kim, D. M., Heo, Y. S., Kim, Y. I., Kim, H. M., Kim, J., Han, Y. M., Kim, I. C., and Yoo, O. J. (2009). New p53 target, phosphatase of regenerating liver 1 (PRL-1) downregulates p53. Oncogene 28, 545–554.
Minnick, M. F., Hicks, L. D., Battisti, J. M., and Raghavan, R. (2010). Pentamidine inhibits Coxiella burnetii growth and 23S rRNA intron splicing in vitro. Int. J. Antimicrob. Agents 36, 380–382.
Ng Yan Hing, J. D., Desjardins, M., and Descoteaux, A. (2004). Proteomic analysis reveals a role for protein kinase C-alpha in phagosome maturation. Biochem. Biophys. Res. Commun. 319, 810–816.
Ninio, S., Celli, J., and Roy, C. R. (2009). A Legionella pneumophila effector protein encoded in a region of genomic plasticity binds to Dot/Icm-modified vacuoles. PLoS Pathog. 5, e1000278. doi: 10.1371/journal.ppat.1000278.
Pan, X., Luhrmann, A., Satoh, A., Laskowski-Arce, M. A., and Roy, C. R. (2008). Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science 320, 1651–1654.
Parent, N., Winstall, E., Beauchemin, M., Paquet, C., Poirier, G. G., and Bertrand, R. (2009). Proteomic analysis of enriched lysosomes at early phase of camptothecin-induced apoptosis in human U-937 cells. J. Proteomics 72, 960–973.
Pearce, L. R., Komander, D., and Alessi, D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22.
Qin, Q. M., Pei, J., Ancona, V., Shaw, B. D., Ficht, T. A., and de Figueiredo, P. (2008). RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1alpha in supporting Brucella replication. PLoS Pathog. 4, e1000110. doi: 10.1371/journal.ppat.1000110.
Ragaz, C., Pietsch, H., Urwyler, S., Tiaden, A., Weber, S. S., and Hilbi, H. (2008). The Legionella pneumophila PtdIns(4)P-binding Icm/Dot substrate SidC recruits ER vesicles to a replication-permissive vacuole. Cell. Microbiol.10, 2416–2433.
Raoult, D., Marrie, T., and Mege, J. (2005). Natural history and pathophysiology of Q fever. Lancet Infect. Dis. 5, 219–226.
Reyland, M. E. (2009). Protein kinase C isoforms: multi-functional regulators of cell life and death. Front. Biosci. 14, 2386–2399.
Schimmer, B., Dijkstra, F., Vellema, P., Schneeberger, P. M., Hackert, V., ter Schegget, R., Wijkmans, C., van Duynhoven, Y., and van der Hoek, W. (2009). Sustained intensive transmission of Q fever in the south of the Netherlands, 2009. Eurosurveillance 14, 1–3.
Schneeberger, P. M., Hermans, M. H., van Hannen, E. J., Schellekens, J. J., Leenders, A. C., and Wever, P. C. (2010). Real-time PCR with serum samples is indispensable for early diagnosis of acute Q Fever. Clin. Vaccine Immunol. 17, 286–290.
Soeiro, M. N., de Castro, S. L., de Souza, E. M., Batista, D. G., Silva, C. F., and Boykin, D. W. (2008). Diamidine activity against trypanosomes: the state of the art. Curr. Mol. Pharmacol. 1, 151–161.
Tapinos, N., and Rambukkana, A. (2005). Insights into regulation of human Schwann cell proliferation by Erk1/2 via a MEK-independent and p56Lck-dependent pathway from leprosy bacilli. Proc. Natl. Acad. Sci. U.S.A. 102, 9188–9193.
Tse, H. M., Josephy, S. I., Chan, E. D., Fouts, D., and Cooper, A. M. (2002). Activation of the mitogen-activated protein kinase signaling pathway is instrumental in determining the ability of Mycobacterium avium to grow in murine macrophages. J. Immunol. 168, 825–833.
Vazquez, C. L., and Colombo, M. I. (2009). Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 17, 421–438.
Voth, D. E., and Heinzen, R. A. (2007). Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell. Microbiol. 9, 829–840.
Voth, D. E., and Heinzen, R. A. (2009a). Coxiella type IV secretion and cellular microbiology. Curr. Opin. Microbiol. 12, 74–80.
Voth, D. E., and Heinzen, R. A. (2009b). Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect. Immun. 77, 205–213.
Voth, D. E., Howe, D., Beare, P. A., Vogel, J. P., Unsworth, N., Samuel, J. E., and Heinzen, R. A. (2009). The Coxiella burnetii ankyrin repeat domain-containing protein family is heterogeneous, with C-terminal truncations that influence Dot/Icm-mediated secretion. J. Bacteriol. 191, 4232–4242.
Keywords: Coxiella burnetii, intracellular, vacuole, kinase, signaling, phosphorylation
Citation: Hussain SK, Broederdorf LJ, Sharma UM and Voth DE (2010) Host kinase activity is required for Coxiella burnetii parasitophorous vacuole formation. Front. Microbio. 1:137. doi: 10.3389/fmicb.2010.00137
Received: 01 November 2010;
Paper pending published: 15 November 2010;
Accepted: 04 December 2010;
Published online: 23 December 2010.
Edited by:
Rey Carabeo, Imperial College London, UKReviewed by:
Jason A. Carlyon, Virginia Commonwealth University School of Medicine, USASanjeev K. Sahni, University of Rochester School of Medicine and Dentistry, USA
Copyright: © 2010 Hussain, Broederdorf, Sharma and Voth. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Daniel E. Voth, Department of Microbiology and Immunology, University of Arkansas for Medical Sciences, 4301 W. Markham Street, Little Rock, AR 72205, USA. e-mail:ZHZvdGhAdWFtcy5lZHU=