 Alexander Skeldon1
Alexander Skeldon1 Maya Saleh1,2,3*
Maya Saleh1,2,3*
- 1 Department of Biochemistry, McGill University, Montreal, QC, Canada
- 2 Department of Medicine, McGill University, Montreal, QC, Canada
- 3 Department of Microbiology and Immunology, McGill University, Montreal, QC, Canada
The inflammasomes are large multi-protein complexes scaffolded by cytosolic pattern recognition receptors (PRRs) that form an important part of the innate immune system. They are activated following the recognition of microbial-associated molecular patterns or host-derived danger signals (danger-associated molecular patterns) by PRRs. This recognition results in the recruitment and activation of the pro-inflammatory protease caspase-1, which cleaves its preferred substrates pro-interleukin-1β (IL-1β) and pro-IL-18 into their mature biologically active cytokine forms. Through processing of a number of other cellular substrates, caspase-1 is also required for the release of “alarmins” and the induction and execution of an inflammatory form of cell death termed pyroptosis. A growing spectrum of inflammasomes have been identified in the host defense against a variety of pathogens. Reciprocally, pathogens have evolved effector strategies to antagonize the inflammasome pathway. In this review we discuss recent developments in the understanding of inflammasome-mediated recognition of bacterial, viral, parasitic, and fungal infections and the beneficial or detrimental effects of inflammasome signaling in host resistance.
Introduction
The innate immune system plays a primary role in the rapid recognition and elimination of invading microorganisms, through different processes such as phagocytosis and the induction of inflammation. It is also vital in priming and activating the adaptive immune system, which provides long-lasting immunity. Host cells, including among others cells of the myeloid and epithelial lineages, express a number of germline-encoded pattern recognition receptors (PRRs) that recognize conserved protein, lipid, polysaccharide, or nucleic acid motifs and activate different inflammatory and antimicrobial pathways. PRRs include, but are not limited to, trans-membrane Toll-like receptors (TLRs), C-type lectin receptors (CLRs), cytosolic Nod-like receptors (NLRs), retinoic acid inducible gene-I (RIG-I)-like helicases and HIN200 proteins (O’Neill and Bowie, 2010; Takeuchi and Akira, 2010). These receptors also sense and respond to endogenous, host-derived, danger signals that are released in response to stress, tissue injury or cell death (Bianchi, 2007; Medzhitov, 2008; Schroder and Tschopp, 2010).
To date, over 20 human NLRs have been identified. NLRs are related to disease resistance R proteins in plants (Belkhadir et al., 2004) and to apoptosis protease-activating factor 1 (Apaf1; Proell et al., 2008). They share with these proteins a tripartite structure consisting of an N-terminal protein–protein interaction module [which is either a pyrin domain (PYD), a caspase recruitment domain (CARD), or a baculovirus IAP repeat (BIR)], a central nucleotide binding and oligomerization domain (NOD or NAHCT) and a C-terminal agonist-sensing domain, which in NLRs is a series of leucine-rich repeats (LRRs), as found in TLRs. The first identified NLRs, NOD1, and NOD2, were determined to recognize bacterial peptidoglycan moieties (Chamaillard et al., 2003; Girardin et al., 2003; Inohara et al., 2003) and trigger inflammation by activating NF-κB and MAP kinase (MAPK) pathways (Shaw et al., 2008). Further study of the NLR family revealed that some members are capable of forming multi-protein complexes, termed inflammasomes that recruit pro-caspase-1 and induce its “activation by proximity,” either directly or through adaptor proteins such as apoptosis-associated speck-like protein containing a CARD (ASC; Martinon et al., 2002).
Multiple inflammasomes have been characterized, including those scaffolded by the NLR family members NLRP1, NLRP3, NLRC4, NAIP5 and the more recently described AIM2 and RIG-I inflammasomes that are stimulated by cytosolic DNA and viral RNA, respectively. The adaptor protein ASC is required for the recruitment of caspase-1 to the NLRP3, AIM2, and RIG-I inflammasomes while its role in the NLRP1, NLRC4, and NIAP5-associated complexes is less clear. Following inflammasome stimulation, the cellular pool of ASC coalesces into a large structure called the pyroptosome, which is the site of caspase-1 activation. Dependent on the associated PRR, inflammasomes are activated by a variety of signals and may have redundant functions during infection. Dysregulation and/or genetic mutations or variations in genes encoding inflammasome components have been linked to genetic disorders in humans, including a continuum of autoinflammatory syndromes ranging from familial cold urticaria and Muckle–Wells syndrome to neonatal onset multisystem inflammatory disease, Crohn’s disease, sepsis as well as susceptibility to infection with certain pathogens (Rodrigue-Gervais and Saleh, 2010).
Caspase-1 processes a number of cellular substrates, which is a prerequisite to the induction of an inflammatory response. Most notably, caspase-1 converts pro-IL-1β and pro-IL-18 into their mature biologically active cytokine forms (Thornberry et al., 1992; Ghayur et al., 1997; Gu et al., 1997). Moreover, caspase-1 is required for the release of a number of pro-inflammatory molecules, which are not necessarily caspase-1 substrates, including IL-1α (Keller et al., 2008). In addition to its pro-inflammatory effects, excessive activation of caspase-1 leads to a form of cell death called pyroptosis, with characteristics of both apoptosis and necrosis (Fink and Cookson, 2005; Labbe and Saleh, 2008). Among the different caspase-1 substrates, we have shown that it cleaves a number of glycolysis enzymes (Shao et al., 2007), which is possibly a primary mechanism to execute cell death. Moreover, caspase-1 has been shown to cleave and activate the pro-apoptotic executioner caspase, caspase-7 (Lamkanfi et al., 2008), suggesting that this enzyme might provide a secondary amplification loop during pyroptosis.
Inflammasome Regulation
Because of the paramount effects of caspase-1 during infection and inflammation, regulation of the inflammasome pathway is tightly controlled through a number of checkpoints (Figure 1). Firstly, inflammasome activation depends on a two-step process, whereby signal 1 “primes” the inflammasome via transcriptional induction of inflammasome components and cytokine proforms (usually mediated by NF-κB downstream of PRRs or cytokine receptors) and signal 2, which “activates” the inflammasome.
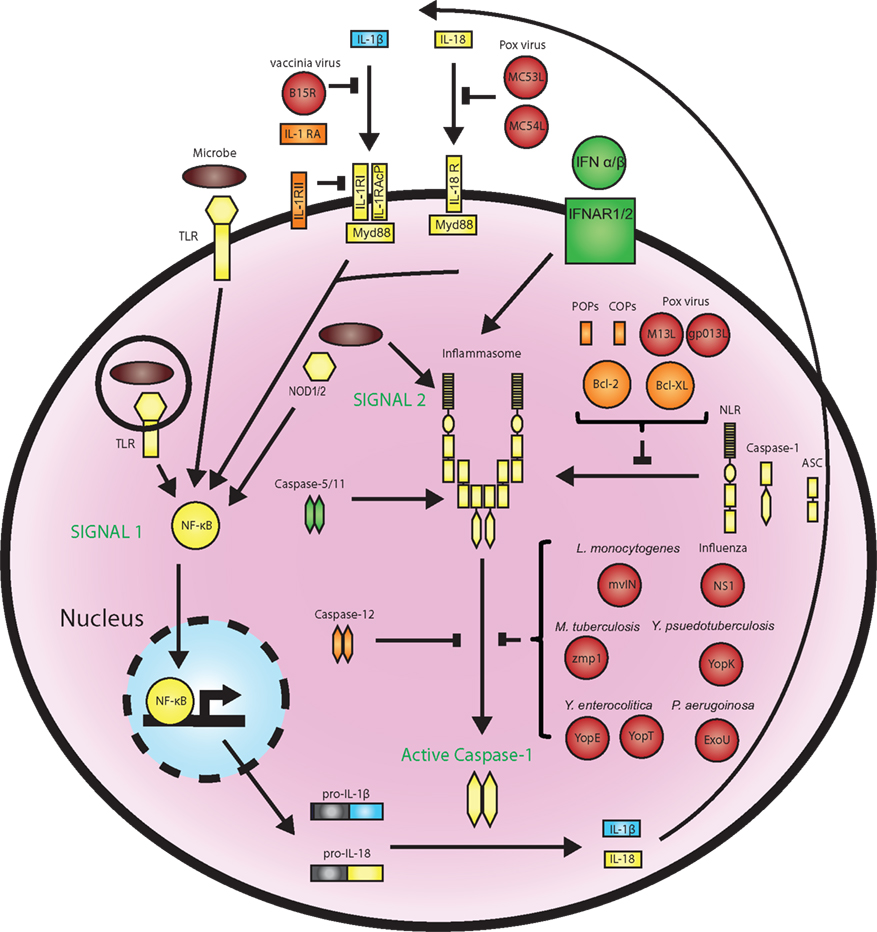
Figure 1. Regulation of the inflammasomes by host factors and pathogen effectors. Inflammasomes are activated in a two-step process beginning with PRR-mediated induction of inflammasome components and pro-IL-1β production through NF-κB, followed by a second signal that activates the inflammasome and caspase-1 catalysis. This process can be regulated at multiple steps by host proteins that function as positive regulators (green) or inhibitors (orange) or targeted by pathogen effectors (red). Host COPs and POPs, the poxvirus proteins M13L and gp013L, and the anti-apoptotic factors Bcl-2 and Bcl-XL inhibit inflammasome assembly. Caspase-1 activation is inhibited by caspase-12, and multiple pathogen effectors, while murine caspase-11 and human caspase-5 are required for caspase-1 activation in response to certain stimuli. Type I IFN is required for AIM2 inflammasome activation in response to cytosolic DNA. The IL-1β and IL-18 pathways are also highly regulated. Endogenous IL-1 receptor antagonist (RA) prevents IL-1 signaling by binding to the IL-1 receptor, while the vaccinia virus proteins B15R and Molluscum contagiosum poxvirus MC53L and MC54L can bind and inhibit IL-1 and IL-18, respectively.
In addition to this bi-modal mechanism of activation, the inflammasome is kept in check via a number of positive and negative regulators (Figure 1). For instance, murine caspase-11 and its closest human homolog caspase-5 (Lin et al., 2000) are required for caspase-1 activation in some contexts (Wang et al., 1998), presumably through direct interaction with the inflammasome (Martinon et al., 2002). Conversely, caspase-12 is a negative regulator of caspase-1 (Saleh et al., 2006). Two groups of proteins termed COPs (CARD-only proteins) and POPs (PYD-only proteins) also inhibit caspase-1 activation by preventing inflammasome assembly (Stehlik and Dorfleutner, 2007). In addition to these dominant negative proteins, the anti-apoptotic factors Bcl-2 and Bcl-XL can bind and suppress NLRP1 oligomerization and caspase-1 activation by inhibiting ATP binding (Bruey et al., 2007; Faustin et al., 2009). The IL-1 pathway is also highly regulated. Secreted IL-1β binds to the IL-1 receptor I (IL-1RI) and recruits the IL-1R accessory protein (IL-1RAcP), which is required for signaling (Gabay et al., 2010). The dominant negative receptor IL-1RII and soluble IL-1 receptor antagonist (IL-RA) are able to inhibit this process (Gabay et al., 2010). Together, these factors add an additional layer of regulation on the inflammasome pathway.
Given the widespread activation of inflammasomes in response to various microbes, it is not surprising that pathogens have devised multiple strategies to alter inflammasome function and downstream signaling. Bacteria utilize effector molecules, injected through secretion systems into the host cytosol, for this purpose. For instance, Y. enterocolitica YopE and YopT (Schotte et al., 2004), Y. pseudotuberculosis YopK (Brodsky et al., 2010), and P. aeruginosa ExoU (Sutterwala et al., 2007) have been reported to blunt inflammasome activation. Viruses also encode proteins that target this pathway including influenza NS1 (Stasakova et al., 2005), Myxoma virus M13L-PYD and Shope fibroma virus gp013L (Johnston et al., 2005; Dorfleutner et al., 2007) that act as POPs. Vaccinia virus encodes a soluble IL-1β receptor, B15R, that blunts IL-1 signaling (Alcami and Smith, 1992), whereas Molluscum contagiosum poxvirus produces two IL-18 inhibitors, MC53L and MC54L (Xiang and Moss, 1999). The active inhibition of the inflammasome by various pathogens supports the notion that its pro-inflammatory effects together with the induction of pyroptosis are deleterious for the pathogen.
Inflammasome Activation
A spectrum of agonists activate the inflammasomes, with some being more specific than others depending on the associated NLR. NLRP3 forms a multi-protein complex with ASC and caspase-1, and is currently the most well characterized inflammasome. It can be activated by various structurally unrelated stimuli including microbial-associated molecular patterns (MAMPs), and danger-associated molecular patterns (DAMPs). For instance, elevated concentrations of ATP (Mariathasan et al., 2006), pore-forming toxins (Mariathasan et al., 2006), UVB irradiation and particulate matter such as crystalline forms of monosodium urate (MSU; Martinon et al., 2006), asbestos and silica (Cassel et al., 2008; Dostert et al., 2008; Hornung et al., 2008), and amyloid β aggregates (Halle et al., 2008) have all been reported to trigger NLRP3 activation. Due to the high disparity of these agonists, it is suggested that a downstream signal is instead sensed by NLRP3. In the case of particulate agonists, disruption of the lysosomal membrane along with cathepsins appear to be upstream of inflammasome activation. For instance, chemical inhibition of cathepsin B, cathepsin B-deficiency, or treatment of cells with inhibitors of the vacuolar H+ ATPase result in reduced caspase-1 activation (Halle et al., 2008; Hornung et al., 2008). On the other hand, inflammasome activation triggered by ATP is not affected by these inhibitors. ATP activates the P2X7 receptor cation channel, which induces potassium efflux and causes the recruitment of the pannexin-1 channel that amplifies this response (Pelegrin and Surprenant, 2006). Treatment of macrophages with nigericin, a Streptomyces-derived potassium ionophore (Mariathasan et al., 2006), or with α-hemolysin, a Staphylococcus aureus pore-forming toxin, similarly triggers NLRP3 inflammasome activation (Craven et al., 2009). It has been further suggested that reactive oxygen species (ROS) may be involved in this process. Depletion of the p22phox subunit of the ROS-generating NADPH complex in the human monocytic cell line THP-1 results in reduced IL-1β processing in response to asbestos, but not MSU crystals (Dostert et al., 2008). The inhibition of cellular autophagy results in the accumulation of damaged, ROS producing mitochondria that also triggers NLRP3 activation (Zhou et al., 2011). Therefore, different ligands appear to require an assortment of mechanisms to activate NLRP3. The precise signal sensed by NLRP3 remains unclear but may be a combination of those mentioned above. Unlike NLRP3, the other known inflammasomes, namely NLRP1, NLRP4, AIM2, and RIG-I, have more defined activators and primarily play a role in the detection of pathogens. Recently, we have gained significant insights into the understanding of how the inflammasomes detect infectious microorganisms and the contribution of inflammasome signaling to the immune response. In this review, we focus our discussion on the role of the inflammasomes in bacterial, viral, parasitic, and fungal infections.
Bacteria
The innate immune system plays an important role in eliminating bacterial pathogens and preventing their replication and damage to the host. The human body is also home to numerous commensal microorganisms and it is therefore imperative that a distinction can be made in the response to these species versus pathogens to prevent the development of inflammatory conditions. During bacterial infection, inflammation is generally beneficial, but if uncontrolled could lead to deleterious effects such as septic shock.
Some pathogenic bacteria have an intracellular lifestyle within the host and are either engulfed by phagocytes or capable of invading non-phagocytic cells. This aids in the avoidance of immune detection and reduces exposure to antimicrobial components of the immune system, such as antimicrobial peptides, complements, and immunoglobulins. Inside the cell, bacteria are capable of manipulating the endocytic pathways that would otherwise result in their degradation within lysosomes. Interfering with these pathways allows the pathogen to survive and in many cases replicate. This is often achieved through bacterial secretion systems, which mediate the translocation of virulence factors or effector molecules into the host cell cytosol, where they become active. Following replication within the cell, the increase in bacterial load triggers cell death, allowing dissemination to new host cells.
Salmonella enterica is the best characterized intracellular bacterium with respect to inflammasome activation (Table 1). The study of Salmonella has greatly expanded our understanding of how the inflammasome recognizes pathogenic intracellular bacteria and how this contributes to the immune response. S. enterica serovar Typhimurium induces gastroenteritis and systemic typhoid-like disease in mice (Valdez et al., 2009). The bacterium employs a Salmonella Pathogenicity Island 1 (SPI-1) Type III Secretion System (T3SS) to invade epithelial cells and cross the intestinal barrier. S. typhimurium is then exposed to cells of the immune system and is capable of surviving within macrophages. The SPI-2 T3SS creates an intracellular niche called the Salmonella-containing vacuole (SCV) by inhibiting intracellular trafficking that would result in bacterial degradation (Haraga et al., 2008).
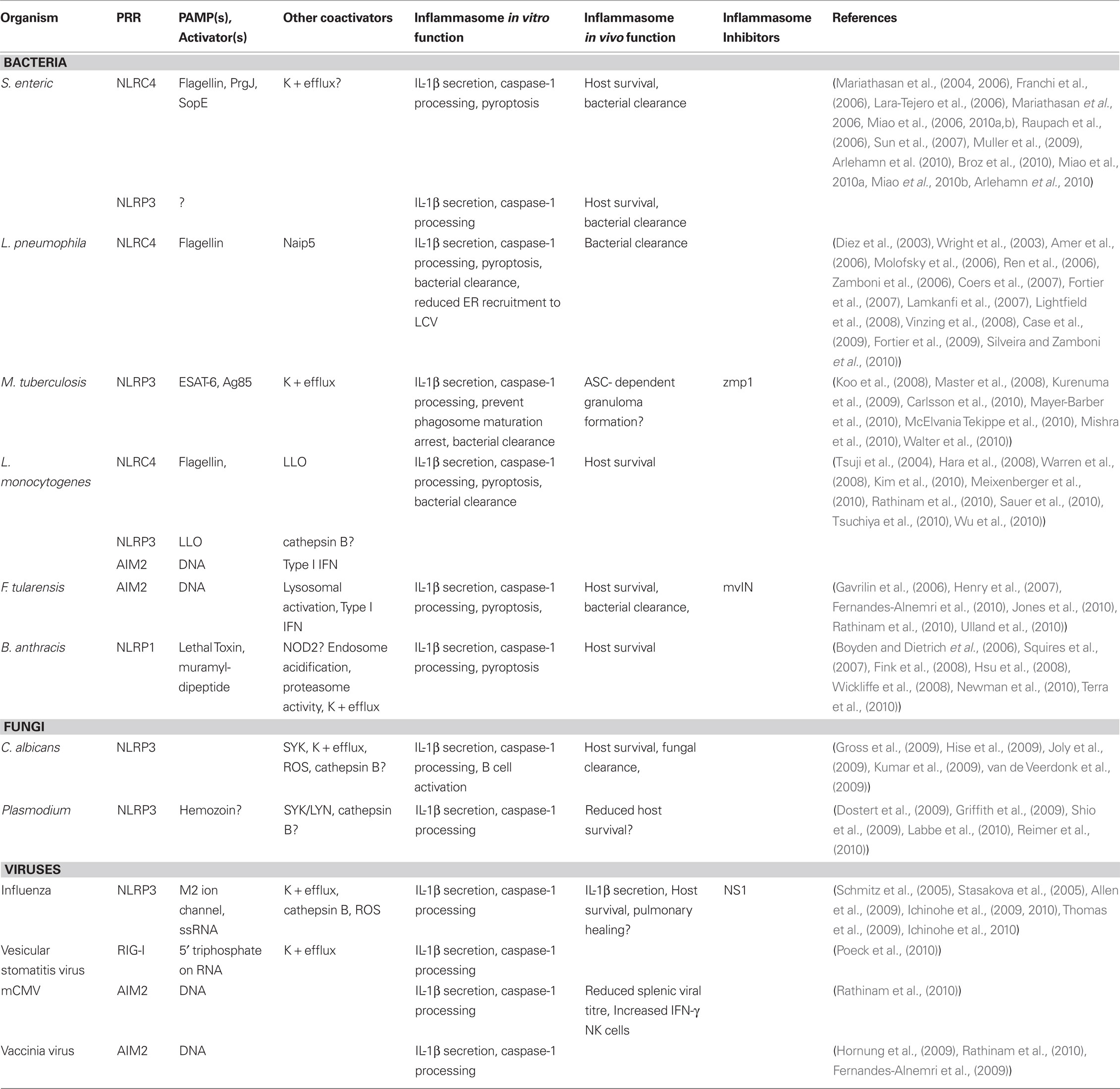
Table 1. Inflammasome-mediated pathogen recognition and response.
Early studies have determined that Salmonella induces caspase-1-dependent cell death in infected macrophages (Hersh et al., 1999). Subsequently, the inflammasome components Asc and Nlrc4 and a Salmonella SPI-1 T3SS component SipB were shown to be required for this response (Hersh et al., 1999; Mariathasan et al., 2004). The Nlrp3 inflammasome was thought to be irrelevant in this context, as Nlrp3-deficient macrophages showed no defect in caspase-1 activation or IL-1β secretion in in vitro studies (Mariathasan et al., 2006). A beneficial role for caspase-1 during S. typhimurium infection was identified in vivo using caspase-1-deficient mice. These mice displayed reduced survival, increased bacterial counts in the spleen, Peyer’s patch, and mesenteric lymph node (MLN), and increased intestinal inflammation after oral infection compared to wild-type animals (Lara-Tejero et al., 2006; Raupach et al., 2006). In contrast, mice singly deficient in one of the inflammasome components Nlrp3, Nlrc4, or Asc were phenotypically equivalent to wild-type mice, suggesting that Salmonella induces redundant pathways to activate caspase-1 in vivo (Lara-Tejero et al., 2006). Consistently, it has been recently demonstrated that both Nlrp3 and Nlrc4 are activated during Salmonella infection (Broz et al., 2010). Earlier in vitro work has demonstrated that Asc−/− macrophages have reduced IL-1β secretion and caspase-1 processing in response to Salmonella compared to wild-type cells (Mariathasan et al., 2004, 2006; Miao et al., 2006). In addition, Asc−/− macrophages have a more blunted response compared to Nlrp3-deficient cells, suggesting that Asc plays a role independent of the Nlrp3 inflammasome (Broz et al., 2010). Notably, deficiency in both Asc and Nlrc4 reduces IL-1β levels to that of caspase-1 null macrophages, suggesting that Asc may work to enhance Nlrc4 activity during Salmonella infection. However, a recent study has demonstrated that flagellin-dependent Nlrc4 inflammasome activation resulted in macrophage pyroptosis, independently of Asc, leading to bacterial release and subsequent killing by neutrophils (Miao et al., 2010a). Nlrc4−/− mice were impaired in clearing the infection, pointing to a critical role of Nlrc4 in bacterial elimination that is independent of IL-1r, IL-18, Nlrp3, and Asc.
Multiple Salmonella motifs trigger inflammasome activation (Figure 2). The SPI-1 (Miao et al., 2006) and SPI-2 (Broz et al., 2010) T3SS and flagellin (Franchi et al., 2006; Miao et al., 2006) stimulate NLRC4, while NLRP3 is activated by a currently unknown signal (Broz et al., 2010). The T3SS effector SopE (Muller et al., 2009; Hoffmann et al., 2010) is also capable of caspase-1 activation through an undetermined inflammasome. Salmonella strains lacking flagellin have reduced ability to activate caspase-1 or induce IL-1β secretion in macrophages, and transfected flagellin can activate the Nlrc4 inflammasome (Franchi et al., 2006; Miao et al., 2006; Broz et al., 2010). In addition to their role in flagellin translocation, T3SSs independently stimulate the Nlrc4 inflammasome. Indeed, a conserved component of the SPI-1 T3SS, the periplasmic rod-forming protein PrgJ, is recognized by Nlrc4 (Miao et al., 2010b). In contrast, The SPI-2 rod protein SsaI is not detected, suggesting that repression of SPI-1 in phagocytes is an adaptation mechanism to evade recognition by NLRC4. Homologous rod proteins from Escherichia coli, Shigella flexneri, and Pseudomonas aeruginosa are similarly sensed by Nlrc4, indicating that this is a conserved mechanism of caspase-1 activation found in a number of bacterial species. This may provide a possible explanation as to why Shigella and Pseudomonas strains that lack flagella are still capable of activating the Nlrc4 inflammasome (Sutterwala et al., 2007; Suzuki et al., 2007). A recent study has suggested that potassium efflux is required, but not sufficient, for NLRC4 activation induced by Salmonella or P. aeruginosa infection (Arlehamn et al., 2010). This is at odds with earlier reports that used lower extracellular potassium concentrations and different infection time points (Franchi et al., 2007; Petrilli et al., 2007; Fink et al., 2008).
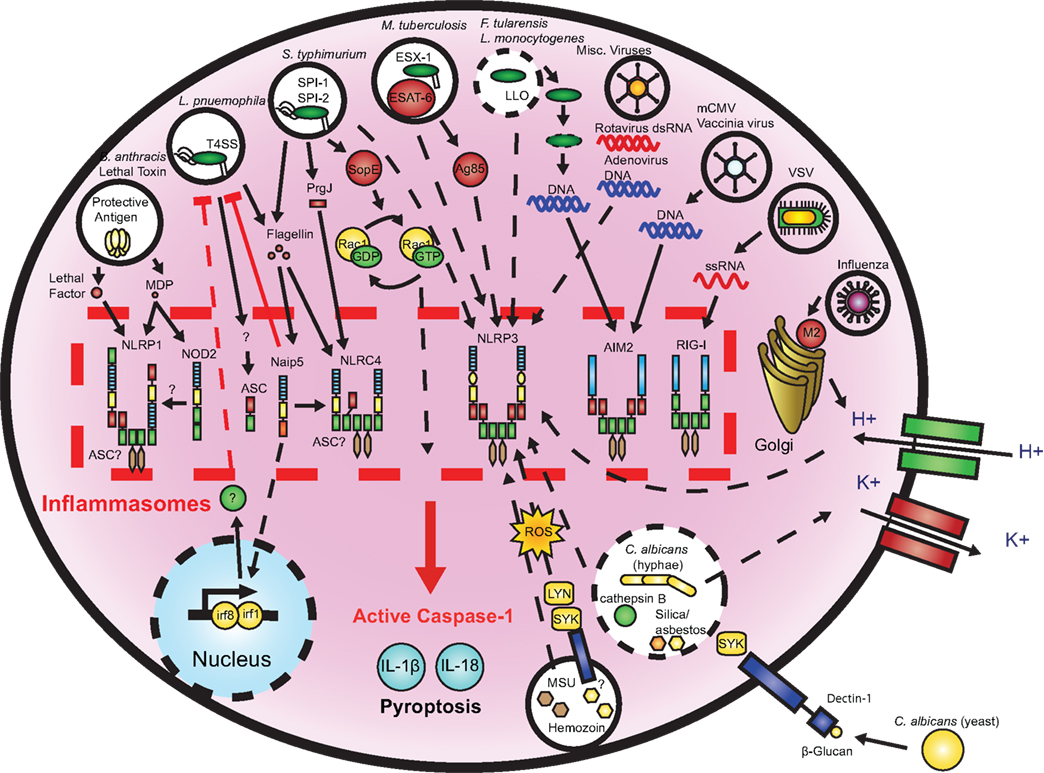
Figure 2. Microbial activation of the inflammasomes. Pathogenic microorganisms activate the inflammasomes through multiple agonists and pathways. S. typhimurium, L. pneumophila, and M. tuberculosis reside within the host cell phagosome and are capable of activating inflammasomes through secreted flagellin, effectors, or undefined NLRP3 agonists. F. tularensis and L. monocytogenes, which escape the phagosome activate AIM2 that senses cytosolic DNA. B. anthracis lethal toxin activates the NLRP1 inflammasome. C. albicans and hemozoin activate NLRP3 through SYK signaling. Viral-mediated inflammasome activation is heavily dependent on the detection of nucleic acids by NLRP3, AIM2, and RIG-I. Dotted lines indicate signaling through an unknown mechanism.
In addition to flagellin, bacterial effector proteins translocated through T3SSs are also capable of inflammasome activation. For instance, SopE, a guanine nucleotide exchange factor (GEF) of Rho GTPases that induces Salmonella invasion into epithelial cells (Hardt et al., 1998), leads to caspase-1 activation in vitro, in a manner dependent on its GEF activity and the Rho GTPases Rac1 and Cdc42 (Muller et al., 2009). This effect appears to be independent of flagellin (Hoffmann et al., 2010). In vivo, SopE triggers the caspase-1-IL-18 axis of inflammation, predominantly in non-hematopoietic cells (Muller et al., 2009). Y. enterocolitica’s effector YopE, a GTPase activating protein (GAP) and therefore an inhibitor of Rho GTPases, conversely inhibits caspase-1 activation (Schotte et al., 2004). It is currently unknown how Rho GTPases impact caspase-1 activation, but this raises the possibility that the large number of pathogen effector molecules targeting Rho GTPase functions might also play an important role in inflammasome signaling. Taking into account the different signals involved in Salmonella-induced caspase-1 activation, it is clear that the T3SS is a conserved inflammasome activator.
Legionella pneumophila, the causative agent of Legionnaires’ disease in humans, is another bacterial pathogen that can survive and replicate within macrophages. This bacterium employs the Dot/Icm Type IV Secretion System (T4SS) to recruit endoplasmic reticulum components to a Legionella-containing vacuole (LCV), inhibiting its acidification and fusion with the lysosome (Isberg et al., 2009). Caspase-1 and Nlrc4, but not IL-1β are required to restrict Legionella growth in macrophages in vitro and in the lungs of mice in vivo (Zamboni et al., 2006). Nlrc4-dependent activation of caspase-1 in this case leads to macrophage pyroptosis through pore formation (Silveira and Zamboni, 2010). The absence of caspase-1 also leads to enhanced recruitment of endoplasmic reticulum to LCV, which inhibits LCV fusion with the lysosome (Amer et al., 2006).
A second NLR, Naip5, encoded by the Birc1e gene, is also required for flagellin recognition and host defense against L. pneumophila (Molofsky et al., 2006; Ren et al., 2006). Unlike human cells, murine macrophages are generally non-permissive to Legionella replication, with the exception of the A/J mouse line that is susceptible to infection due to polymorphisms in Birc1e (Diez et al., 2003; Wright et al., 2003). It has now been determined that Naip5 is involved in the recognition of a 35 amino acid motif in flagellin, and along with Nlrc4 leads to caspase-1 activation, pyroptosis, and phagosomal maturation of the LCV (Lightfield et al., 2008; Figure 2). Flagellin regions outside this motif can activate Nlrc4, independently of Naip5. In addition, Naip5 is only partially required for recognition of Pseudomonas or Salmonella flagellin (Lightfield et al., 2008; Miao et al., 2008). C57BL/6 mice, harboring the restrictive Birc1e allele, display increased bacterial burden in their lungs when infected with a flagellin-deficient strain of Legionella, which correlates with macrophage resistance to cell death and increased bacterial replication (Amer et al., 2006; Molofsky et al., 2006; Ren et al., 2006). The role of Naip5 in LCV acidification seems to depend on the activation of caspase-7 by caspase-1; caspase-7 promotes lysosomal fusion with the LCV and reduces bacterial replication in macrophages in an Nlrc4, Naip5, and caspase-1 dependent manner. Consistently, caspase-7-deficient mice have increased bacterial counts in their lungs after infection (Akhter et al., 2009).
The adaptor Asc is required for IL-1β secretion by Legionella-infected macrophages, although it has no role in the inhibition of Legionella replication in macrophages in vitro and in vivo (Zamboni et al., 2006; Case et al., 2009). Interestingly, Asc-mediated inflammasome activation does not lead to pyroptosis and is independent of the flagellin-Nlrc4 pathway or Nlrp3 (Case et al., 2009). Instead, it can be inhibited by high concentrations of extracellular potassium, suggesting the involvement of a third unknown inflammasome sensor in response to Legionella.
Interestingly, Naip5’s effects in host defense to Legionella may be mediated by both caspase-1 dependent and independent pathways. Notably, the A/J hypomorphic Naip5 allele that is associated with permissiveness to Legionella replication, does not fully impair the caspase-1 pathway (Lamkanfi et al., 2007), whereas macrophages from Naip5−/− mice that are similarly permissive to bacterial replication, exhibit significantly blunted caspase-1 activation (Lightfield et al., 2008). Together, these studies suggest that Naip5’s function in inhibiting bacterial replication is distinct from its role in caspase-1 activation. Notably, Naip5 and Nlrc4 are also able to activate Interferon regulatory factor 1 (Irf1) and Irf8, which inhibit Legionella replication in macrophages (Fortier et al., 2009). The human homologs of NLRC4 and NAIP5 were also demonstrated to inhibit Legionella replication in human macrophages and epithelial cells (Vinzing et al., 2008).
Mycobacterium tuberculosis, the causative agent of tuberculosis in humans, survives and replicates within alveolar macrophages (Flynn and Chan, 2001). M. tuberculosis is capable of inflammasome activation and the Il-1r pathway is critical for the control of infection (Juffermans et al., 2000; Fremond et al., 2007). Caspase-1 and IL-1β prevent the arrest of Mycobacteria phagosome maturation and reduce bacterial survival in macrophages (Master et al., 2008). The ESX-1 or Type 7 Secretion System is required for IL-1β and IL-18 secretion by M. tuberculosis- or M. marinum-infected macrophages (Koo et al., 2008). Nlrp3 and Asc, but not Nlrc4, are implicated in ESX-1-induced caspase-1 activation, and the ESX-1 effector molecule ESAT-6 acts upstream of NLRP3 activation (Mishra et al., 2010; Figure 2). ESAT-6 functions by disrupting cell membranes and allowing for the M. tuberculosis protein Ag85 to enter into the cytosol and stimulate the inflammasome (Mishra et al., 2010). Interestingly, a putative Zn2+ metalloprotease, zmp1, which is required for M. tuberculosis virulence in mice, was found to inhibit inflammasome activation and IL-1β secretion in macrophages (Master et al., 2008). M. bovis BCG zmp1 mutants do not block phagolysosome maturation and are eliminated by a caspase-1, Asc, and Nlrc4 dependent mechanism. Wild-type M. bovis BCG induced low levels of IL-1β in macrophages, which may be explained by the lack of the ESX-1 secretion system. However, the precise molecular mechanism by which zmp1 impacts caspase-1 or whether this inhibition occurs in vivo remain to be determined.
Recent in vivo mouse studies have begun to determine the role of the inflammasome pathway in Mycobacteria infection. In one study, Nlrp3-deficient mice infected with M. tuberculosis did not exhibit enhanced histopathology or increased bacterial burden in the lungs, despite reduced IL-1β and IL-18 levels (Walter et al., 2010). Consistently, a second report demonstrated that ablation of either Nlrp3 or caspase-1 did not affect mouse mortality following M. tuberculosis infection. Surprisingly, however, a third study demonstrated that Asc, but not caspase-1 or Nlrp3, promoted mouse survival (McElvania Tekippe et al., 2010). Asc-mediated protection was independent of IL-1β levels in the lung or bacterial clearance. Instead, Asc was required to contain M. tuberculosis within granulomas, suggesting that Asc has important caspase-1-independent roles during infection. In striking contrast, a study using M. marinum found that Asc promoted lung pathology, as Asc-deficient mice had reduced inflammation and lung histophathology (Carlsson et al., 2010). The presence of the ESX-1 secretion system triggered this inflammatory response through an Asc-dependent mechanism. The different Mycobacteria species combined with a different infectious dose and method (tail injection versus aerosol in all other studies) may explain the differences observed here with regard to Asc. Given the conflicting data in these studies, further work is required to clarify the role of inflammasome signaling during M. tuberculosis infection in vivo. Interestingly, casp1−/− mice retain the ability to produce mature biologically active IL-1β during M. tuberculosis infection, suggesting that other redundant mechanisms are capable of generating IL-1β (Mayer-Barber et al., 2010). Caspase-1-independent IL-1β processing in response to M. tuberculosis was found in vivo, but not in vitro, underlining the importance of animal models when examining responses to infection. Overall, these studies indicate that the inflammasomes and ASC in particular have additional roles during infection with Mycobacteria species, possibly independent of IL-1 and IL-18 secretion. Moreover, enzymes other than caspase-1, presumably neutrophil elastase or other proteases, are also deployed in vivo for the generation of the key IL-1 family cytokines.
Francisella tularensis is an intracellular bacterium responsible for tularaemia in humans. Following, phagocytosis by host macrophages, the bacteria escape from the phagosome into the cytosol and replicate in this compartment (Santic et al., 2006; Figure 2). Early studies have demonstrated that caspase-1 and Asc, but not Nlrp3 and Nlrc4, were essential for macrophage cell death, bacterial clearance in vitro and in vivo and mouse survival (Mariathasan et al., 2005). This suggested that caspase-1 activation was mediated by a different inflammasome that required Asc. It was recently discovered that absent in melanoma 2 (AIM2), a member of the hematopoietic interferon-inducible nuclear protein (HIN-200) family that senses cytosolic DNA was the primary inflammasome triggered during F. tularensis infection (Fernandes-Alnemri et al., 2010; Jones et al., 2010; Rathinam et al., 2010). Lysosomal activation was shown to be required to stimulate this inflammasome during infection, possibly due to lysis of bacteria, which allows the release of DNA into the cytosol (Fernandes-Alnemri et al., 2010; Jones et al., 2010).
In contrast to previously discussed bacterial pathogens, caspase-1 activation in response to F. novicida requires IRF3-dependent Type I IFN signaling (Henry et al., 2007). Salmonella, which resides in vacuoles within the macrophage, does not require Type I IFN, but the cytosolic bacteria L. monocytogenes does (Henry et al., 2007). Type I IFN was demonstrated to increase AIM2 expression and hence AIM2-dependent caspase-1 activation, which was reduced in Irf3−/− macrophages (Fernandes-Alnemri et al., 2010; Jones et al., 2010). Interestingly, while phagosome lysis was required for AIM2 sensing, NLRP3 thought to be activated by vacuole lysis, was not activated in Francisella infection (Fernandes-Alnemri et al., 2010). Notably, the F. tularensis protein mvIN, which promotes virulence, exerts its effects by inhibiting AIM2-mediated caspase-1 activation (Ulland et al., 2010).
Similarly to Francisella, L. monocytogenes escapes from the phagosome into the cytosol, through the membrane-disrupting enzyme listeriolysin O (LLO). In vivo, caspase-1-deficient mice display reduced survival following infection and could be partially rescued through exogenous IL-18 administration (Tsuji et al., 2004), suggesting that inflammasome signaling is protective during infection. Macrophage infection results in caspase-1 and Asc-dependent cell death, with involvement of AIM2, Nlrp3, and Nlrc4 (Kim et al., 2010; Sauer et al., 2010). LLO is required for cell death and LLO-deficient strains, which remain in the phagosome, are unable to activate caspase-1 or induce IL-1β secretion (Mariathasan et al., 2006; Ozoren et al., 2006; Cervantes et al., 2008). Purified LLO can directly induce Nlrp3-dependent IL-1β secretion (Meixenberger et al., 2010; Sauer et al., 2010). Interestingly, however, entry into the cytosol alone is not sufficient to trigger caspase-1 activation. Indeed, LLO substitution with the L. ivanovii homolog ivanolysin O is not permissive for inflammasome activation (Hara et al., 2008), suggesting that LLO may have additional roles in this process.
Different reports have implicated the AIM2, NLRC4, and NLRP3 inflammasomes to varying degrees in the response to Listeria infection, however there have been some conflicting evidence. The earliest study identified Nlrp3 and Asc as essential for caspase-1 activation and IL-1β secretion in macrophages (Mariathasan et al., 2006). The role of these inflammasomes was confirmed along with Nlrc4, which was implicated in the detection of Listeria flagellin, and also dependent on LLO (Warren et al., 2008). Asc-deficient macrophages infected with flagellin deleted strains resulted in reduced IL-1β secretion than similarly infected Nlrp3 null macrophages, suggesting that an unknown inflammasome, signaling through Asc, was required. Similarly to recognition of Francisella, AIM2 was found to activate caspase-1 and induce IL-1β secretion (Kim et al., 2010; Rathinam et al., 2010; Sauer et al., 2010; Wu et al., 2010). Lysis of the bacteria is important for inflammasome recognition as treatment of intracellular bacteria with β-lactam antibiotics or the use of mutant bacteria that undergo rapid lysis in the cytosol resulted in increased inflammasome activation and pyroptosis (Sauer et al., 2010). L. monocytogenes DNA was also found to colocalize with ASC pyroptosomes (Warren et al., 2010). Another study using siRNA depletion in human PBMCs determined that NLRP3 was required for IL-1β secretion, but not AIM2 or NLRC4 (Meixenberger et al., 2010). The Listeria strain used however, expresses lower levels of flagellin, which may have resulted in the lack of requirement for NLRC4. Similarly other studies have implicated Nlrc4 along with Aim2, Nlrp3, and Asc (Tsuchiya et al., 2010; Wu et al., 2010), while others did not find that it played a role (Kim et al., 2010). Inflammasomes appear to have a protective effect during L. monocytogenes infection, through pyroptosis. Interestingly, Listeria attempts to evade inflammasome recognition by downregulating activators such as flagellin at 37°C (Shen and Higgins, 2006).
While some pathogenic organisms evade the immune system through an intracellular lifestyle, other pathogenic bacteria survive outside of the cell. Extracellular species often secrete toxins that can cause tissue damage and inflammation. Upon attachment and adhesion to surfaces, such as epithelial cells, extracellular bacteria often produce a biofilm, a polysaccharide rich matrix that provides protection from leucocytes and components of the immune system. B. anthracis, is a well known extracellular bacterial pathogen that is the cause of anthrax. The bacteria secrete lethal toxin (LeTx), which is composed of the protective antigen (PA) and the zinc-dependent metalloprotease lethal factor (LF) subunits (Collier and Young, 2003). PA mediates toxin entry into the cytosol while LF cleaves MAPK to disrupt cell signaling (Duesbery et al., 1998). Polymorphisms in the gene encoding Nlrp1 confer susceptibility or resistance to LeTx in mouse macrophages. LeTx is responsible for caspase-1-dependent pyroptosis in murine macrophages through activation of the Nlrp1 inflammasome (Boyden and Dietrich, 2006), but the adaptor Asc is not required in this process (Reig et al., 2008). The exact mechanism by which LeTx triggers Nlrp1 activation is currently unknown. Endosome acidification, proteasome activity, and potassium efflux are required for cell death (Squires et al., 2007; Fink et al., 2008; Wickliffe et al., 2008). Nlrp1-mediated cell lysis appears to have a protective effect in rats as it reduces mortality to infection with B. anthracis (Newman et al., 2010). B. anthracis spores are also capable of inducing caspase-1 activation through Nlrp1 in vivo, which confers protection in mice (Terra et al., 2010). Human NLRP1 has also been shown to recognize the bacterial cell wall product muramyl-dipeptide (Faustin et al., 2007).
In summary, multiple inflammasomes have been implicated in response to intracellular and extracellular bacteria. While this can often lead to pyroptotic cell death, the overall effect in mouse models suggests that caspase-1 activation and IL-1β and IL-18 secretion promote bacterial clearance and host survival. Caspase-1-mediated pyroptosis of cells infected with intracellular bacteria may play a beneficial role in eliminating their replicative niche. The discovery of bacterial proteins that inhibit the inflammasomes, such as P. aeruginosa ExoU (Sutterwala et al., 2007) and M. tuberculosis zmp1 (Master et al., 2008) further indicates that the effects of caspase-1 are likely beneficial for the host rather than the pathogen. Activation can be initiated by flagellin, secretion system components, and bacterial effector molecules injected in the host cell cytosol through these secretion systems. These signals are relatively conserved and it is likely that the NLRs have evolved to detect these distinct molecules. The most recent evidence suggests that there is significant redundancy in inflammasome activation during bacterial infections. In addition to promoting host defense against pathogenic bacteria, the inflammasome has been recently implicated in sensing commensal microorganisms in the gut and maintaining intestinal homeostasis and immune tolerance to the microflora. In addition, upon tissue damage, Nlrp3 inflammasome-dependent production of IL-18 was shown to be required for tissue repair and protection from colitis and colitis-associated colorectal cancer (Allen et al., 2010; Dupaul-Chicoine et al., 2010; Saleh and Trinchieri, 2011; Zaki et al., 2010).
Viruses
Viruses infect and replicate within host cells, utilizing the host cellular machinery to manufacture viral particles. In mammals, both the innate and adaptive branches of the immune system play an important role in fighting viral infections. Different PRRs are capable of recognizing viral motifs, primarily those with nucleic acids, and initiate type I interferon production which inhibits viral replication (Saito and Gale Jr., 2007). Multiple inflammasomes including NLRP3, AIM2, and RIG-I have been linked to viral infections (Table 1).
Prior to the discovery of the inflammasome, it has been reported that influenza infection induced IL-1 production by infected macrophages and in mice (Hennet et al., 1992; Pirhonen et al., 1999), which mediated mouse survival by enhancing adaptive immunity to the virus (Schmitz et al., 2005). The Nlrp3 inflammasome was subsequently identified to play a role in influenza and Sendai virus infection in macrophages (Kanneganti et al., 2006). In vitro, the NLRP3 inflammasome is triggered by various viruses and viral PAMPs, including viral RNA analogs poly(I:C) and ssRNA40 (Kanneganti et al., 2006; Allen et al., 2009), transfected adenovirus DNA (Muruve et al., 2008), and rotavirus dsRNA (Kanneganti et al., 2006). However, in vivo, there have been some conflicting findings as to the role of the inflammasome, and the involvement of its different components, in influenza virus infection. Notably, Asc, caspase-1, and Nlrp3, but not Nlrc4 were shown to be essential for mouse survival (Allen et al., 2009; Thomas et al., 2009), but this result was only reproduced for Asc, caspase-1 and IL-1R, but not Nlrp3 (Ichinohe et al., 2009). These discrepancies may be largely due to different infectious doses. Higher infectious doses (>1000 pfu) trigger marked inflammatory cell infiltration to the airways, pulmonary necrosis and fibrosis, that are reduced in Nlrp3 and caspase-1 deficient mice (Allen et al., 2009; Thomas et al., 2009), suggesting a role of the inflammasome in lung tissue healing. Lower infectious doses (10 pfu), on the other hand, are less injurious, which allows for the examination of inflammasome function in immune responses. Under these conditions, casp1−/− and Asc−/− mice exhibit reduced CD4 and CD8 T cell responses and decreased immunoglobulin titers (Ichinohe et al., 2009). A recently discovered influenza M2 ion channel was demonstrated to localize to the host cell Golgi and activate Nlrp3 by inducing cytosolic acidification (Ichinohe et al., 2010).
Vesicular stomatitis virus (VSV), another RNA virus, is recognized by the intracellular receptor with RNA helicase activity RIG-I. RIG-I detects 5′ triphosphate RNA as a viral signature and induces type I IFN production and NF-κB activation through recruitment of the adaptor protein MAVS (Nakhaei et al., 2009). Interestingly, infection with VSV also triggers IL-1β secretion in a RIG-I-dependent manner (Poeck et al., 2010). ASC interacts with RIG-I and is required for VSV-induced caspase-1 activation, which requires potassium efflux, but not Type I IFN. These results indicate that RIG-I serves multiple roles during the innate immune response to viruses, as it triggers Type I IFN expression and activates the NF-κB and caspase-1 pro-inflammatory pathways.
DNA viruses are also capable of activating caspase-1 through the AIM2 inflammasome. As previously discussed, AIM2 detects cytosolic DNA, and is engaged in response to vaccinia virus and mouse cytomegalovirus (mCMV), but interestingly not herpes simplex virus type 1 (HSV-1) (Rathinam et al., 2010). Similarly to its response to bacterial DNA, AIM2 requires ASC, but not NLRP3 (Hornung et al., 2009; Rathinam et al., 2010). mCMV-infected AIM2 deficient mice have reduced IL-18 serum levels that correlate with a reduced population of IFNγ+ NK cells. IL-18 is a known activator of IFNγ production in NK cells (Hyodo et al., 1999; Chaix et al., 2008), which play an important role in eliminating tumor or virus-infected cells, and is likely one of the primary contributors to the higher viral titers found in the spleen of AIM2-deficient mice (Rathinam et al., 2010).
Caspase-1-induced inflammation and pyroptosis are deleterious for virus replication and dissemination. Consequently, viruses have evolved proteins to target caspase-1 function and its downstream signaling, including a poxvirus PYD containing protein (Johnston et al., 2005) and influenza NS1 (Stasakova et al., 2005).
Parasites
The protozoan Plasmodium genus is the causative agent of malaria, which is endemic to the tropics and sub-Saharan Africa. The parasite is transmitted by mosquito bite into the host, where after an incubation period, replicates within erythrocytes (Sachs and Malaney, 2002). The breakdown of hemoglobin by Plasmodium results in the release of heme, which is then converted into the crystalline product hemozoin (Hanscheid et al., 2007). Recent work has suggested that the NLRP3 inflammasome might play a role in the recognition of hemozoin. Hemozoin was reported to activate caspase-1 and induce IL-1β secretion in vitro, in an ASC and NLRP3, but not NLRC4, dependent manner (Dostert et al., 2009; Shio et al., 2009), but this was not reproduced in one study (Griffith et al., 2009). Mechanistically, hemozoin is not likely to be directly recognized by NLRP3, but instead activates the inflammasome through other signals, such as uric acid crystal release (Griffith et al., 2009) and/or activation of cathepsin B downstream of the SYK and LYN kinases (Shio et al., 2009). The role of cathepsin B is controversial however, as cathepsin B-deficient macrophages did not exhibit a defect in caspase-1 activation in response to hemozoin (Dostert et al., 2009). Interestingly, deficiency in Nlrp3 but not Asc or caspase-1 increased mouse survival after injection with red blood cells infected with the mouse virulent strain P. chabaudi adami (Shio et al., 2009). Another mouse model of cerebral malaria using P. berghei similarly showed that Nlrp3−/− mice, but not Asc−/−, casp1−/−, or Il1b−/− mice, had a greater time to death than wild-type animals (Reimer et al., 2010). This suggests that NLRP3 may have a deleterious role in cerebral malaria, independently of caspase-1 and IL-1β. Consistently, we have shown that casp1−/− mice were equivalent to wild-type mice in their response to the P. chabaudi and P. berghei models of malaria (Labbe et al., 2010). In contrast, mice deficient in caspase-12, an inhibitor of caspase-1 and NF-κB, showed enhanced inflammatory and immune response to the Plasmodium parasites and were susceptible to cerebral malaria. This phenotype was, however, independent of caspase-1, but resulted from excessive NF-κB activation and IFNγ production (Labbe et al., 2010). Plasmodium is currently the only parasite characterized with respect to inflammasome activation, and the study of other protozoa and helminths may lead to a better understanding of the immune response to these organisms and to insights into unknown functions of the inflammasome and NLRs.
Fungi
Fungi are eukaryotic organisms that often cause opportunistic infections in immuno-compromised hosts. The most common cause of fungal infections is Candida albicans, a commensal that resides in the majority of the human population. It exists as a unicellular yeast or a filamentous hyphae, the latter associated with invasion (Lo et al., 1997). C. albicans and other opportunistic fungi are cleared in healthy individuals and various PRRs, such as TLRs and CLRs have been implicated as important players in the immune response to fungi (Netea and Marodi, 2010). IL-1β is essential in host defense in candidiasis (Vonk et al., 2006) and recently the inflammasome has been linked to this response.
Caspase-1, IL-1R, ASC, and NLRP3, have been implicated in controlling fungal dissemination and host survival in mice (Gross et al., 2009; Hise et al., 2009; Joly et al., 2009). Caspase-1, Asc, or Il-1r deficient mice have reduced survival and increased organ fungal burden upon C. albicans infection (Hise et al., 2009). Nlrp3−/− mice have increased mortality following infection (Gross et al., 2009) and higher tissue colony counts (Gross et al., 2009; Hise et al., 2009; Joly et al., 2009). The fungal cell wall component β-glucan is recognized by the CLR, dectin-1, which recruits SYK via phosphorylated ITAMs in its cytoplasmic tail leading to synthesis of pro-IL-1β and NLRP3 inflammasome components (Rogers et al., 2005; Gross et al., 2009; Hise et al., 2009; Kumar et al., 2009; van de Veerdonk et al., 2009). TLR2 has also been implicated in this recognition (Hise et al., 2009; van de Veerdonk et al., 2009). While these receptors provide signal 1 toward NLRP3 activation, signal 2 is mediated by potassium efflux and ROS production (Gross et al., 2009). Consistently, Syk−/− dendritic cells display reduced caspase-1 activation and production of IL-1β in response to C. albicans, but not Salmonella, ATP or nigericin (Gross et al., 2009). The role of cathepsin B in C. albicans inflammasome activation is unclear as IL-1β secretion from cathepsin B-deficient dendritic cells was unaffected in one study (Gross et al., 2009), while cell treatment with the cathepsin B cell permeable inhibitor CA-074-Me caused a significant decrease in secreted IL-1β in another (Joly et al., 2009).
Interestingly, the transition from the yeast to the filamentous form of C. albicans is required for Nlrp3 inflammasome activation, as mutant fungi unable to transform to the hyphae stage trigger blunted IL-1β production (Joly et al., 2009). The hyphae form is able to disrupt the phagosome (Lo et al., 1997), and it is hypothesized that this event is sensed as signal 2. The observation that cathepsin B inhibitors reduced IL-1β secretion from macrophages (Joly et al., 2009) supports this hypothesis as vacuole rupture and cathepsin B activity have been linked to NLRP3 activation (Hornung et al., 2008). Notably, UV-inactivated C. albicans yeast cells are unable to activate the inflammasome (Hise et al., 2009; Joly et al., 2009). However, in human PBMCs heat-killed yeast cells could induce IL-1β secretion (van de Veerdonk et al., 2009), presumably due to the ability of these cells to produce excessive levels of ATP which activates the inflammasome in the presence of a signal 1, provided in this case by the heat-killed cells.
Interestingly, treatment of B cells with β-glucan resulted in Nlrp3 activation and antibody production. This occurred independently of IL-1R signaling, as determined by the use of Myd88−/− cells. This is suggestive of a link between NLRP3 and the adaptive immune system in response to fungal infections (Kumar et al., 2009). While the large majority of studies on the role of inflammasomes in fungal infections has been discovered using C. albicans, another opportunistic fungus, Aspergillus fumigates, was also found to activate the NLRP3 inflammasome via similar mechanisms (Said-Sadier et al., 2010). Altogether, these studies indicate that the NLRP3 inflammasome plays a protective role against fungal infections.
Conclusion
Innate immunity and inflammation are important for the host defense against invading microbes. The rapidly expanding number of studies on the inflammasomes has begun to unravel their roles in response to bacterial, parasitic, fungal, and viral infections. Current evidence supports that inflammasome activation during infection confers protection to the host through various mechanisms including pyroptosis, pro-inflammatory cytokine secretion, and stimulation of the adaptive immune system. While pyroptosis of infected macrophages may intuitively seem deleterious for the host, it can permit exposure of intracellular bacteria to other bactericidal host factors. Consequently many pathogens have evolved different mechanisms to inhibit inflammasome activation and function. There have been many recent advances in understanding the different motifs that are recognized by the inflammasomes and how they work. However, much remains to be done for a full understanding of the molecular events linking cellular perturbations and DAMP sensing to inflammasome activation. While the majority of studies researching the inflammasomes have focused on the NLRP3 and NLRC4 complexes, the recent discoveries of AIM2 and RIG-I in caspase-1 activation open a new window of investigation. Moreover, it is likely that additional inflammasomes have not yet been discovered. Importantly, it is becoming clear that while NLRs and ASC assemble inflammasomes, their functions are not always necessarily mediated by the caspase-1 pathway. Similarly, caspase-1 dependent effector cytokines could be produced by other proteases during infection. Additionally, NLR signaling pathways are significantly redundant and overlapping with those of other PRRs. Therefore, a stronger understanding of inflammasome-mediated innate immune responses is warranted toward the development of therapeutic strategies for infectious disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akhter, A., Gavrilin, M. A., Frantz, L., Washington, S., Ditty, C., Limoli, D., Day, C., Sarkar, A., Newland, C., Butchar, J., Marsh, C. B., Wewers, M. D., Tridandapani, S., Kanneganti, T. D., and Amer, A. O. (2009). Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. 5, e1000361. doi: 10.1371/journal.ppat.1000361
Alcami, A., and Smith, G. L. (1992). A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell 71, 153–167.
Allen, I. C., Scull, M. A., Moore, C. B., Holl, E. K., Mcelvania-Tekippe, E., Taxman, D. J., Guthrie, E. H., Pickles, R. J., and Ting, J. P. (2009). The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565.
Allen, I. C., Tekippe, E. M., Woodford, R. M. T., Uronis, J. M., Holl, E. K., Rogers, A. B., Herfarth, H. H., Jobin, C., and Ting, J. P. Y. (2010). The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056.
Amer, A., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Ozoren, N., Brady, G., Meshinchi, S., Jagirdar, R., Gewirtz, A., Akira, S., and Nunez, G. (2006). Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223.
Arlehamn, C. S., Petrilli, V., Gross, O., Tschopp, J., and Evans, T. J. (2010). The role of potassium in inflammasome activation by bacteria. J. Biol. Chem. 285, 10508–10518.
Belkhadir, Y., Subramaniam, R., and Dangl, J. L. (2004). Plant disease resistance protein signaling: NBS-LRR proteins and their partners. Curr. Opin. Plant Biol. 7, 391–399.
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5.
Boyden, E. D., and Dietrich, W. F. (2006). Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 38, 240–244.
Brodsky, I. E., Palm, N. W., Sadanand, S., Ryndak, M. B., Sutterwala, F. S., Flavell, R. A., Bliska, J. B., and Medzhitov, R. (2010). A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387.
Broz, P., Newton, K., Lamkanfi, M., Mariathasan, S., Dixit, V. M., and Monack, D. M. (2010). Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755.
Bruey, J. M., Bruey-Sedano, N., Luciano, F., Zhai, D., Balpai, R., Xu, C., et al. (2007). Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 129, 45–56.
Carlsson, F., Kim, J., Dumitru, C., Barck, K. H., Carano, R. A., Sun, M., Diehl, L., and Brown, E. J. (2010). Host-detrimental role of Esx-1-mediated inflammasome activation in mycobacterial infection. PLoS Pathog. 6, e1000895. doi: 10.1371/journal.ppat.1000895
Case, C. L., Shin, S., and Roy, C. R. (2009). Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect. Immun. 77, 1981–1991.
Cassel, S. L., Eisenbarth, S. C., Iyer, S. S., Sadler, J. J., Colegio, O. R., Tephly, L. A., Carter, A. B., Rothman, P. B., Flavell, R. A., and Sutterwala, F. S. (2008). The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. U.S.A. 105, 9035–9040.
Cervantes, J., Nagata, T., Uchijima, M., Shibata, K., and Koide, Y. (2008). Intracytosolic Listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cell. Microbiol. 10, 41–52.
Chaix, J., Tessmer, M. S., Hoebe, K., Fuseri, N., Ryffel, B., Dalod, M., Alexopoulou, L., Beutler, B., Brossay, L., Vivier, E., and Walzer, T. (2008). Cutting edge: priming of NK cells by IL-18. J. Immunol. 181, 1627–1631.
Chamaillard, M., Hashimoto, M., Horie, Y., Masumoto, J., Qiu, S., Saab, L., Ogura, Y., Kawasaki, A., Fukase, K., Kusumoto, S., Valvano, M. A., Foster, S. J., Mak, T. W., Nunez, G., and Inohara, N. (2003). An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707.
Coers, J., Vance, R. E., Fontana, M. F., and Dietrich, W. F. (2007). Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol. 9, 2344–2357.
Craven, R. R., Gao, X., Allen, I. C., Gris, D., Bubeck Wardenburg, J., Mcelvania-Tekippe, E., Ting, J. P., and Duncan, J. A. (2009). Staphylococcus aureus α-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 4, e7446. doi: 10.1371/journal.pone.0007446
Diez, E., Lee, S. H., Gauthier, S., Yaraghi, Z., Tremblay, M., Vidal, S., and Gros, P. (2003). Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat. Genet. 33, 55–60.
Dorfleutner, A., Bryan, N. B., Talbott, S. J., Funya, K. N., Rellick, S. L., Reed, J. C., Shi, X., Rojanasakul, Y., Flynn, D. C., and Stehlik, C. (2007). Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infect. Immun. 75, 1484–1492.
Dostert, C., Guarda, G., Romero, J. F., Menu, P., Gross, O., Tardivel, A., Suva, M. L., Stehle, J. C., Kopf, M., Stamenkovic, I., Corradin, G., and Tschopp, J. (2009). Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE 4, e6510. doi: 10.1371/journal.pone.0006510
Dostert, C., Petrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677.
Duesbery, N. S., Webb, C. P., Leppla, S. H., Gordon, V. M., Klimpel, K. R., Copeland, T. D., Ahn, N. G., Oskarsson, M. K., Fukasawa, K., Paull, K. D., and Vande Woude, G. F. (1998). Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280, 734–737.
Dupaul-Chicoine, J., Yeretssian, G., Doiron, K., Bergstrom, K. S., Mcintire, C. R., Leblanc, P. M., Meunier, C., Turbide, C., Gros, P., Beauchemin, N., Vallance, B. A., and Saleh, M. (2010). Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378.
Faustin, B., Chen, Y., Zhai, D. Y., Le Negrate, G., Lartigue, L., Satterthwait, A., and Reed, J. C. (2009). Mechanism of Bcl-2 and Bcl-X-L inhibition of NLRP1 inflammasome: loop domain-dependent suppression of ATP binding and oligomerization. Proc. Natl. Acad. Sci. U.S.A. 106, 3935–3940.
Faustin, B., Lartigue, L., Bruey, J. M., Luciano, F., Sergienko, E., Bailly-Maitre, B., Volkmann, N., Hanein, D., Rouiller, I., and Reed, J. C. (2007). Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 25, 713–724.
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513.
Fernandes-Alnemri, T., Yu, J. W., Juliana, C., Solorzano, L., Kang, S., Wu, J., Datta, P., Mccormick, M., Huang, L., Mcdermott, E., Eisenlohr, L., Landel, C. P., and Alnemri, E. S. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393.
Fink, S. L., Bergsbaken, T., and Cookson, B. T. (2008). Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1- dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 105, 4312–4317.
Fink, S. L., and Cookson, B. T. (2005). Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916.
Fortier, A., de Chastellier, C., Balor, S., and Gros, P. (2007). Birc1e/Naip5 rapidly antagonizes modulation of phagosome maturation by Legionella pneumophila. Cell. Microbiol. 9, 910–923.
Fortier, A., Doiron, K., Saleh, M., Grinstein, S., and Gros, P. (2009). Restriction of Legionella pneumophila replication in macrophages requires concerted action of the transcriptional regulators Irf1 and Irf8 and nod-like receptors Naip5 and Nlrc4. Infect. Immun. 77, 4794–4805.
Franchi, L., Amer, A., Body-Malapel, M., Kanneganti, T. D., Ozoren, N., Jagirdar, R., Inohara, N., Vandenabeele, P., Bertin, J., Coyle, A., Grant, E. P., and Nunez, G. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat. Immunol. 7, 576–582.
Franchi, L., Kanneganti, T. D., Dubyak, G. R., and Nunez, G. (2007). Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 282, 18810–18818.
Fremond, C. M., Togbe, D., Doz, E., Rose, S., Vasseur, V., Maillet, I., Jacobs, M., Ryffel, B., and Quesniaux, V. F. (2007). IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J. Immunol. 179, 1178–1189.
Gabay, C., Lamacchia, C., and Palmer, G. (2010). IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 6, 232–241.
Gavrilin, M. A., Bouakl, I. J., Knatz, N. L., Duncan, M. D., Hall, M. W., Gunn, J. S., and Wewers, M. D. (2006). Internalization and phagosome escape required for Francisella to induce human monocyte IL-1beta processing and release. Proc. Natl. Acad. Sci. U.S.A. 103, 141–146.
Ghayur, T., Banerjee, S., Hugunin, M., Butler, D., Herzog, L., Carter, A., Quintal, L., Sekut, L., Talanian, R., Paskind, M., Wong, W., Kamen, R., Tracey, D., and Allen, H. (1997). Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386, 619–623.
Girardin, S. E., Boneca, I. G., Carneiro, L.a.M., Antignac, A., Jehanno, M., Viala, J., Tedin, K., Taha, M. K., Labigne, A., Zahringer, U., Coyle, A. J., Bertin, J., Sansonetti, P. J., and Philpott, D. J. (2003). Nod1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science 300, 1584–1587.
Griffith, J. W., Sun, T., McIntosh, M. T., and Bucala, R. (2009). Pure hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J. Immunol. 183, 5208–5220.
Gross, O., Poeck, H., Bscheider, M., Dostert, C., Hannesschlager, N., Endres, S., Hartmann, G., Tardivel, A., Schweighoffer, E., Tybulewicz, V., Mocsai, A., Tschopp, J., and Ruland, J. (2009). Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459, 433–436.
Gu, Y., Kuida, K., Tsutsui, H., Ku, G., Hsiao, K., Fleming, M. A., Hayashi, N., Higashino, K., Okamura, H., Nakanishi, K., Kurimoto, M., Tanimoto, T., Flavell, R. A., Sato, V., Harding, M. W., Livingston, D. J., and Su, M. S. S. (1997). Activation of interferon-gamma inducing factor mediated by interleukin-1 beta converting enzyme. Science 275, 206–209.
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., Fitzgerald, K. A., Latz, E., Moore, K. J., and Golenbock, D. T. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865.
Hanscheid, T., Egan, T. J., and Grobusch, M. P. (2007). Haemozoin: from melatonin pigment to drug target, diagnostic tool, and immune modulator. Lancet Infect. Dis. 7, 675–685.
Hara, H., Tsuchiya, K., Nomura, T., Kawamura, I., Shoma, S., and Mitsuyama, M. (2008). Dependency of caspase-1 activation induced in macrophages by Listeria monocytogenes on cytolysin, listeriolysin O, after evasion from phagosome into the cytoplasm. J. Immunol. 180, 7859–7868.
Haraga, A., Ohlson, M. B., and Miller, S. I. (2008). Salmonellae interplay with host cells. Nat. Rev. Microbiol. 6, 53–66.
Hardt, W. D., Chen, L. M., Schuebel, K. E., Bustelo, X. R., and Galan, J. E. (1998). S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93, 815–826.
Hennet, T., Ziltener, H. J., Frei, K., and Peterhans, E. (1992). A kinetic study of immune mediators in the lungs of mice infected with influenza A virus. J. Immunol. 149, 932–939.
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J., and Monack, D. M. (2007). Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994.
Hersh, D., Monack, D. M., Smith, M. R., Ghori, N., Falkow, S., and Zychlinsky, A. (1999). The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. U.S.A. 96, 2396–2401.
Hise, A. G., Tomalka, J., Ganesan, S., Patel, K., Hall, B. A., Brown, G. D., and Fitzgerald, K. A. (2009). An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5, 487–497.
Hoffmann, C., Galle, M., Dilling, S., Kappeli, R., Muller, A. J., Songhet, P., Beyaert, R., and Hardt, W. D. (2010). In macrophages, caspase-1 activation by SopE and the type III secretion system-1 of S. typhimurium can proceed in the absence of flagellin. PLoS ONE 5, e12477. doi: 10.1371/journal.pone.0012477
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., Latz, E., and Fitzgerald, K. A. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518.
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., Fitzgerald, K. A., and Latz, E. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856.
Hsu, L. C., Ali, S. R., Mcgillivray, S., Tseng, P. H., Mariathasan, S., Humke, E. W., Eckmann, L., Powell, J. J., Nizet, V., Dixit, V. M., and Karin, M. (2008). A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. U.S.A. 105, 7803–7808.
Hyodo, Y., Matsui, K., Hayashi, N., Tsutsui, H., Kashiwamura, S., Yamauchi, H., Hiroishi, K., Takeda, K., Tagawa, Y., Iwakura, Y., Kayagaki, N., Kurimoto, M., Okamura, H., Hada, T., Yagita, H., Akira, S., Nakanishi, K., and Higashino, K. (1999). IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 162, 1662–1668.
Ichinohe, T., Lee, H. K., Ogura, Y., Flavell, R., and Iwasaki, A. (2009). Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206, 79–87.
Ichinohe, T., Pang, I. K., and Iwasaki, A. (2010). Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11, 404–410.
Inohara, N., Ogura, Y., Fontalba, A., Gutierrez, O., Pons, F., Crespo, J., Fukase, K., Inamura, S., Kusumoto, S., Hashimoto, M., Foster, S. J., Moran, A. P., Fernandez-Luna, J. L., and Nunez, G. (2003). Host recognition of bacterial muramyl dipeptide mediated through NOD2. J. Biol. Chem. 278, 5509–5512.
Isberg, R. R., O’Connor, T. J., and Heidtman, M. (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24.
Johnston, J. B., Barrett, J. W., Nazarian, S. H., Goodwin, M., Ricciuto, D., Wang, G., and McFadden, G. (2005). A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 23, 587–598.
Joly, S., Ma, N., Sadler, J. J., Soll, D. R., Cassel, S. L., and Sutterwala, F. S. (2009). Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J. Immunol. 183, 3578–3581.
Jones, J. W., Kayagaki, N., Broz, P., Henry, T., Newton, K., O’Rourke, K., Chan, S., Dong, J., Qu, Y., Roose-Girma, M., Dixit, V. M., and Monack, D. M. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776.
Juffermans, N. P., Florquin, S., Camoglio, L., Verbon, A., Kolk, A. H., Speelman, P., Van Deventer, S. J., and Van Der Poll, T. (2000). Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J. Infect. Dis. 182, 902–908.
Kanneganti, T. D., Body-Malapel, M., Amer, A., Park, J. H., Whitfield, J., Franchi, L., Taraporewala, Z. F., Miller, D., Patton, J. T., Inohara, N., and Nunez, G. (2006). Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568.
Keller, M., Ruegg, A., Werner, S., and Beer, H. D. (2008). Active caspase-1 is a regulator of unconventional protein secretion. Cell 132, 818–831.
Kim, S., Bauernfeind, F., Ablasser, A., Hartmann, G., Fitzgerald, K. A., Latz, E., and Hornung, V. (2010). Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 40, 1545–1551.
Koo, I. C., Wang, C., Raghavan, S., Morisaki, J. H., Cox, J. S., and Brown, E. J. (2008). ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell. Microbiol. 10, 1866–1878.
Kumar, H., Kumagai, Y., Tsuchida, T., Koenig, P. A., Satoh, T., Guo, Z., Jang, M. H., Saitoh, T., Akira, S., and Kawai, T. (2009). Involvement of the NLRP3 inflammasome in innate and humoral adaptive immune responses to fungal beta-glucan. J. Immunol. 183, 8061–8067.
Kurenuma, T., Kawamura, I., Hara, H., Uchiyama, R., Daim, S., Dewamitta, S. R., Sakai, S., Tsuchiya, K., Nomura, T., and Mitsuyama, M. (2009). The RD1 locus in the Mycobacterium tuberculosis genome contributes to activation of caspase-1 via induction of potassium ion efflux in infected macrophages. Infect. Immun. 77, 3992–4001.
Labbe, K., Miu, J., Yeretssian, G., Serghides, L., Tam, M., Finney, C. A., Erdman, L. K., Goulet, M. L., Kain, K. C., Stevenson, M. M., and Saleh, M. (2010). Caspase-12 dampens the immune response to malaria independently of the inflammasome by targeting NF-kappaB signaling. J. Immunol. 185, 5495–5502.
Labbe, K., and Saleh, M. (2008). Cell death in the host response to infection. Cell Death Differ. 15, 1339–1349.
Lamkanfi, M., Amer, A., Kanneganti, T. D., Munoz-Planillo, R., Chen, G., Vandenabeele, P., Fortier, A., Gros, P., and Nunez, G. (2007). The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J. Immunol. 178, 8022–8027.
Lamkanfi, M., Kanneganti, T. D., Van Damme, P., Vanden Berghe, T., Vanoverberghe, I., Vandekerckhove, J., Vandenabeele, P., Gevaert, K., and Nunez, G. (2008). Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell Proteomics 7, 2350–2363.
Lara-Tejero, M., Sutterwala, F. S., Ogura, Y., Grant, E. P., Bertin, J., Coyle, A. J., Flavell, R. A., and Galan, J. E. (2006). Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 203, 1407–1412.
Lightfield, K. L., Persson, J., Brubaker, S. W., Witte, C. E., Von Moltke, J., Dunipace, E. A., Henry, T., Sun, Y. H., Cado, D., Dietrich, W. F., Monack, D. M., Tsolis, R. M., and Vance, R. E. (2008). Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178.
Lin, X. Y., Choi, M. S. K., and Porter, A. G. (2000). Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. J. Biol. Chem. 275, 39920–39926.
Lo, H. J., Kohler, J. R., DiDomenico, B., Loebenberg, D., Cacciapuoti, A., and Fink, G. R. (1997). Nonfilamentous C. albicans mutants are avirulent. Cell 90, 939–949.
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., Roose-Girma, M., Erickson, S., and Dixit, V. M. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218.
Mariathasan, S., Weiss, D. S., Dixit, V. M., and Monack, D. M. (2005). Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049.
Mariathasan, S., Weiss, D. S., Newton, K., Mcbride, J., O’rourke, K., Roose-Girma, M., Lee, W. P., Weinrauch, Y., Monack, D. M., and Dixit, V. M. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232.
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241.
Master, S. S., Rampini, S. K., Davis, A. S., Keller, C., Ehlers, S., Springer, B., Timmins, G. S., Sander, P., and Deretic, V. (2008). Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3, 224–232.
Mayer-Barber, K. D., Barber, D. L., Shenderov, K., White, S. D., Wilson, M. S., Cheever, A., Kugler, D., Hieny, S., Caspar, P., Nunez, G., Schlueter, D., Flavell, R. A., Sutterwala, F. S., and Sher, A. (2010). Caspase-1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J. Immunol. 184, 3326–3330.
McElvania Tekippe, E., Allen, I. C., Hulseberg, P. D., Sullivan, J. T., McCann, J. R., Sandor, M., Braunstein, M., and Ting, J. P. (2010). Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS ONE 5, e12320. doi: 10.1371/journal.pone.0012320
Meixenberger, K., Pache, F., Eitel, J., Schmeck, B., Hippenstiel, S., Slevogt, H., N’guessan, P., Witzenrath, M., Netea, M. G., Chakraborty, T., Suttorp, N., and Opitz, B. (2010). Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J. Immunol. 184, 922–930.
Miao, E. A., Alpuche-Aranda, C. M., Dors, M., Clark, A. E., Bader, M. W., Miller, S. I., and Aderem, A. (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575.
Miao, E. A., Ernst, R. K., Dors, M., Mao, D. P., and Aderem, A. (2008). Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. U.S.A. 105, 2562–2567.
Miao, E. A., Leaf, I. A., Treuting, P. M., Mao, D. P., Dors, M., Sarkar, A., Warren, S. E., Wewers, M. D., and Aderem, A. (2010a). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1194.
Miao, E. A., Mao, D. P., Yudkovsky, N., Bonneau, R., Lorang, C. G., Warren, S. E., Leaf, I. A., and Aderem, A. (2010b). Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 107, 3076–3080.
Mishra, B. B., Moura-Alves, P., Sonawane, A., Hacohen, N., Griffiths, G., Moita, L. F., and Anes, E. (2010). Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell. Microbiol. 12, 1046–1063.
Molofsky, A. B., Byrne, B. G., Whitfield, N. N., Madigan, C. A., Fuse, E. T., Tateda, K., and Swanson, M. S. (2006). Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203, 1093–1104.
Muller, A. J., Hoffmann, C., Galle, M., Van Den Broeke, A., Heikenwalder, M., Falter, L., Misselwitz, B., Kremer, M., Beyaert, R., and Hardt, W. D. (2009). The S. typhimurium effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation. Cell Host Microbe 6, 125–136.
Muruve, D. A., Petrilli, V., Zaiss, A. K., White, L. R., Clark, S. A., Ross, P. J., Parks, R. J., and Tschopp, J. (2008). The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107.
Nakhaei, P., Genin, P., Civas, A., and Hiscott, J. (2009). RIG-I-like receptors: sensing and responding to RNA virus infection. Semin. Immunol. 21, 215–222.
Netea, M. G., and Marodi, L. (2010). Innate immune mechanisms for recognition and uptake of Candida species. Trends Immunol. 31, 346–353.
Newman, Z. L., Printz, M. P., Liu, S., Crown, D., Breen, L., Miller-Randolph, S., Flodman, P., Leppla, S. H., and Moayeri, M. (2010). Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PLoS Pathog. 6, e1000906. doi: 10.1371/journal.ppat.1000906
O’Neill, L. A., and Bowie, A. G. (2010). Sensing and signaling in antiviral innate immunity. Curr. Biol. 20, R328–R333.
Ozoren, N., Masumoto, J., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Erturk, I., Jagirdar, R., Zhu, L., Inohara, N., Bertin, J., Coyle, A., Grant, E. P., and Nunez, G. (2006). Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J. Immunol. 176, 4337–4342.
Pelegrin, P., and Surprenant, A. (2006). Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 25, 5071–5082.
Petrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., and Tschopp, J. (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589.
Pirhonen, J., Sareneva, T., Kurimoto, M., Julkunen, I., and Matikainen, S. (1999). Virus infection activates IL-1 beta and IL-18 production in human macrophages by a caspase-1-dependent pathway. J. Immunol. 162, 7322–7329.
Poeck, H., Bscheider, M., Gross, O., Finger, K., Roth, S., Rebsamen, M., Hannesschlager, N., Schlee, M., Rothenfusser, S., Barchet, W., Kato, H., Akira, S., Inoue, S., Endres, S., Peschel, C., Hartmann, G., Hornung, V., and Ruland, J. (2010). Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 11, 63–69.
Proell, M., Riedl, S. J., Fritz, J. H., Rojas, A. M., and Schwarzenbacher, R. (2008). The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS ONE 3, e2119. doi: 10.1371/journal.pone.0002119
Rathinam, V. A., Jiang, Z., Waggoner, S. N., Sharma, S., Cole, L. E., Waggoner, L., Vanaja, S. K., Monks, B. G., Ganesan, S., Latz, E., Hornung, V., Vogel, S. N., Szomolanyi-Tsuda, E., and Fitzgerald, K. A. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402.
Raupach, B., Peuschel, S. K., Monack, D. M., and Zychlinsky, A. (2006). Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 74, 4922–4926.
Reig, N., Jiang, A., Couture, R., Sutterwala, F. S., Ogura, Y., Flavell, R. A., Mellman, I., and Van Der Goot, F. G. (2008). Maturation modulates caspase-1-independent responses of dendritic cells to anthrax lethal toxin. Cell. Microbiol. 10, 1190–1207.
Reimer, T., Shaw, M. H., Franchi, L., Coban, C., Ishii, K. J., Akira, S., Horii, T., Rodriguez, A., and Nunez, G. (2010). Experimental cerebral malaria progresses independently of the Nlrp3 inflammasome. Eur. J. Immunol. 40, 764–769.
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F., and Vance, R. E. (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. doi: 10.1371/journal.ppat.0020018
Rodrigue-Gervais, I. G., and Saleh, M. (2010). Genetics of inflammasome-associated disorders: a lesson in the guiding principals of inflammasome function. Eur. J. Immunol. 40, 643–648.
Rogers, N. C., Slack, E. C., Edwards, A. D., Nolte, M. A., Schulz, O., Schweighoffer, E., Williams, D. L., Gordon, S., Tybulewicz, V. L., Brown, G. D., and Reis E. Sousa, C. (2005). Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22, 507–517.
Said-Sadier, N., Padilla, E., Langsley, G., and Ojcius, D. M. (2010). Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS ONE 5, e10008. doi: 10.1371/journal.pone.0010008
Saito, T. and Gale, M. Jr. (2007). Principles of intracellular viral recognition. Curr. Opin. Immunol. 19, 17–23.
Saleh, M., Mathison, J. C., Wolinski, M. K., Bensinger, S. J., Fitzgerald, P., Droin, N., Ulevitch, R. J., Green, D. R., and Nicholson, D. W. (2006). Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440, 1064–1068.
Saleh, M., and Trinchieri, G. (2011). Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat. Rev. Immunol. 11, 9–20.
Santic, M., Molmeret, M., Klose, K. E., and Abu Kwaik, Y. (2006). Francisella tularensis travels a novel, twisted road within macrophages. Trends Microbiol. 14, 37–44.
Sauer, J. D., Witte, C. E., Zemansky, J., Hanson, B., Lauer, P., and Portnoy, D. A. (2010). Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7, 412–419.
Schmitz, N., Kurrer, M., Bachmann, M. F., and Kopf, M. (2005). Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J. Virol. 79, 6441–6448.
Schotte, P., Denecker, G., Van Den Broeke, A., Vandenabeele, P., Cornelis, G. R., and Beyaert, R. (2004). Targeting Rac1 by the Yersinia effector protein YopE inhibits caspase-1-mediated maturation and release of interleukin-1beta. J. Biol. Chem. 279, 25134–25142.
Shao, W., Yeretssian, G., Doiron, K., Hussain, S. N., and Saleh, M. (2007). The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J. Biol. Chem. 282, 36321–36329.
Shaw, M. H., Reimer, T., Kim, Y. G., and Nunez, G. (2008). NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr. Opin. Immunol. 20, 377–382.
Shen, A., and Higgins, D. E. (2006). The MogR transcriptional repressor regulates nonhierarchal expression of flagellar motility genes and virulence in Listeria monocytogenes. PLoS Pathog. 2, e30. doi: 10.1371/journal.ppat.0020030
Shio, M. T., Eisenbarth, S. C., Savaria, M., Vinet, A. F., Bellemare, M. J., Harder, K. W., Sutterwala, F. S., Bohle, D. S., Descoteaux, A., Flavell, R. A., and Olivier, M. (2009). Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 5, e1000559. doi: 10.1371/journal.ppat.1000559
Silveira, T. N., and Zamboni, D. S. (2010). Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome- dependent host cell response that precedes pyroptosis. Infect. Immun. 78, 1403–1413.
Squires, R. C., Muehlbauer, S. M., and Brojatsch, J. (2007). Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J. Biol. Chem. 282, 34260–34267.
Stasakova, J., Ferko, B., Kittel, C., Sereinig, S., Romanova, J., Katinger, H., and Egorov, A. (2005). Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J. Gen. Virol. 86, 185–195.
Stehlik, C., and Dorfleutner, A. (2007). COPs and POPs: modulators of inflammasome activity. J. Immunol. 179, 7993–7998.
Sun, Y. H., Rolan, H. G., and Tsolis, R. M. (2007). Injection of flagellin into the host cell cytosol by Salmonella enterica serotype Typhimurium. J. Biol. Chem. 282, 33897–33901.
Sutterwala, F. S., Mijares, L. A., Li, L., Ogura, Y., Kazmierczak, B. I., and Flavell, R. A. (2007). Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245.
Suzuki, T., Franchi, L., Toma, C., Ashida, H., Ogawa, M., Yoshikawa, Y., Mimuro, H., Inohara, N., Sasakawa, C., and Nunez, G. (2007). Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3, e111. doi: 10.1371/journal.ppat.0030111
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820.
Terra, J. K., Cote, C. K., France, B., Jenkins, A. L., Bozue, J. A., Welkos, S. L., Levine, S. M., and Bradley, K. A. (2010). Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J. Immunol. 184, 17–20.
Thomas, P. G., Dash, P., Aldridge, J. R. Jr., Ellebedy, A. H., Reynolds, C., Funk, A. J., Martin, W. J., Lamkanfi, M., Webby, R. J., Boyd, K. L., Doherty, P. C., and Kanneganti, T. D. (2009). The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30, 566–575.
Thornberry, N. A., Bull, H. G., Calaycay, J. R., Chapman, K. T., Howard, A. D., Kostura, M. J., Miller, D. K., Molineaux, S. M., Weidner, J. R., Aunins, J., Elliston, K. O., Ayala, J. M., Casano, F. J., Chin, J., Ding, G. J. F., Egger, L. A., Gaffney, E. P., Limjuco, G., Palyha, O. C., Raju, S. M., Rolando, A. M., Salley, J. P., Yamin, T. T., Lee, T. D., Shively, J. E., Maccross, M., Mumford, R. A., Schmidt, J. A., and Tocci, M. J. (1992). A novel heterodimeric cysteine protease is required for interleukin-1-beta processing in monocytes. Nature 356, 768–774.
Tsuchiya, K., Hara, H., Kawamura, I., Nomura, T., Yamamoto, T., Daim, S., Dewamitta, S. R., Shen, Y., Fang, R., and Mitsuyama, M. (2010). Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J. Immunol. 185, 1186–1195.
Tsuji, N. M., Tsutsui, H., Seki, E., Kuida, K., Okamura, H., Nakanishi, K., and Flavell, R. A. (2004). Roles of caspase-1 in Listeria infection in mice. Int. Immunol. 16, 335–343.
Ulland, T. K., Buchan, B. W., Ketterer, M. R., Fernandes-Alnemri, T., Meyerholz, D. K., Apicella, M. A., Alnemri, E. S., Jones, B. D., Nauseef, W. M., and Sutterwala, F. S. (2010). Cutting edge: mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J. Immunol. 185, 2670–2674.
Valdez, Y., Ferreira, R. B. R., and Finlay, B. B. (2009). Molecular mechanisms of Salmonella virulence and host resistance. Curr. Top. Microbiol. 337, 93–127.
Van De Veerdonk, F. L., Joosten, L. A., Devesa, I., Mora-Montes, H. M., Kanneganti, T. D., Dinarello, C. A., Van Der Meer, J. W., Gow, N. A., Kullberg, B. J., and Netea, M. G. (2009). Bypassing pathogen-induced inflammasome activation for the regulation of interleukin-1beta production by the fungal pathogen Candida albicans. J. Infect. Dis. 199, 1087–1096.
Vinzing, M., Eitel, J., Lippmann, J., Hocke, A. C., Zahlten, J., Slevogt, H., N’guessan, P, D., Gunther, S., Schmeck, B., Hippenstiel, S., Flieger, A., Suttorp, N., and Opitz, B. (2008). NAIP and Ipaf control Legionella pneumophila replication in human cells. J. Immunol. 180, 6808–6815.
Vonk, A. G., Netea, M. G., van Krieken, J. H., Iwakura, Y., van der Meer, J. W., and Kullberg, B. J. (2006). Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J. Infect. Dis. 193, 1419–1426.
Walter, K., Holscher, C., Tschopp, J., and Ehlers, S. (2010). NALP3 is not necessary for early protection against experimental tuberculosis. Immunobiology 215, 804–811.
Wang, S. Y., Miura, M., Jung, Y. K., Zhu, H., Li, E., and Yuan, J. Y. (1998). Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92, 501–509.
Warren, S. E., Armstrong, A., Hamilton, M. K., Mao, D. P., Leaf, I. A., Miao, E. A., and Aderem, A. (2010). Cutting edge: cytosolic bacterial DNA activates the inflammasome via Aim2. J. Immunol. 185, 818–821.
Warren, S. E., Mao, D. P., Rodriguez, A. E., Miao, E. A., and Aderem, A. (2008). Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 180, 7558–7564.
Wickliffe, K. E., Leppla, S. H., and Moayeri, M. (2008). Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell. Microbiol. 10, 332–343.
Wright, E. K., Goodart, S. A., Growney, J. D., Hadinoto, V., Endrizzi, M. G., Long, E. M., Sadigh, K., Abney, A. L., Bernstein-Hanley, I., and Dietrich, W. F. (2003). Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr. Biol. 13, 27–36.
Wu, J., Fernandes-Alnemri, T., and Alnemri, E. S. (2010). Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 30, 693–702.
Xiang, Y., and Moss, B. (1999). IL-18 binding and inhibition of interferon gamma induction by human poxvirus-encoded proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 11537–11542.
Zaki, M. H., Boyd, K. L., Vogel, P., Kastan, M. B., Lamkanfi, M., and Kanneganti, T. D. (2010). The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391.
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., Kuida, K., Mariathasan, S., Dixit, V. M., Flavell, R. A., Dietrich, W. F., and Roy, C. R. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325.
Keywords: innate immunity, inflammation, Nod-like receptors, inflammasome, caspases, infection
Citation: Skeldon A and Saleh M (2011) The inflammasomes: molecular effectors of host resistance against bacterial, viral, parasitic, and fungal infections. Front. Microbio. 2:15. doi: 10.3389/fmicb.2011.00015
Received: 03 November 2010;
Paper pending published: 04 December 2010;
Accepted: 20 January 2011;
Published online: 17 February 2011.
Edited by:
Amal Amer, The Ohio State University, USAReviewed by:
Amal Amer, The Ohio State University, USACharlene Kahler, University of Western Australia, Australia
Jose A. Bengoechea, Fundacion Caubet-CIMERA Illes Balears, Spain
Copyright: © 2011 Skeldon and Saleh. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Maya Saleh, McGill University, McGill Life Sciences Complex, 3649 Promenade Sir-William Osler Montreal, QC, Canada H3G 0B1. e-mail:bWF5YS5zYWxlaEBtY2dpbGwuY2E=