 Rexford Asare
Rexford Asare Yousef Abu Kwaik*
Yousef Abu Kwaik*
- Department of Microbiology and Immunology, School of Medicine, University of Louisville, Louisville, KY, USA
Francisella tularensis is an intracellular bacterium that infects humans and many small mammals. During infection, F. tularensis replicates predominantly in macrophages but also proliferate in other cell types. Entry into host cells is mediate by various receptors. Complement-opsonized F. tularensis enters into macrophages by looping phagocytosis. Uptake is mediated in part by Syk, which may activate actin rearrangement in the phagocytic cup resulting in the engulfment of F. tularensis in a lipid raft rich phagosome. Inside the host cells, F. tularensis resides transiently in an acidified late endosome-like compartment before disruption of the phagosomal membrane and escape into the cytosol, where bacterial proliferation occurs. Modulation of phagosome biogenesis and escape into the cytosol is mediated by the Francisella pathogenicity island-encoded type VI-like secretion system. Whilst inside the phagosome, F. tularensis temporarily induce proinflammatory cytokines in PI3K/Akt-dependent manner, which is counteracted by the induction of SHIP that negatively regulates PI3K/Akt activation and promotes bacterial escape into the cytosol. Interestingly, F. tularensis subverts CD4 T cells-mediated killing by inhibiting antigen presentation by activated macrophages through ubiquitin-dependent degradation of MHC II molecules on activated macrophages. In the cytosol, F. tularensis is recognized by the host cell inflammasome, which is down-regulated by F. tularensis that also inhibits caspase-1 and ASC activity. During late stages of intracellular proliferation, caspase-3 is activated but apoptosis is delayed through activation of NF-κB and Ras, which ensures cell viability.
Infection by Francisella Tularensis
Tularemia is a zoonotic disease caused by Francisella tularensis, a facultative intracellular pathogen that infects a broad range of small mammals and humans (Ellis et al., 2002; Pechous et al., 2009; Santic et al., 2010a). Four subspecies of F. tularensis have been identified to date (Keim et al., 2007; Nigrovic and Wingerter, 2008; Santic et al., 2009) and they share about 97% genomic identity (Champion et al., 2009; Larsson et al., 2009). These are subspecies tularensis, holarctica, mediasiatica, and novicida. Disease in humans is mostly caused by subspecies tularensis and holarctica. Subspecies tularensis is found in North America and is the most virulent, causing the most severe form of tularemia. In contrast subspecies holarctica is distributed throughout the northern hemisphere and causes a mild form of tularemia (Santic et al., 2006). Subspecies novicida does not cause disease in humans but causes a disease in mice that is similar to the disease in humans.
Francisella tularensis is transmitted to humans through inhalation of contaminated aerosol or ingestion of contaminated food and water, a bite by an arthropod vector, or direct contact with infected animals through skin abrasions (Ellis et al., 2002). Clinical presentation of disease depends on the route of infection and include pneumonic tularemia, oropharyngeal tularemia, and glandular or ulceroglandular tularemia (Ellis et al., 2002). Occasionally, F. tularensis can also infect the eye resulting in oculoglandular tularemia (Harrell and Whitaker, 1985). Ulceroglandular tularemia is characterized by an ulcer at the infected site with swelling of the regional lymph node. Glandular tularemia is similar to ulceroglandular but without the ulcer. In oropharyngeal tularemia the ulcer occurs in the mouth with swelling of the lymph nodes around the neck region. Irrespective of the route of infection the bacteria ultimately enter the blood stream, causing typhoidal tularemia, which leads to septicemia (Oyston et al., 2004; Nigrovic and Wingerter, 2008). Symptoms of the typhoidal tularemia include headache, fever, chills, nausea, diarrhea, and myalgia (Oyston et al., 2004; Nigrovic and Wingerter, 2008). Due to the high morbidity and mortality rate, the ease of dissemination and the fact that inhalation of as few as 10 organisms of subspecies tularensis can cause disease, F. tularensis has been classified by the CDC as a category A select agent.
Once inside the mammalian host, F. tularensis enters and replicates in macrophages (Anthony et al., 1991; Conlan and North, 1992; Fortier et al., 1994). However, there is increasing evidence that the organism can infect other cell types including neutrophils, dendritic cells, hepatocytes, and lung epithelial cells (Pechous et al., 2009). During infection, bacteria migrate from the initial site of infection to the liver and spleen where they replicate (Eigelsbach et al., 1962; Conlan et al., 2003). Although it has been shown that F. tularensis exhibit extracellular phase during in vivo infection in mice (Forestal et al., 2007; Yu et al., 2008), there is no data demonstrating extracellular growth during human or animal infection.
Available data indicate that intracellular trafficking of F. tularensis is similar in macrophages, neutrophils, epithelial cells, and Drosophila melanogaster S2 cells suggesting trafficking might be similar in all cell types (Golovliov et al., 2003a; Clemens et al., 2004; Santic et al., 2005a; McCaffrey and Allen, 2006; Craven et al., 2008; Santic et al., 2009). F. tularensis enters into host cells through binding to surface receptors. This results in the uptake of the bacterium in a spacious loop by a mechanism referred to as looping phagocytosis (Clemens et al., 2005). Uptake by neutrophils and dendritic cells is dependent on opsonization (Lofgren et al., 1983; Ben Nasr et al., 2006) whereas entry into macrophages is through both opsonin dependent and independent mechanisms (Clemens et al., 2005; Balagopal et al., 2006; Pierini, 2006; Schulert and Allen, 2006; Barel et al., 2008). Inside the host cell, the bacteria reside transiently in a phagosome before escaping into the cytosol (Figure 1; Golovliov et al., 2003b; Clemens et al., 2004; Santic et al., 2005a,b; Checroun et al., 2006; Santic et al., 2007; Bonquist et al., 2008; Santic et al., 2008; Qin et al., 2009). Escape is preceded by modification of the phagosome to an acidified late endosome-like compartment (Fortier et al., 1995; Chong et al., 2008; Santic et al., 2008). Within this acidified compartment F. tularensis activates virulence genes that allow it to disrupt the phagosome membrane and escape into the cytosol (Chong et al., 2008; Santic et al., 2008).
Figure 1. Intracellular trafficking of Francisella tularensis within macrophages. F. tularensis enters macrophages using different receptors and resides transiently in the FCP, which acquires EE1 followed Lamp-1, Lamp-2, and Rab7 but excludes Cathepsin D. Within 30 min of infection the FCP acquires vATPase enabling F. tularensis to acidify the FCP and escape into the cytosol. Within the cytosol, F. tularensis activates caspase-1 and caspase-3 but delays pyropoptosis and apoptosis and maintain cell viability till late stages of infection when the bacteria exit the spent cell.
Once inside the cytosol, the bacteria is recognized by the host cell inflammasome resulting in the cleavage of IL-1 and IL-18 (Figure 1; Mariathasan et al., 2005; Gavrilin et al., 2006; Henry et al., 2007; Fernandes-Alnemri et al., 2010; Jones et al., 2010). Similarly, there is activation of caspase-3 through both the extrinsic and intrinsic pathways between 6 and 12 post-infection. Although caspases are activated early during infection (Lai and Sjostedt, 2003; Mariathasan et al., 2005; Santic et al., 2010b), F. tularensis is able to delay death of the cells for its survival and replication by activating NF-κB and Ras both of which stimulate cells survival (Al-Khodor and Abu Kwaik, 2010; Santic et al., 2010b). During late stages of infection of mouse macrophages, F. tularensis is taken up in an autophagy-like compartment (Checroun et al., 2006). However, this re-entry of the F. tularensis into the endosomal–lysosomal pathway through autophagy does not occur in human macrophages, and therefore is not relevant to infection of humans (Akimana et al., 2010). Toward the end of the infectious cycle, the induction of apoptosis allows the bacteria to disrupt the cytoplasmic membrane and escape the spent cell to begin new infectious cycle (Figure 1).
Entry into and Replication within Host Cells
Francisella tularensis enters primary macrophages through both opsonin dependent and independent mechanisms. Complement-opsonized bacteria enter macrophages either through complement receptor 3 (CR3) or the scavenger receptor A (SRA)(Clemens et al., 2005; Pierini, 2006). Antibody-opsonized F. tularensis enters macrophages through FC gamma receptor (Balagopal et al., 2006) in contrast to unopsonized bacteria that enter macrophages through binding to the mannose receptor and surface nucleolin (Balagopal et al., 2006; Schulert and Allen, 2006; Barel et al., 2008). It has also been shown that opsonization of F. tularensis with lung collectin surfactant protein A (SP-A) enhance bacterial uptake by primary macrophages but the host cell receptor is not known (Balagopal et al., 2006). Similarly, the bacterial ligand for mannose receptor has not been identified. Elongation factor E2 is expressed on the surface of F. tularensis and bind to surface nucleolin expressed on the surface of macrophages (Barel et al., 2008). It is however not known which of these receptors are used predominantly in vivo.
When opsonized F. tularensis binds macrophages, it is engulfed in a unique asymmetric spacious pseudopod loops (Clemens et al., 2005). This unique mechanism of uptake has been shown to be dependent on intact CR3 and complement factor 3 (Clemens et al., 2005). Syk is important for Fcγ-mediated phagocytosis in macrophages and neutrophils (Greenberg et al., 1994; Raeder et al., 1999). Activation of Syk results in the activation of MAP kinase (ERKs) through Protein kinase C (PKC) leading to actin polymerization and induction of phagocytosis (Cox et al., 1996; Raeder et al., 1999). Syk has been shown to be important for the uptake of F. tularensis but the upstream receptor required for activation of Syk has not been identified (Parsa et al., 2008). Activation of Syk leads to subsequent activation of the Erk pathway but the direct binding partner of Syk is yet to be identified (Figure 2; Parsa et al., 2008). Interestingly, actin microfilament has been shown to be important for this process (Clemens et al., 2005).
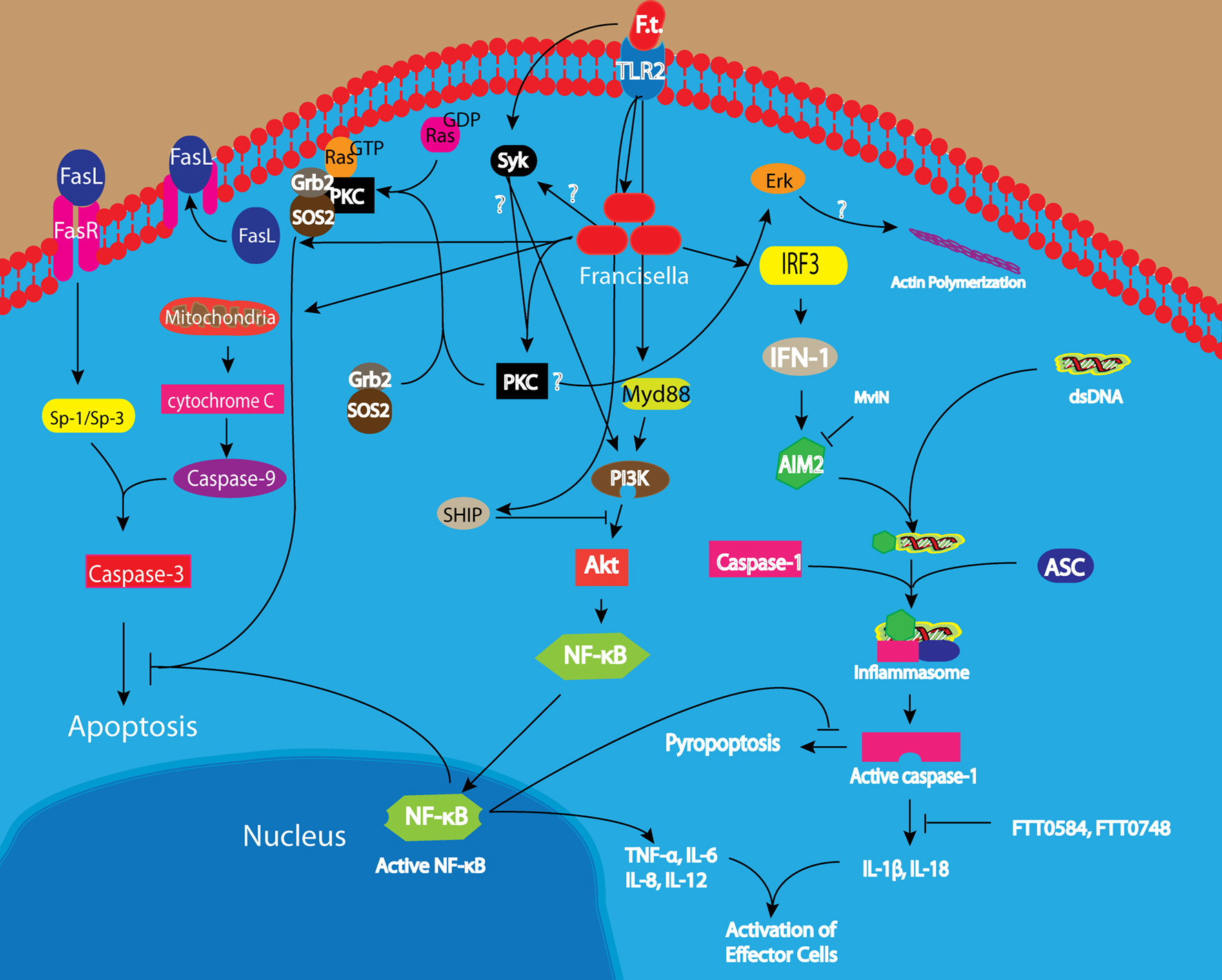
Figure 2. Entry into and evasion of host cell innate immune response by Francisella tularensis. Phagocytosis of F. tularensis by macrophages is mediated by Syk-dependent activation of Erk, which likely triggers actin polymerization at the phagocytic cup. In addition, there is TLR2 dependent activation of Akt leading to induction of proinflammatory cytokines and phagosomal maturation. Akt activation is counteracted by SHIP activation, but the balance between the two opposing process is tilted toward escape of F. tularensis into the cytosol. Within the cytosol, F. tularensis activates both caspase-1 and caspase-3 but it is able to delay induction of apoptosis and pyropoptosis through Ras and NF-κB dependent anti-apoptotic mechanisms as well as AIM2-dependent inhibition of caspase-1 activation.
In contrast to other intracellular bacteria such as Salmonella typhimurium, which requires PI3K to form the phagocytic cup, the uptake of F. tularensis is not affected by inhibition of the PI3K pathway (Parsa et al., 2006, 2008). This is consistent with a different mechanism used by F. tularensis to enter into host cells (Clemens et al., 2005). In addition to actin microfilament, the entry of F. tularensis into macrophages has been shown to be dependent on cholesterol-rich lipid domains known as lipid rafts since lipid rafts-associated components such as cholesterol and caveolin-1 are incorporated into the Francisella-containing phagosome (FCP) membrane upon its biogenesis from the macrophage plasma membrane (Tamilselvam and Daefler, 2008). The recruitment of lipid rafts to the FCP may act as a platform for linking the entry process of F. tularensis at the cell membrane to the cytoskeleton and the intracellular signaling pathways (Tamilselvam and Daefler, 2008).
To date, at least 268 gene products have been identified, that are important for replication of F. tularensis within mammalian cells (Table 1; Anthony et al., 1994; Baron and Nano, 1998; Gray et al., 2002; Golovliov et al., 2003a; Nano et al., 2004; Santic et al., 2005b, 2007; Twine et al., 2005; Deng et al., 2006; Pechous et al., 2006, 2008; Tempel et al., 2006; Charity et al., 2007; de Bruin et al., 2007; Maier et al., 2007; Mohapatra et al., 2007a,b, 2008; Raynaud et al., 2007; Bonquist et al., 2008; Brotcke and Monack, 2008; Fuller et al., 2008; Meibom et al., 2008; Sammons-Jackson et al., 2008; Alkhuder et al., 2009; Buchan et al., 2009; Dean et al., 2009; Mahawar et al., 2009; Santiago et al., 2009; Schulert et al., 2009; Ahlund et al., 2010; Asare and Abu Kwaik, 2010; Jia et al., 2010; Sen et al., 2010). These include the Francisella pathogenicity Island (FPI) proteins IglA, IglB, IglC, IglD, pdpA, pdpB, pdpD and their regulators, MglA, SspA, FevR, MigR, RipA, PigR, and PmrA (Baron and Nano, 1998; Gray et al., 2002; Golovliov et al., 2003a; Charity et al., 2007, 2009; de Bruin et al., 2007; Mohapatra et al., 2007b; Bonquist et al., 2008; Brotcke and Monack, 2008; Fuller et al., 2008; Buchan et al., 2009). The FPI is composed of 17 genes and recent mutagenesis experiments have shown that most of the genes are important for survival within the host cell (Golovliov et al., 2003a; de Bruin et al., 2007; Barker et al., 2009; Broms et al., 2009; Schmerk et al., 2009). Some of the gene products on the FPI form a type VI-like secretion system through which effector proteins are injected into the host cell cytosol to modulate biogenesis of the FCP and to enable the bacterium to disrupt the phagosome membrane and escape into the cytosol (Golovliov et al., 2003a; Santic et al., 2007; Barker et al., 2009; Broms et al., 2009; Schmerk et al., 2009). MglA, SspA, and PmrA bind cooperatively with RNA polymerase to regulate a large number of genes including those of the FPI (Brotcke et al., 2006; Charity et al., 2007; Mohapatra et al., 2007b; Bell et al., 2010) that are important for survival within the host cell. This regulation is mediated in part by FevR which is important for escape and replication within the cytosol (Brotcke and Monack, 2008). FevR is also independently regulated by MigR (Buchan et al., 2009) indicating that FevR plays a central role in the regulation of virulence in F. tularensis. Independent of FevR, MglA, and SspA also regulate virulence genes through cooperative interaction with PigR and the alamone ppGpp (Charity et al., 2009). Whereas most of the genes regulated by the different pathways are common, there are subsets of genes that are regulated independently by the different pathways (Brotcke et al., 2006; Charity et al., 2007; Mohapatra et al., 2007b). A large number of these gene products and most of the proteins that are necessary for escape and replication are hypothetical proteins. It is conceivable to speculate that some of these gene products constitute effector proteins that are secreted by the type VI secretion-like system. Also important for intracellular replication are genes involved in the transport of metabolic intermediates and different metabolic pathways including amino acid metabolism, nucleotide metabolism, and carbohydrate metabolism (Pechous et al., 2006; Alkhuder et al., 2009; Mahawar et al., 2009; Schulert et al., 2009; Asare and Abu Kwaik, 2010). The large number of metabolic genes that is required for replication indicates that the FCP is replete of nutrients and F. tularensis require de novo synthesis in order to survive and replicate within the host cells. Once in the cytosol where nutrient is readily available F. tularensis may acquire nutrients through the importation of metabolic intermediates from the host cell cytosol. This may explain why mutations in a large number of metabolic intermediate transporters block bacterial escape into the cytosol (Table 1; Qin and Mann, 2006; Maier et al., 2007; Asare and Abu Kwaik, 2010).

Table 1. List of intracellular growth defective mutants.
Modulation of Phagosome Biogenesis
Phagosomal maturation involves sequential interaction between the nascent phagosome and the endocytic and lysosomal vesicles resulting in the conversion of the phagosome to a phagolysosome within which the bacterium or a particle is degraded (Duclos and Desjardins, 2000; Hackstadt, 2000; Kahn et al., 2002). After biogenesis from the plasma membrane, the nascent phagosome fuses with vesicles from the early endosome in a process that is regulated by Rab5 GTPase and the downstream effector early endosomal antigen 1 (EEA1). This is followed by interaction with the late endosome that is controlled by Rab7 GTPase. The late endosome-like phagosome becomes acidified through acquisition of the vacuolar ATPase, which pumps protons into the lumen of the phagosome resulting in acidification of the lumen. The acidified phagosome subsequently fuses to the lysosomes to form a phagolysosome, which is very rich in acid hydrolases. Within this compartment the microbe or particle is degraded (Duclos and Desjardins, 2000; Hackstadt, 2000; Kahn et al., 2002; Figure 1). The maturation process is very rapid and is completed within 15–30 min of phagosome biogenesis from the plasma membrane (Duclos and Desjardins, 2000; Hackstadt, 2000; Kahn et al., 2002).
Different intracellular pathogens have evolved different mechanisms to subvert the default endocytic maturation to create permissive niches that allow intracellular replication (Duclos and Desjardins, 2000; Hackstadt, 2000; Kahn et al., 2002). The strategies include (i) Arrest of phagosome maturation at a distinct stage in the endosomal–lysosomal degradation pathway, as occurs in infection with Legionella pneumophila; (ii) Survival and replication within an acidic environment of a mature phagolysosomes, as exemplified by Coxiella burnetii; and (iii) Replication within the cytosol after degradation of the phagosomal membrane, as occurs in infection with Listeria monocytogenes and Shigella flexneri (Duclos and Desjardins, 2000; Hackstadt, 2000; Kahn et al., 2002). Although the endocytic maturation stage of phagosome harboring vacuolar pathogens has been classified into early or late endosome, maturation of phagosome harboring intracellular pathogens is aberrant and is not a classical full maturation of any of the defined endocytic stages (Santic et al., 2010a). For example, the Mycobacterium tuberculosis phagosome acquires Rab5 but lacks several downstream effectors of Rab5 that are present on mature early endosome. It also acquires procathepsin D, which is the immature form of the lysosomal enzyme cathepsin D (Sturgill-Koszycki et al., 1996; Derre and Isberg, 2004). Therefore, it might be more accurate to classify phagosome of intracellular vacuolar pathogens as early endosome-like or late endosome-like phagosome (Santic et al., 2010a).
The FCP transiently acquires the EEA1 followed by the acquisition of the late endosomal markers, Lamp1/2, Cd63, and Rab7 as well as the vacuolar ATPase, which acidifies the phagosome (Figure 1; Golovliov et al., 2003b; Clemens et al., 2004; Santic et al., 2005a,b, 2007, 2008; Checroun et al., 2006; Bonquist et al., 2008; Qin et al., 2009). The FCP does not however co-localize with the lysosomal acid hydrolase cathepsin D and the fluid face marker, lysotraker (Figure 1; Chong et al., 2008; Santic et al., 2008). Within 30–60 min, the bacterium disrupts the phagosomal membrane and escapes into the host cell cytosol (Figure 1; Chong et al., 2008; Santic et al., 2008). Acidification of the vacuole is important for the ability of the bacteria to escape into the cytosol, since inhibition of the vATPase results in a delay in bacterial escape into the cytosol (Chong et al., 2008; Santic et al., 2008). F. tularensis has been shown to escape into the cytosol in different cell types including macrophages and neutrophils (Figure 1; Golovliov et al., 2003a; Clemens et al., 2004; Santic et al., 2005a; McCaffrey and Allen, 2006). It has not been determined if F. tularensis arrest phagosome maturation before escaping into cytosol or if the bacterium manages to escape before the phagosome fuses to the lysosome. However, trafficking of the migR and fevR mutants of F. tularensis in macrophages suggests that there is arrest of phagosome biogenesis prior to bacterial escape into the cytosol (Buchan et al., 2009). Comparison of trafficking of the migR and fevR mutants showed that whereas the fevR mutant is trapped in a LAMP1-positive compartment, the phagosome containing the migR mutant matures into a phagolysosome enriched in LAMP1 and cathepsin D (Buchan et al., 2009). Conversely, data from studies of F. tularensis trafficking in neutrophils suggest that a fraction of the phagosome of wild-type bacteria that are unable to escape end up in a phagolysosome. This may suggest that F. tularensis does not inhibit phagosome maturation in neutrophils but rather escape into the cytosol before the phagosome matures into a phagolysosome (McCaffrey and Allen, 2006). Interestingly, arrest in phagosome biogenesis and rapid escape of F. tularensis into the cytosol is also exhibited in arthropod vector-derived cells, indicating exploitation of conserved eukaryotic processes by F. tularensis to infect and proliferate within arthropod and mammalian cells (Santic et al., 2009).
Escape into the Cytosol
Like other intracellular pathogens, F. tularensis must overcome the host innate immune response to successfully colonize the intracellular niche. Since the primary host defense is centered on the antimicrobial properties of the phagosome, F. tularensis like other cytosolic bacteria escapes from the phagosome into the cytosol where it replicates (Goebel and Kuhn, 2000; Golovliov et al., 2003a; Clemens et al., 2004; Santic et al., 2005a; McCaffrey and Allen, 2006; Ray et al., 2009). In order to escape into the cytosol, the FCP transiently acquires the vacuolar ATPase, which acidifies the phagosome followed by rapid escape of F. tularensis to the cytosol (Figure 1; Golovliov et al., 2003b; Clemens et al., 2004; Santic et al., 2005a,b, 2007, 2008; Checroun et al., 2006; Bonquist et al., 2008; Qin et al., 2009). The acidification is important since inhibition of the vATPase by bafilomycin A delays escape of the bacterium into the host cell cytosol indicating that there is a factor involved in disruption of the phagosome that is expressed or activated at acidic pH. Between 15 and 30 min of residence in the phagosome in human macrophages, the bacteria begin to escape into the cytosol (Figure 1; Santic et al., 2010a). It is within the cytosol that the bacteria replicate (Figure 1). The mechanism by which the bacterium escapes into the cytosol is not well understood.
Unlike L. monocytogenes, a large number of genes have been shown to be important for escape of F. tularensis into the host cell cytosol (Table 2; Golovliov et al., 2003a; Santic et al., 2005b; Qin and Mann, 2006; Mohapatra et al., 2008; Barker et al., 2009; Broms et al., 2009; Buchan et al., 2009; Schmerk et al., 2009; Schulert et al., 2009; Asare and Abu Kwaik, 2010). Recent mutagenesis experiments have shown that most of the genes of the FPI that form the type VI-like secretion system, affect escape of the bacterium into the cytosol and subsequent replication (Golovliov et al., 2003a; de Bruin et al., 2007; Barker et al., 2009; Broms et al., 2009; Schmerk et al., 2009). In contrast, IglD has been shown to be important for replication of the bacteria within the cytosol without any effect on phagosomal escape of the bacterium (Santic et al., 2007). The FPI protein VgrG has been shown to be a component of the secretary system as well as a substrate of the system (Barker et al., 2009). Unlike VgrG, IglI is a substrate of the type VI secretion system with no effect on the secretion apparatus. Both genes are important for escape of F. tularensis into the host cell cytosol but the mechanism of action has not been elucidated (Barker et al., 2009). The FPI proteins IglA, IglC, and pdpA are also required for escape of F. tularensis into the host cells cytosol but it has not been determined if they are secreted substrates or component of the type VI-like secretion apparatus.
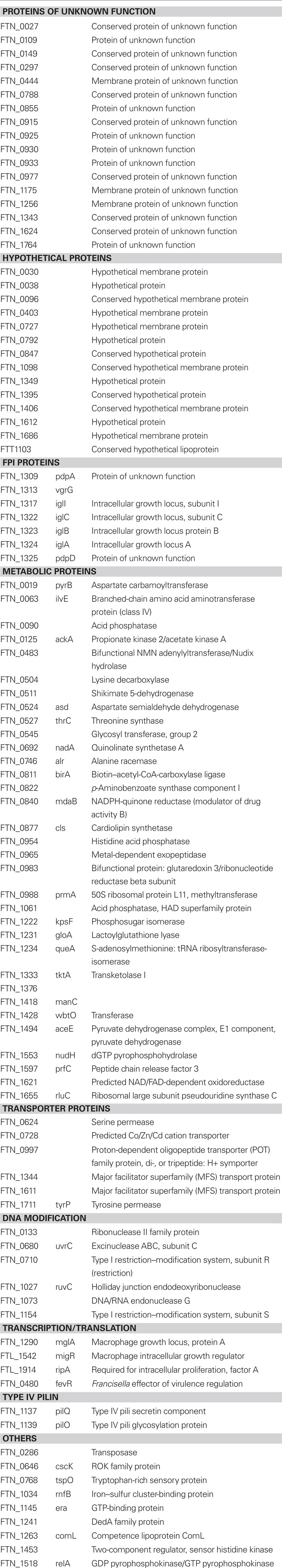
Table 2. List of escape defective mutants.
Mutations in MglA and FevR negatively affect the ability of F. tularensis to escape from the phagosome into the cytosol (Santic et al., 2005b; Bonquist et al., 2008; Buchan et al., 2009). In contrast, MigR mutant behaves similar to the IglD mutant, which escapes but is unable to replicate within the cytosol indicating that MigR regulate genes that are important for replication within the cytosol (Santic et al., 2007; Buchan et al., 2009). Other genes that play critical roles in the escape of bacteria into the host cell cytosol include genes involved in DNA modification, transcription and translation, type II secretion, metabolic genes as well as genes involved in the transport of nutrients (Schulert et al., 2009; Asare and Abu Kwaik, 2010).
Although hemolytic activity has been observed in F. tularensis subspecies novicida and F. philomiragia (Lai et al., 2003), no hemolysin homolog has been identified in all the sequenced Francisella genome to date including those of novicida and philomiragia. There are between four and eight acid phosphatases in the Francisella genome depending on the subspecies. There are eight acid Phosphatases in the subspecies novicida genome, four (AcpA, AcpB, AcpC, and HAP) of which are also found in the virulent subspecies tularensis genome (Mohapatra et al., 2008). AcpA has been shown to possess lipase activity (Mohapatra et al., 2007a), but all three Acp molecules are predicted to possess phosphoric ester hydrolase activity. Independent studies have shown that mutations in AcpA, AcpC, and HAP result in delay or inhibition of escape into the cytosol and reduced replication within human macrophages (Mohapatra et al., 2007a; Asare and Abu Kwaik, 2010), and that combined deletion of AcpA, AcpB, AcpC, and HAP results in complete inhibition of phagosomal escape and replication of subspecies novicida in the cytosol (Mohapatra et al., 2008). However, there is contradictory data on the role of these acid phosphatases in escape and intracellular replication. For example, Baron et al. (1999) have shown that AcpA in subspecies novicida is not important for replication within mouse macrophages. The difference between the role of AcpA in various subspecies may be due to the difference in the macrophages used, since trafficking of F. tularensis has been shown to be slightly different in mouse and human macrophages (Clemens et al., 2004; Checroun et al., 2006). Similarly, Child et al. (2010) have shown that combined deletion of AcpA-C does not affect the phagosomal escape or replication of the virulent subspecies tularensis within human macrophages. This indicates that there may be subtle differences in the mechanisms used by the different subspecies to escape into the host cell cytosol. There are numerous genes identified to be important for escape of F. tularensis that are designated as hypothetical proteins or proteins with unknown functions (Asare and Abu Kwaik, 2010). Some of these may be potential substrates for the type VI-like secretion system and identifying and characterizing them will help us to understand how F. tularensis modulates biogenesis of its phagosome and escape into the cytosol.
Modulation of Inflammatory Response to Infection by F. Tularensis
The transcription factor NF-κB is involved in the regulation of inflammation by activating the induction of different proinflammatory cytokines (Lawrence, 2009). NF-κB represents a family of homo and heterodimer transcription factors, and the p65/p50 heterodimer is the most predominant active complex in mammalian cells (Burstein and Duckett, 2003). In resting cells, NF-κB proteins are predominantly sequestered in the cytoplasm by the NF-κB inhibitory proteins (IκBs; Karin and Ben-Neriah, 2000). The IκB kinase mediates phosphorylation of IκBs, followed by ubiquitination and proteasomal degradation, which is crucial to the activation and nuclear translocation of NF-κB (Karin and Ben-Neriah, 2000).
Early during infection when F. tularensis is localized within the phagosome, it activates the inflammatory response in macrophages by inducing the secretion of TNF-α in TLR-2 dependent manner (Figure 2; Telepnev et al., 2005; Katz et al., 2006). Induction of TNF-α secretion is mediated by the PI3K/Akt pathway, which also leads to activation of NF-κB (Telepnev et al., 2003; Katz et al., 2006; Parsa et al., 2006; Rajaram et al., 2006). Activation of NF-κB results in the induction and secretion of proinflammatory cytokines that restrict the escape of F. tularensis from the phagosome into the cytosol and promotes fusion of the FCP with the lysosome (Figure 2; Rajaram et al., 2009). Concomitant with escape into the cytosol, F. tularensis down-regulates NF-κB activation and TNF-α, IL-6, IL-8, and IL-12 secretion within 5 h post-infection, since the IglC mutant which is unable to escape into the cytosol, does not down-regulate TNF-α, IL-6, IL-8, and IL-12 secretion (Figure 2; Telepnev et al., 2003, 2005).
The activation of PI3K/Akt pathway is negatively regulated by the Src homology 2 (SH2) domain-containing inositol-5′-phosphatase (SHIP) protein, since deficiency in SHIP expression results in enhanced Akt activation and NF-κB-driven transcription of proinflammatory cytokines, which promote fusion of the FCP with the Lysosome (Figure 2; Parsa et al., 2006; Rajaram et al., 2009). Conversely, over expression of SHIP leads to a decrease in NF-κB activation (Parsa et al., 2006). It is unknown how the delicate balance of Akt and SHIP activation is tilted toward SHIP promoted escape of F. tularensis into the cytosol. It will be interesting to determine how F. tularensis activates SHIP and the relations between SHIP activation and the disruption of the phagosome membrane that allow F. tularensis to escape into the cytosol (Figure 2). Once inside the cytosol F. tularensis induces Sp-1/Sp-3 dependent Fas expression that results in activation of caspase-3 and host cell death (Rajaram et al., 2009).
Cytosolic localization of F. tularensis in mouse macrophages results in type I interferon (IFN-I) and AIM2 dependent activation of the inflammasome (Figure 2; Mariathasan et al., 2005; Gavrilin et al., 2006; Henry et al., 2007; Fernandes-Alnemri et al., 2010; Jones et al., 2010). Cytosolic bacteria induce IRF-3 dependent activation of IFN-I, which in turn increases the expression of AIM2 (Figure 2; Jones et al., 2010). AIM2 recognize F. tularensis (Fernandes-Alnemri et al., 2010), likely through binding to dsDNA from the bacteria since F. tularensis DNA co-localizes with AIM2 (Jones et al., 2010). Upon binding to F. tularensis DNA, AIM2 forms a complex with the adapter protein ASC and caspase-1 known as the inflammasome (Mariathasan et al., 2005; Fernandes-Alnemri et al., 2010; Jones et al., 2010). Inflammasome activation by F. tularensis results in the formation of pyropoptosome, which is critical for innate host defense and leads to pyropoptotic death of infected cells and the concomitant release of the proinflammatory cytokines IL-1β and IL-18 (Figure 2; Mariathasan et al., 2005; Gavrilin et al., 2006). The F. tularensis lipid/polysaccharide (MOP) transporter protein, MviN, which is homologous to the E. coli putative lipid II flippase, has recently been shown to suppress the induction of AIM2 (Ulland et al., 2010), since a mutation in the gene results in increase induction of AIM2 inflammasome-dependent IL-1β secretion and cytotoxicity in macrophages (Ulland et al., 2010). In addition to MviN, two other genes FTT_0584, with no characterized orthologs, and FTT_0748, which is homologous to the IclR family of transcriptional regulators, have been shown to suppress caspase-1 and ASC dependent secretion of IL-1β, since mutations in these genes resulted in hyper secretion of IL-1β (Weiss et al., 2007). Since AIM2 is not present in human macrophages, inflammasome mediators are likely to be different from the one described for mouse macrophages.
Activation and Control of Host Cell Apoptosis
Between 6 and 12 h post-infection, F. tularensis induce caspase-3 activation within the host cells, which culminate in the induction of apoptosis (Lai and Sjostedt, 2003; Santic et al., 2010b). F. tularensis LVS induces apoptosis in the J774A.1 murine macrophage cell line through a pathway partly resembling the intrinsic apoptotic pathway (Lai and Sjostedt, 2003). The induction of apoptosis involves the release of mitochondrial cytochrome C into the cytosol with concomitant activation of caspase-9 and caspase-3 but not caspase-1, caspase-8, Bcl-2, or Bid (Lai and Sjostedt, 2003). In contrast, another study has shown that F. tularensis induces Sp-1/Sp-3 activation of Fas in RAW 264.7 murine macrophage cells line, which results in activation of caspase-3, suggesting that F. tularensis induce apoptosis through the extrinsic pathway (Figures 1 and 2; Rajaram et al., 2009). Interestingly, infection of murine macrophages by F. tularensis has been shown to induce apoptotic cell death through down-regulation of activation of p38 MAPK compared to uninfected cells, but the mechanism of induction is yet to be defined (Hrstka et al., 2005). Although caspase-3 activation occurs early during infection in non-activated macrophages, it is not until about 18 h post-infection before there is induction of apoptosis, which is likely due to triggering anti-apoptotic processes (Figures 1 and 2; Lai and Sjostedt, 2003; Al-Khodor and Abu Kwaik, 2010; Santic et al., 2010b).
Host Factors Required Intracellular Growth of F. Tularensis
NF-κB plays a crucial role in regulation of apoptosis by triggering expression of various anti-apoptotic genes (Burstein and Duckett, 2003). We have shown that in order to maintain viability of the infected cell and allow F. tularensis to survive, there is simultaneous activation of caspases and NF-κB creating a delicate balance between them to maintain cell viability that is necessary for proliferation of the bacterium. Activation of NF-κB involves IκB kinase-mediated phosphorylation of IκBs, followed by ubiquitination and proteasomal degradation (Karin and Ben-Neriah, 2000). Interestingly, in activated macrophages, F. tularensis elicit ubiquitin-dependent MHC class II down-regulation and degradation, thus compromising antigen presentation by macrophages to CD4 T cells (Wilson et al., 2009). It is not surprising that two ubiquitin proteins, the ubiquitin hydrolase USP22, and the ubiquitin ligase CDC27 has been shown to be important for replication of F. tularensis in human macrophages (Akimana et al., 2010).
It has been shown that F. tularensis triggers activation of Ras through the recruitment of PKCα and PKCβ-I to the SOS2/GrB2 complex (Figure 2; Al-Khodor and Abu Kwaik, 2010). Silencing of SOS2, GrB2, PKCα, and PKCβ-1 is associated with rapid early activation of caspase-3 but does not affect phosphorylation of Akt or Erk (Al-Khodor and Abu Kwaik, 2010). This indicates that F. tularensis utilizes two independent mechanisms to modulate caspase-3 activity in order to survive inside host cells till the terminal stages of infection when induction of apoptosis leads to cell lysis and release of bacteria to the extracellular milieu. The bacterial factor necessary for the activation of NF-κB and Ras are yet to be identified.
Concluding Remarks and Future Directions
Upon infection with F. tularensis, the host cells employ a myriad of arsenals to try to limit proliferation of the bacteria. The host cells activate signaling pathways to try to restrict escape of F. tularensis into the cytosol. Once the bacteria escape into the cytosol, a new arsenal is put into motion by the host cells through activation of caspase-1 and caspase-3, geared toward pyropoptosis and apoptosis of the infected cells. Concomitantly, there is activation of NF-κB geared toward triggering pro-survival signals and the induction of proinflammatory cytokines. Intuitively, F. tularensis has devised different strategies to counteract the innate host defense mechanisms. These include inhibition of components of the host defense mechanism and hijacking the cells own defense system and other signaling pathways through bacterial effectors that are likely exported through a type VI-like secretion system. For example, F. tularensis co-opts the host cell NF-κB transcription factor, which is used to activate proinflammatory cytokines, to induce the expression of anti-apoptotic genes to maintain cell viability. Similarly, F. tularensis co-opts the host cell Ras signaling pathway to inhibit caspase-3-induced apoptosis. Finally, F. tularensis utilizes the host cell ubiquitin-dependent proteasome degradation system to degrade MHC class II molecules on activated macrophages to inhibit antigen presentation to effector T cells.
Many virulence factors have been identified that are required for bacterial escape and replication within the cytosol. Although some of these are involved in known pathways, majority of these have no known functions. The roles of some of these factors in the virulence mechanisms exhibited by F. tularensis are beginning to be defined but the functions are largely unknown. Cytosolic F. tularensis activates PKC leading to activation of Ras and inhibition of apoptosis. MigR regulates genes that are important of phagosome biogenesis. Identifying and characterizing MigR-regulated genes will lead to an understanding of how F. tularensis arrest phagosome maturation. Delineating how MviN, FTT0584, and FTT0748 inhibit caspase-1 activity will shed light on how F. tularensis modulate pyropoptosis and proinflammatory cytokine induction. Since NF-κB is required for both cytokine induction and inhibition of apoptosis, its activation must be tightly controlled. It will be interesting to identify the bacterial factors important for activation of Ras and diversion of NF-κB to the expression of anti-apoptotic effectors. It is not known how F. tularensis tilts the balance of power between Akt and SHIP toward SHIP activation and bacterial escape and the mechanism by which this is achieved. Unlike L. monocytogenes, no hemolysin-like molecule have been identified in F. tularensis and there is contradictory data on role of AcpA in escape, which may be partly due to the studies being done using different species of Francisella and different sources of macrophages. It will be interesting to know if SHIP does not only inhibit cytokine activation but also activate a host cell factor that leads to disruption of the phagosome membrane and escape of the bacteria. It will be interesting to determine how F. tularensis modulate the ubiquitin ligase in activated macrophages leading to degradation of MHC II molecules and evasion of adaptive immunity. There is little doubt that F. tularensis employs various strategies to modulate cellular processes that have evolved to degrade invading microbes in addition to evasion of various innate and adaptive immune processes to inflict disease in the mammalian host. It is just as interesting to uncover the molecular and cellular bases of the interaction of F. tularensis with the arthropod vector and its role in pathogenic evolution and infection of the mammalian host. It is likely that the pathogen exploits conserved eukaryotic processes to infect evolutionarily distant hosts as well as processes unique to the infection of mammals.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahlund, M. K., Ryden, P., Sjostedt, A., and Stoven, S. (2010). Directed screen of Francisella novicida virulence determinants using Drosophila melanogaster. Infect. Immun. 78, 3118–3128.
Akimana, C., Al-Khodor, S., and Abu Kwaik, Y. (2010). Host factors required for modulation of phagosome biogenesis and proliferation of Francisella tularensis within the cytosol. PLoS ONE 5, e11025. doi: 10.1371/journal.pone.0011025
Al-Khodor, S., and Abu Kwaik, Y. (2010). Triggering Ras signalling by intracellular Francisella tularensis through recruitment of PKCalpha and betaI to the SOS2/GrB2 complex is essential for bacterial proliferation in the cytosol. Cell Microbiol. 12, 1604–1621.
Alkhuder, K., Meibom, K. L., Dubail, I., Dupuis, M., and Charbit, A. (2009). Glutathione provides a source of cysteine essential for intracellular multiplication of Francisella tularensis. PLoS Pathog. 5, e1000284. doi: 10.1371/journal.ppat.1000284
Anthony, L. D., Burke, R. D., and Nano, F. E. (1991). Growth of Francisella spp. in rodent macrophages. Infect Immun. 59, 3291–3296.
Anthony, L. S., Cowley, S. C., Mdluli, K. E., and Nano, F. E. (1994). Isolation of a Francisella tularensis mutant that is sensitive to serum and oxidative killing and is avirulent in mice: correlation with the loss of MinD homologue expression. FEMS Microbiol. Lett. 124, 157–165.
Asare, R., and Abu Kwaik, Y. (2010). Molecular complexity orchestrates modulation of phagosome biogenesis and escape to the cytosol of macrophages by Francisella tularensis. Environ Microbiol. 12, 2559–2586.
Balagopal, A., MacFarlane, A. S., Mohapatra, N., Soni, S., Gunn, J. S., and Schlesinger, L. S. (2006). Characterization of the receptor–ligand pathways important for entry and survival of Francisella tularensis in human macrophages. Infect. Immun. 74, 5114–5125.
Barel, M., Hovanessian, A. G., Meibom, K., Briand, J. P., Dupuis, M., and Charbit, A. (2008). A novel receptor – ligand pathway for entry of Francisella tularensis in monocyte-like THP-1 cells: interaction between surface nucleolin and bacterial elongation factor Tu. BMC Microbiol. 8, 145. doi: 10.1186/1471-2180-8-145
Barker, J. R., Chong, A., Wehrly, T. D., Yu, J. J., Rodriguez, S. A., Liu, J., Celli, J., Arulanandam, B. P., and Klose, K. E. (2009). The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol. Microbiol. 74, 1459–1470.
Baron, G. S., and Nano, F. E. (1998). MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol. Microbiol. 29, 247–259.
Baron, G. S., Reilly, T. J., and Nano, F. E. (1999). The respiratory burst-inhibiting acid phosphatase AcpA is not essential for the intramacrophage growth or virulence of Francisella novicida. FEMS Microbiol. Lett. 176, 85–90.
Bell, B. L., Mohapatra, N. P., and Gunn, J. S. (2010). Regulation of virulence gene transcripts by the Francisella novicida orphan response regulator PmrA: role of phosphorylation and evidence of MglA/SspA interaction. Infect. Immun. 78, 2189–2198.
Ben Nasr, A., Haithcoat, J., Masterson, J. E., Gunn, J. S., Eaves-Pyles, T., and Klimpel, G. R. (2006). Critical role for serum opsonins and complement receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in phagocytosis of Francisella tularensis by human dendritic cells (DC): uptake of Francisella leads to activation of immature DC and intracellular survival of the bacteria. J. Leukoc. Biol. 80, 774–786.
Bonquist, L., Lindgren, H., Golovliov, I., Guina, T., and Sjostedt, A. (2008). MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect. Immun. 76, 3502–3510.
Broms, J. E., Lavander, M., and Sjostedt, A. (2009). A conserved alpha-helix essential for a type VI secretion-like system of Francisella tularensis. J. Bacteriol. 191, 2431–2446.
Brotcke, A., and Monack, D. M. (2008). Identification of fevR, a novel regulator of virulence gene expression in Francisella novicida. Infect. Immun. 76, 3473–3480.
Brotcke, A., Weiss, D. S., Kim, C. C., Chain, P., Malfatti, S., Garcia, E., and Monack, D. M. (2006). Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect. Immun. 74, 6642–6655.
Buchan, B. W., McCaffrey, R. L., Lindemann, S. R., Allen, L. A., and Jones, B. D. (2009). Identification of migR, a regulatory element of the Francisella tularensis live vaccine strain iglABCD virulence operon required for normal replication and trafficking in macrophages. Infect. Immun. 77, 2517–2529.
Burstein, E., and Duckett, C. S. (2003). Dying for NF-kappaB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr. Opin. Cell Biol. 15, 732–737.
Champion, M. D., Zeng, Q., Nix, E. B., Nano, F. E., Keim, P., Kodira, C. D., Borowsky, M., Young, S., Koehrsen, M., Engels, R., Pearson, M., Howarth, C., Larson, L., White, J., Alvarado, L., Forsman, M., Bearden, S. W., Sjostedt, A., Titball, R., Michell, S. L., Birren, B., and Galagan, J. (2009). Comparative genomic characterization of Francisella tularensis strains belonging to low and high virulence subspecies. PLoS Pathog. 5, e1000459. doi: 10.1371/journal.ppat.1000459
Charity, J. C., Blalock, L. T., Costante-Hamm, M. M., Kasper, D. L., and Dove, S. L. (2009). Small molecule control of virulence gene expression in Francisella tularensis. PLoS Pathog. 5, e1000641. doi: 10.1371/journal.ppat.1000641
Charity, J. C., Costante-Hamm, M. M., Balon, E. L., Boyd, D. H., Rubin, E. J., and Dove, S. L. (2007). Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog. 3, e84. doi: 10.1371/journal.ppat.0030084
Checroun, C., Wehrly, T. D., Fischer, E. R., Hayes, S. F., and Celli, J. (2006). Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. U.S.A. 103, 14578–14583.
Child, R., Wehrly, T. D., Rockx-Brouwer, D., Dorward, D. W., and Celli, J. (2010). Acid phosphatases do not contribute to the pathogenesis of type A Francisella tularensis. Infect. Immun. 78, 59–67.
Chong, A., Wehrly, T. D., Nair, V., Fischer, E. R., Barker, J. R., Klose, K. E., and Celli, J. (2008). The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect. Immun. 76, 5488–5499.
Clemens, D. L., Lee, B. Y., and Horwitz, M. A. (2004). Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 72, 3204–3217.
Clemens, D. L., Lee, B. Y., and Horwitz, M. A. (2005). Francisella tularensis enters macrophages via a novel process involving pseudopod loops. Infect. Immun. 73, 5892–5902.
Conlan, J. W., Chen, W., Shen, H., Webb, A., and KuoLee, R. (2003). Experimental tularemia in mice challenged by aerosol or intradermally with virulent strains of Francisella tularensis: bacteriologic and histopathologic studies. Microb. Pathog. 34, 239–248.
Conlan, J. W., and North, R. J. (1992). Early pathogenesis of infection in the liver with the facultative intracellular bacteria Listeria monocytogenes, Francisella tularensis, and Salmonella typhimurium involves lysis of infected hepatocytes by leukocytes. Infect. Immun. 60, 5164–5171.
Cox, D., Chang, P., Kurosaki, T., and Greenberg, S. (1996). Syk tyrosine kinase is required for immunoreceptor tyrosine activation motif-dependent actin assembly. J. Biol. Chem. 271, 16597–16602.
Craven, R. R., Hall, J. D., Fuller, J. R., Taft-Benz, S., and Kawula, T. H. (2008). Francisella tularensis invasion of lung epithelial cells. Infect. Immun. 76, 2833–2842.
de Bruin, O. M., Ludu, J. S., and Nano, F. E. (2007). The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol. 7, 1. doi: 10.1186/1471-2180-7-1
Dean, R. E., Ireland, P. M., Jordan, J. E., Titball, R. W., and Oyston, P. C. (2009). RelA regulates virulence and intracellular survival of Francisella novicida. Microbiology 155, 4104–4113.
Deng, K., Blick, R. J., Liu, W., and Hansen, E. J. (2006). Identification of Francisella tularensis genes affected by iron limitation. Infect. Immun. 74, 4224–4236.
Derre, I., and Isberg, R. R. (2004). Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect. Immun. 72, 3048–3053.
Duclos, S., and Desjardins, M. (2000). Subversion of a young phagosome: the survival strategies of intracellular pathogens. Cell. Microbiol. 2, 365–377.
Eigelsbach, H. T., Tulis, J. J., McGavran, M. H., and White, J. D. (1962). Live tularemia vaccine I. Host–parasite relationship in monkeys caccinated intracutaneously or aerogenically. J. Bacteriol. 84, 1020–1027.
Ellis, J., Oyston, P. C., Green, M., and Titball, R. W. (2002). Tularemia. Clin. Microbiol. Rev. 15, 631–646.
Fernandes-Alnemri, T., Yu, J. W., Juliana, C., Solorzano, L., Kang, S., Wu, J., Datta, P., McCormick, M., Huang, L., McDermott, E., Eisenlohr, L., Landel, C. P., and Alnemri, E. S. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393.
Forestal, C. A., Malik, M., Catlett, S. V., Savitt, A. G., Benach, J. L., Sellati, T. J., and Furie, M. B. (2007). Francisella tularensis has a significant extracellular phase in infected mice. J. Infect. Dis. 196, 134–137.
Fortier, A. H., Green, S. J., Polsinelli, T., Jones, T. R., Crawford, R. M., Leiby, D. A., Elkins, K. L., Meltzer, M. S. and Nacy, C. A. (1994). Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol. Ser. 60, 349–361.
Fortier, A. H., Leiby, D. A., Narayanan, R. B., Asafoadjei, E., Crawford, R. M., Nacy, C. A., and Meltzer, M. S. (1995). Growth of Francisella tularensis LVS in macrophages: the acidic intracellular compartment provides essential iron required for growth. Infect. Immun. 63, 1478–1483.
Fuller, J. R., Craven, R. R., Hall, J. D., Kijek, T. M., Taft-Benz, S., and Kawula, T. H. (2008). RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect. Immun. 76, 4934–4943.
Gavrilin, M. A., Bouakl, I. J., Knatz, N. L., Duncan, M. D., Hall, M. W., Gunn, J. S., and Wewers, M. D. (2006). Internalization and phagosome escape required for Francisella to induce human monocyte IL-1beta processing and release. Proc. Natl. Acad. Sci. U.S.A. 103, 141–146.
Goebel, W., and Kuhn, M. (2000). Bacterial replication in the host cell cytosol. Curr. Opin. Microbiol. 3, 49–53.
Golovliov, I., Baranov, V., Krocova, Z., Kovarova, H., and Sjostedt, A. (2003a). An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 71, 5940–5950.
Golovliov, I., Sjostedt, A., Mokrievich, A., and Pavlov, V. (2003b). A method for allelic replacement in Francisella tularensis. FEMS Microbiol. Lett. 222, 273–280.
Gray, C. G., Cowley, S. C., Cheung, K. K., and Nano, F. E. (2002). The identification of five genetic loci of Francisella novicida associated with intracellular growth. FEMS Microbiol. Lett. 215, 53–56.
Greenberg, S., Chang, P., and Silverstein, S. C. (1994). Tyrosine phosphorylation of the gamma subunit of Fc gamma receptors, p72syk, and paxillin during Fc receptor-mediated phagocytosis in macrophages. J. Biol. Chem. 269, 3897–3902.
Hackstadt, T. (2000). Redirection of host vesicle trafficking pathways by intracellular parasites. Traffic 1, 93–99.
Harrell, R. E. Jr., and Whitaker, G. R. (1985). Tularemia: emergency department presentation of an infrequently recognized disease. Am. J. Emerg. Med. 3, 415–418.
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J., and Monack, D. M. (2007). Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994.
Hrstka, R., Stulik, J., and Vojtesek, B. (2005). The role of MAPK signal pathways during Francisella tularensis LVS infection-induced apoptosis in murine macrophages. Microbes Infect. 7, 619–625.
Jia, Q., Lee, B. Y., Bowen, R., Dillon, B. J., Som, S. M., and Horwitz, M. A. (2010). A Francisella tularensis live vaccine strain (LVS) mutant with a deletion in capB, encoding a putative capsular biosynthesis protein, is significantly more attenuated than LVS yet induces potent protective immunity in mice against F. tularensis challenge. Infect. Immun. 78, 4341–4355.
Jones, J. W., Kayagaki, N., Broz, P., Henry, T., Newton, K., O’Rourke, K., Chan, S., Dong, J., Qu, Y., Roose-Girma, M., Dixit, V. M., and Monack, D. M. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776.
Kahn, R. A., Fu, H., and Roy, C. R. (2002). Cellular hijacking: a common strategy for microbial infection. Trends Biochem. Sci. 27, 308–314.
Karin, M., and Ben-Neriah, Y. (2000). Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 18, 621–663.
Katz, J., Zhang, P., Martin, M., Vogel, S. N., and Michalek, S. M. (2006). Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect. Immun. 74, 2809–2816.
Keim, P., Johansson, A., and Wagner, D. M. (2007). Molecular epidemiology, evolution, and ecology of Francisella. Ann. N. Y. Acad. Sci. 1105, 30–66.
Lai, X. H., and Sjostedt, A. (2003). Delineation of the molecular mechanisms of Francisella tularensis-induced apoptosis in murine macrophages. Infect. Immun. 71, 4642–4646.
Lai, X. H., Wang, S. Y., Edebro, H., and Sjostedt, A. (2003). Francisella strains express hemolysins of distinct characteristics. FEMS Microbiol. Lett. 224, 91–95.
Larsson, P., Elfsmark, D., Svensson, K., Wikstrom, P., Forsman, M., Brettin, T., Keim, P., and Johansson, A. (2009). Molecular evolutionary consequences of niche restriction in Francisella tularensis, a facultative intracellular pathogen. PLoS Pathog. 5, e1000472. doi: 10.1371/journal.ppat.1000472
Lawrence, T. (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1, a001651.
Lofgren, S., Tarnvik, A., Bloom, G. D., and Sjoberg, W. (1983). Phagocytosis and killing of Francisella tularensis by human polymorphonuclear leukocytes. Infect. Immun. 39, 715–720.
Mahawar, M., Kirimanjeswara, G. S., Metzger, D. W., and Bakshi, C. S. (2009). Contribution of citrulline ureidase to Francisella tularensis strain Schu S4 pathogenesis. J. Bacteriol. 191, 4798–4806.
Maier, T. M., Casey, M. S., Becker, R. H., Dorsey, C. W., Glass, E. M., Maltsev, N., Zahrt, T. C., and Frank, D.W. (2007). Identification of Francisella tularensis Himar1-based transposon mutants defective for replication in macrophages. Infect. Immun. 75, 5376–5389.
Mariathasan, S., Weiss, D. S., Dixit, V. M. and Monack, D. M. (2005). Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049.
McCaffrey, R. L., and Allen, L. A. (2006). Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J. Leukoc. Biol. 80, 1224–1230.
Meibom, K. L., Dubail, I., Dupuis, M., Barel, M., Lenco, J., Stulik, J., Golovliov, I., Sjostedt, A., and Charbit, A.(2008). The heat-shock protein ClpB of Francisella tularensis is involved in stress tolerance and is required for multiplication in target organs of infected mice. Mol. Microbiol. 67, 1384–1401.
Mohapatra, N. P., Balagopal, A., Soni, S., Schlesinger, L. S., and Gunn, J. S. (2007a). AcpA is a Francisella acid phosphatase that affects intramacrophage survival and virulence. Infect. Immun. 75, 390–396.
Mohapatra, N. P., Soni, S., Bell, B. L., Warren, R., Ernst, R. K., Muszynski, A., Carlson, R. W., and Gunn, J. S. (2007b). Identification of an orphan response regulator required for the virulence of Francisella spp. and transcription of pathogenicity island genes. Infect. Immun. 75, 3305–3314.
Mohapatra, N. P., Soni, S., Reilly, T. J., Liu, J., Klose, K. E., and Gunn, J. S. (2008). Combined deletion of four Francisella novicida acid phosphatases attenuates virulence and macrophage vacuolar escape. Infect. Immun. 76, 3690–3699.
Nano, F. E., Zhang, N., Cowley, S. C., Klose, K. E., Cheung, K. K., Roberts, M. J., Ludu, J. S., Letendre, G. W., Meierovics, A. I., Stephens, G., and Elkins, K. L. (2004). A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 186, 6430–6436.
Nigrovic, L. E., and Wingerter, S. L. (2008). Tularemia. Infect. Dis. Clin. North Am. 22, 489–504, ix.
Oyston, P. C., Sjostedt, A., and Titball, R. W. (2004). Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2, 967–978.
Parsa, K. V., Butchar, J. P., Rajaram, M. V., Cremer, T. J., and Tridandapani, S. (2008). The tyrosine kinase Syk promotes phagocytosis of Francisella through the activation of Erk. Mol. Immunol. 45, 3012–3021.
Parsa, K. V., Ganesan, L. P., Rajaram, M. V., Gavrilin, M. A., Balagopal, A., Mohapatra, N. P., Wewers, M. D., Schlesinger, L. S., Gunn, J. S., and Tridandapani, S. (2006). Macrophage pro-inflammatory response to Francisella novicida infection is regulated by SHIP. PLoS Pathog. 2, e71. doi: 10.1371/journal.ppat.0020071
Pechous, R., Celli, J., Penoske, R., Hayes, S. F., Frank, D. W., and Zahrt, T. C. (2006). Construction and characterization of an attenuated purine auxotroph in a Francisella tularensis live vaccine strain. Infect. Immun. 74, 4452–4461.
Pechous, R. D., McCarthy, T. R., Mohapatra, N. P., Soni, S., Penoske, R. M., Salzman, N. H., Frank, D. W., Gunn, J. S., and Zahrt, T. C. (2008). A Francisella tularensis Schu S4 purine auxotroph is highly attenuated in mice but offers limited protection against homologous intranasal challenge. PLoS ONE 3, e2487. doi: 10.1371/journal.pone.0002487
Pechous, R. D., McCarthy, T. R., and Zahrt, T. C. (2009). Working toward the future: insights into Francisella tularensis pathogenesis and vaccine development. Microbiol. Mol. Biol. Rev. 73, 684–711.
Pierini, L. M. (2006). Uptake of serum-opsonized Francisella tularensis by macrophages can be mediated by class A scavenger receptors. Cell. Microbiol. 8, 1361–1370.
Qin, A., and Mann, B. J. (2006). Identification of transposon insertion mutants of Francisella tularensis tularensis strain Schu S4 deficient in intracellular replication in the hepatic cell line HepG2. BMC Microbiol. 6, 69. doi: 10.1186/1471-2180-6-69
Qin, A., Scott, D. W., Thompson, J. A., and Mann, B. J. (2009). Identification of an essential Francisella tularensis subsp. tularensis virulence factor. Infect Immun. 77, 152–161.
Raeder, E. M., Mansfield, P. J., Hinkovska-Galcheva, V., Shayman, J. A., and Boxer, L. A. (1999). Syk activation initiates downstream signaling events during human polymorphonuclear leukocyte phagocytosis. J. Immunol. 163, 6785–6793.
Rajaram, M. V., Butchar, J. P., Parsa, K. V., Cremer, T. J., Amer, A., Schlesinger, L. S., and Tridandapani, S. (2009). Akt and SHIP modulate Francisella escape from the phagosome and induction of the Fas-mediated death pathway. PLoS ONE 4, e7919. doi: 10.1371/journal.pone.0007919
Rajaram, M. V., Ganesan, L. P., Parsa, K. V., Butchar, J. P., Gunn, J. S., and Tridandapani, S. (2006). Akt/Protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J. Immunol. 177, 6317–6324.
Ray, K., Marteyn, B., Sansonetti, P. J., and Tang, C. M. (2009). Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat. Rev. Microbiol. 7, 333–340.
Raynaud, C., Meibom, K. L., Lety, M. A., Dubail, I., Candela, T., Frapy, E., and Charbit, A. (2007). Role of the wbt locus of Francisella tularensis in lipopolysaccharide O-antigen biogenesis and pathogenicity. Infect. Immun. 75, 536–541.
Sammons-Jackson, W. L., McClelland, K., Manch-Citron, J. N., Metzger, D. W., Bakshi, C. S., Garcia, E., Rasley, A., and Anderson, B. E. (2008). Generation and characterization of an attenuated mutant in a response regulator gene of Francisella tularensis live vaccine strain (LVS). DNA Cell Biol. 27, 387–403.
Santiago, A. E., Cole, L. E., Franco, A., Vogel, S. N., Levine, M. M., and Barry, E. M. (2009). Characterization of rationally attenuated Francisella tularensis vaccine strains that harbor deletions in the guaA and guaB genes. Vaccine 27, 2426–2436.
Santic, M., Akimana, C., Asare, R., Kouokam, J. C., Atay, S., and Kwaik, Y. A. (2009). Intracellular fate of Francisella tularensis within arthropod-derived cells. Environ. Microbiol. 11, 1473–1481.
Santic, M., Al-Khodor, S., and Abu Kwaik, Y. (2010a). Cell biology and molecular ecology of Francisella tularensis. Cell. Microbiol. 12, 129–139.
Santic, M., Pavokovic, G., Jones, S., Asare, R., and Kwaik, Y. A. (2010b). Regulation of apoptosis and anti-apoptosis signalling by Francisella tularensis. Microbes Infect. 12, 126–134.
Santic, M., Asare, R., Skrobonja, I., Jones, S., and Abu Kwaik, Y. (2008). Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect. Immun. 76, 2671–2677.
Santic, M., Molmeret, M., and Abu Kwaik, Y. (2005a). Modulation of biogenesis of the Francisella tularensis subsp. novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN-gamma. Cell Microbiol. 7, 957–967.
Santic, M., Molmeret, M., Klose, K. E., Jones, S., and Kwaik, Y. A. (2005b). The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell. Microbiol. 7, 969–979.
Santic, M., Molmeret, M., Barker, J. R., Klose, K. E., Dekanic, A., Doric, M., and Abu Kwaik, Y. (2007). A Francisella tularensis pathogenicity island protein essential for bacterial proliferation within the host cell cytosol. Cell. Microbiol. 9, 2391–2403.
Santic, M., Molmeret, M., Klose, K. E., and Abu Kwaik, Y. (2006). Francisella tularensis travels a novel, twisted road within macrophages. Trends Microbiol. 14, 37–44.
Schmerk, C. L., Duplantis, B. N., Howard, P. L., and Nano, F. E. (2009). A Francisella novicida pdpA mutant exhibits limited intracellular replication and remains associated with the lysosomal marker LAMP-1. Microbiology 155, 1498–1504.
Schulert, G. S., and Allen, L. A. (2006). Differential infection of mononuclear phagocytes by Francisella tularensis: role of the macrophage mannose receptor. J. Leukoc. Biol. 80, 563–571.
Schulert, G. S., McCaffrey, R. L., Buchan, B. W., Lindemann, S. R., Hollenback, C., Jones, B. D., and Allen, L. A. (2009). Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of strain LVS. Infect. Immun. 77, 1324–1336.
Sen, B., Meeker, A., and Ramakrishnan, G. (2010). The fslE homolog, FTL_0439 (fupA/B), mediates siderophore-dependent iron uptake in Francisella tularensis LVS. Infect. Immun. 78, 4276–4285.
Sturgill-Koszycki, S., Schaible, U. E., and Russell, D. G. (1996). Mycobacterium-containing phagosomes are accessible to early endosomes and reflect a transitional state in normal phagosome biogenesis. EMBO J. 15, 6960–6968.
Tamilselvam, B., and Daefler, S. (2008). Francisella targets cholesterol-rich host cell membrane domains for entry into macrophages. J. Immunol. 180, 8262–8271.
Telepnev, M., Golovliov, I., Grundstrom, T., Tarnvik, A., and Sjostedt, A. (2003). Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-alpha and IL-1 from murine macrophages. Cell. Microbiol. 5, 41–51.
Telepnev, M., Golovliov, I., and Sjostedt, A. (2005). Francisella tularensis LVS initially activates but subsequently down-regulates intracellular signaling and cytokine secretion in mouse monocytic and human peripheral blood mononuclear cells. Microb. Pathog. 38, 239–247.
Tempel, R., Lai, X. H., Crosa, L., Kozlowicz, B., and Heffron, F. (2006). Attenuated Francisella novicida transposon mutants protect mice against wild-type challenge. Infect. Immun. 74, 5095–5105.
Twine, S., Bystrom, M., Chen, W., Forsman, M., Golovliov, I., Johansson, A., Kelly, J., Lindgren, H., Svensson, K., Zingmark, C., Conlan, W., and Sjostedt, A. (2005). A mutant of Francisella tularensis strain SCHU S4 lacking the ability to express a 58-kilodalton protein is attenuated for virulence and is an effective live vaccine. Infect. Immun. 73, 8345–8352.
Ulland, T. K., Buchan, B. W., Ketterer, M. R., Fernandes-Alnemri, T., Meyerholz, D. K., Apicella, M. A., Alnemri, E. S., Jones, B. D., Nauseef, W. M., and Sutterwala, F. S. (2010). Cutting edge: mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J. Immunol. 185, 2670–2674.
Weiss, D. S., Brotcke, A., Henry, T., Margolis, J. J., Chan, K. and Monack, D. M. (2007). In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 6037–6042.
Wilson, J. E., Katkere, B., and Drake, J. R. (2009). Francisella tularensis induces ubiquitin-dependent major histocompatibility complex class II degradation in activated macrophages. Infect. Immun. 77, 4953–4965.
Keywords: tularemia, ASC, caspase, apoptosis, Ras, Akt, SHIP
Citation: Asare R and Abu Kwaik Y (2011) Exploitation of host cell biology and evasion of immunity by Francisella tularensis. Front. Microbio. 1:145. doi: 10.3389/fmicb.2010.00145
Received: 12 November 2010;
Accepted: 21 December 2010;
Published online: 13 January 2011.
Edited by:
Anders Sjostedt, Umeå University, SwedenReviewed by:
Tonyia Eaves-Pyles, University of Texas Medical Branch, USASusheela Tridandapani, Ohio State University, USA
Copyright: © 2011 Asare and Abu Kwaik. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Yousef Abu Kwaik, Department of Microbiology and Immunology, School of Medicine, University of Louisville, Room 412A, Louisville, KY 40202, USA. e-mail:YWJ1a3dhaWtAbG91aXN2aWxsZS5lZHU=