
- Department of Molecular Microbiology and Immunology, Oregon Health Sciences University, Portland, OR, USA
Tuberculosis remains a significant global health concern. The hallmark of Mycobacterium tuberculosis pathogenicity is its ability to infect resting macrophages and establish an intracellular niche. Activated and autophagic macrophages control mycobacterial infections through bactericidal mechanisms ranging from reactive oxygen and nitrogen intermediates to the delivery of the bacterium to the acidified, hydrolytically active lysosome. The mycobactericidal activity of the lysosome is due in part to the action of ubiquitin-derived peptides (Ub-peptides). In this review we discuss the trafficking events that result in delivery M. tuberculosis to the lysosome, the source and lysosomal generation of Ub-peptides and their role in macrophage control of M. tuberculosis infection.
Introduction
Despite the availability of antibiotics to combat Mycobacterium tuberculosis infections, tuberculosis (TB) is one of the leading causes of death due to infectious disease. The World Health Organization estimates that M. tuberculosis infects one-third of the world population, and 9.4 million new cases of TB were reported in 2009 (WHO, 2010). The bacterium’s ability to infect and replicate within the host macrophage is key to M. tuberculosis success as a pathogen. It does so by arresting phagosome maturation and preventing fusion of the bacteria-containing vacuole with the lysosome. In resting macrophages, M. tuberculosis resides in a vacuole resembling the early endosome with a pH of 6.4 and markers such as the Rab5 GTPase (Sturgill-Koszycki et al., 1994; Clemens and Horwitz, 1996; Via et al., 1997; Clemens et al., 2000). The pathogen successfully modulates phagosome trafficking to establish a niche in resting macrophages, but immune activation or the induction of autophagy in infected macrophages shifts the balance toward mycobacterial clearance (Schaible et al., 1998; Via et al., 1998; Gutierrez et al., 2004; Alonso et al., 2007). In activated macrophages, M. tuberculosis is exposed to reactive oxygen and reactive nitrogen intermediates (ROI and RNI), and the bacterial compartment fuses with the acidic and hydrolytic lysosome. Recently, we showed that ubiquitin and ubiquitin-derived peptides (Ub-peptides) contribute to the mycobactericidal activity of the lysosome (Alonso et al., 2007; Purdy et al., 2009). In this review we discuss the role of Ub-peptides in lysosomal killing of M. tuberculosis. We will review the trafficking events that result in delivery of M. tuberculosis and ubiquitin to the lysosome, and we describe our current working model for the mycobactericidal activity of Ub-peptides.
M. Tuberculosis Phagosome Maturation Arrest vs. Trafficking to the Lysosome
The arrest of M. tuberculosis phagosome maturation in resting macrophages is a complex process that depends upon both cell wall constituents and protein effectors (Rohde et al., 2007). The bioactive mycobacterial lipid trehalose 6,6′-dimycolate (TDM) along with the mannosylated lipoglycans lipoarabinomannan (ManLAM) and higher-order phosphatidyl-myo-inositol mannosides (PIMs) play a role. When purified and coated onto inert particles, these surface molecules can prevent lysosomal fusion of the bead-containing phagosome (Fratti et al., 2003; Indrigo et al., 2003; Vergne et al., 2004; Kang et al., 2005; Torrelles et al., 2006). Recent work indicates that recognition of these mannosylated lipoglycans by specific receptors promotes phagosome maturation arrest and likely prevents a pro-inflammatory response (reviewed in Schlesinger et al., 2008). During macrophage infection M. tuberculosis engages the mannose receptor, and blocking bacterial entry via the mannose receptor increases co-localization of the pathogen with lysosomal markers (Kang et al., 2005). Mycobacterial proteins also contribute to phagosome maturation arrest, and bacterial mutants with reduced ability to arrest phagosome maturation in macrophages were identified in genetic screens (Pethe et al., 2004; MacGurn and Cox, 2007). The incomplete elimination of these mutants by cultured macrophages points to the extensive host-resistance repertoire of M. tuberculosis (reviewed recently by Ehrt and Schnappinger, 2009; Stallings and Glickman, 2010).
Recent work has examined the means by which immune activation and the induction of autophagy promote mycobacterial clearance. Activated macrophages are mycobactericidal through both oxidative and non-oxidative mechanisms (Figure 1). IFN-γ activated macrophages express phagocyte NADPH oxidase (NOX2) and inducible nitric oxide synthase (NOS2) that produce ROI and RNI, respectively. Studies in mice indicate that ROI and RNI play a key role in control of M. tuberculosis infection (Chan et al., 1992; MacMicking et al., 1997; Darwin et al., 2003). However, IFN-γ knockout mice are substantially more susceptible to M. tuberculosis infection than Nos2 knockout mice, indicating the presence of an IFN-γ regulated, NO-independent killing mechanism (Cooper et al., 1993, 2000; Flynn et al., 1993). Activated macrophages are also mycobactericidal because of enhanced fusion between the mycobacteria-containing vacuole with the lysosome. The mechanisms underlying delivery of mycobacterium to the lysosome are beginning to be elucidated. Work by MacMicking et al. (2003) demonstrated a role for the IFN-γ and LPS – responsive GTPase Irgm1 (also known as LRG-47) in host control of mycobacterial infections. Irgm−/− mice are more susceptible than wild type to M. tuberculosis, similar to IFNR−/− mice. Further investigation of Irgm−/− macrophages indicated that these cells were defective in phagosome–lysosome fusion. The presence of PI(3,4)P2 and PI(3,4,5)P3 on the M. tuberculosis phagosome recruits Irgm1. Upon binding of Irgm1 to these lipid moieties, Irgm1 interacts with trafficking molecules such as Snapin to promote SNARE complex assembly and fusion of the M. tuberculosis phagosome with late endosomal and lysosomal compartments (Tiwari et al., 2009).
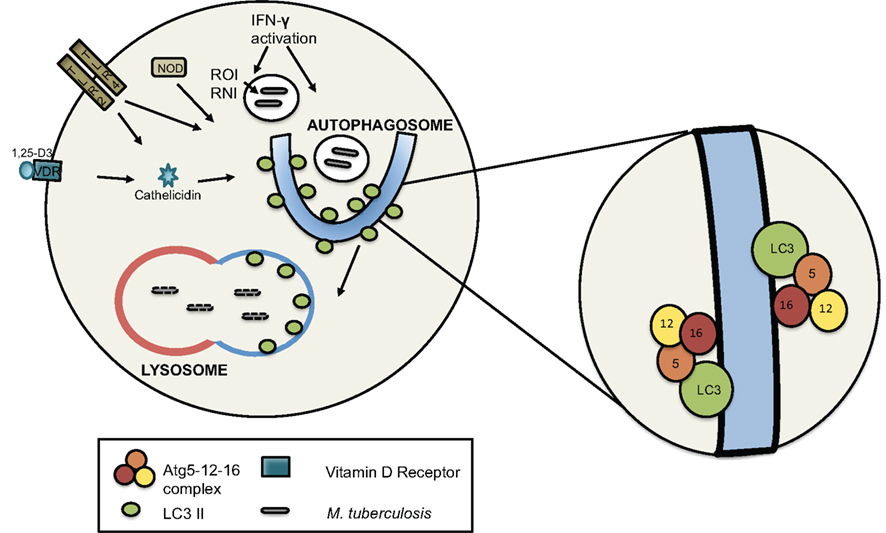
Figure 1. The induction of autophagy in macrophages. Autophagy can be induced via IFN-γ stimulation, TLR and Nod-like receptor signaling, and vitamin D3 (1,25-D3)-mediated signaling. IFN-γ activated macrophages generate RNI and ROI in the phagosome that are associated with mycobacterial killing. Phagosome-associated ROI also promotes co-localization of autophagic proteins with the phagosome. Autophagosome formation involves two Atg5-mediated conjugation events: Atg5 is covalently attached to Atg12. Atg5-12 then non-covalently associates with Atg16 to form the Atg5-12-16 E3-like enzyme complex that ligates LC3 to the lipid phosphatidylethanolamine (PE) in the isolation membrane. LC3 conjugated to PE (LC3-II) is attached with the inner and outer leaflet of the autophagosome and is important for both cargo capture and the membrane fusion events that result in the double-membrane autophagosome. When mycobacteria are autophagosomal cargo, they are delivered to the lysosome for degradation.
Deretic and colleagues demonstrated that IFN-γ activation also induces autophagy and promotes delivery of mycobacteria to the lysosome via autophagosomes. In these experiments, Irgm localized with autophagosomes upon IFN-γ activation. This observation suggests a close relationship between IFN-γ-mediated and autophagic mycobacterial control (Gutierrez et al., 2004). Autophagy facilitates protein and organelle degradation by the lysosome and has recently emerged as a key component of the innate immune response (see Deretic and Levine, 2009 for a comprehensive review). An autophagosome is formed when an isolation membrane expands and engulfs cytoplasmic material and organelles, then fuses to form a morphologically distinct double-membrane vacuole. Autophagosome formation requires two Atg5-dependent conjugation systems that generate Atg5–Atg12, which complexes with Atg16 on the isolation membrane and ligates LC3 (also known as Atg8) to phosphatidylethanolamine (PE) thereby localizing LC3 to the nascent autophagosome. PE-conjugated LC3 (LC3-II) decorates both the inner and outer leaflet of the autophagosome membrane. Proteolysis by Atg4 can cleave LC3-II from the cytoplasmic facing outer leaflet, and LC3-II on the inner leaflet is trafficked to the lysosome and degraded (Kirisako et al., 2000; Tanida et al., 2004). LC3 has been routinely used as an autophagosome-specific marker for microscopy and to quantify autophagic activity biochemically. Since LC3 mediates membrane tethering and hemifusion in vitro and in vivo, it likely plays an integral role in driving autophagosome formation and expansion (Nakatogawa et al., 2007). Once formed, autophagosomes mature into autophagolysosomes via a late endosomal intermediate.
Autophagy contributes to innate immunity by engulfing cytoplasmic or vacuolar pathogens and delivering them to the lysosome for elimination. The importance of autophagy in controlling infections has been illustrated using murine models with tissue-specific autophagy deficiency. Atg5−/− macrophages are impaired for autophagosome formation and have reduced mycobactericidal activity (Ponpuak et al., 2010). Impaired control of M. bovis BCG in these experiments may be independent of autophagosome formation itself. Zhao et al. showed that in activated Atg5−/− macrophages the p47 GTPase Irga6 was not recruited to the T. gondii-vacuole as in wild type macrophages. As a result these macrophages failed to clear the parasite, whereas wild type macrophages promote parasite killing (Zhao et al., 2008). Autophagosome-independent functions of the autophagy machinery, including potential roles in recruitment of p47 GTPase members, are a current focus of research in several laboratories.
Autophagy is induced in macrophages by nutrient starvation, pharmacologically via the drug rapamycin, or through immune signaling. Work by Deretic’s group showed that IFN-γ activation stimulates autophagy in primary bone marrow-derived macrophages and macrophage-like cell lines (Gutierrez et al., 2004). Subsequently several groups have demonstrated induction of autophagy via stimulation of pattern recognition receptors (PRRs) including Toll like receptors (TLRs) and nucleotide-binding oligomerization domain-containing (NOD) proteins. LPS induces autophagy via TLR4 in a TRIF-dependent manner, and LPS-induced autophagy in RAW264.7 macrophages results in localization of M. tuberculosis in autophagosomes (Sanjuan et al., 2007; Xu et al., 2007). TLR2 signaling also induces autophagy. Moreover LC3 levels were reduced in macrophages from Tlr2−/− mice, consistent with a potential role for TLR signaling in enhancing autophagy (Sanjuan et al., 2007). The TLR7 ligands ssRNA and imiquimod strongly induced autophagosome formation in RAW 264.7 and bone marrow-derived macrophages in a MyD88-dependent manner (Delgado et al., 2008). While TLR7 is not implicated in mycobacterial infections, the authors showed that artificially stimulating TLR7-mediated autophagy promoted M. bovis BCG clearance in infected macrophages. Signaling through the cytoplasmic PRRs also stimulates autophagy. Activation of the NOD2 receptors by bacterial cell wall constituents induced autophagy in murine macrophages in vitro and in vivo (Travassos et al., 2010). Recently Brooks et al. showed that human macrophages respond to M. tuberculosis in a NOD2-dependent manner, which implicates this cytoplasmic sensor in control of mycobacterial infections. Lack of functional NOD2 increased bacterial survival, but the mechanism of mycobacterial control was not addressed experimentally (Brooks et al., 2010). In light of the above reports, one possible scenario is that NOD2 participates in recruitment of the autophagic machinery to the M. tuberculosis vacuole and autophagic clearance.
Extensive literature demonstrates ROI-regulation of autophagy, and this was recently reviewed elsewhere (Scherz-Shouval and Elazar, 2010). ROI generated by mitochondria upon nutrient starvation and exogenously added hydrogen peroxide both promote autophagy (Scherz-Shouval et al., 2007; Chen et al., 2009). In macrophages, ROI generated by the phagocyte NADPH oxidase NOX2 also induced autophagy and LC3 accumulation at the phagosome. The phagocytosis of IgG beads and zymosan was associated with LC3 accumulation on the phagosome, and this was blocked using the NOX2 inhibitor diphenyleneiodonium chloride (Huang et al., 2009). While the mechanism by which localized ROI activates autophagy is unclear, ROI generation appears to be key for Fcγ – receptor and TLR-mediated autophagic clearance.
Finally, the immune mediator vitamin D promotes mycobactericidal activity in human macrophages, and the mechanisms underlying this control were elucidated recently. Work by Liu et al. (2006, 2007) showed that the active form of vitamin D, 1,25-dihydroxyvitamin D3 (1,25-D3) induces the generation of the mycobactericidal cathelicidin-derived peptide LL-37 in human macrophages. Data from Jo and colleagues subsequently demonstrated that 1,25-D3 induced autophagy in a cathelicidin-dependent manner (Yuk et al., 2009). The cathelicidin-derived peptide LL-37 also directly contributes to the mycobactericidal properties of autophagic macrophages and will be discussed further below.
While our understanding of autophagy as an antimicrobial process has expanded dramatically in the last decade, a number of questions remain with regard to how autophagic clearance initiates and whether this is a directed process. Work by several investigators suggests a link between the phagocytic and autophagic machinery. LC3 was previously considered a marker exclusively for autophagosomes, but it has recently been associated with phagocytic vacuoles that lacked the defining double-membrane structure of autophagosomes: Sanjuan et al. (2007) reported that LC3 recruitment to the phagosomal membrane upon uptake of zymosan. Proteomic analysis of latex bead phagosomes from RAW264.7 macrophages revealed the presence of LC3, and LC3 levels increased upon starvation-induced autophagy (Shui et al., 2008). Finally, using both immunofluorescence microscopy and biochemical approaches Huang et al. (2009) showed LC3 and Atg12 were recruited to nascent phagosomes during Fcγ-receptor and TLR-mediated phagocytosis. Since Atg12 in complex with Atg5 and Atg16 determine the site of LC3 lipidation (Fujita et al., 2008), it is tempting to speculate that LC3 is directly targeted to phagosomal membranes. Combined, these studies suggest closer examination of LC3-associated phagocytosis and phagosome maturation is warranted. Post-phagocytosis, PRRs may recognize bacterial pathogens and recruit the autophagy machinery to cytoplasmic and vacuolar pathogens. In support of this model, NOD2 co-localized with autophagosome-specific proteins (Travassos et al., 2010). However, it remains to be shown experimentally that NOD2 directly targets intracellular bacteria for clearance by the autophagic pathway.
Lysosomal Killing of Mycobacteria
Autophagic and immune responsive control of mycobacterial infections promotes trafficking of the bacterium to the lysosome, however there are a number of remaining questions regarding the mechanism of mycobacterial killing. We found that solubilized lysosomes isolated from resting bone marrow-derived macrophages are bactericidal toward both M. tuberculosis and M. smegmatis (Alonso et al., 2007). Solubilized lysosomal material was fractionated by HPLC, and a single fraction retained bactericidal activity. Mass spec analysis of this fraction revealed that bactericidal activity is associated with ubiquitin peptides. Full-length purified ubiquitin lacks bactericidal activity, but Ub-peptides obtained from digest of purified ubiquitin with cathepsin proteases or a synthesized peptide Ub2 (STLHLVLRLRGG) are bactericidal against Mycobacteria. We subsequently showed that immunodepletion of ubiquitin from the lysosomal extract with a polyclonal antibody against ubiquitin results in diminished bactericidal activity (Purdy et al., 2009). These data suggest that ubiquitylated proteins are delivered to the lysosomal compartment, where cathepsin proteinases release Ub-peptides that possess antimicrobial activity. To establish a mechanism for bacterial killing by Ub-peptides, it was key that the ubiquitin be localized to the lumen of lysosomes where they would come in direct contact with the bacteria. We performed immunoelectron microscopy and found that ubiquitin localizes in the lumen of electron dense lysosomal compartments. Microscopy of M. tuberculosis-infected macrophages indicated that the induction of autophagy enhances localization of ubiquitin to the lumen of bacteria-containing vacuoles (Alonso et al., 2007). To confirm that autophagy resulted in increased lysosomal ubiquitin, we performed immunoblotting on lysosomal lysates isolated from resting, autophagic, and activated macrophages. Lysates from autophagic and activated macrophages have increased levels of ubiquitin relative to lysates from resting macrophages, and this correlates with increased bactericidal activity of these lysosomal lysates when tested in vitro.
The antimicrobial peptide repertoire of macrophages is not limited to Ub-peptides. Other peptides with demonstrated mycobactericidal activity include hepcidin, cathelicidin, and Fau-derived peptides. Hepcidin is expressed in the RAW264.7 macrophage-like cell line and co-localizes with M. tuberculosis in IFN-γ activated macrophages (Sow et al., 2007). In human-derived macrophages, Vitamin D and TLR2 stimulation induce transcription of cathelicidin. In autophagic macrophages, cathelicidin co-localizes in vacuoles containing M. tuberculosis, and this is associated with decreased bacterial viability (Liu et al., 2006, 2007; Yuk et al., 2009). Deretic and colleagues recently reported the lysosomal generation of Fau-derived antimycobacterial peptides (Ponpuak et al., 2010). Fau is a precursor protein comprised of a 74-amino acid (aa) polypeptide with 38% identity to ubiquitin fused to the 59-aa ribosomal protein S30. The S30 domain, designated ubiquicidin, was identified as a component of IFN-γ stimulated RAW 264.7 macrophages that possess antimicrobial activity against Listeria monocytogenes (Hiemstra et al., 1999). Fau co-localizes with M. bovis BCG phagosomes in autophagic RAW 264.7 macrophages. Digestion of S30/ubiquicidin in vitro with cathepsin L-generated mycobactericidal peptides, and depletion of Fau from phagosomal extracts is associated with reduced bactericidal capacity. Since Fau also contributes mycobactericidal peptides to lysosomal extracts, our data indicating that some bactericidal activity remained upon ubiquitin immunodepletion of lysosomal extracts is not unexpected. In the lysosome, it is likely that Ub-peptides and Fau-derived peptides act synergistically with the complex mixture of hydrolytic enzymes present in the lysosomal lumen, including proteases and lipases, to promote bacterial killing.
Trafficking of Proteins that are Cleaved into Antimicrobial Peptides
As outlined below, our data suggest both the multivesicular body (MVB)/endocytic pathway and autophagy are the source for lysosomal ubiquitin (Figure 2). Ubiquitin is best known as a post-translational addition to proteins targeting them for degradation through the proteasome (Ciechanover, 2009). While polyubiquitylation via Lys48-linked conjugation targets proteins for proteasomal degradation, mono-ubiquitylation, and Lys63-linked polyubiquitylation specifies which proteins should be recycled or sequestered by MVBs and the endocytic pathway (Thrower et al., 2000; Lauwers et al., 2009). The formation of MVBs is important for the recycling and turnover of membrane proteins such as activated growth factor, hormone, and cytokine receptors. MVBs also traffic misfolded membrane proteins for lysosomal degradation and thus contribute to cellular protein quality control. Autophagy also participates in the clearance of ubiquitylated proteins in a proteasome-independent manner. There is some overlap in the two pathways since both MVBs and autophagosomes deliver their contents to the lysosome via a late endosomal intermediate.
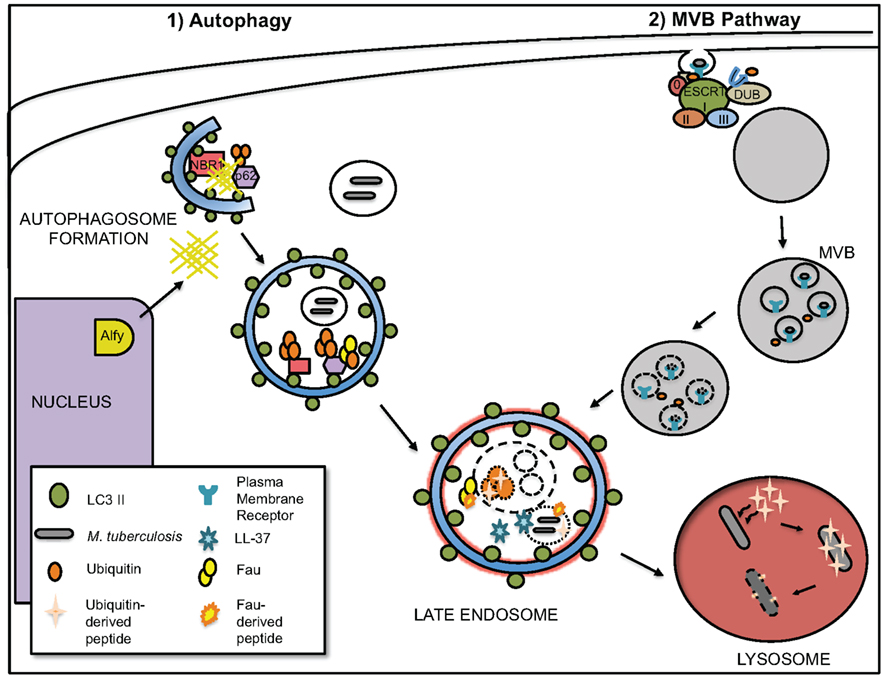
Figure 2. Autophagy and multivesicular body (MVB) pathway are main sources of lysosomal ubiquitin. The autophagosome and MVB trafficking pathways that likely deliver ubiquitylated proteins to the lysosome are depicted. (1) Ubiquitylated proteins may be directly targeted for degradation via the autophagic pathway. The adaptor proteins p62 and NBR1 possess ubiquitin- and LC3-binding domains, and they may tether ubiquitylated cytoplasmic proteins and aggregates to the nascent autophagosome. Upon the induction of autophagy, Alfy relocates from the nucleus to associate with Atg5, LC3, p62, and aggregated proteins. Alfy may provide a scaffold (indicated by hashes) for the assembly of the autophagy machinery. (2) Ubiquitylated membrane proteins are recognized by the ESCRT complex for lysosomal degradation via MVBs. Unlike degradation of ubiquitylated proteins via the proteasome, deubiquitination is not required for lysosomal degradation. In the lysosome, ubiquitin-derived peptides are generated by cathepsin proteolysis of ubiquitylated proteins. Induction of autophagy by starvation or IFN-γ activation increases the concentration of lysosomal ubiquitin, which correlated with increased mycobactericidal activity of the solubilized lysosomal extracts. Upon the induction of autophagy, the antimicrobial peptide LL-37 and Fau also localize to the mycobacteria-containing vacuole.
Multivesicular Body
Immunoelectron microscopy of bone marrow-derived macrophages using anti-ubiquitin antibody revealed ubiquitin label was associated with the internal membranes of structures resembling MVBs or late endosomes (Alonso et al., 2007). The biogenesis and maturation of MVBs requires the endosomal sorting complex required for transport (ESCRT) machinery that has been studied extensively in both the yeast and mammalian systems (Raiborg and Stenmark, 2009). Ubiquitylated proteins destined for the MVB are recognized by Hrs and STAM (ESCRT 0), which subsequently recruit the ESCRT I, ESCRT II, and ESCRT III complexes. The ESCRT I and ESCRT II complexes both contain subunits that bind ubiquitylated proteins. The ESCRT III complex orchestrates the formation of the inward budding vesicles of the MVB, where cargo destined for the lysosome is sorted. Unlike proteasomal degradation, deubiquitylation is not necessary for lysosomal sorting other than to maintain free ubiquitin. In yeast, the de-ubiquitinating enzyme (DUB) Doa4 is associated with the ESCRT III complex and facilitates the removal of ubiquitin from proteins destined for vacuolar degradation. Doa4 null mutations result in the depletion of free ubiquitin and defects in the proteolysis of both proteasomal and vacuolar substrates (Swaminathan et al., 1999; Reggiori and Pelham, 2001). While the yeast and mammalian endocytic pathways are highly conserved, the role of deubiquitylation of MVB cargo proteins in mammalian cells remains a matter of debate (McCullough et al., 2004; Bowers et al., 2006; Mizuno et al., 2006; Row et al., 2006). The two mammalian DUBs AMSH and UBPY, are associated with both ESCRT 0 and ESCRT III and appear to exert regulatory control on MVB-mediated protein degradation. Our immunoelectron microscopy data showing ubiquitin present in MVB and electron dense lysosomal compartments suggest that deubiquitylation of cargo proteins is not necessary.
Autophagosomes
Our data demonstrate induction of autophagy by starvation or IFN-γ activation results in an increase in the ubiquitin concentration of the solubilized lysosomes. This correlates with the increased bacterial killing capacity of isolated lysosomes (Alonso et al., 2007). Using immunoelectron microscopy we localized ubiquitin to LAMP1-positive lysosomal vacuoles, and we found increasing levels of ubiquitin in the lumen of lysosomal structures upon the induction of autophagy. In autophagic macrophages, ubiquitin-positive vacuoles were also positive for the autophagic marker LC3, suggesting that ubiquitylated proteins were present in autophagosomes that matured into autophagolysosomes. Deretic and colleagues subsequently showed that ubiquitin co-localization with mycobacteria depends upon Atg5 (Ponpuak et al., 2010). Therefore, the autophagic pathway is a valid source of lysosomal ubiquitin and mycobactericidal Ub-peptides.
Our work and that of the Deretic group demonstrate that autophagic macrophages traffic cytosolic material to the lysosome where cathepsin proteases generate mycobactericidal fragments. A growing body of literature links clearance of ubiquitylated proteins present in cytoplasmic inclusions termed aggresomes or aggresome-like induced structures (ALIS) to the autophagic pathway. The formation of aggregates containing ubiquitylated proteins is thought to reduce the cytotoxic effects of such misfolded proteins. As such, aggresomes accumulate upon treatment with proteasome inhibitors or in neurological disorders like Parkinson’s and Huntington’s when the cell is unable to efficiently degrade ubiquitylated proteins. Aggresomes form by the active transport of ubiquitylated proteins to the microtubule organizing center (MTOC; Johnston et al., 1998). HDAC6, a cytoplasmic histone deacetylase, has been implicated in this process as it binds both dynein and ubiquitylated proteins (Kawaguchi et al., 2003; Iwata et al., 2005). It appears that HDAC6 plays a role in the microtubule-dependent concentration of ubiquitylated proteins as well as localization of the autophagic machinery to the aggresomes to promote efficient bulk protein degradation (Pandey et al., 2007). The work of Brumell and colleagues recently documented the presence of ubiquitylated protein aggregates termed ALIS that are unassociated with the MTOC in immune and non-immune cells upon stress conditions (Canadien et al., 2005; Szeto et al., 2006). The proteasome is recruited to these ALIS, but they are degraded in the presence of proteasome inhibitors and are positive for LC3, suggesting an autophagic clearance mechanism. Mice lacking the autophagy genes Atg5 or Atg7 exhibit signs of neurodegeneration, and despite normal proteasome function, ubiquitylated proteins aggregates accumulate in the cytosol of neural cells (Komatsu et al., 2005; Hara et al., 2006). Finally, both free ubiquitin and ubiquitin-modified proteins have been found within autophagosomes (Schwartz et al., 1988, 1992; Lenk et al., 1992; Low et al., 1995).
Extensive data connects ubiquitylation with lysosomal degradation via autophagosomes, but only recently the mechanism by which ubiquitylated proteins are recognized by the autophagic machinery have been elucidated. Bjorkoy et al. (2005) showed that the ubiquitin binding protein p62 interacted with ubiquitin and the autophagic marker LC3. Their data suggested that autophagosomes recognize and selectively sequester ubiquitylated protein aggregates for lysosomal degradation. Subsequent analysis demonstrated that p62 directly interacted with LC3 via a short LC3 interacting region (LIR) located between its zinc finger domain and UBA ubiquitin binding domain (Komatsu et al., 2007; Pankiv et al., 2007). NBR1 is a second protein associated with selective autophagy of ubiquitylated protein aggregates. Microscopy demonstrated that NBR1 is recruited to LC3-positive, ubiquitin-positive protein aggregates. Like p62, NBR1 contains a UBA domain and an LIR domain that are required for binding of ubiquitylated proteins and autophagic degradation, respectively (Kirkin et al., 2009). NBR1 appears to play a compensatory role in p62-knockout tissues to prevent the accumulation of ubiquitylated protein aggregates. However, recent data suggests p62 and NBR1 have distinct functions in innate immunity. p62, but not NBR1, co-localized with M. bovis BCG in autophagic macrophages (Ponpuak et al., 2010). p62 complexed with Fau and ubiquitylated proteins via its UBA domain and appeared to directly deliver these proteins to the lysosome where antimycobacterial peptides can be generated. Therefore, p62 plays an integral role in the generating the mycobactericidal capacity of the lysosome.
Upon binding of ubiquitylated proteins by p62 and NBR1, the autophagic machinery must be recruited to promote autophagosome formation and clearance. The FYVE-domain-containing protein Alfy appears to play a role in this process. Upon the induction of autophagy, Alfy relocates from the nucleus and multimerizes to form a meshwork that associates with ubiquitylated proteins and partially localized with vacuoles positive for the autophagic markers Atg5 and LC3 (Simonsen et al., 2004). Further analysis indicates that Alfy directly interacts with Atg5 via a WD40 domain and forms a complex with LC3, p62, and NBR1. Overexpression of Alfy suppresses the toxic effects of protein aggregation, and Johansen and colleagues propose that Alfy acts as a scaffold to promote macroautophagic clearance (Filimonenko et al., 2010). Combined these data provide the foundation for our understanding of the recognition and autophagic delivery of ubiquitylated proteins to the lysosome.
Autophagic clearance of M. tuberculosis depends on the p62-mediated delivery of cytoplasmic proteins with bactericidal potential to the mycobacteria-containing vacuole. However, it is worth noting that other pathogens that are cytosolic or residing in damaged vacuoles can co-localize with ubiquitin and are thereby targeted for autophagic clearance. Cytosolic Listeria monocytogenes actA mutants are associated with polyubiquitin and p62 and cleared by an autophagic mechanism (Yoshikawa et al., 2009). During infection of epithelial cells, a small population of Salmonella enterica access the cytosol, and subsequently co-localize with ubiquitin, LC3 and p62 (Birmingham et al., 2006; Zheng et al., 2009). Further analysis demonstrated that recruitment of LC3 and efficient autophagic clearance of S. enterica requires the protein NDP52 (Thurston et al., 2009). Like p62 and NBR1, NDP52 is an adaptor protein that directly binds ubiquitin and LC3. Knockdown studies indicate that p62 and NDP52 are recruited independently to S. enterica, and that the two proteins do not co-localize but surround the bacterium in distinct microdomains (Cemma et al., 2011). Ubiquitin also associates with a proportion of intracellular M. marinum, a mycobacterial species that escapes the phagosome to reside in the cytosol (Collins et al., 2009). Within 24 h, ubiquitin-associated M. marinum localizes to LAMP1-positive double-membrane vacuoles. Bacterial sequestration occurs in an Atg5-independent manner, but the fate of these bacteria was not determined. A number of key questions remain regarding the mechanisms underlying ubiquitin-mediated autophagic clearance of bacteria, including the identity of the ubiquitin ligase(s), the ubiquitylated substrate(s) and the adaptor proteins required for this process.
Mechanism of Ub-Peptide Action
We have used biochemical and genetic approaches to better understand the bactericidal activity of lysosomal Ub-peptides and their interactions with mycobacteria. Many of our studies utilize the synthetic model Ub-peptide Ub2, which was originally identified by Kieffer et al. (2003) as an antifungal peptide that also had activity against some Gram-positive bacteria. Our model that Ub-peptides contribute to the mycobactericidal capacity of the lysosome is dependent on the ability of these peptides to function at low pH. Our in vitro experiments with the synthetic bactericidal ubiquitin peptide Ub2 are performed in standard mycobacterial 7H9 media at pH 6.6. Immature and mature mycobacteria-containing phagosomes are at pH 6.2 and pH 5–5.5, respectively (Sturgill-Koszycki et al., 1994; Schaible et al., 1998; Via et al., 1998; de Chastellier and Thilo, 2006). To determine whether pH affected the bactericidal capacity of Ub2, we treated bacteria in 7H9 media at pH 5, pH 5.5, pH 6, and pH 6.6. Ub2 was functional at each of these pH and exhibited the greatest bactericidal activity at pH 5.5 and pH 6 (Purdy et al., 2009). These data demonstrate that Ub-peptides are functional at the physiological relevant pH of the phagolysosomal pathway.
Since Ub2 is a 12-aa positively charged peptide, we predicted that it might behave like a cationic antimicrobial peptide. These conserved components of innate immunity are thought to act by disrupting the bacterial membrane. To directly measure mycobacterial membrane integrity, an assay based upon a pH-sensitive fluorophore was performed on intact bacteria (Figure 3). The cytoplasm of M. smegmatis was labeled with the membrane-permeable probe 5-chloromethylfluorescein diacetate (CMFDA), which is processed into a membrane-impermeable form following uptake. The fluorescence emission of fluorescein at 520 nm is pH-sensitive upon excitation at 490 nm, but is pH-insensitive when excited at 450 nm. The intracellular pH of the labeled bacteria was neutral, and the bacteria were placed in suspension buffered at pH 5.5. The loss of membrane integrity was therefore measured as a shift in fluorescence upon exposure of the intracellular fluorophore to the extracellular pH. As a positive control bacteria were treated for 5 min with the ionophore nigericin. This resulted in a fluorescence emission shift indicating that the intracellular pH had dropped from pH 7 to pH 5.5 (Figure 3). A 5-min treatment with Ub2 resulted in a drop of intracellular pH to pH 5.8. These experiments show that Ub2 treatment impairs membrane function and results in exposure to external pH (Figure 3 and Purdy et al., 2009). Ongoing work in our lab indicates that Ub2 interacts with membranes and adopts a stable structure in membrane mimetic environments. Peptide charge and primary sequence is important for both stable insertion into membranes and bactericidal activity. While there is no significant homology between ubiquitin and the S30/ubiquicidin domain of Fau, both proteins are highly composed of positively charged amino acids. Therefore, the peptides generated from ubiquitin and Fau are likely to be cationic, a property that is likely to be important for their mycobactericidal function. Our data on Ub2 are consistent with the properties of cationic antimicrobial peptides that are thought to mediate bacterial killing through multimerizing and inserting into membranes. Some antimicrobial peptides may also directly inhibit intracellular processes such as nucleic acid or protein synthesis (Brogden, 2005), but we do not have any evidence to suggest that Ub2 functions in this manner.
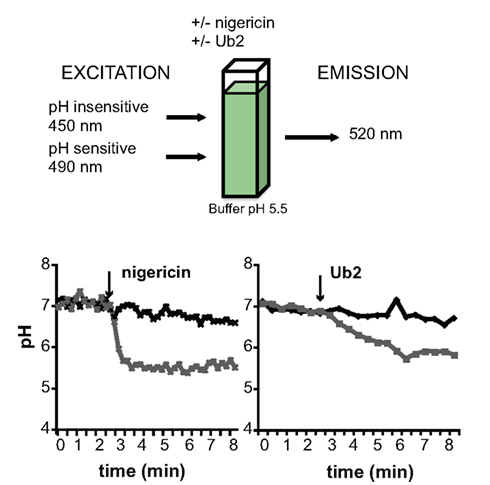
Figure 3. Mycobactericidal activity of Ub-peptides. A kinetic assay was developed that depends upon the pH-sensitive vs. pH-insensitive excitation wavelengths of carboxyfluorescein. CMFDA-labeled M. smegmatis mc2 155 was resuspended in buffer (pH 5.5). The fluorescence emission at 520 nm measured at excitation wavelengths of 450 and 490 nm over time. At the time point indicated by the arrow, the bacteria were treated with either 10 mM nigericin (left) or 100 μM Ub2 (right). Treated and untreated samples are shown in black diamonds and gray squares, respectively. The excitation ratio was converted to pH by regression. Figure adapted from Purdy et al. (2009).
We hypothesized that isolation of bacterial mutants with increased resistance to the bactericidal ubiquitin-derived peptide Ub2 would provide insight into its mechanism of action. We screened a transposon mutant library for M. smegmatis mutants that exhibited increased resistance to the bactericidal activity of the synthetic ubiquitin-derived peptide Ub2 (Purdy et al., 2009). The Ub2-resistant mutants were more resistant to macrophage killing, consistent with our observation that Ub-peptides contribute to lysosomal killing. A unifying feature of the Ub2-resistant mutants was that they exhibited reduced outer membrane permeability relative to wild type bacteria. To determine whether reduced membrane permeability in Ub2-resistant mutants rendered these bacteria more resistant to other antimicrobial peptides, the M. smegmatis Ub2-resistant mutants were exposed to the cathelicidin LL-37, an LL-37-derived peptide with increased mycobactericidal activity (LL-37 18–37), hepcidin, and granulysin-derived peptide granF2. The four Ub2-resistant M. smegmatis mutants were also resistant to the bactericidal action of these other antimicrobial peptides (Purdy et al., 2009). Our data support a model where the access of host antimicrobial peptides, including Ub-peptides, to the mycobacterial inner membrane depends on outer membrane permeability. Once these peptides gain access to the inner membrane we propose that they multimerize, insert into the membrane and form pores or otherwise disrupt membrane integrity of the bacterium. Membrane damage likely exposes the bacterium to external conditions such as the low pH of the lysosome and promotes cytoplasmic leakage over time that contributes to bacterial death. The contribution of low membrane permeability to the intrinsic resistance of mycobacteria has long been appreciated (Barry, 2001). Our findings emphasize the contribution of this intrinsic resistance to host antimicrobial compounds and the intracellular environment.
Conclusion
Autophagy has emerged as a key component in the host defense against infection by intracellular pathogens. In the context of M. tuberculosis infections, autophagy plays two roles. First, autophagy provides a mechanism by which cytoplasmic material is delivered to the lysosome where digest of some proteins releases mycobactericidal peptides. Second, autophagy promotes fusion of the M. tuberculosis-containing phagosome with the lysosome and thus delivers the pathogen to a compartment enriched with mycobactericidal peptides. Our work is focused on the mycobactericidal properties of Ub-peptides, namely their ability to disrupt mycobacterial membranes. In the lysosome, it is likely that Ub-peptides act synergistically with other mycobactericidal peptides generated in the lysosome and the complex mixture of lysosomal hydrolases to promote bacterial killing. We anticipate that future studies will fully elucidate the Ub-peptide mechanism of action and determine whether this mechanism is shared among lysosomal mycobactericidal peptides.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the Purdy lab is supported by grants to Georgiana E. Purdy from the NIH (AI079399), the Collins Medical Trust and the Medical Research Foundation at OHSU.
References
Alonso, S., Pethe, K., Russell, D. G., and Purdy, G. E. (2007). Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. U.S.A. 104, 6031–6036.
Barry, C. E. (2001). Interpreting cell wall “virulence factors” of Mycobacterium tuberculosis. Trends Microbiol. 9, 237–241.
Birmingham, C. L., Smith, A. C., Bakowski, M. A., Yoshimori, T., and Brumell, J. H. (2006). Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281, 11374–11383.
Bjorkoy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., Stenmark, H., and Johansen, T. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614.
Bowers, K., Piper, S. C., Edeling, M. A., Gray, S. R., Owen, D. J., Lehner, P. J., and Luzio, J. P. (2006). Degradation of endocytosed epidermal growth factor and virally ubiquitinated major histocompatibility complex class I is independent of mammalian ESCRTII. J. Biol. Chem. 281, 5094–5105.
Brogden, K. A. (2005). Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250.
Brooks, M. N., Rajaram, M. V., Azad, A. K., Amer, A. O., Valdivia-Arenas, M. A., Park, J. H., Nunez, G., and Schlesinger, L. S. (2010). NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cell. Microbiol. doi: 10.1111/j.1462-5822.2010.01544.x. (Accepted for publication).
Canadien, V., Tan, T., Zilber, R., Szeto, J., Perrin, A. J., and Brumell, J. H. (2005). Cutting edge: microbial products elicit formation of dendritic cell aggresome-like induced structures in macrophages. J. Immunol. 174, 2471–2475.
Cemma, M., Kim, P. K., and Brumell, J. H. (2011). The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy 7, 22–26.
Chan, J., Xing, Y., Magliozzo, R. S., and Bloom, B. R. (1992). Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 175, 1111–1122.
Chen, Y., Azad, M. B., and Gibson, S. B. (2009). Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 16, 1040–1052.
Ciechanover, A. (2009). Tracing the history of the ubiquitin proteolytic system: the pioneering article. Biochem. Biophys. Res. Commun. 387, 1–10.
Clemens, D. L., and Horwitz, M. A. (1996). The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J. Exp. Med. 184, 1349–1355.
Clemens, D. L., Lee, B. Y., and Horwitz, M. A. (2000). Deviant expression of Rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infect. Immun. 68, 2671–2684.
Collins, C. A., De Maziere, A., van Dijk, S., Carlsson, F., Klumperman, J., and Brown, E. J. (2009). Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog. 5, e1000430. doi: 10.1371/journal.ppat.1000430
Cooper, A. M., Dalton, D. K., Stewart, T. A., Griffin, J. P., Russell, D. G., and Orme, I. M. (1993). Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 178, 2243–2247.
Cooper, A. M., Segal, B. H., Frank, A. A., Holland, S. M., and Orme, I. M. (2000). Transient loss of resistance to pulmonary tuberculosis in p47(phox−/−) mice. Infect. Immun. 68, 1231–1234.
Darwin, K. H., Ehrt, S., Gutierrez-Ramos, J. C., Weich, N., and Nathan, C. F. (2003). The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302, 1963–1966.
de Chastellier, C., and Thilo, L. (2006). Cholesterol depletion in Mycobacterium avium-infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome-derived autophagic vacuoles. Cell. Microbiol. 8, 242–256.
Delgado, M. A., Elmaoued, R. A., Davis, A. S., Kyei, G., and Deretic, V. (2008). Toll-like receptors control autophagy. EMBO J. 27, 1110–1121.
Deretic, V., and Levine, B. (2009). Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5, 527–549.
Ehrt, S., and Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 11, 1170–1178.
Filimonenko, M., Isakson, P., Finley, K. D., Anderson, M., Jeong, H., Melia, T. J., Bartlett, B. J., Myers, K. M., Birkeland, H. C., Lamark, T., Krainc, D., Brech, A., Stenmark, H., Simonsen, A., and Yamamoto, A. (2010). The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 38, 265–279.
Flynn, J. L., Chan, J., Triebold, K. J., Dalton, D. K., Stewart, T. A., and Bloom, B. R. (1993). An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178, 2249–2254.
Fratti, R. A., Chua, J., Vergne, I., and Deretic, V. (2003). Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc. Natl. Acad. Sci. U.S.A. 100, 5437–5442.
Fujita, N., Itoh, T., Omori, H., Fukuda, M., Noda, T., and Yoshimori, T. (2008). The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19, 2092–2100.
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766.
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., Yokoyama, M., Mishima, K., Saito, I., Okano, H., and Mizushima, N. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889.
Hiemstra, P. S., van den Barselaar, M. T., Roest, M., Nibbering, P. H., and van Furth, R. (1999). Ubiquicidin, a novel murine microbicidal protein present in the cytosolic fraction of macrophages. J. Leukoc. Biol. 66, 423–428.
Huang, J., Canadien, V., Lam, G. Y., Steinberg, B. E., Dinauer, M. C., Magalhaes, M. A., Glogauer, M., Grinstein, S., and Brumell, J. H. (2009). Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. U.S.A. 106, 6226–6231.
Indrigo, J., Hunter, R. L. J., and Actor, J. K. (2003). Cord factor trehalose 6,6′-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology 149, 2049–2059.
Iwata, A., Riley, B. E., Johnston, J. A., and Kopito, R. R. (2005). HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280, 40282–40292.
Johnston, J. A., Ward, C. L., and Kopito, R. R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898.
Kang, P. B., Azad, A. K., Torrelles, J. B., Kaufman, T. M., Beharka, A., Tibesar, E., DesJardin, L. E., and Schlesinger, L. S. (2005). The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J. Exp. Med. 202, 987–999.
Kawaguchi, Y., Kovacs, J. J., McLaurin, A., Vance, J. M., Ito, A., and Yao, T. P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738.
Kieffer, A. E., Goumon, Y., Ruh, O., Chasserot-Golaz, S., Nullans, G., Gasnier, C., Aunis, D., and Metz-Boutigue, M. H. (2003). The N- and C-terminal fragments of ubiquitin are important for the antimicrobial activities. FASEB J. 17, 776–778.
Kirisako, T., Ichimura, Y., Okada, H., Kabeya, Y., Mizushima, N., Yoshimori, T., Ohsumi, M., Takao, T., Noda, T., and Ohsumi, Y. (2000). The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 151, 263–276.
Kirkin, V., Lamark, T., Sou, Y. S., Bjorkoy, G., Nunn, J. L., Bruun, J. A., Shvets, E., McEwan, D. G., Clausen, T. H., Wild, P., Bilusic, I., Theurillat, J. P., Overvatn, A., Ishii, T., Elazar, Z., Komatsu, M., Dikic, I., and Johansen, T. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516.
Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., Mizushima, N., Iwata, J., Ezaki, J., Murata, S., Hamazaki, J., Nishito, Y., Iemura, S., Natsume, T., Yanagawa, T., Uwayama, J., Warabi, E., Yoshida, H., Ishii, T., Kobayashi, A., Yamamoto, M., Yue, Z., Uchiyama, Y., Kominami, E., and Tanaka, K. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163.
Komatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., Ezaki, J., Mizushima, N., Ohsumi, Y., Uchiyama, Y., Kominami, E., Tanaka, K., and Chiba, T. (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434.
Lauwers, E., Jacob, C., and Andre, B. (2009). K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J. Cell Biol. 185, 493–502.
Lenk, S. E., Dunn, W. A. J., Trausch, J. S., Ciechanover, A., and Schwartz, A. L. (1992). Ubiquitin-activating enzyme, E1, is associated with maturation of autophagic vacuoles. J. Cell Biol. 118, 301–308.
Liu, P. T., Stenger, S., Li, H., Wenzel, L., Tan, B. H., Krutzik, S. R., Ochoa, M. T., Schauber, J., Wu, K., Meinken, C., Kamen, D. L., Wagner, M., Bals, R., Steinmeyer, A., Zugel, U., Gallo, R. L., Eisenberg, D., Hewison, M., Hollis, B. W., Adams, J. S., Bloom, B. R., and Modlin, R. L. (2006). Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311, 1770–1773.
Liu, P. T., Stenger, S., Tang, D. H., and Modlin, R. L. (2007). Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J. Immunol. 179, 2060–2063.
Low, P., Doherty, F. J., Fellinger, E., Sass, M., Mayer, R. J., and Laszlo, L. (1995). Related organelles of the endosome-lysosome system contain a different repertoire of ubiquitinated proteins in Sf9 insect cells. FEBS Lett. 368, 125–131.
MacGurn, J. A., and Cox, J. S. (2007). A genetic screen for Mycobacterium tuberculosis mutants defective for phagosome maturation arrest identifies components of the ESX-1 secretion system. Infect. Immun. 75, 2668–2678.
MacMicking, J. D., North, R. J., LaCourse, R., Mudgett, J. S., Shah, S. K., and Nathan, C. F. (1997). Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 94, 5243–5248.
MacMicking, J. D., Taylor, G. A., and McKinney, J. D. (2003). Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302, 654–659.
McCullough, J., Clague, M. J., and Urbe, S. (2004). AMSH is an endosome-associated ubiquitin isopeptidase. J. Cell Biol. 166, 487–492.
Mizuno, E., Kobayashi, K., Yamamoto, A., Kitamura, N., and Komada, M. (2006). A deubiquitinating enzyme UBPY regulates the level of protein ubiquitination on endosomes. Traffic 7, 1017–1031.
Nakatogawa, H., Ichimura, Y., and Ohsumi, Y. (2007). Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178.
Pandey, U. B., Nie, Z., Batlevi, Y., McCray, B. A., Ritson, G. P., Nedelsky, N. B., Schwartz, S. L., DiProspero, N. A., Knight, M. A., Schuldiner, O., Padmanabhan, R., Hild, M., Berry, D. L., Garza, D., Hubbert, C. C., Yao, T. P., Baehrecke, E. H., and Taylor, J. P. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863.
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., Overvatn, A., Bjorkoy, G., and Johansen, T. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145.
Pethe, K., Swenson, D. L., Alonso, S., Anderson, J., Wang, C., and Russell, D. G. (2004). Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc. Natl. Acad. Sci. U.S.A. 101, 13642–13647.
Ponpuak, M., Davis, A. S., Roberts, E. A., Delgado, M. A., Dinkins, C., Zhao, Z., Virgin, H. W. t., Kyei, G. B., Johansen, T., Vergne, I., and Deretic, V. (2010). Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32, 329–341.
Purdy, G. E., Niederweis, M., and Russell, D. G. (2009). Decreased outer membrane permeability protects mycobacteria from killing by ubiquitin-derived peptides. Mol. Microbiol. 73, 844–857.
Raiborg, C., and Stenmark, H. (2009). The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452.
Reggiori, F., and Pelham, H. R. (2001) Sorting of proteins into multivesicular bodies: ubiquitin-dependent and -independent targeting. EMBO J. 20, 5176–5186.
Rohde, K., Yates, R. M., Purdy, G. E., and Russell, D. G. (2007). Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219, 37–54.
Row, P. E., Prior, I. A., McCullough, J., Clague, M. J., and Urbe, S. (2006). The ubiquitin isopeptidase UBPY regulates endosomal ubiquitin dynamics and is essential for receptor down-regulation. J. Biol. Chem. 281, 12618–12624.
Sanjuan, M. A., Dillon, C. P., Tait, S. W., Moshiach, S., Dorsey, F., Connell, S., Komatsu, M., Tanaka, K., Cleveland, J. L., Withoff, S., and Green, D. R. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257.
Schaible, U. E., Sturgill-Koszycki, S., Schlesinger, P. H., and Russell, D. G. (1998). Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 160, 1290–1296.
Scherz-Shouval, R., and Elazar, Z. (2010). Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. doi: 10.1016/j.tibs.2010.07.007. (Accepted for publication).
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760.
Schlesinger, L. S., Azad, A. K., Torrelles, J. B., Roberts, E., Vergne, I., and Deretic, V. (2008). “Determinants of Phagocytosis, Phagosome Biogenesis and Autophagy for Mycobacterium tuberculosis,” in Handbook of Tuberculosis: Immunology and Cell Biology, eds S. H. E. Kaufmann and W. J. Britton (Weinheim: Wiley-VCH), 1–22.
Schwartz, A. L., Brandt, R. A., Geuze, H., and Ciechanover, A. (1992). Stress-induced alterations in autophagic pathway: relationship to ubiquitin system. Am. J. Physiol. 262, C1031–C1038.
Schwartz, A. L., Ciechanover, A., Brandt, R. A., and Geuze, H. J. (1988). Immunoelectron microscopic localization of ubiquitin in hepatoma cells. EMBO J. 7, 2961–2966.
Shui, W., Sheu, L., Liu, J., Smart, B., Petzold, C. J., Hsieh, T. Y., Pitcher, A., Keasling, J. D., and Bertozzi, C. R. (2008). Membrane proteomics of phagosomes suggests a connection to autophagy. Proc. Natl. Acad. Sci. U.S.A. 105, 16952–16957.
Simonsen, A., Birkeland, H. C., Gillooly, D. J., Mizushima, N., Kuma, A., Yoshimori, T., Slagsvold, T., Brech, A., and Stenmark, H. (2004) Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 117, 4239–4251.
Sow, F. B., Florence, W. C., Satoskar, A. R., Schlesinger, L. S., Zwilling, B. S., and Lafuse, W. P. (2007). Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J. Leukoc. Biol. 82, 934–945.
Stallings, C. L., and Glickman, M. S. (2010). Is Mycobacterium tuberculosis stressed out? A critical assessment of the genetic evidence. Microbes Infect. 12, 1091–1101.
Sturgill-Koszycki, S., Schlesinger, P. H., Chakraborty, P., Haddix, P. L., Collins, H. L., Fok, A. K., Allen, R. D., Gluck, S. L., Heuser, J., and Russell, D. G. (1994). Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263, 678–681.
Swaminathan, S., Amerik, A. Y., and Hochstrasser, M. (1999). The Doa4 deubiquitinating enzyme is required for ubiquitin homeostasis in yeast. Mol. Biol. Cell 10, 2583–2594.
Szeto, J., Kaniuk, N. A., Canadien, V., Nisman, R., Mizushima, N., Yoshimori, T., Bazett-Jones, D. P., and Brumell, J. H. (2006). ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2, 189–199.
Tanida, I., Sou, Y. S., Ezaki, J., Minematsu-Ikeguchi, N., Ueno, T., and Kominami, E. (2004). HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 279, 36268–36276.
Thrower, J. S., Hoffman, L., Rechsteiner, M., and Pickart, C. M. (2000). Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102.
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221.
Tiwari, S., Choi, H. P., Matsuzawa, T., Pypaert, M., and MacMicking, J. D. (2009). Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P(2) and PtdIns(3,4,5)P(3) promotes immunity to mycobacteria. Nat. Immunol. 10, 907–917.
Torrelles, J. B., Azad, A. K., and Schlesinger, L. S. (2006). Fine discrimination in the recognition of individual species of phosphatidyl-myo-inositol mannosides from Mycobacterium tuberculosis by C-type lectin pattern recognition receptors. J. Immunol. 177, 1805–1816.
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhaes, J. G., Yuan, L., Soares, F., Chea, E., Le Bourhis, L., Boneca, I. G., Allaoui, A., Jones, N. L., Nunez, G., Girardin, S. E., and Philpott, D. J. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62.
Vergne, I., Fratti, R. A., Hill, P. J., Chua, J., Belisle, J., and Deretic, V. (2004). Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol. Biol. Cell 15, 751–760.
Via, L. E., Deretic, D., Ulmer, R. J., Hibler, N. S., Huber, L. A., and Deretic, V. (1997). Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J. Biol. Chem. 272, 13326–13331.
Via, L. E., Fratti, R. A., McFalone, M., Pagan-Ramos, E., Deretic, D., and Deretic, V. (1998). Effects of cytokines on mycobacterial phagosome maturation. J. Cell Sci. 111, 897–905.
WHO. (2010). Fact Sheet No. 104, Available at: http://www.who.int/mediacentre/factsheets/fs104/en/
Xu, Y., Jagannath, C., Liu, X. D., Sharafkhaneh, A., Kolodziejska, K. E., and Eissa, N. T. (2007). Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27, 135–144.
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., Mimuro, H., Nakagawa, I., Yanagawa, T., Ishii, T., Kakizuka, A., Sztul, E., Chakraborty, T., and Sasakawa, C. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240.
Yuk, J. M., Shin, D. M., Lee, H. M., Yang, C. S., Jin, H. S., Kim, K. K., Lee, Z. W., Lee, S. H., Kim, J. M., and Jo, E. K. (2009). Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6, 231–243.
Zhao, Z., Fux, B., Goodwin, M., Dunay, I. R., Strong, D., Miller, B. C., Cadwell, K., Delgado, M. A., Ponpuak, M., Green, K. G., Schmidt, R. E., Mizushima, N., Deretic, V., Sibley, L. D., and Virgin, H. W. (2008). Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 4, 458–469.
Keywords: Mycobacterium, tuberculosis, autophagy, ubiquitin, Ub-peptides, lysosome
Citation: Purdy GE (2011) Taking out TB – lysosomal trafficking and mycobactericidal ubiquitin-derived peptides. Front. Microbio. 2:7. doi: 10.3389/fmicb.2011.00007
Received: 06 December 2010;
Accepted: 13 January 2011;
Published online: 31 January 2011.
Edited by:
Daoguo Zhou, Purdue University, USAReviewed by:
Jordi Torrelles, Ohio State University, USAJean Celli, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USA
Srinand Sreevastan, University of Minnesota, USA
Martin Voskuil, University of Colorado Denver, USA
Copyright: © 2011 Purdy. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Georgiana E. Purdy, Department of Molecular Microbiology and Immunology, Oregon Health and Sciences University, 3181 S.W. Sam Jackson Park Road, Mail Code L220, Portland, OR 97239, USA. e-mail:cHVyZHlnQG9oc3UuZWR1