


- Department of Microbiology and Immunology, School of Medicine, Stanford University, Stanford, CA, USA
Francisella tularensis is an intracellular pathogen that can cause severe disease in a wide range of mammalian hosts. Primarily residing in host macrophages, F. tularensis escapes phagosomal degradation, and replicates in the macrophage cytosol. The macrophage uses a series of pattern recognition receptors to detect conserved microbial molecules from invading pathogens, and initiates an appropriate host response. In the cytosol, F. tularensis is recognized by the inflammasome, a multiprotein complex responsible for the activation of the cysteine protease caspase-1. Caspase-1 activation leads to processing and release of proinflammatory cytokines and host cell death. Here we review recent work on the molecular mechanisms of inflammasome activation by F. tularensis, and its consequences both in vitro and in vivo. Finally, we discuss the coordination between the inflammasome and other cytosolic host responses, and the evidence for F. tularensis virulence factors that suppress inflammasome activation.
Francisella Tularensis
Francisella tularensis is a facultative intracellular pathogen that has evolved the capacity to successfully colonize eukaryotic hosts, sometimes causing disease, even in the face of a robust immune response. The primary intracellular niche for F. tularensis in the mammalian host is the macrophage, though it has been shown to replicate inside dendritic cells, hepatocytes, neutrophils, and type II alveolar epithelial cells in the host (Buddingh and Womack, 1941; White et al., 1964; Long et al., 1993; McCaffrey and Allen, 2006; Hall et al., 2007). Intracellular replication is crucial to the bacterium’s pathogenesis as mutants that fail to replicate intracellularly are avirulent in mice. Because F. tularensis is found primarily in macrophages in vivo and because the macrophage represents an important mediator of host defense, the macrophage serves as the most widely used in vitro model to study F. tularensis infection.
After uptake, F. tularensis initially resides in a membrane bound vacuole termed the Francisella-containing phagosome (FCP). The FCP acquires the early endosomal antigen 1 (EEA1) within 5 min of uptake, but this marker rapidly dissociates from the phagosome followed by the acquisition of late endosomal markers Lamp1, Lamp2, and the Rab7 GTPase within 15–30 min (Clemens et al., 2004; Santic et al., 2005b; Checroun et al., 2006). The FCP does not significantly fuse with lysosomes and though not absolutely required, acidification of the phagosome acts as a cue for F. tularensis to escape this compartment and enter into the host cell cytosol (Chong et al., 2008; Santic et al., 2008). Phagosomal escape is rapid, occurring within 60 min of macrophage infection. Once in the cytosol F. tularensis replicates to high numbers. Both phagosomal escape and intracellular replication are mediated by a locus of F. tularensis genes known as the Francisella pathogenicity island (FPI; Nano et al., 2004). FPI mutants remain in the initial phagosome, which progresses to lysosomes (Bonquist et al., 2008). FPI mutants are also avirulent in vivo (Brotcke et al., 2006; Weiss et al., 2007). At late stages of infection F. tularensis can be found again inside a membrane bound vacuole that exhibits characteristics of autophagosomes (Checroun et al., 2006). The role of autophagy in F. tularensis pathogenesis is unknown, but F. tularensis downregulates several host proteins required for the formation of autophagosomes (Butchar et al., 2008).
The intracellular lifestyle of F. tularensis brings it in contact with distinct environments of the macrophage, namely the surface during uptake, the initial phagosome, and the host cell cytosol. The macrophage is armed with innate immune defenses in each of these compartments that can respond to the presence of F. tularensis. Furthermore, there is crosstalk between signaling pathways that link sensing in one compartment to innate responses in other compartments.
At the macrophage surface, host TLRs engage F. tularensis. Unlike LPS from enteric bacteria, F. tularensis LPS is only mildly inflammatory and stimulates a low level of proinflammatory cytokine production (Hajjar et al., 2006). This likely is due to the unique structure of F. tularensis lipid A. Unlike the hexa-acylated lipid A from Escherichia coli and other gram-negative enterics, lipid A from F. tularensis is tetra-acylated (Raetz et al., 2009). It is thought that this altered structure makes it unrecognizable to LPS-binding protein, and therefore subverts TLR4 recognition (Barker et al., 2006; Cole et al., 2006; Hajjar et al., 2006). Consistent with this observation, TLR4 deficient mice are not more susceptible to infection with F. tularensis than wild-type mice (Chen et al., 2004; Collazo et al., 2006). F. tularensis does significantly stimulate TLR2 signaling resulting in proinflammatory cytokine production, and intracellular bacteria colocalize with TLR2 and MyD88 (Katz et al., 2006; Malik et al., 2006; Cole et al., 2007). In vivo, TLR2 does not seem to play a role in host protection during intradermal challenge, but is important in an intranasal model of infection (Collazo et al., 2006; Malik et al., 2006). However, Myd88 deficient mice are completely susceptible to sublethal doses of the live vaccine strain (LVS) when given intradermally (Collazo et al., 2006).
The ability of F. tularensis to escape the initial phagosome and reside in the macrophage cytosol is essential for the organism’s pathogenesis. Paradoxically, cytosolic localization is also essential for innate immune recognition by a cytosolic surveillance system known as the inflammasome. Here we review recent work that has shed light on the molecular mechanisms that lead to cytosolic detection of F. tularensis in the macrophage, and host defense in vivo.
Caspase-1
Some of the key regulators of inflammation are aspartate-specific cysteine proteases known as inflammatory caspases. These include caspase-1, -4, -5, -11 (which exists in rodents), and -12 (Scott and Saleh, 2007). Caspase-1, the best studied of these inflammatory caspases, is activated in intracellular complexes known as inflammasomes, which are located in the cytosol of certain immune cells and assembled in response to danger signals. Activation of caspase-1 involves autoproteolytic processing of the 45-kDa pro-caspase-1 into 20- and 10-kDa subunits (p20 and p10; Thornberry et al., 1992; Ayala et al., 1994; Wilson et al., 1994). In the cytosol, active caspase-1 processes pro-IL-1β (Black et al., 1989; Kostura et al., 1989) and pro-IL-18 (Ghayur et al., 1997) into their mature, bio-active forms that are secreted and regulate inflammation. When regulated properly, IL-1β is critical for the host response to infection, but excessive levels of Il-1β are associated with several inflammatory diseases such as rheumatoid arthritis, inflammatory bowel disease, and septic shock to name a few (Dinarello and Wolff, 1993). Clearly inflammation can be a double-edged sword but it is essential to combat infection and restore tissue homeostasis after infection (Medzhitov, 2010).
The Inflammasome Responds to Intracellular Pathogens and Danger Signals
In 2002, it was discovered that a multiprotein complex termed the “inflammasome” was responsible for activating caspase-1 (Martinon et al., 2002). The inflammasome, in its simplest form, is composed of a NOD-like receptor (NLR), and the adaptor protein apoptosis associated speck-like protein containing a caspase recruitment domain (ASC). The NLRs are a family of cytosolic pattern recognition receptors (PRRs) that activate inflammatory and antimicrobial responses by sensing “danger signals” or danger-associated molecular patterns (DAMPs; Matzinger, 2002) and conserved microbial products termed pathogen associated molecular patterns (PAMPs; Tschopp et al., 2003; Inohara et al., 2005). There are 22 members of the NLR subfamily in humans and 34 in mice, which are characterized by a C-terminal leucine rich repeat (LRR) domain, a central oligomerization domain (NACHT), and an N-terminus that is either a caspase activation and recruitment domain (CARD; as in NOD1, NOD2, and the NLRC family), three baculovirus IAP repeats (BIR; as in the NAIPs), or a pyrin domain (PYD; as in the NLRP family; Kanneganti, 2010). The adaptor ASC (also known as PYCARD) has an N-terminal PYD that facilitates interactions with the PYD domain of NLRs, and a C-terminal CARD domain that recruits caspase-1 through CARD-CARD interactions (Martinon et al., 2001). Interestingly, besides its role as an adaptor, ASC is required to induce autoproteolysis of pro-caspase-1 in the inflammasome complex, a pre-requirement for efficient cytokine processing (Broz et al., 2010b). LRRs are also found in the toll-like receptors (TLRs), which sense danger signals and microbial patterns on the cell surface and in endosomes (Iwasaki and Medzhitov, 2004; Takeda and Akira, 2005). Interestingly, the TLRs cannot distinguish between pathogenic and non-pathogenic microbes because they sense conserved microbial patterns that are present in both and they are located extracellularly and in endosomes, where both pathogenic and non-pathogenic microbes reside. The NLRs also sense conserved microbial patterns, but the only way these microbial patterns can reach the cytosol is if they are delivered there by disruption of cell membranes by toxins, specialized secretion systems of pathogenic microbes, and/or cytosolic pathogens. Therefore the location of the sensors and not the ligands they sense makes the NLRs specific for detecting pathogens (Brodsky and Monack, 2009).
The vast number of NLR proteins allows the inflammasome to respond to numerous pathogens and danger signals. The cytosolic DAMP or PAMP sensed determines which NLR forms the complex. An inflammasome is named after the NLR that forms it (i.e., the NLRP3 inflammasome, the NLRC4 inflammasome, etc.), and so the inflammasome’s components can vary. As shown in Figure 1, the inflammasome has been implicated in the host response to numerous pathogens and danger signals, as well as a number of autoimmune and auto-inflammatory diseases (Brodsky and Monack, 2009; Rodrigue-Gervais and Saleh, 2010).
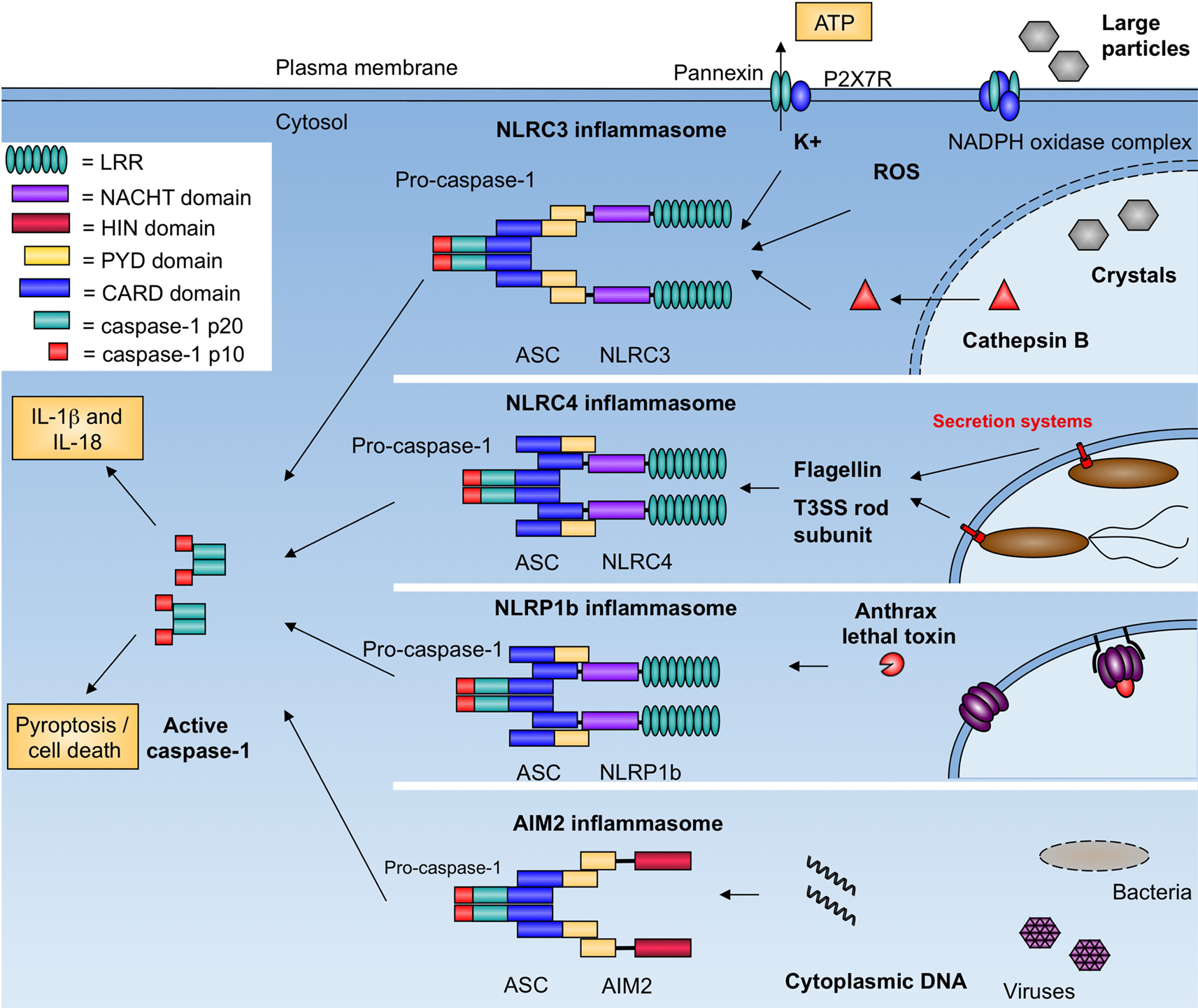
Figure 1. Inflammasomes activate caspase-1 in response to pathogens and danger signals. NLRC3 responds to numerous stimuli including pore-forming toxins, extracellular ATP, and crystalline compounds. The exact mechanism of activation is unknown but may involve potassium efflux, membrane damage, and generation of reactive oxygen species. NLRC4 responds to bacterial flagellin and the T3SS rod subunit. NLRP1b responds to anthrax lethal factor. AIM2 binds cytosolic dsDNA from bacterial, viral, mammalian, and synthetic sources. Complexes are formed through homotypic interactions (LRR, leucine rich repeats; HIN, hemopoietic IFN-inducible nuclear proteins; PYD, pyrin; CARD, caspase activation and recruitment domain). All inflammasomes activate caspase-1, which leads to processing of pro-IL-1β and pro-IL-18, and host cell death.
Conserved microbial structures, such as bacterial cell wall components are potent activators of innate immunity. Muramyl dipeptide (MDP) is a breakdown product of bacterial peptidoglycan that is recognized by NOD2 and induces transcriptional activation of proinflammatory cytokines through the adaptor RIP2. MDP is also a potent activator of the inflammasome, which results in release of mature IL-1β. Inflammasome activation by MDP involves both NOD2 and NLRP3, suggesting that NLRP3 is an additional sensor of MDP (Martinon et al., 2004; Pan et al., 2007; Marina-Garcia et al., 2008). NOD2 has also been shown to cooperate with NLRP1b in inflammasome activation in response to MDP and anthrax lethal toxin (Bruey et al., 2007; Hsu et al., 2008). These results suggest that inflammasome complexes may contain of multiple NLRs that act synergistically to activate caspase-1 in response to PAMPs.
Many bacteria employ pore-forming toxins in their pathogenic arsenal to establish infections. NLRP3 has been implicated in the inflammasome response to listeriolysin O form Listeria monocytogenes, α-toxin from Staphylococcus aureus, and aerolysin from Aeromonas hydrophila (Mariathasan et al., 2006; Fink et al., 2008). The precise mechanism by which NLRP3 recognizes these toxins is unknown. NLRP3 also activates the inflammasome in response to high extracellular concentrations of ATP (Mariathasan et al., 2006), various crystalline compounds (Martinon et al., 2006; Cassel et al., 2008; Dostert et al., 2008; Halle et al., 2008), as well as viral and bacterial nucleic acids (Kanneganti et al., 2006; Muruve et al., 2008). With such a diverse array of stimuli, the hypothesis is that these ligands potentiate a common terminal signal that activates NLRP3. Potassium efflux, membrane damage, and stimulation of reactive oxygen species have all been implicated as the terminal signal for NLRP3, but each has caveats. Therefore, the precise mechanism of NLRP3 activation remains a mystery.
NLRC4 was the first NLR shown to activate caspase-1 in response to bacterial infection with Salmonella typhimurium in a type-III secretion system (T3SS)-dependent manner (Mariathasan et al., 2004). It was later revealed that NLRC4 sensed bacterial flagellin that was secreted into the host cytosol through the T3SS, likely due to the evolutional similarity of the T3SS with the flagellar biosynthesis machinery (Franchi et al., 2006; Miao et al., 2006). Detection of cytosolic flagellin was later linked to the inflammasome response to Pseudomonas aeruginosa, L. monocytogenes, and Legionella pneumophila (Molofsky et al., 2006; Ren et al., 2006; Miao et al., 2008; Warren et al., 2008), though in the case of Legionella the type-IV secretion system (T4SS) was required and Naip5 was required in addition to NLRC4 (Ren et al., 2006; Zamboni et al., 2006). NLRC4 was also implicated in recognition of Shigella flexneri (Suzuki et al., 2007), which encodes a T3SS but does not express flagellin (Tominaga et al., 1994), as well as non-flagellated strains of Pseudomonas (Sutterwala et al., 2007), suggesting that NLRC4 must recognize other ligands besides flagellin. The answer was recently provided when it was demonstrated that NLRC4 could also the rod component of the T3SS needle apparatus, which shares a sequence motif with flagellin (Miao et al., 2010). This motif is essential to NLRC4-mediated recognition and explains how NLRC4 can respond to diverse pathogens that express either a T3SS or flagellin.
Studies of NLRP1b, NLRP3, and NLRC4 have expanded our understanding of the role of the NLR family and inflammasomes in the host response to pathogens. However, transfection of synthetic, bacterial, viral, or mammalian dsDNA triggers caspase-1 activation in a manner that requires ASC, but not any of the known NLRs (Muruve et al., 2008). Recently a member of the family of hemopoietic IFN-inducible nuclear proteins with a 200-amino acid motif (HIN-200), Absent in melanoma 2 (AIM2), was shown to activate the inflammasome in response to cytosolic double stranded DNA (dsDNA; Burckstummer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Roberts et al., 2009). AIM2 contains a HIN domain that facilitates dsDNA binding, and a PYD domain that recruits ASC, allowing for caspase-1 activation. This was the first example of a non-NLR family protein triggering inflammasome activation. Moreover, AIM2 is interferon-inducible, establishing a link between two cytosolic innate immune responses.
Francisella Tularensis Activates the AIM2 Inflammasome
After F. tularensis escapes the phagosome, it is subject to cytosolic innate immune recognition (Figure 2). In murine macrophages, F. tularensis induced inflammasome activation was shown to be independent of known receptors, like NLRP1b, NLRC4, and NLRP3, but required the adaptor protein ASC (Mariathasan et al., 2005, 2006). In addition, inflammasome activation was also dependent on interferon signaling suggesting that an interferon-inducible inflammasome sensor, such as AIM2, might be involved in sensing cytosolic F. tularensis (Henry et al., 2007). Infection of primary macrophages from AIM2-deficient mice demonstrated that AIM2 was required for caspase-1 processing, release of mature IL-1β, and host cell death in response to F. tularensis (Fernandes-Alnemri et al., 2010; Jones et al., 2010; Rathinam et al., 2010). In addition, confocal microscopy demonstrated that cytosolic F. tularensis release bacterial DNA into the macrophage cytosol and that AIM2 colocalized with the bacterial DNA (Fernandes-Alnemri et al., 2010; Jones et al., 2010). Interestingly, if several lysing bacteria were observed in a single cell, their DNA colocalized with AIM2, however only one of these sites served as a nucleation point for inflammasome assembly, as visualized by the formation of the so-called ASC focus (Jones et al., 2010). This observation represented the first visualization of an endogenous inflammasome in complex with its ligand. Formation of similar ASC foci was also observed during Salmonella infections, and the focus was subsequently shown to recruit and activate caspase-1 and to serve as a major site of cytokine processing (Broz et al., 2010a).
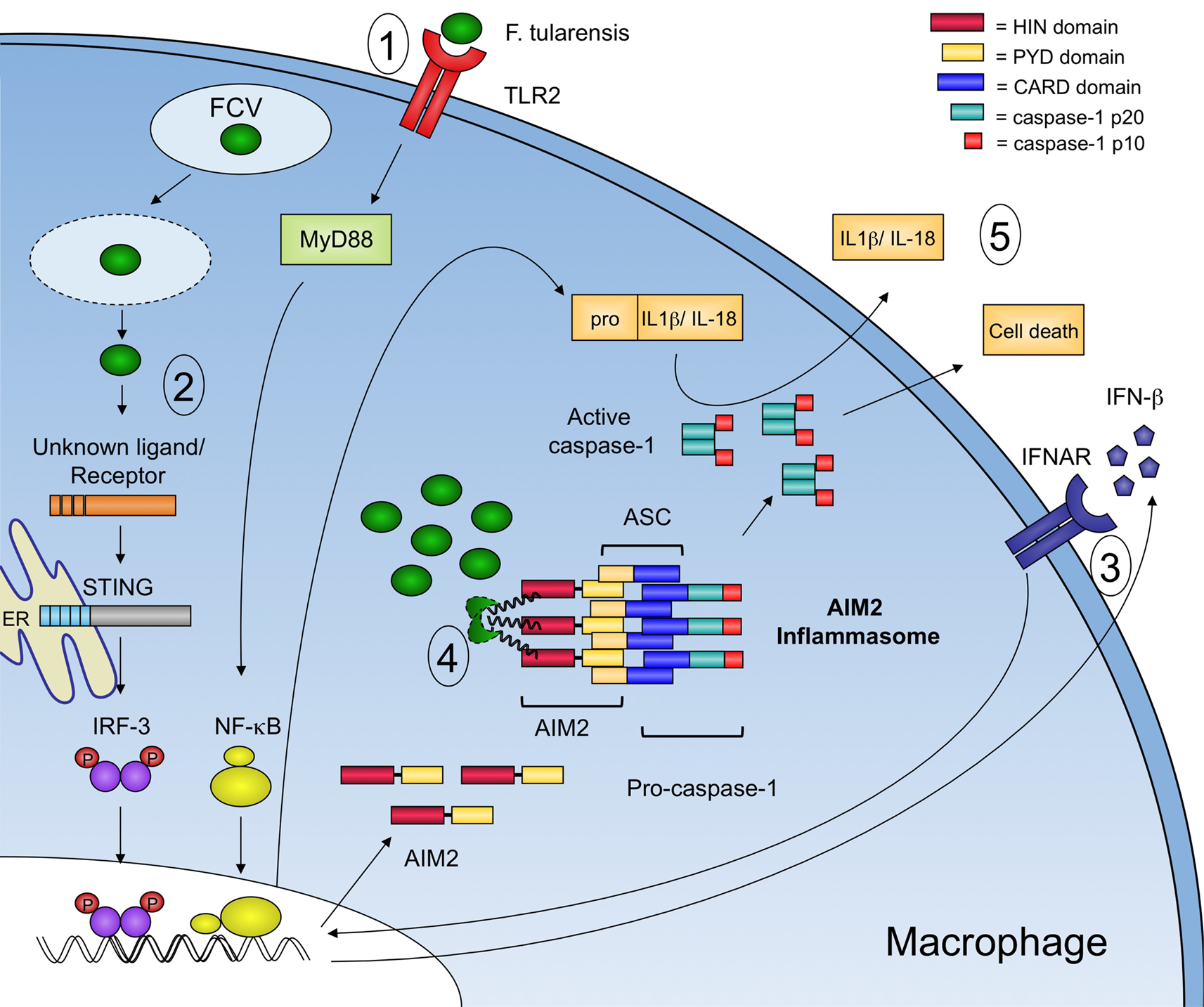
Figure 2. Model of innate immune recognition of F. tularensis in the macrophage. Signaling events involved in the innate immune response to F. tularensis are highlighted. 1 – At the surface of the macrophage, TLR2 induces a MyD88-dependent transcriptional response that leads to the expression of proinflammatory cytokines including pro-IL-1β. F. tularensis then enters the macrophage in a membrane bound vacuole known as the FCV. 2 – Upon escape into the cytosol, an unknown receptor recognizes an unknown F. tularensis ligand that leads to the STING-dependent and IRF-3-dependent production of type-I IFNs. 3 – Autocrine and paracrine signaling through the type-I interferon receptor (IFNAR) leads to an increase in AIM2 protein expression. 4 – Cytosolic F. tularensis lyse, releasing bacterial DNA that is recognized by AIM2, which in turn recruits ASC and procaspase-1 to form an inflammasome complex. 5 – Active capsase-1 processes pro-IL-1β and pro-IL-18 into their mature forms and triggers host cell death.
Inflammasome activation was also shown to be critical to host defense in vivo as mice lacking the inflammasome components AIM2, caspase-1, or ASC have increased bacterial burden and succumb to infection much faster than wild-type mice (Mariathasan et al., 2005; Fernandes-Alnemri et al., 2010; Jones et al., 2010).
As mentioned above, F. tularensis activates a cytosolic surveillance pathway that is characterized by the production of type-I IFNs (Henry et al., 2007; Cole et al., 2008). The host cytosolic sensor responsible for type-I IFN production remains unknown. However, the adaptor protein stimulator of interferon genes (STING) is required for type-I IFN production in response to F. tularensis in murine macrophages (Jones et al., 2010). Autocrine and paracrine signaling through IFNAR leads to an increase in AIM2 protein expression in the macrophage, effectively priming the cell for recognition of cytosolic dsDNA. Although other roles for type-I IFN signaling during F. tularensis infection exist (Henry et al., 2010), the expression of AIM2 is sufficient for inflammasome activation irrespective of other IFN-induced genes. Type-I IFN signaling is critical to inflammasome activation in the macrophage as IFNAR-deficient macrophages do not process capsase-1, release proinflammatory cytokines, or die in response to F. tularensis (Henry et al., 2007).
A positive feedback loop between type-I IFN signaling and inflammasome activation in macrophages exists, but the link in vivo seems to be less clear. Previous reports show that mice deficient in inflammasome components are more susceptible to infection with F. tularensis (Mariathasan et al., 2005). However, mice deficient in type-I IFN signaling are more resistant to infection (Henry et al., 2010). Similar results were also obtained for L. monocytogenes infections (Henry et al., 2010). This apparent discrepancy between the in vitro results and the in vivo results is partially explained by the control of type-I IFN signaling on IL-17 production (Henry et al., 2010). Mice deficient in type-I IFN signaling produce more IL-17, which leads to a greater influx of neutrophils that can better control bacterial infections (Henry et al., 2010). A possible explanation for the in vivo phenotype is that Type-IFN has been shown to promote apoptosis of immune cells such as macrophages, neutrophils, and lymphocytes during Listeria and Francisella infections (O’Connell et al., 2004; Carrero et al., 2006; Navarini et al., 2006; Henry et al., 2007). Additionally, IFN-γ can restore inflammasome activation in vivo in an IFNAR-deficient mouse by signaling through the IFN-γ receptor, which would result in increased expression of AIM2 and subsequent inflammasome activation (our unpublished results). Thus, an interesting paradox exists, where type-I IFN is beneficial to the host in vitro and detrimental in vivo during bacterial infections.
The Inflammasome and F. Tularensis virulence Factors
During a mammalian infection, F. tularensis must establish a replicative niche in the presence of a robust innate immune system. The inflammasome is of critical importance to mount an effective innate immune response against intracellular F. tularensis. Investigation into the interaction of F. tularensis and the inflammasome has led to the discovery of several bacterial genes that seem to suppress inflammasome activation. The first genes to be identified were oppB and the zinc metalloprotease pepO (also FTT1209c). Mutants in these mglA-regulated genes exhibit increased kinetics of replication in bone marrow-derived macrophages and increased macrophage cytotoxicity compared to wild-type F. tularensis, although it was not shown that the increased cytotoxicity of these mutants was mediated by inflammasome activation (Brotcke et al., 2006). Furthermore, both oppB and pepO showed reduced fitness in mice in intradermal competitive index experiments (Brotcke et al., 2006). However, in single infections via the intradermal route, mice infected with a pepO mutant had higher bacterial burdens in the spleen and, when infected by aerosol, displayed increased neutrophil influx into the lung (Hager et al., 2006). It is unclear if either oppB or pepO interacts directly with the inflammasome to suppress macrophage cytotoxicity.
Two additional F. tularensis genes involved in suppressing the macrophage inflammasome, FTT0748 and FTT0584, were identified in a microarray-based in vivo negative selection screen where they displayed reduced fitness in competitive index experiments in mice. Subsequent characterization of FTT0748 and FTT0584 in bone marrow-derived macrophages showed that these mutants induce increased macrophage cytotoxicity and IL-1β release compared to wild-type F. tularensis and these phenotypes were dependent on both the ASC and caspase-1 inflammasome components, as no cell death or IL-1β release was observed in macrophages lacking ASC or caspase-1 (Weiss et al., 2007). Interestingly, and in contrast to the oppB and pepO mutants, neither FTT0748 nor FTT0584 displayed altered intracellular replication kinetics in bone marrow-derived macrophages. These mutants were the first to disassociate F. tularensis intracellular replication from inflammasome activation. FTT0748 has homology to IclR family transcriptional regulators and FTT0584 is a protein of unknown function. The mechanism(s) by which they suppress inflammasome activation are not known.
Investigation into the role of type I secretion systems in F. tularensis virulence led to the identification of TolC, an outer membrane protein that participates in multidrug resistance (Gil et al., 2006). A tolC mutant in the LVS strain of F. tularensis was hypercytotoxic to both murine bone marrow-derived macrophages as well as human monocyte-derived macrophages (huMDM) when compared to wild-type LVS (Platz et al., 2010). The tolC mutant also caused higher levels of IL-1β secretion from huMDM, implicating a role for tolC in suppressing the inflammasome in human cells. However, the hypercytotoxicity observed in muBMDM was inflammasome independent, as hypercytotoxicity was still observed in caspase-1 deficient macrophages and was comparable to IL-1β secretion in macrophages infected with wild-type LVS (Platz et al., 2010). Unlike in human cells, the hypercytotoxicity of the tolC mutant seems to be the result of increased caspase-3/7 activity, resulting in cell death by apoptosis. Thus tolC might play a role in suppressing several different immune pathways, both inflammasome-dependent and -independent. Like FTT0748 and FTT0584, tolC was not required for replication in macrophages but is required for full virulence in mice (Gil et al., 2006; Platz et al., 2010).
Required for intracellular proliferation, factor A (RipA) is a cytoplasmic membrane protein conserved across F. tularensis subspecies that is required for intracellular replication in both macrophages and epithelial cells, and is required for virulence in a mouse model of tularemia (Fuller et al., 2008). A ripA deletion mutant elicits increased IL-1β and IL-18 production from bone marrow-derived macrophages relative to wild-type F. tularensis and induces more macrophage cell death (Huang et al., 2010). The hypercytotoxic and hyperinflammatory phenotype of the ripA mutant was abolished in BMDM deficient for ASC, caspase-1, or MyD88, suggesting that a ripA mutant could elicit higher transcriptional levels of proinflammatory genes among them pro-IL-1β, which would also explain the elevated levels of mature IL-1β. Consistently, levels of TNF-α, which is secreted independently of the inflammasome, were also elevated in infections with a ripA mutant (Fuller et al., 2008). These phenotypes were also absent in THP-1 cells treated with a caspase-1 inhibitor, Y-VAD, or expressing ShRNA against ASC (Huang et al., 2010). Furthermore, intranasal infections with the ripA mutant led to an increase in IL-1β secretion into the bronchial alveolar lavage fluid of mice relative to mice infected with wild-type F. tularensis, supporting a role for ripA in suppressing inflammasome activation during pneumonic tularemia.
Francisella tularensis LPS genes have also been implicated in suppression of the inflammasome. A mutant in a putative lipid II flippase, mviN, induces an increase in the kinetics of caspase-1 processing, proinflammatory cytokine secretion, and cell death in macrophages compared to wild-type F. tularensis. This phenotype is completely dependent on AIM2, caspase-1, and ASC (Ulland et al., 2010). The mechanism by which mviN suppresses inflammasome activation is not known but the deletion mutant exhibits a striking change in bacterial morphology, suggesting that an altered membrane may play a role in the bacteria’s recognition.
Similarly, several more genes involved in LPS and capsule biosynthesis play a role in limiting macrophage cell death. These genes include lpcC, manB, manC, kdtA, FTT1236, FTT1237, and FTT1238 (Lai et al., 2010; Lindemann et al., 2010). Mutants in these genes express a shortened LPS and are phagocytosed by macrophages at a higher frequency than wild-type bacteria. These mutants induce greater macrophage cell death, though this is incompletely explained by the higher rate of phagocytosis (Lai et al., 2010; Lindemann et al., 2010). Furthermore, the cell death pathway involved was not explored in this study, though the AIM2 inflammasome is a strong candidate.
Unanswered Questions and Future Directions
The recent studies demonstrating the role of AIM2 in innate immunity to F. tularensis suggest that bacterial lysis in the cytosol leads to release of F. tularensis DNA, induction of the type-I IFN pathway, and activation of the AIM2 inflammasome. The molecular mechanism that causes bacterial lysis is unclear but several hypotheses emerge from this work. When bacteria are cultured in rich media, they exhibit a life cycle that is characterized by a lag phase of no bacterial replication, a log phase with a net increase in bacterial multiplication, a stationary phase of limited nutrient availability where replication plateaus, and a death phase with a net decrease in bacterial numbers. During this death phase many bacteria lyse, releasing their contents into the culture media. Therefore, bacterial lysis is a natural part of the bacterial life cycle and this could be the mechanism of DNA release in the host cytosol. Additionally, the macrophage phagosome is a professional microbe-killing machine, and although F. tularensis is well equipped to escape with its life that does not mean that it is not wounded in the battle. Studies to date on the intracellular trafficking of the Francisella containing vacuole suggest that the bacteria escape before acquiring markers of lysosomes or degradative enzymes (Santic et al., 2005a,b; Checroun et al., 2006). However, slight perturbations in the bacterial envelope during the vacuolar stage may be enough to induce lysis once the bacteria reach the cytosol. The large number of mutants identified in genes encoding membrane proteins and proteins involved in and LPS synthesis that result in an increased inflammasome activation in the macrophage support this hypothesis. If these mutants have an unstable outer membrane they may lyse at a higher frequency than wild-type F. tularensis and lead to increased cytosolic sensing by the DNA receptors. In support of this idea, recent reports show that L. monocytogenes lyses at a low frequency in the macrophage cytosol and induces AIM2-dependent inflammasome activation (Sauer et al., 2010). Furthermore, L. monocytogenes mutants that induced higher levels of inflammasome activation were shown to lyse with increased frequency compared to wild-type L. monocytogenes.
Yet another possible hypothesis, is the existence of antimicrobial defenses in the cytosol itself. Little is known about the cytosolic environment except that it is pH neutral. In addition, little is known about changes to this environment after macrophages are stimulated with proinflammatory cytokines such as interferons. The interferons were first described for their ability to induce and antiviral state in cells, but whether or not this includes defenses in the cytosol is unknown. However, IFN-γ treatment of primary mouse- and human-derived macrophages has been shown to restrict cytosolic growth of F. tularensis subsp. tularensis (Edwards et al., 2010), and INF-β induces a similar transcriptional response as IFN-γ in macrophages so we hypothesize that type-I IFNs may induce bacterial lysis in the cytosol. These hypotheses are not mutually exclusive and it is likely that multiple mechanisms contribute to bacterial lysis.
Although mainly studied as an antiviral mechanism, a number of recent reports demonstrate that type-I IFN is induced by a number of intracellular bacteria such as Mycobaterium tuberculosis (Weiden et al., 2000; Giacomini et al., 2001), L. monocytogenes (O’Riordan et al., 2002), and Le. pneumophila (Opitz et al., 2006). In the case of L. monocytogenes it was recently reported that cyclic diadenosine monophosphate (c-di-AMP) is released by the bacterium into the macrophage cytosol during infection and can trigger the type-I IFN response (Woodward et al., 2010). The bacterial PAMP that stimulates type-IFN production in the other aforementioned bacterial infections is unknown, though in the case of F. tularensis infection it is suspected that a cytosolic DNA sensor could be involved (Stetson and Medzhitov, 2006; Henry et al., 2007). Similar to its effects on viral infections, type-I IFN can act in concert with other cytosolic sensing pathways like the inflammasome to trigger a protective host cell death (Coers et al., 2007; Henry et al., 2007).
The mechanism linking the type-I IFN pathway and the inflammasome is currently unknown. To better understand the molecular mechanisms of the coordination between these two pathways we need to identify the host receptors that lead to type-I IFN production. One such receptor, DAI (Takaoka et al., 2007), is either not active in macrophages, or there are redundant receptors since DAI-deficient macrophages and mice respond normally to stimulation with dsDNA (Ishii et al., 2008). However, two new cytosolic DNA sensors have recently been identified. One, LRRFIP1, mediates type-I IFN production in macrophages in response to L. monocytogenes and vesicular stomatitus virus (Yang et al., 2010). The other, IFI16, contains a pryin domain and an HIN domain, similar to AIM2, and mediates type-I IFN production in response to DNA and herpes simplex virus-1 (HSV-1; Unterholzner et al., 2010). In this report, the authors found that IFI16 was able to associate with STING. If LRRFIP1 and/or IFI16 were also involved in the macrophage type-I IFN response to F. tularensis, we would have a system to study the coordination of the IFN pathway and inflammasome pathway in a biologically relevant model.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Jonathan W. Jones and Denise M. Monack are supported by grants AI063302 and AI065359 from the NIHNIAID. Petr Broz holds a Stanford ITI Young Investigator Award and was supported by postdoctoral fellowships from the Swiss National Science Foundation (SNSF) and the Human Frontiers in Science Program (HFSP).
References
Ayala, J. M., Yamin, T. T., Egger, L. A., Chin, J., Kostura, M. J., and Miller, D. K. (1994). IL-1 beta-converting enzyme is present in monocytic cells as an inactive 45-kDa precursor. J. Immunol. 153, 2592–2599.
Barker, J. H., Weiss, J., Apicella, M. A., and Nauseef, W. M. (2006). Basis for the failure of Francisella tularensis lipopolysaccharide to prime human polymorphonuclear leukocytes. Infect. Immun. 74, 3277–3284.
Black, R. A., Kronheim, S. R., Merriam, J. E., March, C. J., and Hopp, T. P. (1989). A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J. Biol. Chem. 264, 5323–5326.
Bonquist, L., Lindgren, H., Golovliov, I., Guina, T., and Sjostedt, A. (2008). MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect. Immun. 76, 3502–3510.
Brodsky, I. E., and Monack, D. (2009). NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens. Semin. Immunol. 21, 199–207.
Brotcke, A., Weiss, D. S., Kim, C. C., Chain, P., Malfatti, S., Garcia, E., and Monack, D. M. (2006). Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect. Immun. 74, 6642–6655.
Broz, P., Newton, K., Lamkanfi, M., Mariathasan, S., Dixit, V. M., and Monack, D. M. (2010a). Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755.
Broz, P., von Moltke, J., Jones, J. W., Vance, R. E., and Monack, D. M. (2010b). Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 8, 471–483.
Bruey, J. M., Bruey-Sedano, N., Luciano, F., Zhai, D., Balpai, R., Xu, C., Kress, C. L., Bailly-Maitre, B., Li, X., Osterman, A., Matsuzawa, S., Terskikh, A. V., Faustin, B., and Reed, J. C. (2007). Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 129, 45–56.
Buddingh, G. J., and Womack, F. C. (1941). Observations on the Infection of chick embryos with Bacterium Tularense, Brucella, and Pasteurella Pestis. J. Exp. Med. 74, 213–222.
Burckstummer, T., Baumann, C., Bluml, S., Dixit, E., Durnberger, G., Jahn, H., Planyavsky, M., Bilban, M., Colinge, J., Bennett, K. L., and Superti-Furga, G. (2009). An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272.
Butchar, J. P., Cremer, T. J., Clay, C. D., Gavrilin, M. A., Wewers, M. D., Marsh, C. B., Schlesinger, L. S., and Tridandapani, S. (2008). Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS One 3, e2924. doi: 10.1371/journal.pone.0002924
Carrero, J. A., Calderon, B., and Unanue, E. R. (2006). Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J. Exp. Med. 203, 933–940.
Cassel, S. L., Eisenbarth, S. C., Iyer, S. S., Sadler, J. J., Colegio, O. R., Tephly, L. A., Carter, A. B., Rothman, P. B., Flavell, R. A., and Sutterwala, F. S. (2008). The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. U.S.A. 105, 9035–9040.
Checroun, C., Wehrly, T. D., Fischer, E. R., Hayes, S. F., and Celli, J. (2006). Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. U.S.A. 103, 14578–14583.
Chen, W., KuoLee, R., Shen, H., Busa, M., and Conlan, J. W. (2004). Toll-like receptor 4 (TLR4) does not confer a resistance advantage on mice against low-dose aerosol infection with virulent type A Francisella tularensis. Microb. Pathog. 37, 185–191.
Chong, A., Wehrly, T. D., Nair, V., Fischer, E. R., Barker, J. R., Klose, K. E., and Celli, J. (2008). The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect. Immun. 76, 5488–5499.
Clemens, D. L., Lee, B. Y., and Horwitz, M. A. (2004). Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 72, 3204–3217.
Coers, J., Vance, R. E., Fontana, M. F., and Dietrich, W. F. (2007). Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell Microbiol. 9, 2344–2357.
Cole, L. E., Elkins, K. L., Michalek, S. M., Qureshi, N., Eaton, L. J., Rallabhandi, P., Cuesta, N., and Vogel, S. N. (2006). Immunologic consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J. Immunol. 176, 6888–6899.
Cole, L. E., Santiago, A., Barry, E., Kang, T. J., Shirey, K. A., Roberts, Z. J., Elkins, K. L., Cross, A. S., and Vogel, S. N. (2008). Macrophage proinflammatory response to Francisella tularensis live vaccine strain requires coordination of multiple signaling pathways. J. Immunol. 180, 6885–6891.
Cole, L. E., Shirey, K. A., Barry, E., Santiago, A., Rallabhandi, P., Elkins, K. L., Puche, A. C., Michalek, S. M., and Vogel, S. N. (2007). Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect. Immun. 75, 4127–4137.
Collazo, C. M., Sher, A., Meierovics, A. I., and Elkins, K. L. (2006). Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect. 8, 779–790.
Dinarello, C. A., and Wolff, S. M. (1993). The role of interleukin-1 in disease. N. Engl. J. Med. 328, 106–113.
Dostert, C., Petrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677.
Edwards, J. A., Rockx-Brouwer, D., Nair, V., and Celli, J. (2010). Restricted cytosolic growth of Francisella tularensis subsp. tularensis by IFN-gamma activation of macrophages. Microbiology 156, 327–339.
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513.
Fernandes-Alnemri, T., Yu, J. W., Juliana, C., Solorzano, L., Kang, S., Wu, J., Datta, P., McCormick, M., Huang, L., McDermott, E., Eisenlohr, L., Landel, C.P., and Alnemri, E. S. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393.
Fink, S. L., Bergsbaken, T., and Cookson, B. T. (2008). Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 105, 4312–4317.
Franchi, L., Amer, A., Body-Malapel, M., Kanneganti, T. D., Ozoren, N., Jagirdar, R., Inohara, N., Vandenabeele, P., Bertin, J., Coyle, A., Grant, E. P., and Nunez, G. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582.
Fuller, J. R., Craven, R. R., Hall, J. D., Kijek, T. M., Taft-Benz, S., and Kawula, T. H. (2008). RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect. Immun. 76, 4934–4943.
Ghayur, T., Banerjee, S., Hugunin, M., Butler, D., Herzog, L., Carter, A., Quintal, L., Sekut, L., Talanian, R., Paskind, M., Wong, W., Kamen, R., Tracey, D., and Allen, H. (1997). Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386, 619–623.
Giacomini, E., Iona, E., Ferroni, L., Miettinen, M., Fattorini, L., Orefici, G., Julkunen, I., and Coccia, E. M. (2001). Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J. Immunol. 166, 7033–7041.
Gil, H., Platz, G. J., Forestal, C. A., Monfett, M., Bakshi, C. S., Sellati, T. J., Furie, M. B., Benach, J. L., and Thanassi, D. G. (2006). Deletion of TolC orthologs in Francisella tularensis identifies roles in multidrug resistance and virulence. Proc. Natl. Acad. Sci. U.S.A. 103, 12897–12902.
Hager, A. J., Bolton, D. L., Pelletier, M. R., Brittnacher, M. J., Gallagher, L. A., Kaul, R., Skerrett, S. J., Miller, S. I., and Guina, T. (2006). Type IV pili-mediated secretion modulates Francisella virulence. Mol. Microbiol. 62, 227–237.
Hajjar, A. M., Harvey, M. D., Shaffer, S. A., Goodlett, D. R., Sjostedt, A., Edebro, H., Forsman, M., Bystrom, M., Pelletier, M., Wilson, C. B., Miller, S. I., Skerrett, S. J., and Ernst, R. K. (2006). Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infect. Immun. 74, 6730–6738.
Hall, J. D., Craven, R. R., Fuller, J. R., Pickles, R. J., and Kawula, T. H. (2007). Francisella tularensis replicates within alveolar type II epithelial cells in vitro and in vivo following inhalation. Infect. Immun. 75, 1034–1039.
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., Fitzgerald, K. A., Latz, E., Moore, K. J., and Golenbock, D. T. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865.
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J., and Monack, D. M. (2007). Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994.
Henry, T., Kirimanjeswara, G. S., Ruby, T., Jones, J. W., Peng, K., Perret, M., Ho, L., Sauer, J. D., Iwakura, Y., Metzger, D. W., and Monack, D. M. (2010). Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J. Immunol. 184, 3755–3767.
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., Latz, E., and Fitzgerald, K. A. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518.
Hsu, L. C., Ali, S. R., McGillivray, S., Tseng, P. H., Mariathasan, S., Humke, E. W., Eckmann, L., Powell, J. J., Nizet, V., Dixit, V. M., and Karin, M. (2008). A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. U.S.A. 105, 7803–7808.
Huang, M. T., Mortensen, B. L., Taxman, D. J., Craven, R. R., Taft-Benz, S., Kijek, T. M., Fuller, J. R., Davis, B. K., Allen, I. C., Brickey, W. J., Gris, D., Wen, H., Kawula, T. H., and Ting, J. P. (2010). Deletion of ripA alleviates suppression of the inflammasome and MAPK by Francisella tularensis. J. Immunol. 185, 5476–5485.
Inohara, C., McDonald, C., and Nunez, G. (2005). NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu. Rev. Biochem. 74, 355–383.
Ishii, K. J., Kawagoe, T., Koyama, S., Matsui, K., Kumar, H., Kawai, T., Uematsu, S., Takeuchi, O., Takeshita, F., Coban, C., and Akira, S. (2008). TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729.
Iwasaki, A., and Medzhitov, R. (2004). Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995.
Jones, J. W., Kayagaki, N., Broz, P., Henry, T., Newton, K., O’Rourke, K., Chan, S., Dong, J., Qu, Y., Roose-Girma, M., Dixit, V. M., and Monack, D. M. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776.
Kanneganti, T. D. (2010). Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10, 688–698.
Kanneganti, T. D., Body-Malapel, M., Amer, A., Park, J. H., Whitfield, J., Franchi, L., Taraporewala, Z. F., Miller, D., Patton, J. T., Inohara, N., and Nunez, G. (2006). Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568.
Katz, J., Zhang, P., Martin, M., Vogel, S. N., and Michalek, S. M. (2006). Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect. Immun. 74, 2809–2816.
Kostura, M. J., Tocci, M. J., Limjuco, G., Chin, J., Cameron, P., Hillman, A. G., Chartrain, N. A., and Schmidt, J. A. (1989). Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc. Natl. Acad. Sci. U.S.A. 86, 5227–5231.
Lai, X. H., Shirley, R. L., Crosa, L., Kanistanon, D., Tempel, R., Ernst, R. K., Gallagher, L. A., Manoil, C., and Heffron, F. (2010). Mutations of Francisella novicida that alter the mechanism of its phagocytosis by murine macrophages. PLoS One 5, e11857. doi: 10.1371/journal.pone.0011857
Lindemann, S. R., Peng, K., Long, M. E., Hunt, J. R., Apicella, M. A., Monack, D. M., Allen, L. A., and Jones, B. D. (2010). F. tularensis Schu S4 mutants in O-antigen and capsule biosynthesis genes induce early cell death in human macrophages. Infect Immun. 79, 581–594.
Long, G. W., Oprandy, J. J., Narayanan, R. B., Fortier, A. H., Porter, K. R., and Nacy, C. A. (1993). Detection of Francisella tularensis in blood by polymerase chain reaction. J. Clin. Microbiol. 31, 152–154.
Malik, M., Bakshi, C. S., Sahay, B., Shah, A., Lotz, S. A., and Sellati, T. J. (2006). Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect. Immun. 74, 3657–3662.
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., Roose-Girma, M., Erickson, S., and Dixit, V. M. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218.
Mariathasan, S., Weiss, D. S., Dixit, V. M., and Monack, D. M. (2005). Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049.
Mariathasan, S., Weiss, D. S., Newton, K., McBride, J., O’Rourke, K., Roose-Girma, M., Lee, W. P., Weinrauch, Y., Monack, D. M., and Dixit, V. M. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232.
Marina-Garcia, N., Franchi, L., Kim, Y. G., Miller, D., McDonald, C., Boons, G. J., and Nunez, G. (2008). Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J. Immunol. 180, 4050–4057.
Martinon, F., Agostini, L., Meylan, E., and Tschopp, J. (2004). Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr. Biol. 14, 1929–1934.
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Martinon, F., Hofmann, K., and Tschopp, J. (2001). The pyrin domain: a possible member of the death domain-fold family implicated in apoptosis and inflammation. Curr. Biol. 11, R118–R120.
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241.
McCaffrey, R. L., and Allen, L. A. (2006). Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J. Leukoc. Biol. 80, 1224–1230.
Miao, E. A., Alpuche-Aranda, C. M., Dors, M., Clark, A. E., Bader, M. W., Miller, S. I., and Aderem, A. (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575.
Miao, E. A., Ernst, R. K., Dors, M., Mao, D. P., and Aderem, A. (2008). Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. U.S.A. 105, 2562–2567.
Miao, E. A., Mao, D. P., Yudkovsky, N., Bonneau, R., Lorang, C. G., Warren, S. E., Leaf, I. A., and Aderem, A. (2010). Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 107, 3076–3080.
Molofsky, A. B., Byrne, B. G., Whitfield, N. N., Madigan, C. A., Fuse, E. T., Tateda, K., and Swanson, M. S. (2006). Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203, 1093–1104.
Muruve, D. A., Petrilli, V., Zaiss, A. K., White, L. R., Clark, S. A., Ross, P. J., Parks, R. J., and Tschopp, J. (2008). The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107.
Nano, F. E., Zhang, N., Cowley, S. C., Klose, K. E., Cheung, K. K., Roberts, M. J., Ludu, J. S., Letendre, G. W., Meierovics, A. I., Stephens, G., and Elkins, K. L. (2004). A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 186, 6430–6436.
Navarini, A. A., Recher, M., Lang, K. S., Georgiev, P., Meury, S., Bergthaler, A., Flatz, L., Bille, J., Landmann, R., Odermatt, B., Hengartner, H., and Zinkernagel, R. M. (2006). Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc. Natl. Acad. Sci. U.S.A. 103, 15535–15539.
O’Connell, R. M., Saha, S. K., Vaidya, S. A., Bruhn, K. W., Miranda, G. A., Zarnegar, B., Perry, A. K., Nguyen, B. O., Lane, T. F., Taniguchi, T., Miller, J.F., and Cheng, G. (2004). Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 200, 437–445.
O’Riordan, M., Yi, C. H., Gonzales, R., Lee, K. D., and Portnoy, D. A. (2002). Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 13861–13866.
Opitz, B., Vinzing, M., van Laak, V., Schmeck, B., Heine, G., Gunther, S., Preissner, R., Slevogt, H., N’Guessan, P. D., Eitel, J., Goldmann, T., Flieger, A., Suttorp, N., and Hippenstiel, S. (2006). Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J. Biol. Chem. 281, 36173–36179.
Pan, Q., Mathison, J., Fearns, C., Kravchenko, V. V., Da Silva Correia, J., Hoffman, H. M., Kobayashi, K. S., Bertin, J., Grant, E. P., Coyle, A. J., Sutterwala, F. S., Ogura, Y., Flavell, R. A., and Ulevitch, R. J. (2007). MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J. Leukoc. Biol. 82, 177–183.
Platz, G. J., Bublitz, D. C., Mena, P., Benach, J. L., Furie, M. B., and Thanassi, D. G. (2010). A tolC mutant of Francisella tularensis is hypercytotoxic compared to the wild type and elicits increased proinflammatory responses from host cells. Infect. Immun. 78, 1022–1031.
Raetz, C. R., Guan, Z., Ingram, B. O., Six, D. A., Song, F., Wang, X., and Zhao, J. (2009). Discovery of new biosynthetic pathways: the lipid A story. J. Lipid Res. 50(Suppl.) S103–S108.
Rathinam, V. A., Jiang, Z., Waggoner, S. N., Sharma, S., Cole, L. E., Waggoner, L., Vanaja, S. K., Monks, B. G., Ganesan, S., Latz, E., Hornung, V., Vogel, S. N., Szomolanyi-Tsuda, E., and Fitzgerald, K. A. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402.
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F., and Vance, R. E. (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. doi: 10.1371/journal.ppat.0020018
Roberts, T. L., Idris, A., Dunn, J. A., Kelly, G. M., Burnton, C. M., Hodgson, S., Hardy, L. L., Garceau, V., Sweet, M. J., Ross, I. L., Hume, D. A., and Stacey, K. J. (2009). HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060.
Rodrigue-Gervais, I. G., and Saleh, M. (2010). Genetics of inflammasome-associated disorders: a lesson in the guiding principals of inflammasome function. Eur. J. Immunol. 40, 643–648.
Santic, M., Asare, R., Skrobonja, I., Jones, S., and Abu Kwaik, Y. (2008). Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect. Immun. 76, 2671–2677.
Santic, M., Molmeret, M., and Abu Kwaik, Y. (2005a). Modulation of biogenesis of the Francisella tularensis subsp. novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN-gamma. Cell Microbiol. 7, 957–967.
Santic, M., Molmeret, M., Klose, K. E., Jones, S., and Kwaik, Y. A. (2005b). The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol. 7, 969–979.
Sauer, J. D., Witte, C. E., Zemansky, J., Hanson, B., Lauer, P., and Portnoy, D. A. (2010). Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe. 7, 412–419.
Scott, A. M., and Saleh, M. (2007). The inflammatory caspases: guardians against infections and sepsis. Cell Death Differ. 14, 23-31.
Stetson, D. B., and Medzhitov, R. (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103.
Sutterwala, F. S., Mijares, L. A., Li, L., Ogura, Y., Kazmierczak, B. I., and Flavell, R. A. (2007). Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245.
Suzuki, T., Franchi, L., Toma, C., Ashida, H., Ogawa, M., Yoshikawa, Y., Mimuro, H., Inohara, N., Sasakawa, C., and Nunez, G. (2007). Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3, e111. doi: 10.1371/journal.ppat.0030111
Takaoka, A., Wang, Z., Choi, M. K., Yanai, H., Negishi, H., Ban, T., Lu, Y., Miyagishi, M., Kodama, T., Honda, K., Ohba, Y., and Taniguchi, T. (2007). DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505.
Thornberry, N. A., Bull, H. G., Calaycay, J. R., Chapman, K. T., Howard, A. D., Kostura, M. J., Miller, D. K., Molineaux, S. M., Weidner, J. R., Aunins, J., Elliston, K. O., Ayala, J. M., Casano, F. J., Chin, J., Ding, G. J.-F., Egger, L. A., Gaffney, E. P., Limjuco, G., Palyha, O. C., Raju, S. M., Rolando, A. M., Salley, J. P., Yamin, T.-T., Lee, T. D., Shively, J. E., Maccross, M., Mumford, R. A., Schmidt, J. A., and Tocci, M. J. (1992). A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774.
Tominaga, A., Mahmoud, M. A., Mukaihara, T., and Enomoto, M. (1994). Molecular characterization of intact, but cryptic, flagellin genes in the genus Shigella. Mol. Microbiol. 12, 277–285.
Tschopp, J., Martinon, F., and Burns, K. (2003). NALPs: a novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 4, 95–104.
Ulland, T. K., Buchan, B. W., Ketterer, M. R., Fernandes-Alnemri, T., Meyerholz, D. K., Apicella, M. A., Alnemri, E. S., Jones, B. D., Nauseef, W. M., and Sutterwala, F. S. (2010). Cutting edge: mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J. Immunol. 185, 2670–2674.
Unterholzner, L., Keating, S. E., Baran, M., Horan, K. A., Jensen, S. B., Sharma, S., Sirois, C. M., Jin, T., Latz, E., Xiao, T. S., Fitzgerald, K. A., Paludan, S. R., and Bowie, A. G. (2010). IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004.
Warren, S. E., Mao, D. P., Rodriguez, A. E., Miao, E. A., and Aderem, A. (2008). Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 180, 7558–7564.
Weiden, M., Tanaka, N., Qiao, Y., Zhao, B. Y., Honda, Y., Nakata, K., Canova, A., Levy, D. E., Rom, W. N., and Pine, R. (2000). Differentiation of monocytes to macrophages switches the Mycobacterium tuberculosis effect on HIV-1 replication from stimulation to inhibition: modulation of interferon response and CCAAT/enhancer binding protein beta expression. J. Immunol. 165, 2028–2039.
Weiss, D. S., Brotcke, A., Henry, T., Margolis, J. J., Chan, K., and Monack, D. M. (2007). In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 6037–6042.
White, J. D., Rooney, J. R., Prickett, P. A., Derrenbacher, E. B., Beard, C. W., and Griffith, W. R. (1964). Pathogenesis of experimental respiratory tularemia in monkeys. J. Infect. Dis. 114, 277–283.
Wilson, K. P., Black, J. A., Thomson, J. A., Kim, E. E., Griffith, J. P., Navia, M. A., Murcko, M. A., Chambers, S. P., Aldape, R. A., Raybuck, S. A., and Livingston, D. J. (1994). Structure and mechanism of interleukin-1 beta converting enzyme. Nature 370, 270–275.
Woodward, J. J., Iavarone, A. T., and Portnoy, D. A. (2010). c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705.
Yang, P., An, H., Liu, X., Wen, M., Zheng, Y., Rui, Y., and Cao, X. (2010). The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat. Immunol. 11, 487–494.
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., Kuida, K., Mariathasan, S., Dixit, V. M., Flavell, R. A., Dietrich, W. F., and Roy, C. R. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325.
Keywords: Francisella, inflammasome, AIM2, ASC, caspase-1, interferon, STING, interleukin-1b
Citation: Jones JW, Broz P and Monack DM (2011) Innate immune recognition of Francisella tularensis: activation of type-I interferons and the inflammasome. Front. Microbio. 2:16. doi: 10.3389/fmicb.2011.00016
Received: 03 December 2010;
Accepted: 20 January 2011;
Published online: 03 February 2011.
Edited by:
Anders Sjostedt, Umeå University, SwedenCopyright: © 2011 Jones, Broz and Monack. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Denise M. Monack, Department of Microbiology and Immunology, School of Medicine, Stanford University, Stanford, CA 94305, USA. e-mail:ZG1vbmFja0BzdGFuZm9yZC5lZHU=