
- Department of Pathology and Comprehensive Cancer Center, University of Michigan Medical School, Ann Arbor, MI, USA
Pattern recognition receptors (PRRs) play a crucial role in both the detection of pathogens and the activation of the innate immune system. Nod-like receptors (NLR) family members are cytosolic PRRs that sense bacterial products or endogenous danger signals. Recent evidence suggests that NLRs contribute to the detection of Salmonella through the activation of inflammasomes, molecular platforms that promotes the maturation of the proinflammatory cytokines IL-1β and IL-18. During enteric Salmonella infection the activation of caspase-1 and the production of IL-1β and IL-18 result in a protective host response. In macrophages, the activation of caspase-1 induced by Salmonella is mainly mediated by the NLR family member NLRC4 that senses cytosolic flagellin. Recent data suggest that an effective innate immune response against Salmonella requires the engagement of multiple inflammasomes in both hematopoietic and non-hematopoietic cell lineages. Further understanding of the innate immune response mediated by inflammasomes should provide new insights into the mechanisms of host defense and the pathogenesis of inflammatory diseases.
Salmonella
Salmonella enterica consists of more than 2500 serovars. S. enterica serovar Typhi and Paratyphi cause typhoid in humans, a disease characterized by fever, systemic infection, and gastroenteritis. S. enterica serovar Typhi and Paratyphi are estimated to affect more than 16 million people worldwide and cause 600,000 deaths/year. Several non-typhoidal S. enterica serovars (NTS) can cause enterocolitis in humans. In infants and immunocompromised patients, NTS can disseminate and cause potentially lethal bacteremia. In the United States, NTS are the leading cause of foodborne disease with an estimated cost of 2 billion dollars/year (Grassl et al., 2008; Santos et al., 2009).
Mouse Model of Salmonella Infections
Salmonella enterica serovar Typhimurium (herein referred to as Salmonella) causes enterocolitis in humans. In mice, Salmonella cause a systemic infection that resembles human typhoid fever. During the natural infection, Salmonella enters the host via the oral route and overcome the defense provided by mucus and the endogenous microbiota. Then Salmonella crosses the epithelial barrier, mainly by invading M cells (Hase et al., 2009; Knoop et al., 2009), an intestinal epithelial cell type specialized in the transport of particles from the gut lumen to the lamina propria. Once in the lamina propria, Salmonella can be taken up by dendritic cells (Uematsu and Akira, 2009; DC) and transported to Peyer patches and other gut-associated lymphoid tissues, from which Salmonella disseminates to other organs such as the liver and/or spleen where they can survive within phagocytic cells. An alternative route for Salmonella uptake via DCs that sample the intestinal lumen has been described (Rescigno et al., 2001; Niess et al., 2005). In resistant mouse strains, Salmonella causes chronic infections (Monack et al., 2004; Grassl et al., 2008) while in susceptible strains Salmonella is lethal. Commensal bacteria have an important role in preventing Salmonella from colonizing the intestine and causing acute intestinal inflammation (Hapfelmeier and Hardt, 2005). Pretreatment with a single dose of the antibiotic streptomycin reduces the presence of commensal bacteria and renders otherwise resistant mice susceptible to Salmonella (Hapfelmeier and Hardt, 2005). These findings have opened the way to studies aimed at investigating both the bacterial virulence factors and the host response in the acute intestinal inflammation triggered by Salmonella in mice.
Bacteria Virulence Factors
Bacterial secretion systems are molecular machineries used by pathogenic bacteria to deliver effectors proteins inside the host cell and manipulate cell behavior. Salmonella encodes two type III protein secretion systems which manipulate host cell behavior through the delivery of specific effectors proteins. One of the systems is encoded within the Salmonella pathogenicity island 1 (SPI-1) and is important in the initial phase of infection (Grassl and Finlay, 2008); in agreement effectors proteins encoded within the SPI-1 have been implicated in cytoskeletal rearrangement responsible for bacterial uptake by epithelial cells, a step required for the bacteria to breach the intestinal barrier (Grassl and Finlay, 2008). The other is encoded within the SPI-2 and has an essential role in bacterial survival during the systemic phase of infection (Monack et al., 2004). Effector proteins encoded within the SPI-2 have been shown to be essential for intracellular growth and survival inside macrophages (Grassl and Finlay, 2008).
The Host Response
Initial studies aimed at identifying host factors responsible for susceptibility to pathogens identified natural resistance-associated macrophage protein 1 (NRAMP1) as a crucial gene in natural resistance to infection by Salmonella, Mycobacterium tuberculosis, and Leishmania donovani. Successive studies have greatly expanded our knowledge about the host factors responsible for defense against Salmonella, a topic that has been reviewed in detail elsewhere (Raupach and Kaufmann, 2001). Here, I will focus on the role of pattern recognition receptors (PRRs) belonging to the Toll-like receptors (TLR) and Nod-like receptors (NLR) in mounting protective host responses against Salmonella.
Role of TLR4 and Tlr5 in Salmonella Infection
The initial detection of microbes is achieved though the detection of conserved microbial structures by PRRs. PRRs comprise an array of sensors present in different cell types and cellular location that control the activation of signaling pathways ultimately aimed at the elimination of invading microbes (Takeuchi and Akira, 2010). TLRs localize either at the cell surface or within endosomes, in contrast to NLRs that are located in the host cytosol.
Toll-like receptor 4 recognizes bacterial lipopolysaccharide (LPS), a major constituent of the outer membrane of Gram-negative bacteria (Takeuchi and Akira, 2010), such as Salmonella. The recognition of LPS by TLR4 is assisted by several proteins such as CD14, LPS-binding protein (LBP), and MD-2. Knockout mice revealed a crucial role of TLR4, LBP, and CD14 in host defense during the systemic phase of Salmonella infection (Bernheiden et al., 2001). The increased susceptibility to Salmonella was attributed to a delayed recruitment of neutrophils allowing increased bacterial growth in liver macrophages (Vazquez-Torres et al., 2004), and not to defects in activation of the adaptive immune response (Ko et al., 2009).
Toll-like receptor 5 is a sensor of extracellular flagellin, the main component of the flagella, an appendage that is used by bacteria for movement. TLR5 is expressed on the basal surface of intestinal epithelial cells and in a specific population of intestinal DCs (Uematsu et al., 2006). Initial experiments using intestinal epithelial cell lines indicate that flagellin, but not LPS, is a powerful inducer of the proinflammatory cytokine IL-8 (Zeng et al., 2003). Experiments with polarized intestinal epithelial cell demonstrate that stimulation with flagellin induces an inflammatory response only when in contact with the basolateral surface (Gewirtz et al., 2001a,b). Together these data suggest that in the intestine, flagellin can activate TLR5 only if it breaches the intestinal epithelial barrier, thus potentially preventing the induction of an inflammatory response against flagellated intestinal commensal bacteria (Vijay-Kumar et al., 2008).
While there is evidence that flagellin can trigger the recruitment of immature DCs and other leukocytes through TLR5 in human epithelial cell lines (Sierro et al., 2001), the expression of TLR5 in mouse intestinal epithelial cells is low. This is in contrast to specific populations of lamina propria DCs (CD11chi CD11bhi) isolated from the small intestine that express high levels of TLR5 (Uematsu et al., 2006). Importantly, while conventional DCs, macrophages, and lamina propria macrophages do not express TLR5 and fail to respond to flagellin, lamina propria DCs stimulated with flagellin produce elevated levels of IL-6 and IL-12 in a TLR5-dependent manner (Uematsu et al., 2006). The response of lamina propria DCs was specific for flagellin, in that lamina propria DCs did not respond to stimulation with other TLR ligands such as LPS (Uematsu et al., 2006). So it seems that some lamina propria DCs are equipped to mount a proinflammatory response against infectious flagellated bacteria such as Salmonella. Surprisingly, TLR5-deficient mice are less susceptible than wild-type mice to infection with Salmonella (Uematsu et al., 2006). Given that DCs are important for transporting bacteria from the lamina propria to the mesenteric lymph nodes (Uematsu and Akira, 2009), it has been suggested that in response to Salmonella infection TLR5 plays an important role in the maturation of lamina propria DCs and subsequent migration to mesenteric lymph nodes (Uematsu et al., 2006). These data suggest that during Salmonella infection the host response mediated by TLR5 in lamina propria DCs is hijacked by the bacterium using intestinal DCs as Trojan horses to cause systemic infection.
Role of NLRs in Salmonella Infection
Nucleotide-binding oligomerization domain 1 (NOD1) and NOD2 sense bacterial molecules produced during the synthesis, degradation, and remodeling of peptidoglycan (PGN). NOD2 is expressed in a variety of different cells types including macrophages, DCs, and Paneth cells. NOD2 senses muramyl dipeptide (MDP), a moiety found in PGN of nearly all Gram-positive and Gram-negative organisms. Genetic studies revealed that several NOD2 variants are associated with susceptibility to Crohn’s disease (CD), a chronic disorder characterized by transmural inflammation of the intestine, particularly in the distal ileum. NOD1 is ubiquitously expressed and recognizes γ-D-glutamyl-meso-diaminopimelic acid that is found in PGN from most Gram-negative bacteria and certain Gram-positive bacteria. Stimulation of NOD1 or NOD2 results in the activation of the adaptor protein receptor-interacting protein 2 (RIP2; also called RICK) which promotes the activation of NF-κB and MAPKs pathway leading to transcription of numerous genes involved in inflammation and host defense. NOD1 and NOD2 are involved in the sensing of several pathogenic bacteria, including Salmonella, and has been reviewed in detail elsewhere (Inohara and Nunez, 2003; Franchi et al., 2009b). The first attempt to characterize the role of NOD2 in Salmonella infection was performed in human DCs carrying a homozygous loss-of-function NOD2 L1007fs mutation and showed a decreased production of proinflammatory cytokines compared to DCs from healthy individuals (Salucci et al., 2008). Subsequent studies investigated the role of NOD1 and NOD2 in epithelial cells and found that the proinflammatory response induced by Salmonella was dependent on the SPI-1 and a set of effectors proteins (including SopE and SopE2), but was independent of Rip2 (Bruno et al., 2009). However, recent evidence suggests that NOD1 and NOD2 may restrict bacterial replication independent of NF-κB activation. For example, NOD1 and NOD2 can sense certain bacteria through the stimulation of the autophagic response in a Rip2- and NF-κB independent fashion (Travassos et al., 2010). In addition, exogenous stimulation of NOD2 via MDP further enhances both the autophagic response and Salmonella killing (Homer et al., 2010). In mice, infection with wild-type Salmonella failed to reveal a major role for NOD1 or NOD2 in host defense (Le Bourhis et al., 2009; Geddes et al., 2010). However, a role for NOD1 and NOD2 was revealed when mice were infected with a bacterial mutant deficient in SPI-1 or when Salmonella was cultured in a manner that favors the expression of SPI-2 (Le Bourhis et al., 2009; Geddes et al., 2010), suggesting that the contribution of NOD1 and NOD2 in the host defense response against Salmonella is dependent on the expression of Salmonella virulence factors.
Inflammasomes in Salmonella Infection
Inflammasomes are molecular complexes that regulate the activation of the proteolytic enzyme caspase-1 (Franchi et al., 2009a). Four different inflammasome has been functionally characterized. The NLRP1 inflammasome is activated by MDP and anthrax lethal toxin. The NLRC4 is activated by cytosolic flagellin. The NLRP3-inflammasome is activated by a variety of stimuli of microbial origins as well as particulate matter and danger signals. The AIM2 inflammasome is activated by cytosolic DNA (Franchi et al., 2009a).
In macrophages, Salmonella induces the activation of caspase-1 necessary for the maturation of the proinflammatory cytokines IL-1β and IL-18. The activation of caspase-1 also induces a form of cell death called pyroptosis that has feature of both apoptosis (such as the activation of caspases) and necrosis (such as the permeabilization of the cellular membrane; Fink and Cookson, 2007) and that may play a role in host defense independent of cytokine production (Miao et al., 2010a).
Initial studies aimed at identifying the inflammasome responsible for the activation of caspase-1 in macrophages infected with Salmonella revealed a crucial role of NLRC4 (also called IPAF; Mariathasan et al., 2004). Subsequently, we and others have found that NLRC4 senses cytosolic flagellin. In fact, macrophages infected with Salmonella mutants lacking flagellin possess an impaired ability to induce caspase-1 activation, IL-1β production, and cell death (Amer et al., 2006; Franchi et al., 2006; Miao et al., 2006). In addition, macrophages infected with a Salmonella mutant carrying a point mutation in the C-terminal region of flagellin are impaired in their ability to induce the activation of the NLRC4-inflammasome (Franchi et al., 2006). Furthermore, cytosolic delivery of purified flagellin, but not other microbial molecules, activates caspase-1 in an NLRC4-dependent manner (Amer et al., 2006; Franchi et al., 2006; Miao et al., 2006). Consistent with the role of cytosolic flagellin, TLR5-deficient macrophages show no impairment in the activation of caspase-1 induced by Salmonella or cytosolic flagellin (Franchi et al., 2006; Miao et al., 2006). Subsequent studies using macrophages infected with Legionella pneumophila (Amer et al., 2006; Zamboni et al., 2006) or Pseudomonas aeruginosa (Franchi et al., 2007; Miao et al., 2008) and their mutants lacking flagellin confirmed the role of NLRC4 in the activation of caspase-1 induced by cytosolic flagellin. In the case of L. pneumophila, but not P. aeruginosa or Salmonella, the recognition of flagellin by NLRC4 has been suggested to be mediated by the NLR family member NAIP5 (Lightfield et al., 2008). However NAIP5 can restrict L. pneumophila growth independently of caspase-1 activation suggesting that the host defense response mediated by NAIP5 proceeds independently of the NLRC4-inflammasome (Lamkanfi et al., 2007). NLRC4 is also important in the activation of caspase-1 induced by Shigella, a non-flagellated bacteria, indicating that the existence of a flagellin-independent pathway that leads to the activation of the NLRC4-inflammasome (Suzuki et al., 2007). There is also evidence suggesting that other proteins of the type III secretion system which share homology with flagellin can stimulate the NLRC4-inflammasome (Miao et al., 2010b).
The activation of caspase-1 induced by Salmonella, L. pneumophila, or P. aeruginosa requires a functional bacterial secretion system (Franchi et al., 2008). The observation that during Salmonella infection the type III secretion system encoded by SPI-1 is required for both the translocation of flagellin into the cytosol and the activation of caspase-1 (Sun et al., 2007), suggests that small amounts of flagellin may inadvertently leak into the cytosol when bacterial secretion systems puncture cellular membranes.
The role of inflammasomes in Salmonella infection in vivo has been investigated in caspase-1 deficient mice. Initial work with caspase-1 deficient mice in a B10.RIII background revealed a dramatic phenotype suggesting that Salmonella require caspase-1 for bacterial dissemination (Monack et al., 2000). These results are at odds with the role of caspase-1 in other models of bacterial infection (Franchi et al., 2008). More recently, the role of caspase-1 in Salmonella infection has been reexamined in caspase-1 deficient mice in the B6 background (Lara-Tejero et al., 2006; Raupach et al., 2006). These experiments showed an increased susceptibility of caspase-1 knockout mice which was associated with increased bacterial loads in the spleen and the mesenteric lymph nodes (Lara-Tejero et al., 2006; Raupach et al., 2006). In agreement with a role for caspase-1 in the systemic phase of the infection, caspase-1 deficient mice were more susceptible than wild-type mice when challenged with Salmonella intraperitoneally (Raupach et al., 2006). Using IL-1β and IL-18 knockout mice, IL-18 was revealed as the main cytokine responsible for host defense (Raupach et al., 2006). While caspase-1 has a protective role against Salmonella infection, there is also evidence that caspase-1 has a detrimental role in septic shock. Furthermore, in a mouse model in which septic shock was induced by intraperitoneal injection of high doses of an attenuated Salmonella strain, caspase-1 and IL-18 deficient mice, but not IL-1β deficient mice, were protected (Raupach et al., 2006). Furthermore, NLRC4-deficient mice have been shown to be equally susceptible as wild-type mice (Lara-Tejero et al., 2006), a result that is not surprising considering that Salmonella is known to down regulate the expression of flagellin during the systemic phase of the infection (Alaniz et al., 2006). In contrast, NLRC4 plays an important role in the host defense response toward Salmonella mutants that express flagellin during the systemic phase of the infection (Miao et al., 2010a).
Another inflammasome involved in both microbial and non-microbial signaling pathways is the NLRP3-inflammasome. The mechanism of activation of the NLRP3-inflammasome is complex, in that its activation requires two different signals. One signal provided by microbial ligands or endogenous cytokines is necessary to prime macrophages through the upregulation of NLRP3 and pro-IL-1β. The second signal directly triggers caspase-1 activation, and can be mediated by at least four separate pathways that include ATP-P2X7R-pannexin-1, Syk signaling, particulate matters (such as silica and urate crystals), and bacterial exotoxins (Franchi et al., 2009a, 2010). Although disruption of the lysosomal membrane and/or production of reactive oxygen species (ROS) and/or decrease in K+ cytosolic concentration have been implicated the activation of the NLRP3-inflammasome, the molecular mechanisms responsible for the activation of the NLRP3-inflammasome remain elusive.
A role of the NLRP3-inflammasome in the control of Salmonella infection was initially excluded based on the fact that NLRP3-deficient mice were as susceptible as wild-type mice to Salmonella infection (Lara-Tejero et al., 2006). More recently, Broz et al. (2010) investigated whether the lack of phenotype in NLRC4- and NLRP3 deficient mice was due to redundant functions of these two inflammasomes and found that mice deficient in both NLRP3 and NLRC4 are more susceptible than wild-type mice to Salmonella infection similar to caspase-1 deficient mice. Using a combination of bacterial mutants and specific experimental procedures to maximize the expression of SPI-2, Broz et al. found that in macrophages infected with Salmonella that do not express SPI-1 the activation of caspase-1 occurs via the NLRP3-inflammasome and is dependent on SPI-2. These data suggest the existence of two different pathways (Figure 1), one dependent on SPI-1, NLRC4, and flagellin that triggers a robust activation of caspase-1 within minutes after infection, and a second pathway dependent on SPI-2 and NLRP3 that triggers the activation of caspase-1 with slower kinetics. The molecular mechanisms by which SPI-2 activates the NLRP3-inflammasome remain unknown. Surprisingly Apoptosis-associated Speck-like protein containing a CARD (ASC), a common adaptor protein used by both NLRC4 and NLRP3 to activate caspase-1 and to trigger the production of IL-1β, has no effect on susceptibility to Salmonella infection (Lara-Tejero et al., 2006).
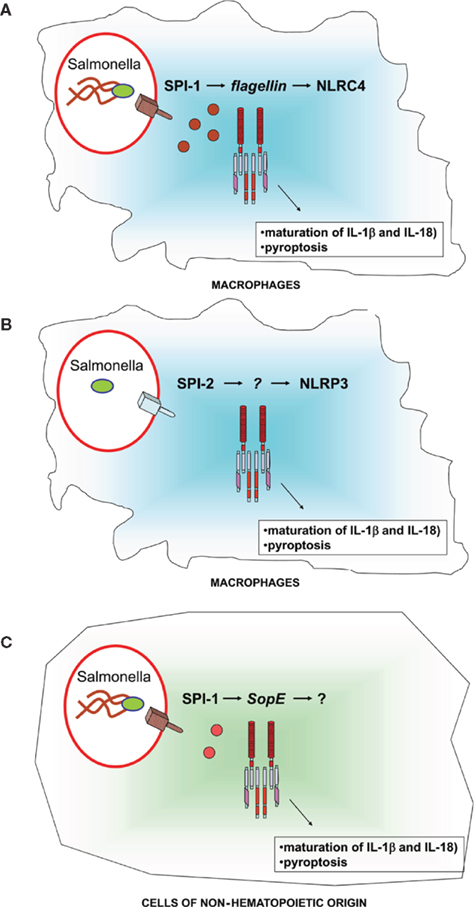
Figure 1. Schematic representation of the pathways leading to caspase-1 activation in cells infected with Salmonella. In macrophages (A,B) Salmonella induces the activation of the NLRC4-inflammasome via flagellin and SPI-1 (A) and the NLRP3-inflammasome via SPI-2. (C) In cells of non-hematopoietic origin Salmonella induces the activation of the inflammasome via SopE and SPI-1. See text for more detail.
A third pathway (Figure 1) of caspase-1 activation has recently been identified. Experiments using epithelial cell lines suggest that the virulence factor SopE encoded by the SPI-1 is able to induce caspase-1 activation (Muller et al., 2009). Furthermore, bone-marrow chimera experiments showed that Salmonella mutants that selectively express the virulence factor SopE could induce the activation of caspase-1 in cells of non-hematopoietic origin (Muller et al., 2009). The SopE-dependent activation of caspase-1 in cells of non-hematopoietic origin was found to play a significant role in gut inflammation, but was not sufficient to restrict bacterial replication (Muller et al., 2009). It remains to be determined whether the SopE-dependent activation of caspase-1 is NLRC4 or NLRP3 dependent.
Concluding Remarks
In this review, I have summarized recent advances in our understanding of the role of NLRs in the innate immune response to Salmonella. The available data suggest that the main role of inflammasomes is to restrict the replication of the bacteria during the systemic phase of the infection. In macrophages, the activation of the NLRC4-inflammasome requires SPI-1 and cytosolic flagellin. In macrophages infected with bacteria that do not express SPI-1 and flagellin a second pathway dependent on NLRP3 and the expression of SPI-2 is revealed. A third pathway that can lead to the activation of caspase-1 in non-hematopoietic cells is dependent on SPI-1 and the virulence factor SopE. In vivo both the NLRC4- and NLRP3-inflammasome are required for the innate immune response to Salmonella. It will be interesting in future studies to explore the role of inflammasomes in the activation of adaptive immune responses elicited by Salmonella.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author is grateful to Neil Warner and Gabriel Nunez for critical review of the manuscript. The authors’ research is supported by a Career Development Award from Crohn’s and Colitis Foundation of America (CCFA).
References
Alaniz, R. C., Cummings, L. A., Bergman, M. A., Rassoulian-Barrett, S. L., and Cookson, B. T. (2006). Salmonella typhimurium coordinately regulates FliC location and reduces dendritic cell activation and antigen presentation to CD4+ T cells. J. Immunol. 177, 3983–3993.
Amer, A., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Ozoren, N., Brady, G., Meshinchi, S., Jagirdar, R., Gewirtz, A., Akira, S., and Nunez, G. (2006). Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223.
Bernheiden, M., Heinrich, J. M., Minigo, G., Schutt, C., Stelter, F., Freeman, M., Golenbock, D., and Jack, R. S. (2001). LBP, CD14, TLR4 and the murine innate immune response to a peritoneal Salmonella infection. J. Endotoxin Res. 7, 447–450.
Broz, P., Newton, K., Lamkanfi, M., Mariathasan, S., Dixit, V. M., and Monack, D. M. (2010). Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755.
Bruno, V. M., Hannemann, S., Lara-Tejero, M., Flavell, R. A., Kleinstein, S. H., and Galan, J. E. (2009). Salmonella typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog. 5, e1000538. doi: 10.1371/journal.ppat.1000538
Fink, S. L., and Cookson, B. T. (2007). Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol. 9, 2562–2570.
Franchi, L., Amer, A., Body-Malapel, M., Kanneganti, T. D., Ozoren, N., Jagirdar, R., Inohara, N., Vandenabeele, P., Bertin, J., Coyle, A., Grant, E. P., and Nunez, G. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat. Immunol. 7, 576–582.
Franchi, L., Eigenbrod, T., Munoz-Planillo, R., and Nunez, G. (2009a). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247.
Franchi, L., Warner, N., Viani, K., and Nunez, G. (2009b). Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128.
Franchi, L., Munoz-Planillo, R., Reimer, T., Eigenbrod, T., and Nunez, G. (2010). Inflammasomes as microbial sensors. Eur. J. Immunol. 40, 611–615.
Franchi, L., Park, J. H., Shaw, M. H., Marina-Garcia, N., Chen, G., Kim, Y. G., and Nunez, G. (2008). Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 10, 1–8.
Franchi, L., Stoolman, J., Kanneganti, T. D., Verma, A., Ramphal, R., and Nunez, G. (2007). Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur. J. Immunol. 37, 3030–3039.
Geddes, K., Rubino, S., Streutker, C., Cho, J. H., Magalhaes, J. G., Le Bourhis, L., Selvanantham, T., Girardin, S. E., and Philpott, D. J. (2010). Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect. Immun. 78, 5107–5115.
Gewirtz, A. T., Navas, T. A., Lyons, S., Godowski, P. J., and Madara, J. L. (2001a). Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 167, 1882–1885.
Gewirtz, A. T., Simon, P. O. Jr., Schmitt, C. K., Taylor, L. J., Hagedorn, C. H., O’Brien, A. D., Neish, A. S., and Madara, J. L. (2001b). Salmonella typhimurium translocates flagellin across intestinal epithelia, inducing a proinflammatory response. J. Clin. Invest. 107, 99–109.
Grassl, G. A., and Finlay, B. B. (2008). Pathogenesis of enteric Salmonella infections. Curr. Opin. Gastroenterol. 24, 22–26.
Grassl, G. A., Valdez, Y., Bergstrom, K. S., Vallance, B. A., and Finlay, B. B. (2008). Chronic enteric Salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology 134, 768–780.
Hapfelmeier, S., and Hardt, W. D. (2005). A mouse model for S. typhimurium-induced enterocolitis. Trends Microbiol. 13, 497–503.
Hase, K., Kawano, K., Nochi, T., Pontes, G. S., Fukuda, S., Ebisawa, M., Kadokura, K., Tobe, T., Fujimura, Y., Kawano, S., Yabashi, A., Waguri, S., Nakato, G., Kimura, S., Murakami, T., Iimura, M., Hamura, K., Fukuoka, S., Lowe, A. W., Itoh, K., Kiyono, H., and Ohno, H. (2009). Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune response. Nature 462, 226–230.
Homer, C. R., Richmond, A. L., Rebert, N. A., Achkar, J. P., and McDonald, C. (2010). ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139, 1630–1641.
Inohara, N., and Nunez, G. (2003). NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3, 371–382.
Knoop, K. A., Kumar, N., Butler, B. R., Sakthivel, S. K., Taylor, R. T., Nochi, T., Akiba, H., Yagita, H., Kiyono, H., and Williams, I. R. (2009). RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 183, 5738–5747.
Ko, H. J., Yang, J. Y., Shim, D. H., Yang, H., Park, S. M., Curtiss, R., 3rd, and Kweon, M. N. (2009). Innate immunity mediated by MyD88 signal is not essential for induction of lipopolysaccharide-specific B cell responses but is indispensable for protection against Salmonella enterica serovar Typhimurium infection. J. Immunol. 182, 2305–2312.
Lamkanfi, M., Amer, A., Kanneganti, T. D., Munoz-Planillo, R., Chen, G., Vandenabeele, P., Fortier, A., Gros, P., and Nunez, G. (2007). The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J. Immunol. 178, 8022–8027.
Lara-Tejero, M., Sutterwala, F. S., Ogura, Y., Grant, E. P., Bertin, J., Coyle, A. J., Flavell, R. A., and Galan, J. E. (2006). Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 203, 1407–1412.
Le Bourhis, L., Magalhaes, J. G., Selvanantham, T., Travassos, L. H., Geddes, K., Fritz, J. H., Viala, J., Tedin, K., Girardin, S. E., and Philpott, D. J. (2009). Role of Nod1 in mucosal dendritic cells during Salmonella pathogenicity island 1-independent Salmonella enterica serovar Typhimurium infection. Infect. Immun. 77, 4480–4486.
Lightfield, K. L., Persson, J., Brubaker, S. W., Witte, C. E., J. von Moltke, Dunipace, E. A., Henry, T., Sun, Y. H., Cado, D., Dietrich, W. F., Monack, D. M., Tsolis, R. M., and Vance, R. E. (2008). Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178.
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., Roose-Girma, M., Erickson, S., and Dixit, V. M. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218.
Miao, E. A., Alpuche-Aranda, C. M., Dors, M., Clark, A. E., Bader, M. W., Miller, S. I., and Aderem, A. (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575.
Miao, E. A., Ernst, R. K., Dors, M., Mao, D. P., and Aderem, A. (2008). Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. U.S.A. 105, 2562–2567.
Miao, E. A., Leaf, I. A., Treuting, P. M., Mao, D. P., Dors, M., Sarkar, A., Warren, S. E., Wewers, M. D., and Aderem, A. (2010a). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142.
Miao, E. A., Mao, D. P., Yudkovsky, N., Bonneau, R., Lorang, C. G., Warren, S. E., Leaf, I. A., and Aderem, A. (2010b). Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 107, 3076–3080.
Monack, D. M., Bouley, D. M., and Falkow, S. (2004). Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+ mice and can be reactivated by IFNgamma neutralization. J. Exp. Med. 199, 231–241.
Monack, D. M., Hersh, D., Ghori, N., Bouley, D., Zychlinsky, A., and Falkow, S. (2000). Salmonella exploits caspase-1 to colonize Peyer’s patches in a murine typhoid model. J. Exp. Med. 192, 249–258.
Muller, A. J., Hoffmann, C., Galle, M., Van Den Broeke, A., Heikenwalder, M., Falter, L., Misselwitz, B., Kremer, M., Beyaert, R., and Hardt, W. D. (2009). The S. Typhimurium, effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation. Cell Host Microbe 6, 125–136.
Niess, J. H., Brand, S., Gu, X., Landsman, L., Jung, S., McCormick, B. A., Vyas, J. M., Boes, M., Ploegh, H. L., Fox, J. G., Littman, D. R., and Reinecker, H. C. (2005). CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307, 254–258.
Raupach, B., and Kaufmann, S. H. (2001). Bacterial virulence, proinflammatory cytokines and host immunity: how to choose the appropriate Salmonella vaccine strain? Microbes Infect. 3, 1261–1269.
Raupach, B., Peuschel, S. K., Monack, D. M., and Zychlinsky, A. (2006). Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 74, 4922–4926.
Rescigno, M., Urbano, M., Valzasina, B., Francolini, M., Rotta, G., Bonasio, R., Granucci, F., Kraehenbuhl, J. P., and Ricciardi-Castagnoli, P. (2001). Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2, 361–367.
Salucci, V., Rimoldi, M., Penati, C., Sampietro, G. M., van Duist, M. M., Matteoli, G., Saibeni, S., Vecchi, M., Ardizzone, S., Porro, G. B., and Rescigno, M. (2008). Monocyte-derived dendritic cells from Crohn patients show differential NOD2/CARD15-dependent immune responses to bacteria. Inflamm. Bowel Dis. 14, 812–818.
Santos, R. L., Raffatellu, M., Bevins, C. L., Adams, L. G., Tukel, C., Tsolis, R. M., and Baumler, A. J. (2009). Life in the inflamed intestine, Salmonella style. Trends Microbiol. 17, 498–506.
Sierro, F., Dubois, B., Coste, A., Kaiserlian, D., Kraehenbuhl, J. P., and Sirard, J. C. (2001). Flagellin stimulation of intestinal epithelial cells triggers CCL20-mediated migration of dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 98, 13722–13727.
Sun, Y. H., Rolan, H. G., and Tsolis, R. M. (2007). Injection of flagellin into the host cell cytosol by Salmonella enterica serotype Typhimurium. J. Biol. Chem. 282, 33897–33901.
Suzuki, T., Franchi, L., Toma, C., Ashida, H., Ogawa, M., Yoshikawa, Y., Mimuro, H., Inohara, N., Sasakawa, C., and Nunez, G. (2007). Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3, e111. doi: 10.1371/journal.ppat.0030111
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820.
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhaes, J. G., Yuan, L., Soares, F., Chea, E., Le Bourhis, L., Boneca, I. G., Allaoui, A., Jones, N. L., Nunez, G., Girardin, S. E., and Philpott, D. J. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62.
Uematsu, S., and Akira, S. (2009). Immune responses of TLR5+ lamina propria dendritic cells in enterobacterial infection. J. Gastroenterol. 44, 803–811.
Uematsu, S., Jang, M. H., Chevrier, N., Guo, Z., Kumagai, Y., Yamamoto, M., Kato, H., Sougawa, N., Matsui, H., Kuwata, H., Hemmi, H., Coban, C., Kawai, T., Ishii, K. J., Takeuchi, O., Miyasaka, M., Takeda, K., and Akira, S. (2006). Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat. Immunol. 7, 868–874.
Vazquez-Torres, A., Vallance, B. A., Bergman, M. A., Finlay, B. B., Cookson, B. T., Jones-Carson, J., and Fang, F. C. (2004). Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J. Immunol. 172, 6202–6208.
Vijay-Kumar, M., Aitken, J. D., and Gewirtz, A. T. (2008). Toll like receptor-5: protecting the gut from enteric microbes. Semin. Immunopathol. 30, 11–21.
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., Kuida, K., Mariathasan, S., Dixit, V. M., Flavell, R. A., Dietrich, W. F., and Roy, C. R. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325.
Keywords: Salmonella, inflammation, innate immunity, inflammasome, IL-1β
Citation: Franchi L (2011) Role of inflammasomes in Salmonella infection. Front. Microbio. 2:8. doi: 10.3389/fmicb.2011.00008
Received: 10 November 2010;
Paper pending published: 03 December 2010;
Accepted: 16 January 2011;
Published online: 31 January 2011.
Edited by:
Amal Amer, The Ohio State University, USAReviewed by:
Amal Amer, The Ohio State University, USALee-Ann H. Allen, University of Iowa, USA
Yufeng Yao, Shanghai Jiao Tong University School of Medicine, China
Copyright: © 2011 Franchi. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Luigi Franchi, Department of Pathology, University of Michigan Medical School, 4131 CCGC, 1500 East Medical Center Drive, Ann Arbor, MI 48109, USA. e-mail:bHVpZ2lmQHVtaWNoLmVkdQ==