
 Jorge Valdés2,3
Jorge Valdés2,3
- 1 Department of Molecular Biology, Umeå University, Umeå, Sweden
- 2 Center for Bioinformatics and Genome Biology, Fundación Ciencia para Vida, Santiago, Chile
- 3 Departamento de Ciencias Biologicas, Andrés Bello University, Santiago, Chile
Given the challenges to life at low pH, an analysis of inorganic sulfur compound (ISC) oxidation was initiated in the chemolithoautotrophic extremophile Acidithiobacillus caldus. A. caldus is able to metabolize elemental sulfur and a broad range of ISCs. It has been implicated in the production of environmentally damaging acidic solutions as well as participating in industrial bioleaching operations where it forms part of microbial consortia used for the recovery of metal ions. Based upon the recently published A. caldus type strain genome sequence, a bioinformatic reconstruction of elemental sulfur and ISC metabolism predicted genes included: sulfide–quinone reductase (sqr), tetrathionate hydrolase (tth), two sox gene clusters potentially involved in thiosulfate oxidation (soxABXYZ), sulfur oxygenase reductase (sor), and various electron transport components. RNA transcript profiles by semi quantitative reverse transcription PCR suggested up-regulation of sox genes in the presence of tetrathionate. Extensive gel based proteomic comparisons of total soluble and membrane enriched protein fractions during growth on elemental sulfur and tetrathionate identified differential protein levels from the two Sox clusters as well as several chaperone and stress proteins up-regulated in the presence of elemental sulfur. Proteomics results also suggested the involvement of heterodisulfide reductase (HdrABC) in A. caldus ISC metabolism. A putative new function of Hdr in acidophiles is discussed. Additional proteomic analysis evaluated protein expression differences between cells grown attached to solid, elemental sulfur versus planktonic cells. This study has provided insights into sulfur metabolism of this acidophilic chemolithotroph and gene expression during attachment to solid elemental sulfur.
Introduction
Inorganic sulfur compounds (ISCs) in acidic, sulfide mineral environments are produced as a result of abiotic Fe(III) oxidation of sulfide minerals such as pyrite (FeS2; initial ISC product is thiosulfate) or chalcopyrite (CuFeS2; initial product is polysulfide sulfur). Their subsequent biooxidation produces sulfuric acid as the final product (Schippers and Sand, 1999; Johnson and Hallberg, 2009). As a result of sulfuric acid production, sulfide mineral environments are typically inhabited by acidophilic microorganisms. These microorganisms are exploited in the biotechnological process of “Biomining” whereby dissolution of sulfide minerals is catalyzed by the action of ISC oxidizing microorganisms as well as Fe(II) oxidizers that regenerate the Fe(III) required for the abiotic attack of sulfide minerals (Rawlings and Johnson, 2007).
A diverse range of acidophilic or neutrophilic photo- and chemolithotrophs can oxidize ISCs from sulfide (oxidation state of −2) to sulfate (+6; reviewed in Ghosh and Dam, 2009). Microorganisms utilize several systems for ISC oxidation including the Paracoccus pantotrophus 15 gene sulfur oxidizing (sox) cluster. This cluster encodes the multiple substrate Sox system that catalyzes oxidation of thiosulfate, elemental sulfur (S0), sulfide, and sulfite to sulfate. SoxAX is composed of the dihemic and monohemic cytochromes SoxA and SoxX, respectively. SoxYZ is predicted to be able to bind ISCs in different oxidation states by the cysteine contained in the V-K-V-T-I-G-G-C-G-G conserved motif on the carboxy terminus of the protein. The SoxB subunit is predicted to have two manganese ions in the active site and works as a sulfate thiohydrolase interacting with SoxYZ (Friedrich et al., 2005). Several sulfur oxidizing bacteria (e.g., green and purple sulfur bacteria) only contain the core thiosulfate oxidizing multi-enzyme system (TOMES). The TOMES lacks the sulfur dehydrogenase Sox(CD)2 and oxidizes thiosulfate to sulfate and S0 (reviewed in Friedrich et al., 2001; Ghosh and Dam, 2009, and Sakurai et al., 2010). Due to the lack of Sox(CD)2, S0 is polymerized to form globules which can be further oxidized by proteins encoded in the dissimilatory sulfite reductase (dsr) gene cluster (Hensen et al., 2006).
In contrast to the well studied Sox enzyme complex, the ISC oxidations pathways in acidophiles are not very well understood. As described in recent reviews (Rohwerder and Sand, 2007; Johnson and Hallberg, 2009), S0 is thought to be oxidized by acidophilic bacteria via sulfur dioxygenase (SDO) and by archaea via sulfur oxygenase reductase (Sor). The SDO has not been characterized although its enzyme activity has been shown (Suzuki, 1999; Rohwerder and Sand, 2003). However, the archaeal Sor has been well studied and the corresponding gene has been identified in Acidianus tengchongensis, Aquifex aeolicus, Picrophilus torridus, “Ferroplasma acidarmanus,” and Sulfolobus tokodaii (Urich et al., 2006). In the presence of oxygen, Sor simultaneously catalyzes oxidation and reduction of S0 generating sulfite, thiosulfate, and sulfide (Urich et al., 2006). The enzyme does not require cofactors or external electron donors for S0 reduction. Due to its cytoplasmic location it is believed that it does not play a role in formation of the transmembrane electron gradient but rather provide substrates for other membrane bound enzymes. Another enzyme which has recently been suggested to be involved in Acidithiobacillus ferrooxidans S0 metabolism is heterodisulfide reductase (Hdr; Quatrini et al., 2009). So far no biochemical evidence for A. ferrooxidans S0 oxidation by Hdr has been reported, however, transcriptomics (Quatrini et al., 2009) and proteomics data (unpublished data) strongly suggests its involvement. Hdr of methanogenic archaea has been studied (Hedderich et al., 2005) and it catalyzes the reversible reduction of the disulfide bond in heterodisulfide accompanied by the extrusion of electrons and the formation of a transmembrane electron gradient. Quatrini et al. (2009) hypothesize that Hdr works in reverse in acidophiles by utilizing the naturally existing proton gradient to oxidize disulfide intermediates originating from S0 and donating electrons to the quinone pool. Other enzymes involved in acidophilic ISC oxidation pathways are thiosulfate:quinone oxidoreductase (Tqr) which oxidizes thiosulfate to tetrathionate, tetrathionate hydrolase (Tth), and sulfide oxidoreductase (Rohwerder and Sand, 2007; Johnson and Hallberg, 2009). Recently, the analysis of gene context has highlighted differences in ISC oxidation strategies in A. ferrooxidans, Acidithiobacillus thiooxidans, and Acidithiobacillus caldus (Cardenas et al., 2010). Microarray analysis suggests the petII (prosthetic group-containing subunits of the cytochrome bc1 complex), cyo (cytochrome o ubiquinol oxidase), cyd (cytochrome bd ubiquinol oxidase), and doxII (encoding thiosulfate quinol reductase) gene clusters are up-regulated during growth on S0 compared to Fe(II) grown cells (Quatrini et al., 2006). From these data, a model for A. ferrooxidans ISC metabolism has been created (Quatrini et al., 2009). A. ferrooxidans proteins with increased expression during growth on S0 include an outer membrane protein (Omp40) and a thiosulfate sulfur transferase protein (Ramirez et al., 2004). Also, a high throughput study of periplasmic proteins identified 41 and 14 proteins uniquely expressed in S0 and thiosulfate grown cells, respectively (Valenzuela et al., 2008). The genome context of these proteins suggests they are involved in ISC metabolism and possibly S0 oxidation and Fe–S cluster construction. Secreted proteins from a pure culture of A. thiooxidans and from co-culture with A. ferrooxidans were studied by proteomics (Bodadilla Fazzini and Parada, 2009). An Omp40 like protein was identified which is suggested to be involved in attachment. Finally, S0 induced genes in the acidophilic archaeon Sulfolobus metallicus include Sor (Bathe and Norris, 2007).
A. caldus is an ISC oxidizing acidophile (Hallberg et al., 1996b) often identified in biomining environments (Okibe et al., 2003; Dopson and Lindström, 2004). A. caldus aids in metal dissolution by removal of solid S0 that may form a passivating layer on the mineral surface (Dopson and Lindström, 1999). The A. caldus draft genome contains genes for ISC oxidation (Valdes et al., 2009). The gene cluster containing the A. caldus tetrathionate hydrolase (tth) and the doxD component (thiosulfate:quinol oxidoreductase) has been characterized (Rzhepishevska et al., 2007). The Tth is a periplasmic homo-dimer with an optimum pH of 3 (Bugaytsova and Lindström, 2004). Previously Tth was also studied in A. ferrooxidans (de Jong et al., 1997).
Owing to the fact that A. caldus is ubiquitous in both natural and anthropogenic sulfide mineral environments, its importance in generating sulfuric acid, and in mitigating mineral passivation we have investigated its ISC metabolism. An in depth bioinformatic analysis revealed the putative genes responsible for sulfuric acid generation, that have then been verified by proteomic comparison between growth with tetrathionate and S0 and via transcript profiling. This has generated insights into the ISC metabolism of this microorganism. Such knowledge might help to better understand the industrial processes.
Materials and Methods
Bioinformatic Reconstruction of A. caldus ISC Metabolism
Genes and metabolic pathways involved in ISC and S0 oxidation/reduction were obtained from Metacyc1 and Kegg2. Amino acid sequences derived from selected genes previously identified to be involved in ISC metabolism were used as a query to conduct BlastP and tBlastN (Altschul et al., 1997) searches to interrogate the A. caldusT (ATCC51756) draft genome sequence (NZ_ACVD00000000.1). Potential gene candidates were further characterized employing the following bioinformatic tools: ClustalW (Larkin et al., 2007) for primary structure similarity relations, PSI-PRED (Bryson et al., 2005) for secondary structure predictions, Prosite (Hulo et al., 2006) for motif predictions, and ProDom (Bru et al., 2005) and Pfam (Finn et al., 2008) for domain predictions. Selected gene candidates were assigned to putative orthologous groups in order to determine potential evolutionary associations3 (Tatusov et al., 2003). Information regarding the organization of genes in other S0 metabolizing microorganisms was obtained from NCBI4 and IMG–JGI5.
Media and Culture Conditions
A. caldusT was cultured in mineral salts medium (MSM) with trace elements (Dopson and Lindström, 1999) at 45°C and pH 2.5 whilst sparging with 2% (vol/vol) CO2 enriched air. Tetrathionate (5 mM; Sigma) and 5 g/L hydrophilic, biologically produced S0 (provided by PAQUES B.V., the Netherlands) were used as substrate. Stock solutions of tetrathionate were sterile filtered and added to the autoclaved (121°C for 15 min) MSM, whereas the finely ground S0 was added to MSM prior to autoclaving at 105°C for 30 min. The medium containing solid S0 was stirred for 24 h to ensure fine dispersion of the S0 particles. Biomass for the transcript profiling and proteomic comparison of growth on tetrathionate versus S0 was produced in continuous culture with a dilution rate of 0.06 h−1. The homogeneous delivery of solid substrate was ensured by continuous stirring of the feed vessel and an appropriate flow rate which did not allow S0 to settle in the tubing. In contrast, cell mass for proteomic comparison of A. caldus sessile versus planktonic growth was grown in 1 L batch cultures with initial pH 2.5. Sessile and planktonic bacteria from batch cultures were harvested in mid exponential growth phase according to planktonic cell counts (pH at collection was 1.3–1.4). Planktonic cells from batch or continuous culture were harvested at 4750 g for 10 min followed by a washing step in MSM pH 2.5. For the continuous culture grown on S0, the remaining S0 particles were separated from planktonic cells by pelleting at 450 g for 30 s which only removed the S0 but left planktonic cells in the supernatant (the solid S0 and any attached cells were discarded). Solid S0 with sessile bacteria attached was collected from batch cultures and washed four times with MSM pH 2.5 until no planktonic bacteria were detected in the supernatant by microscopic investigation before pelleting at 450 g for 30 s. All cell and S0 pellets were stored at −80°C.
Semi Quantitative RNA Transcript Profiles
Primers targeting selected genes putatively involved in ISC metabolism were designed for semi quantitative reverse transcription (RT-) PCR amplification (product sizes 98–530 bp; Table 1). RNA was extracted from 100 to 200 mL medium from the continuous cultures with TRI reagent (Ambion) according to the manufacturer’s recommendations. Contaminating DNA was digested with RNase-free DNase I (Fermentas) followed by a second extraction with TRI reagent. RNA concentration and purity was measured with a NanoDrop 2000 spectrophotometer (ThermoScientific). In order to rule out DNA contamination, the RNA samples were subjected to PCR using illustra PuRE Taq Ready-To-Go PCR Beads (GE Healthcare). RT-PCR was performed in a two step reaction using RevertAid First Strand cDNA Synthesis Kit (Fermentas) for RT and AccessQuick Master Mix (Promega) for PCR. The RT reaction primed with random hexamers was performed with 1 μg total RNA for 1 h at 45°C using two independent biological samples for each condition. PCR primed with specific primers (Table 1) was carried out with 1 μL of RT reaction as template in 25 μL reaction volume in PTC-100 Programmable Thermal Controller (MJ Research Inc.). PCR cycling was started with initial denaturation at 95°C for 5 min, followed by 30 amplification cycles consisting of denaturation at 95°C for 30 s, annealing at primer specific temperatures (Table 1) for 30 s, and elongation at 72°C for 2 min 30 s and concluded with 5 min elongation at 72°C (no further increase of PCR product was observed after 31 cycles). A transcription control gene, DNA gyrase (gyrA), was used (Takle et al., 2007). PCR products were separated by agarose electrophoresis and quantified with QuantityOne (BioRad). For final analysis, the 2 independent RNA samples (biological replicates) were used as templates for duplicate RT-PCR reactions (technical replicates) which yielded a total of four replications. In a few cases one replicate was not considered for analysis.
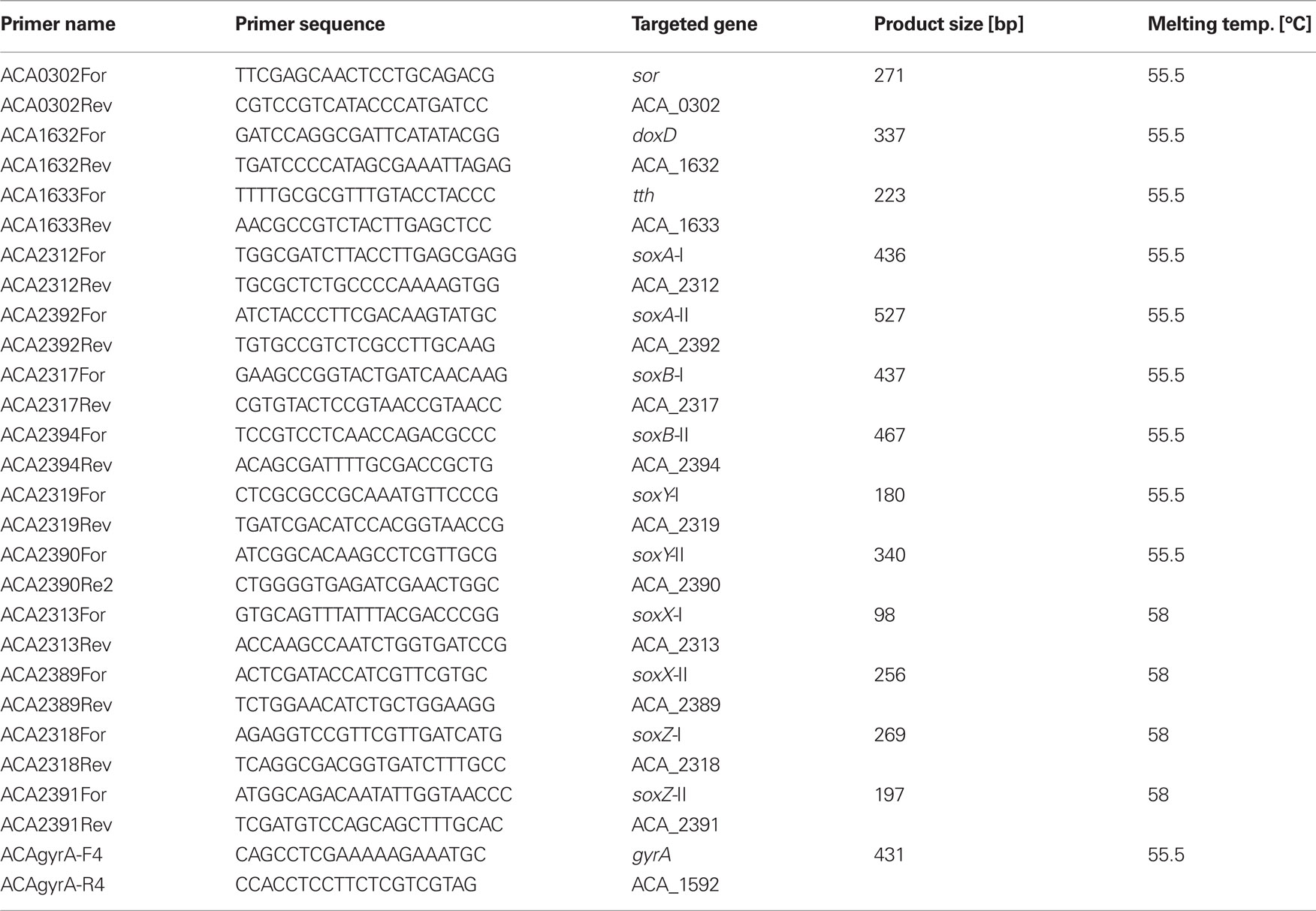
Table 1. Primers used for RNA transcript profiles.
A. caldus Proteomics Analysis
For the preparation of the total soluble proteome, cell pellets from 200 mL culture were re-suspended in lysis buffer (7 M urea, 2 M thiourea, 30 mM Tris, 1 mM EDTA, 1.5% Triton X-100, pH 8.5), broken by sonication (2 min, 5 s pulse, 5 s break, 30% amplitude), cell debris removed by centrifugation (10 min, 10 000 rpm, 4°C), and the lysate stored at −80°C. Isoelectric focusing (IEF) was performed using pre-cast, 18 cm, Immobiline DryStrip IPG gels (GE Healthcare) with a non-linear pH gradient from 3 to 10 in an Ettan IPGphor IEF unit (GE Healthcare). Protein samples (200 μg) were applied to the IPG strip in rehydration buffer (7 M urea, 2 M thiourea, and 1.5% Triton X-100) with 1.45% dithiothreitol (DTT) and 0.5% IPG buffer (GE Healthcare). After passive rehydration for 16 h at 25°C, IEF was run for a total of 42 kVh with stepwise increasing voltage according to supplier’s recommendation. Following IEF, the gel strips were equilibrated in two steps using equilibration buffer (75 mM Tris, 6 M urea, 30% glycerol, 2% SDS) with additional 100 mM DTT in the first step and equilibration buffer with 2.5% iodoacetamide in the second step. The gel strips were then applied to 12% Duracryl (NextGene Genomic Solutions) SDS-polyacrylamide gels and sealed with 1.5% (wt/vol) agarose solution containing bromophenol blue. Electrophoresis was run in an Ettan DALTsix apparatus (GE Healthcare). The gels were fixed and stained to saturation with colloidal Coomassie (Anderson, 1991). After staining was completed the gels were scanned using Image Scanner (GE Healthcare) and analyzed using image analysis software Melanie 7.03 (Genebio). The membrane enriched fractions were prepared according to Molloy (2008). In brief, crude cell extracts were obtained by sonication in Tris buffer (50 mM Tris pH 8.0, 0.5 mM EDTA) and enriched for bacterial membranes by incubation at 4°C for 1 h in 100 mM Na2CO3 and subsequent ultra-centrifugation at 170 000 g for 70 min. This carbonate extraction in combination with membrane solubilization and 2D gel electrophoresis has been used for the recovery of outer membrane proteins of Gram-negative bacteria (Molloy et al., 2000, 2001; Phadke et al., 2001). The pellets were washed with 50 mM Tris buffer pH 8.0 and re-suspended overnight at 4°C in rehydration buffer. The protocol for 2D gels was the same as above except that 24 cm IPG strips were used and IEF was run for a total of 59 kVh. For the preparation of the soluble protein fraction from sessile bacteria it was necessary to detach the cells from the S0 prior to protein extraction and 2D gel electrophoresis (as above). The method for cell detachment by Gehrke et al. (1998) was modified. In brief, the S0 with sessile cells was incubated with a detergent solution pH 7.0 (0.01 mM 3-(Decyldimethylammonio)propanesulfonate inner salt (SB 3–10; Sigma), 10 mM Tris, 1 mM EDTA) for 5 min at room temperature with occasional vortexing before pelleting the S0 by centrifuging at 450 g for 30 s. The treatment was repeated four times and supernatants containing the detached cells pooled and cells collected before proteomic analysis (as above). All gels of the same condition were run in triplicate.
Protein spots were regarded as differentially expressed if they showed the following characteristics: (i) reproducibility in all three gels of the same condition; (ii) the fold change between the two conditions was ≥2.0; and (iii) the fold change between conditions was significant with a probability of 95% according to one-way ANOVA testing. Spots of interest were excised from the gel, destained, and digested with sequencing grade modified trypsin (Promega) according to standard procedures for matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry (Shevchenko et al., 1996; Pandey et al., 2000). Subsequently, tryptic digests were spotted on a MALDI target plate and co-crystallized with α-cyana-4-hydroxy-cinnamic acid solution (Agilent Technologies). Mass spectra were acquired with a Voyager DE-STR mass spectrometer (Applied Biosystems) and analyzed using DataExplorer (Applied Biosystems). External calibration with premixed standard peptides (Sequenzyme Peptide Mass Standard Kit, Applied Biosystems) was performed. Peptide mass fingerprints (PMFs) of mass spectra were searched against a local database containing the A. caldusT draft genome sequence (NZ_ACVD00000000.1) using Mascot with the following search parameters: (i) two missed cleavages; (ii) peptide mass tolerance of 50 ppm; and (iii) variable modifications [carbamidomethyl (C), oxidation (M), propionamide (C)]. Hits in the local database with a Mowse score > 47 were significant at a confidence level of 95%. Two samples were analyzed by Edman degradation performed at the Protein Analysis Center, Karolinska Institute, Stockholm, Sweden. For more detailed functional information of identified proteins the Biocyc database6 was queried and InterProScan signature recognition search7 was performed.
In order to rule out any contaminating proteins originating from the biological S0, 2D gels were run from samples prepared by suspending the S0 in lysis buffer and sonicating. Control gels were stained with silver (Blum et al., 1987) and no protein spots were detected (data not shown).
Results
Bioinformatic Reconstruction of A. caldusisc Metabolism
A detailed analysis of the genes present in the draft genome sequence of A. caldusT revealed genes for ISC oxidation that are common to A. ferrooxidans [sulfide quinone reductase (sqr), doxD, and tth] (Valdes et al., 2008) and other microbial representatives from extreme acidic environments. A. caldus also has gene candidates potentially encoding components of the Sox sulfur oxidizing system and Sor, which are not present in its close relative A. ferrooxidans. The gene clusters are presented in Figure 1A while the bioinformatic reconstruction of A. caldus ISC metabolism is presented in Figure 1B.
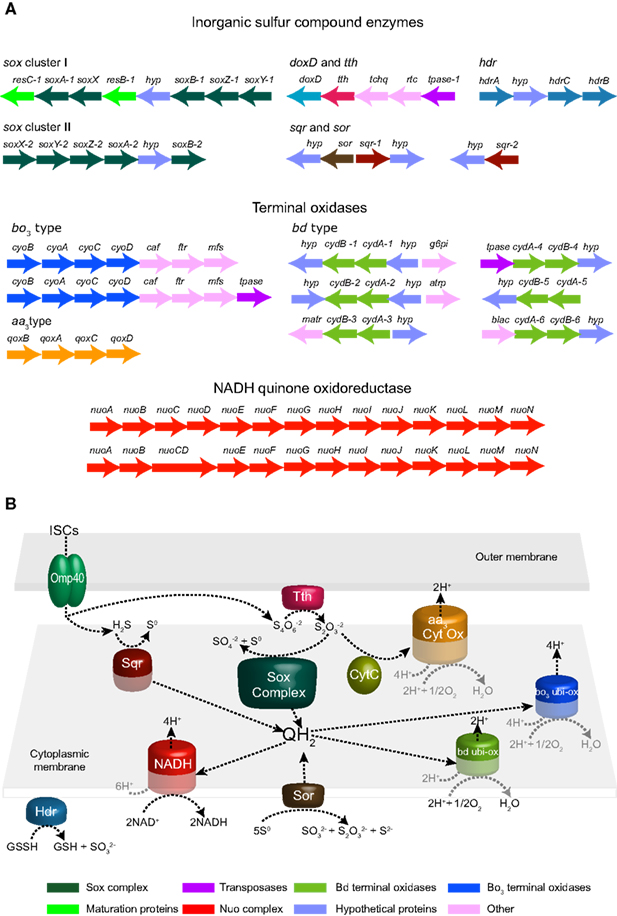
Figure 1. Gene clusters of enzymes potentially involved in inorganic sulfur compound (ISC) metabolism as well as gene clusters of terminal oxidases and the NADH quinone-oxidoreductase complex as predicted by bioinformatic analysis of the A. caldus genome sequence (A). Suggested model for ISC metabolism in A. caldus(B). Abbreviations: sox, sulfur oxidation system; doxD, thiosulfate:quinol oxidoreductase; tth, tetrathionate hydrolase; hdr, heterodisulfide reductase; sqr, sulfide quinone reductase; sor, sulfur oxygenase reductase; hyp, hypothetical protein; caf, cytochrome oxidase assembly factor; ftr, heme O synthase, protoheme IX farnesyltranferase; g6pi, glucose 6-p isomerase; mfs, major facilitator superfamily protein; atrp, putative amino acid transporter; matr, putative malic acid transporter; blac, beta lactamase; tpase, transposase; tchq, two component system – histidine quinase; rtc, two component system – response regulator.
The first documented step in ISC oxidation is the transition of sulfide to Sº. In Gram-negative bacteria, this reaction is carried out by the usually membrane bound Sqr. This reaction can also be catalyzed by membrane bound FCC sulfide dehydrogenase. The enzymatic activity of Sqr (EC 1.8.5.-) has been purified from A. ferrooxidans membranes (Wakai et al., 2007). A sulfide oxidizing activity has been identified in A. caldus but the enzyme has not been identified (Hallberg et al., 1996b). Furthermore, one of the three sqr copies present in the A. ferrooxidans genome is reported to be involved in ISC metabolism (Quatrini et al., 2006, 2009). In A. caldus, an ortholog of the sqr (sqr-1) was identified that was divergently oriented from a gene potentially encoding Sor, that participates in the utilization of S0 as energy source in several organisms (Urich et al., 2004). The product of the A. caldus sqr-1 shares 40% similarity with another putative sqr ortholog (sqr-2) identified in the draft genome sequence. This latter copy has a similar gene context to sqr-3 from A. ferrooxidans and shares a 62% similarity to this protein. Although Sqr representatives belong to a large and considerably variable gene family of the pyridine nucleotide-disulfide oxido-reductases (Pfam: PF07992), the high similarity plus the conservation of gene context observed with A. ferrooxidans strongly suggests similar functional properties. Another enzyme reported to be involved in ISC metabolism is Tth. The activity of this enzyme has been detected in several acidithiobacilli (Hallberg et al., 1996b; Brasseur et al., 2004). An inspection of the draft genome of A. caldus reveals the presence of a candidate tth upstream of doxD (Figure 1A). The putative Tth of A. caldus shares 71% similarity with the Tth of A. ferrooxidans, indicating their high similarity orthologous relationship. The Tth of both sequenced acidithiobacilli have a conserved pyrrolo-quinoline quinone (PQQ) domain (Pfam: PF01011). Although Tth’s are predicted to be external membrane proteins, experimental evidence showed that the A. caldus Tth is a soluble periplasmic protein with maximum activity at pH 3 (Bugaytsova and Lindström, 2004). Furthermore, the tth gene cluster has been recently studied (Rzhepishevska et al., 2007).
In contrast to the enzymes described above, the Sox sulfur oxidizing system has not been found in any microorganisms from the Acidithiobacillus genus. However, the bioinformatics analyses identified the presence of two gene clusters potentially encoding components of the Sox system. The A. caldus Sox complex is predicted to be formed by three periplasmic components (referred to as core TOMES): SoxAX, SoxYZ, and SoxB (Figure 1A). Our bioinformatics inspection did not identify soxCD orthologs in the draft genome sequence of A. caldus; however we cannot disregard the presence of these genes until the A. caldus genome sequence is finished. Nevertheless, based on the model for ISC metabolism in the green sulfur bacterium Chlorobaculum tepidum proposed by Sakurai et al. (2010) it can be predicted that the A. caldus core TOMES is able to oxidize thiosulfate with the possible accumulation of S0 globules in the periplasm and it may be able to oxidize sulfite (Friedrich et al., 2001). No candidates for the enzymes adenylylphosphosulfate (APS) reductase or sulfate adenylyltransferases, which consecutively oxidize sulfite via an indirect pathway, have been found in the genome of A. caldus. However, an oxidoreductase molybdopterin-binding protein (ACA_1585) was identified as a possible sulfite oxidizing enzyme catalyzing the direct oxidation of sulfite to sulfate. ACA_1585 has a molybdopterin-binding domain (PF00174) and a twin-arginine translocation pathway signal sequence. It lacks a dimerization domain and an N-terminal heme domain. Furthermore, no additional putative subunits containing heme domains have been detected. ACA_1585 has 26% similarity with the sulfite oxidizing enzyme DraSO from Deinococcus radiodurans which was proposed to be a novel sulfite oxidizing enzyme without a heme domain (D’Errico et al., 2006).
Another enzyme involved in A. caldus ISC metabolism not found in A. ferrooxidans is Sor (one copy of sor was identified). The predicted protein shows a characteristic domain of the Sor family (Pfam: PF07682) and all activity-critical residues based on structural and experimental data (Urich et al., 2006). Furthermore, the predicted cytoplasmic location of the A. caldus Sor enzyme agrees with experimental data obtained from A. tengchongensis and Escherichia coli transformants (Chen et al., 2005). The sor was found adjacent and divergently arranged from sqr-1 (Figure 1A). This suggests the presence of potentially shared regulatory regions that could be co-regulating both enzymes, catalyzing successive reactions, and thus providing an adaptive strategy. Recently, the enzyme activity of Sor has been reported to be present in A. caldus-like strains (Janosch et al., 2009). However, it is still unknown how its substrate (S8) is incorporated in to the cell or how the resulting products (S0, H2S, and S2O3−2) are excreted. Furthermore, heterodisulfide reductase (Hdr) has been postulated to be involved in A. ferrooxidans S0 oxidation (Quatrini et al., 2009) and is up-regulated during aerobic growth on solid S0 (unpublished data). In A. caldus two orthologs of each HdrABC subunit have been found. Both putative HdrA subunits (ACA_1473, ACA_2418) are flavoproteins with a FAD binding site and they also contain the conserved four cysteine residues (CXGXRDX6–8CSX2CC) for binding of a Fe–S cluster. The putative HdrB, ACA_2421, contains two typical cysteine rich regions whereas the second putative HdrB subunit, ACA_2417, contains only one such region. The remaining subunit, HdrC, is represented by ACA_2376 and ACA_2420 both containing the 4F–4S ferredoxin iron–sulfur binding domain. ACA_2418, ACA_2421, and ACA_2420 are embedded in the gene cluster hdrA hyp hdrC hdrB (Figure 1A). All putative A. caldus Hdr subunits showed >90% similarity to the respective A. ferrooxidans Hdr subunits. As previously shown for A. ferrooxidans (Quatrini et al., 2009), the similarity to Hdr of other acidophilic sulfur oxidizers is significant whereas the similarity of the A. caldus putative HdrABC to their respective subunits of the methanogenic archaea Methanothermobacter marburgensis is only around 30%.
It has been shown that sulfur oxidizing bacteria which lack Sox(CD)2 utilize the reverse dissimilatory sulfite reductase (DsrAB) for the oxidation of S0 to sulfite (reviewed in Friedrich et al., 2001; Ghosh and Dam, 2009, and Sakurai et al., 2010). In Allochromatium vinsum, DsrAB is encoded with 13 other Dsr proteins in a cluster (Dahl et al., 2005) also containing DsrEFH and DsrC. The latter are proposed to be involved in S0 substrate binding and transport of S0 from the periplasmic S0 globules to the cytoplasm (Cort et al., 2008). Escherichia coli DsrEFH and DsrC homologs (TusBCD and TusE) interact in a S0 relay system during 2-thiouridine biosynthesis (Ikeuchi et al., 2006). Although no homologs of dsr were found in the A. caldus draft genome sequence, several potential DsrE-like proteins containing the characteristic DsrE/DsrF-like family features (Pfam 02635) were detected. All of those were annotated as hypothetical proteins (ACA_0867, ACA_0091, ACA_2522, ACA_0556, ACA_1583, ACA_1441, and ACA_2423) and putatively play a role in S0 binding and transport. Additionally, several candidates for the transport of extracellular S0 to the cytoplasm have been proposed for green sulfur bacteria (Frigaard and Bryant, 2008a,b; Sakurai et al., 2010). One possibility is that the thioredoxin SoxW acts together with thiol:disulfide interchange protein DsbD within the periplasm in transferring S0 across the inner membrane (Sakurai et al., 2010). One candidate gene was predicted that potentially encodes a DsbC ortholog (ACA_2033) with similar functions as DsbD. DsbC thiol:disulfide interchange protein ACA_2033 exhibits a conserved N-terminal domain of the disulfide bond isomerase DsbC family (Pfam10411). It has been reported that members of this protein family are responsible for the formation of disulfide bonds and function as a disulfide bond isomerase during oxidative protein-folding in the bacterial periplasm (Hiniker et al., 2005). No homolog of SoxW has been found in A. caldus; however, other thioredoxins might fulfill the same function.
A. caldus is also predicted to contain two gene clusters potentially encoding components of the NADH quinone-oxidoreductase complex (EC 1.6.5.3) as has been observed in Azotobacter vinelandii (Bertsova et al., 2001). In addition, six cydAB copies possibly encoding subunits of Qox-bd (EC 1.10.3.-) terminal oxidase and one gene cluster that might code for a putative aa3-type terminal oxidase were detected. The analysis also revealed the presence of two copies of the cytochrome o (cyoBACD-caf-ftr-mfs) gene cluster (Cyo-1 and Cyo-2), sharing 89 and 75% similarity with orthologs in A. ferrooxidans. This gene redundancy could have several explanations including: (i) to provide regulatory and pathway flexibility to confront environmental changes such as oxygen availability (bo and bd complexes have different O2 affinities); or (ii) to manage ISC oxidizing complexes that introduce electrons at variable places in the electron transport chain (e.g., quinol-level for SQR and cytochrome c-level for Sox).
RNA Transcript Profiles of Selected ISC Genes
Reverse transcription-PCR showed that all assayed genes were transcribed during growth on tetrathionate and S0 (Figure 2). No significant changes in transcript levels were detected for sor, doxD, tth, soxZ-I, and soxZ-II. The remaining sox cluster genes were significantly up-regulated during growth on tetrathionate. The similar RNA levels for soxZ-I and soxZ-II during growth on tetrathionate and S0 was unexpected as both genes would be expected to be co-transcribed with their respective gene clusters. With the exception of the soxA transcript, the transcript levels of both sox clusters followed the same trends indicating that both were involved in ISC metabolism. The control gene, gyrA, displayed similar transcription levels under both conditions.

Figure 2. RNA transcript profiles of selected genes predicted to be involved in A. caldus inorganic sulfur compound metabolism. Shown are the means of relative band intensities from semi quantitative RT-PCR products of three to four replicates with standard deviations. RNA was extracted from cells grown on tetrathionate (gray columns) or S0 (white columns).
Protein Expression During Growth on S0 Versus Tetrathionate
Proteomic analysis yielded 115 identifications of differentially expressed protein spots (Figures 3 and 4; Table A1 in Appendix). Forty-three proteins were up-regulated on tetrathionate (12 in soluble and 31 in membrane fraction) and 30 uniquely found on tetrathionate gels (21 in soluble and 9 in membrane fraction). During growth on S0, 23 protein spots were up-regulated (15 in soluble and 8 in membrane fraction) and 19 were unique (11 in soluble and 8 in membrane fraction). Several protein spots yielded the same identification which indicated protein fragmentation, post translational modification, or the protein was up-regulated in both the soluble and membrane enriched fractions. Protein designations are given in parenthesis with their match ID as designated by Melanie including a capital letter S specifying soluble fraction and M for membrane enriched fractions.
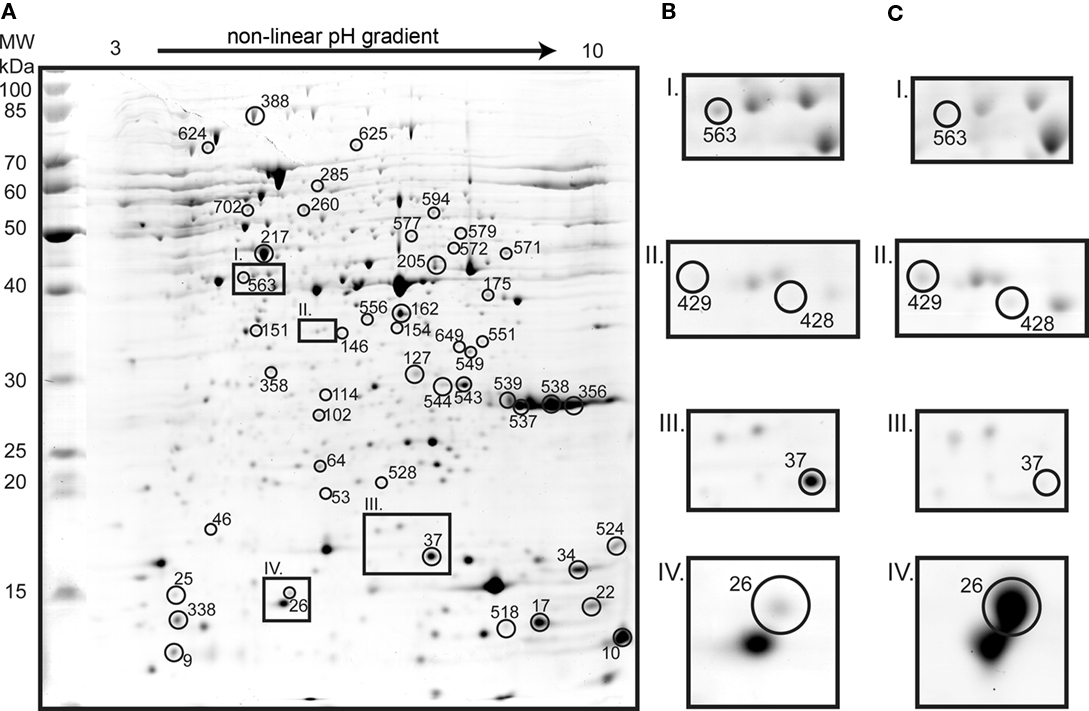
Figure 3. Two dimension gel of the A. caldus soluble proteome from cells grown on tetrathionate (A; identified spots are marked with circles and the numbers indicate match IDs given without S in the figure). Inset frames show tetrathionate (B) and S0 (C) conditions: Radical SAM domain protein (spot S563) unique in tetrathionate grown cells (I); heat shock protein GroEL (S429 and S428) unique in S0 grown cells (II); twin-arginine translocation protein TatA (S37) which was 3.8-fold up-regulated in tetrathionate (III); and heat shock protein Hsp20 (S26) that was 8.6-fold up-regulated in S0 grown cells (IV).
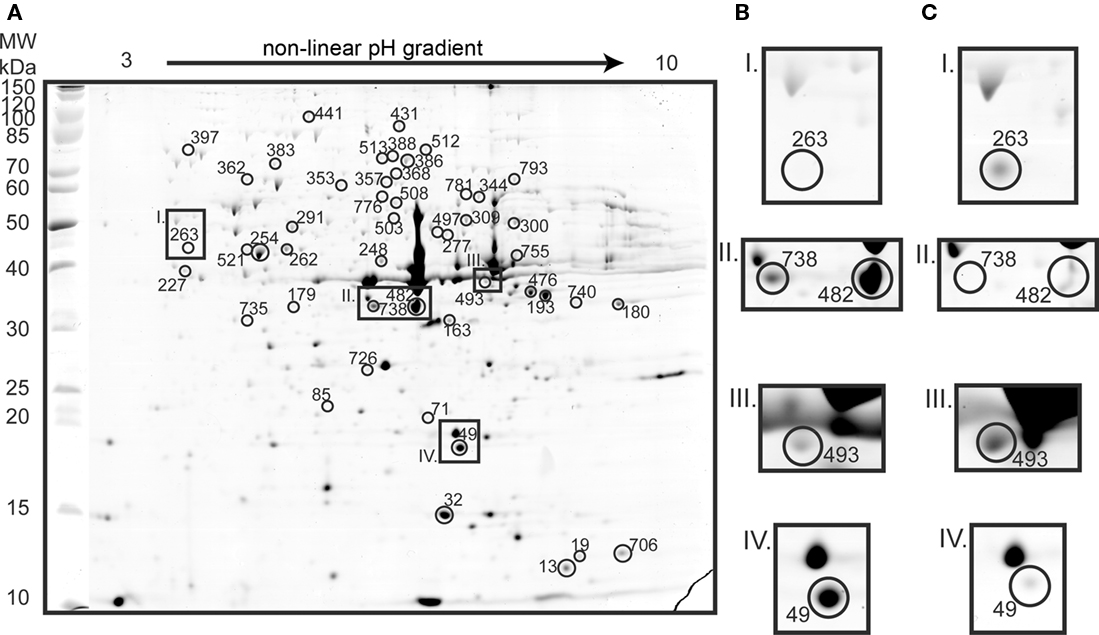
Figure 4. Two dimension gel of the A. caldus membrane enriched proteome on tetrathionate (A; identified proteins indicated with circles and match IDs given without M in the figure). Inset frames are presented for tetrathionate (B) and for S0 (C) conditions: Carboxysome shell protein CsoS1 (spot M263) that was 6.1-fold up-regulated in S0 grown cells (I); two spots identified as hypothetical protein ACA_1144 that were unique (M738) and 10.7-fold up-regulated (M482) in gels from tetrathionate grown cells (II); type 1 secretion outer membrane protein (M493) that was 2.4-fold up-regulated in S0 grown cells (III); and a putative lipoprotein (M49) which was 10.3-fold up-regulated on tetrathionate (IV).
No putative proteins belonging to NADH quinine-oxidoreductase or terminal oxidases were found differentially expressed in the various conditions tested. However, based on the bioinformatic reconstruction the involvement of NADH quinone-oxidoreductase complex and terminal oxidases in the A. caldus ISC metabolism was suggested. Therefore, these complexes were represented in the proposed model (Figure 1B).
Proteins involved in ISC metabolism
Proteins encoded within the two Sox clusters were up-regulated in gels from tetrathionate grown bacteria including SoxB-I (M793) and hypothetical protein ACA_2320 (S524) encoded in sox cluster I; as well as SoxA-II (S356, S538, S539), SoxZ-II (S10), and hypothetical protein ACA_2393 (S34) from sox cluster II. These findings clearly suggest an involvement of proteins encoded by both Sox clusters in ISC metabolism during growth on tetrathionate. Furthermore, both Sqr-1 (S260) and Sqr-2 (M300) were identified as up-regulated on tetrathionate. A DsrE/F-like hypothetical protein ACA_0867 (S17, S518, M13), potentially involved in S0 binding and transport, was up-regulated on tetrathionate and subunit C of CoB–CoM heterodisulfide reductase (S543, S544; ACA_2420) was a unique protein spot on tetrathionate gels. In contrast, HdrA (S462; ACA_2418) was found as a unique protein spot in S0 gels while the DsrE/F-like hypothetical protein ACA_1583 (S9) and DsbC thiol:disulfide interchange protein ACA_2033 (S358) were up-regulated on S0.
General trends in the proteome of tetrathionate grown A. caldus
Several proteins relevant in signal transduction were up-regulated in tetrathionate grown cells, i.e., chemotaxis protein CheV (S151, M735), putative sensory histidine kinase YfhA (S577), nitrogen regulation protein NRI (M 776), and hypothetical protein ACA_1270 (M277) containing a PAS domain fold involved in signaling proteins. However, the signature feature of the tetrathionate grown proteome was up-regulation of proteins involved in central carbon metabolism, cell division, amino acid biosynthesis, fatty acid biosynthesis, translation, and DNA repair. The up-regulated central carbon metabolism proteins included aspartate aminotransferase (S571), dihydrolipoamide dehydrogenase (S594), fructose bisphosphate aldolase (M248), and phophoglucomutase (M368). A few proteins of central carbon metabolism were up-regulated in S0 grown cells those including 6-phosphogluconate dehydrogenase (S154) and HAD-superfamily hydrolase (M71). Proteins involved in cell division were solely found up-regulated on gels of tetrathionate grown cells such as FtsA (M262), FtsZ (M227), and FtsH (M386, M388, M513). Four proteins involved in amino acid biosynthesis and degradation (S549, S694, M353, M755) and three involved in fatty acid biosynthesis (M179, M309, M726) were up-regulated in tetrathionate grown cells. No proteins of this group were up-regulated in S0 grown cells. Another characteristic group consisted of translation related proteins which included two different translation elongation factors (S217, S388, M254, M521), four different tRNA synthetases (S572, S625, M431, M508), and one amidotransferase (S702) up-regulated on tetrathionate; whereas only one ribosomal protein (S338) was detected up-regulated in S0 grown cells. Proteins involved in DNA repair included a MutS2 family protein identified in two protein spots (M344, M781) and DNA repair protein RecN (M362).
Increased expression of proteins with central functions is commonly attributed to an increased growth rate. However, the samples originated from continuous cultures grown at identical dilution rates, meaning that bacteria were theoretically maintained at the same growth rate. Therefore, it was more likely that the observed trends signified down-regulation of central functions in the S0 grown cells due to a stress response. Nevertheless, the observed results motivated the proteomic investigation of sessile and planktonic A. caldus grown in S0 batch cultures as only the planktonic sub-population was investigated for the comparison of growth on tetrathionate and S0.
General trends in the proteome of S0 grown A. caldus
The most striking characteristic of the proteome of S0 grown cells was the high number of up-regulated chaperones and proteases: GroEL (S114, S146, S409, S413, S416, S425, S428, S429, S435, S436), heat shock protein Hsp20 (S26, S46), DnaK (M397), ClpB (M441), and protease Do (M605). In contrast, the only chaperone up-regulated during growth on tetrathionate was HscA (S624). This indicated a stress response during growth on S0. It should be noted that GroEL has been detected in many protein spots most of them with low molecular weight in 2D gels (Figure 3, Table A1 in Appendix) indicating fragmentation of the protein. The cause of this fragmentation remains unknown. A second distinguishing feature of the S0 grown proteome was up-regulation of proteins involved in CO2 fixation, namely ribulose bisphosphate carboxylase large chain (M503) and carboxysome shell protein CsoS1 (M263, M530). However, the reasons for up-regulation of proteins related to CO2 fixation remain unclear. Additionally, stringent starvation protein A (S102) and flagella basal-body rod protein FlgC (S205) were up-regulated on S0. Stringent starvation protein A is believed to be important for stress response during stationary phase and nutrient limitation in E. coli (Williams et al., 1994). The up-regulation of FlgC which is part of the basal body in flagella points at an increase of flagella and the importance of cell motility in S0 grown cells to be able to attach to the solid substrate.
Proteins up-regulated under both conditions
In one case, a two component protein was identified in both conditions whereas in other cases proteins sharing a similar function could not be clearly attributed to either condition. Those included proteins involved in protein transport such as an efflux transporter (M180, M740) and Twin-arginine translocation protein TatA (S37, M32) which were up-regulated in tetrathionate grown cells. While in S0 grown cells protein export chaperone (S25) and Type I secretion outer membrane protein, TolC precursor (M497) were up-regulated. A two component protein was found up-regulated on tetrathionate (M291) and as a unique spot in S0 gels (M607). The two spots were in close proximity on the gels; however the unique spot traveled with slightly larger molecular weight and more basic isoelectric point suggesting post translational modification. This protein is encoded together with the signal transduction histidine kinase up-stream of sox cluster II indicating that this two component system might be involved in its regulation.
Protein Expression During Planktonic Versus Sessile Growth on S0
In 2D gels of planktonic A. caldus, three up-regulated and three unique protein spots were identified. Based on characteristic peaks in mass spectra an additional six protein spots up-regulated in planktonic cells were revealed to be the same protein. The identification of this protein could neither be determined by MALDI-ToF nor by Edman degradation. From gels of sessile cells, 22 up-regulated protein spots were identified (Figure 5; Table A2 in Appendix).
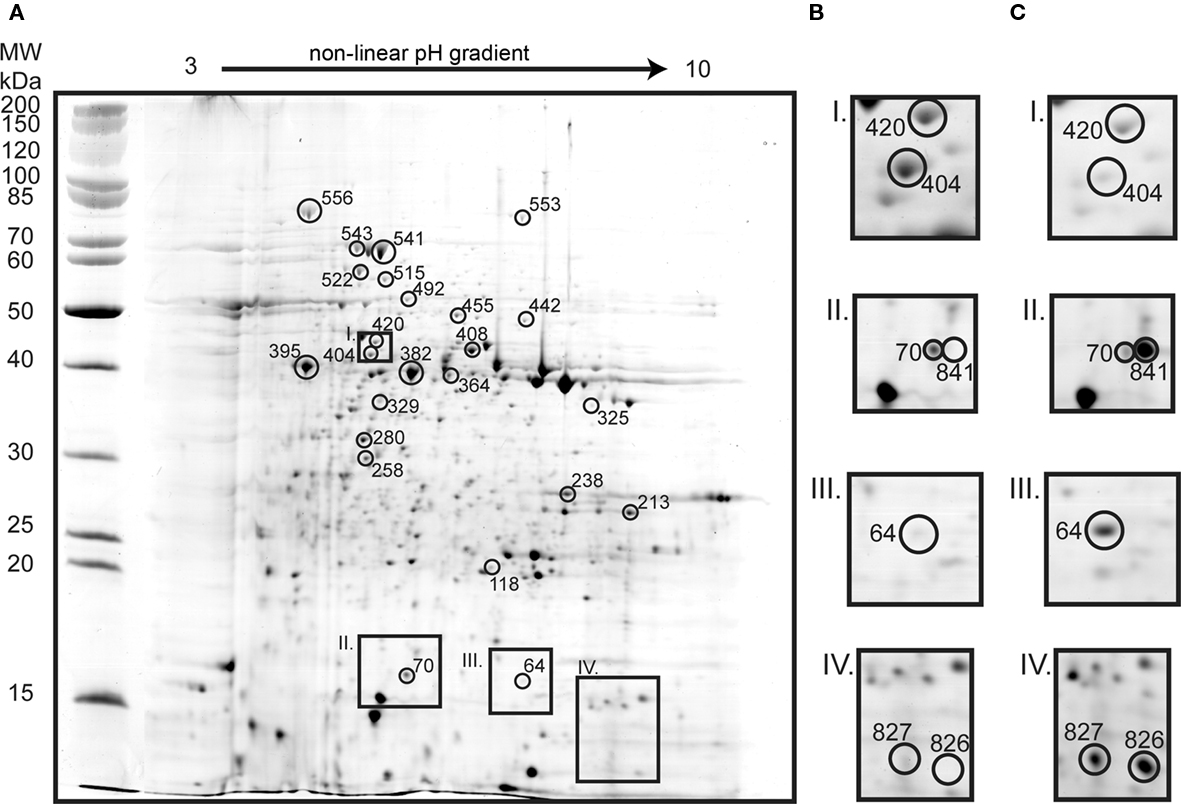
Figure 5. Two dimension gel of the A. caldus soluble proteome of sessile cells grown on S0 (A; identified proteins marked with circles and match IDs). Inset frames show sessile (B) and planktonic (C) conditions: S-adenosylmethionine synthetase (spot 420) and YVTN family beta-propeller repeat protein (404) that were 2.4 and spot 7.7-fold up-regulated in sessile cells (I); single-stranded DNA-binding protein (70) 3.1-fold up-regulated in sessile cells and sulfur oxidation protein SoxY (841) unique in gels from planktonic cells (II); twin-arginine translocation protein TatA (64) was up-regulated 7.9-fold in planktonic cells (III); and hypothetical protein ACA_0867 (spots 827 and 826) uniquely found in planktonic cells (IV).
Several of the proteins identified from planktonic cells were also found up-regulated in tetrathionate grown cells, i.e., TatA (64), Sqr-1 (492), and hypothetical protein ACA_0867 (826 and 827). In addition, SoxY-I (841) and hypothetical protein ACA_2219 (118) were identified. Proteins up-regulated in gels of sessile cells comprised characteristic proteins from tetrathionate grown and S0 grown proteomes as well as proteins not identified from other gels. S0 characteristic proteins included the chaperones GroEL and DnaK (329, 541, 543, and 556), HdrA (382; ACA_2418), and proteins involved in CO2 fixation such as ribulose bisphosphate carboxylase large chain (258) and rubisco activation protein CbbQ (280). Tetrathionate associated proteins consisted of peptidyl-prolyl cis-trans isomerase ppiD (213), CoB–CoM HdrC (238; ACA_2420), proteins involved in amino acid biosynthesis (442, 447, 455, 515), two central carbon metabolism proteins (364, 408), and a single-stranded DNA-binding protein involved in DNA replication (70). The up-regulation of proteins involved in amino acid biosynthesis and central carbon metabolism suggests that sessile cells are less starved than planktonic cells. CoB–CoM HdrB (395; ACA_2421) was not previously detected but was found up-regulated in gels of sessile bacteria. Additionally, twitching motility protein (325) and 40-residue YVTN family β-propeller repeat protein (404) were up-regulated in gels of sessile cells. Twitching motility protein is required for twitching motility and social gliding which allows Gram-negative bacteria to move along surfaces (Merz et al., 2000). The YVTN domain is present in surface layer proteins of archaea (Jing et al., 2002) which protect cells from the environment and have been shown to be involved in cell to cell association in Methanosarcina mazei (Mayerhofer et al., 1998).
Discussion
ISC Metabolism in A. caldus
A model has been constructed for ISC oxidation and electron transport based upon gene predictions and proteomics data (Figure 1B). Tetrathionate is hydrolyzed by a periplasmic Tth with a DoxD component (Hallberg et al., 1996b; Bugaytsova and Lindström, 2004; Rzhepishevska et al., 2007). Previously, the genes encoding Tth and DoxD were shown to be up-regulated during growth on tetrathionate as compared to growth on S0. However, this study showed the same expression levels of tth and doxD in the semi quantitative RT-PCR. Additionally, neither protein was detected in the proteomics investigation suggesting that protein levels were also similar. A possible explanation for the discrepancy between this and the previous report (Rzhepishevska et al., 2007) is that gene transcription of tth and doxD might be different in the sub-populations of S0 grown sessile and planktonic cells (potentially explaining the previously reported large standard deviations Rzhepishevska et al., 2007). The role of the DoxD component is unknown as it lacks the DoxA of the thiosulfate quinine-oxidoreductase and Tth is thought to be a hydrolase (Bugaytsova and Lindström, 2004; Rzhepishevska et al., 2007). Therefore, the main product of Tth in the proposed model, thiosulfate, was suggested to be oxidized by the A. caldus SoxABXYZ system. The experimental data supports this view with up-regulated transcripts of both sox clusters as well as up-regulation of several gene products during growth on tetrathionate. As homologs of neither Sox(CD)2 nor DsrAB were detected in the genome sequence of A. caldus to date it is proposed that the products of its core TOMES are sulfate and S0. This aspect is further strengthened by the observation that thiosulfate oxidation has a S0 intermediate (Hallberg et al., 1996b) and that S0 globules have been detected in A. caldus when under sub-optimal conditions (Hallberg et al., 1996a). Also, sulfur globules are an obligate intermediate in A. vinosum thiosulfate oxidation (Pott and Dahl, 1998; Dahl et al., 2005).
The metabolism of S0 is complicated by its hydrophobic nature which makes an activation of S0 prior to its oxidation necessary. Potentially the DsbC (up-regulated in S0 grown cells) was involved in transferring the S0 equivalent from the membrane to the S0 oxidizing enzyme as suggested for green sulfur bacteria (Sakurai et al., 2010). Additionally, it has also been suggested that sulfane sulfur is the actual substrate of the sulfur oxidizing enzyme (SDO or Hdr) in A. ferrooxidans (Rohwerder and Sand, 2003; Quatrini et al., 2009). The substrate for Sor is believed to be sulfur in a linear form, probably as a polysulfide. Furthermore, two hypothetical DsrE/F-like proteins were detected in the proteomics, one up-regulated on tetrathionate and the other on S0 which might be involved in the transfer of sulfane sulfur in the course of S0 oxidation (Dahl et al., 2005). One candidate for S0 oxidation is Sor that catalyzes the disproportionation of S0 to sulfite, thiosulfate, and sulfide (potentially explaining the up-regulation of Sqr in cells grown on less reduced forms of sulfur). In this study, RT-PCR data demonstrated similar levels of sor transcripts for growth on tetrathionate and S0 suggesting it may be involved in ISC oxidation. A second candidate for S0 oxidation is the trimeric complex HdrABC suggested to be involved in A. ferrooxidans S0 metabolism by utilizing the proton gradient to oxidize disulfide intermediates originating from S0 oxidation to sulfite (Quatrini et al., 2009). To date, this reverse reaction of HdrABC is purely speculative and awaits biochemical evidence. Yet, the similarities of HdrABC within acidophilic sulfur oxidizers are striking and might point to this new function of the enzyme. Additionally, A. ferrooxidans Hdr has been shown to be up-regulated during growth on S0 (Quatrini et al., 2009 and unpublished data). In this study HdrA was up-regulated in S0 grown A. caldus, whereas HdrC was up-regulated in tetrathionate grown cells. However, all subunits HdrABC were up-regulated in sessile compared to planktonic cells. In our model both Sor and Hdr can oxidize S0 and are hypothesized to comprise multiple pathways. The potential use of Hdr and/or Sor in A. caldus S0 oxidation remains to be resolved. Sor or Hdr might be employed in variable growth conditions or for different (internal or external) sources of S0. For instance, the Sor enzyme identified in A. caldus-like strains has an increased activity at 65°C (Janosch et al., 2009) suggesting it may be used at higher growth temperatures. The observation that DsbE-like proteins and different subunits of Hdr were up-regulated in both conditions might point to the fact that tetrathionate oxidation has a S0 intermediate (Hallberg et al., 1996b). Following the reaction of either Sor or Hdr, the produced sulfite is oxidized to sulfate. Although no candidate for a sulfite oxidizing enzyme was detected in the proteomics a sulfite oxidase activity has been reported for A. caldus (Hallberg et al., 1996b) and a candidate for a sulfite oxidizing enzyme without heme domain was detected in the genome sequence. No biochemical evidence (Dopson et al., 2002) or putative gene candidates were found for involvement of the adenosine monophosphate system.
Clear trends in the protein expression of cells grown on tetrathionate versus S0 were observed. In S0 grown cells this included up-regulation of chaperones and a protease and down-regulation of proteins involved in central carbon metabolism, amino acid biosynthesis, fatty acid biosynthesis, cell division proteins, and DNA repair. The latter changes can be attributed to the cell’s effort to conserve energy which is a feature of the general stress response.
Sessile Versus Planktonic A. caldus
It is believed that the stress response observed in S0 grown sessile cells (compared to planktonic) was due to a biological phenomenon and not due to sample treatment. Several points argue in favor of this view: (i) the stress response was also apparent in S0 grown cells when compared to tetrathionate grown cells although the treatment for both conditions was identical; and (ii) sessile cells were frozen prior to detachment which probably killed most cells conserving their proteome and making changes in the proteome of sessile cells during detachment treatment unlikely.
The A. caldus planktonic proteome contained up-regulated proteins similar to the expression pattern of tetrathionate grown cells; whereas, the sessile proteome contained up-regulated proteins that were described to be signature proteins of tetrathionate and S0 grown cells. Possibly cells attached to S0 are less starved than planktonic cells (up-regulation of proteins from central carbon metabolism) but generally more stressed than planktonic cells (up-regulation of chaperones). All three Hdr subunits were up-regulated in the sessile cells suggesting they oxidize S0. In contrast, the planktonic sub-population may oxidize soluble ISCs (e.g., tetrathionate) potentially released from sessile cells. The growth medium of A. caldus grown on S0 was tested for tetrathionate and thiosulfate. Neither compound could be detected during growth for 10 days (data not shown) potentially because the soluble ISCs were oxidized by planktonic cells before they accumulated to a detectable concentration. In addition, proteins were detected, such as a twitching motility protein involved in sessile cell motility (Merz et al., 2000) and a YVTN family beta-propeller repeat protein possibly involved in cell-to-cell interactions (Mayerhofer et al., 1998) that might also be involved in attachment to the solid substrate. Although no flagellar proteins were found up-regulated in the proteomic comparison of sessile versus planktonic cells the up-regulation of flagellar basal-body rod protein FlgC in the comparison between tetrathionate and S0 grown cells indicated the importance of cell motility to be able to attach to the solid substrate.
Typically, planktonic and sessile sub-populations are stable (Vilain et al., 2004b) and their proteomes include many differentially expressed proteins (Vilain et al., 2004a). The proteomes and transcriptomes of biofilms have been widely studied in several neutrophilic organisms (Sauer and Camper, 2001; Oosthuizen et al., 2002; Sauer et al., 2002; Planchon et al., 2009). A transcriptomic study of biofilm and planktonic Leptospirillum spp. suggests acidophile biofilms are, similarly to neutrophilic biofilms, dynamic structures with distinct metabolic differences between planktonic and biofilm cells (Moreno-Paz et al., 2010). The reported differences in the proteomes between sessile and planktonic A. caldus sub-populations comprised a comparatively small number of differentially expressed proteins. In this case study, it may be possible that planktonic and sessile sub-populations interchanged (possibly due to vigorous stirring and shear forces exerted by the solid S0 particles) that resulted in relatively similar proteomes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Mark Dopson wishes to thank the Swedish Research Council for financial support (Vetenskapsrådet contract number 621-2007-3537). David S. Holmes acknowledges Fondecyt 1050063, DI-UNAB 34-06, DI-UNAB 15-06/I, and a Microsoft Sponsored Research Award.
Footnotes
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Anderson, N. L. (1991). Two Dimensional Gel Electrophoresis: Operation of the ISO-DALT System. Rockville, MD: Large Scale Biology Press.
Bathe, S., and Norris, P. R. (2007). Ferrous iron- and sulfur-induced genes in Sulfolobus metallicus. Appl. Environ. Microbiol. 73, 2491–2497.
Bertsova, Y. V., Bogachev, A. V., and Skulachev, V. P. (2001). Noncoupled NADH:ubiquinone oxidoreductase of Azotobacter vinelandii is required for diazotrophic growth at high oxygen concentrations. J. Bacteriol. 183, 6869–6874.
Blum, H., Beier, H., and Gross, H. J. (1987). Improved silver staining of plant-proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8, 93–99.
Bodadilla Fazzini, R. A., and Parada, P. (2009). Analysis of sulfur metasecretome in mixed cultures of Acidithiobacillus thiooxidans and Acidithiobacillus ferrooxidans. Adv. Mat. Res. 71–73, 151–154.
Brasseur, G., Levican, G., Bonnefoy, V., Holmes, D., Jedlicki, E., and Lemesle-Meunier, D. (2004). Apparent redundancy of electron transfer pathways via bc1 complexes and terminal oxidases in the extremophilic chemolithoautotrophic Acidithiobacillus ferrooxidans. Biochim. Biophys. Acta 1656, 114–126.
Bru, C., Courcelle, E., Carrère, S., Beausse, Y., Dalmar, S., and Kahn, D. (2005). The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res. 33, D212–D215.
Bryson, K., McGuffin, L. J., Marsden, R. L., Ward, J. J., Sodhi, J. S., and Jones, D. T. (2005). Protein structure prediction servers at University College London. Nucleic Acids Res. 33, W36–W38.
Bugaytsova, Z., and Lindström, E. B. (2004). Localization, purification and properties of a tetrathionate hydrolase from Acidithiobacillus caldus. Eur. J. Biochem. 271, 272–280.
Cardenas, J. P., Valdes, J., Quatrini, R., Duarte, F., and Holmes, D. S. (2010). Lessons from the genomes of extremely acidophilic bacteria and archaea with special emphasis on bioleaching microorganisms. Appl. Microbiol. Biotechnol. 88, 605–620.
Chen, Z. W., Jiang, C. Y., She, Q., Liu, S. J., and Zhou, P. J. (2005). Key role of cysteine residues in catalysis and subcellular localization of sulfur oxygenase-reductase of Acidianus tengchongensis. Appl. Environ. Microbiol. 71, 621–628.
Cort, J. R., Selan, U., Schulte, A., Grimm, F., Kennedy, M. A., and Dahl, C. (2008). Allochromatium vinosum DsrC: solution-state NMR structure, redox properties, and interaction with DsrEFH, a protein essential for purple sulfur bacterial sulfur oxidation. J. Mol. Biol. 382, 692–707.
D’Errico, G., Di Salle, A., La Cara, F., Rossi, M., and Cannio, R. (2006). Identification and characterization of a novel bacterial sulfite oxidase with no heme binding domain from Deinococcus radiodurans. J. Bacteriol. 188, 694–701.
Dahl, C., Engels, S., Pott-Sperling, A. S., Schulte, A., Sander, J., Lubbe, Y., Deuster, O., and Brune, D. C. (2005). Novel genes of the Dsr gene cluster and evidence for close interaction of Dsr proteins during sulfur oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. J. Bacteriol. 187, 1392–1404.
de Jong, G. A. H., Hazeu, W., Bos, P., and Kuenen, J. G. (1997). Polythionate degradation by tetrathionate hydrolase of Thiobacillus ferrooxidans. Microbiology 143, 499–504.
Dopson, M., and Lindström, E. B. (1999). Potential role of Thiobacillus caldus in arsenopyrite bioleaching. Appl. Environ. Microbiol. 65, 36–40.
Dopson, M., and Lindström, E. B. (2004). Analysis of community composition during moderately thermophilic bioleaching of pyrite, arsenical pyrite and chalcopyrite. Microb. Ecol. 48, 19–28.
Dopson, M., Lindstrom, E. B., and Hallberg, K. B. (2002). ATP generation during reduced inorganic sulfur compound oxidation by Acidithiobacillus caldus is exclusively due to electron transport phosphorylation. Extremophiles 6, 123–129.
Finn, R. D., Tate, J., Mistry, J., Coggill, P. C., Sammut, S. J., Hotz, H.-R., Ceric, G., Forslund, K., Eddy, S. R., Sonnhammer, E. L. L., and Bateman, A. (2008). The Pfam protein families database. Nucleic Acids Res. 36, D281–D288.
Friedrich, C. G., Bardischewsky, F., Rother, D., Quentmeier, A., and Fischer, J. (2005). Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8, 253–259.
Friedrich, C. G., Rother, D., Bardischewsky, F., Quentmeier, A., and Fischer, J. (2001). Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 67, 2873–2882.
Frigaard, N. U., and Bryant, D. A. (2008a). “Genomic and evolutionary perspectives on sulfur metabolism in green sulfur bacteria,” in Microbial Sulfur Metabolism, eds C. Dahl and C. G. Friedrich (Berlin: Springer), 60–76.
Frigaard, N. U., and Bryant, D. A. (2008b). “Genomic insights into the sulfur metabolism of phototrophic green sulfur bacteria,” in Sulfur Metabolism in Phototrophic Organisms, eds R. Hell, C. Dahl, D. B. Knaff, and T. Leustek (Berlin: Springer), 337–355.
Gehrke, T., Telegdi, J., Thierry, D., and Sand, W. (1998). Importance of extracellular polymeric substances from Thiobacillus ferrooxidans for bioleaching. Appl. Environ. Microbiol. 64, 2743–2747.
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Ecol. 33, 999–1043.
Hallberg, K. B., Dopson, M., and Lindstrom, E. B. (1996a). Arsenic toxicity is not due to a direct effect on the oxidation of reduced inorganic sulfur compounds by Thiobacillus caldus. FEMS Microbiol. Lett. 145, 409–414.
Hallberg, K. B., Dopson, M., and Lindstrom, E. B. (1996b). Reduced sulfur compound oxidation by Thiobacillus caldus. J. Bacteriol. 178, 6–11.
Hedderich, R., Hamann, N., and Bennati, M. (2005). Heterodisulfide reductase from methanogenic archaea: a new catalytic role for an iron sulfur cluster. Biol. Chem. 386, 961–970.
Hensen, D., Sperling, D., Truper, H. G., Brune, D. C., and Dahl, C. (2006). Thiosulphate oxidation in the phototrophic sulphur bacterium Allochromatium vinosum. Mol. Microbiol. 62, 794–810.
Hiniker, A., Collet, J. F., and Bardwell, J. C. (2005). Copper stress causes an in vivo requirement for the Escherichia coli disulfide isomerase DsbC. J. Biol. Chem. 280, 33785–33791.
Hulo, N., Bairoch, A., Bulliard, V., Cerutti, L., De Castro, E., Langendijk-Genevaux, P. S., Pagni, M., and Sigrist, C. J. (2006). The PROSITE database. Nucleic Acids Res. 34, D227–D230.
Ikeuchi, Y., Shigi, N., Kato, J., Nishimura, A., and Suzuki, T. (2006). Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Mol. Cell 21, 97–108.
Janosch, C., Thyssen, C., Vera, M., Bonnefoy, V., Rohwerder, T., and Sand, W. (2009). Sulfur oxygenase reductase in different Acidithiobacillus caldus-like strains. Adv. Mat. Res. 71–73, 239–242.
Jing, H., Takagi, J., Liu, J. H., Lindgren, S., Zhang, R. G., Joachimiak, A., Wang, J. H., and Springer, T. A. (2002). Archaeal surface layer proteins contain beta propeller, PKD, and beta helix domains and are related to metazoan cell surface proteins. Structure 10, 1453–1464.
Johnson, D. B., and Hallberg, K. B. (2009). Carbon, iron and sulfur metabolism in acidophilic micro-organisms. Adv. Microb. Physiol. 54, 201–255.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948.
Mayerhofer, L. E., Conway de Macario, E., Yao, R., and Macario, A. J. (1998). Structure, organization, and expression of genes coding for envelope components in the archaeon Methanosarcina mazei S-6. Arch. Microbiol. 169, 339–345.
Merz, A. J., So, M., and Sheetz, M. P. (2000). Pilus retraction powers bacterial twitching motility. Nature 407, 98–102.
Molloy, M. D. (2008). Isolation of bacterial cell membrane proteins using carbonate extraction. Methods Mol. Biol. 424, 397–401.
Molloy, M. P., Herbert, B. R., Slade, M. B., Rabilloud, T., Nouwens, A. S., Williams, K. L., and Gooley, A. A. (2000). Proteomic analysis of the Escherichia coli outer membrane. Eur. J. Biochem. 267, 2871–2881.
Molloy, M. P., Phadke, N. D., Maddock, J. R., and Andrews, P. C. (2001). Two-dimensional electrophoresis and peptide mass fingerprinting of bacterial outer membrane proteins. Electrophoresis 22, 1686–1696.
Moreno-Paz, M., Gomez, M. J., Arcas, A., and Parro, V. (2010). Environmental transcriptome analysis reveals physiological differences between biofilm and planktonic modes of life of the iron oxidizing bacteria Leptospirillum spp. in their natural microbial community. BMC Genomics 11, 404. doi: 10.1186/1471-2164-11-404
Okibe, N., Gericke, M., Hallberg, K. B., and Johnson, D. B. (2003). Enumeration and characterization of acidophilic microorganisms isolated from a pilot plant stirred-tank bioleaching operation. Appl. Environ. Microbiol. 69, 1936–1943.
Oosthuizen, M. C., Steyn, B., Theron, J., Cosette, P., Lindsay, D., von Holy, A., and Brozel, V. S. (2002). Proteomic analysis reveals differential protein expression by Bacillus cereus during biofilm formation. Appl. Environ. Microbiol. 68, 2770–2780.
Pandey, A., Andersen, J. S., and Mann, M. (2000). Use of mass spectrometry to study signaling pathways. Sci. STKE 37, pl1.
Phadke, N. D., Molloy, M. P., Steinhoff, S. A., Ulintz, P. J., Andrews, P. C., and Maddock, J. R. (2001). Analysis of the outer membrane proteome of Caulobacter crescentus by two-dimensional electrophoresis and mass spectrometry. Proteomics 1, 705–720.
Planchon, S., Desvaux, M., Chafsey, I., Chambon, C., Leroy, S., Hebraud, M., and Talon, R. (2009). Comparative subproteome analysis of planktonic and sessile Staphylococcus xylosus C2a: new insight in cell physiology of a coagulase-negative Staphylococcus in biofilm. J. Proteome Res. 8, 1797–1809.
Pott, A. S., and Dahl, C. (1998). Sirohaem sulfite reductase and other proteins encoded by genes at the Dsr locus of Chromatium vinosum are involved in the oxidation of intracellular sulfur. Microbiology 144, 1881–1894.
Quatrini, R., Appia-Ayme, C., Denis, Y., Jedlicki, E., Holmes, D., and Bonnefoy, V. (2009). Extending the models for iron and sulfur oxidation in the extreme acidophile Acidithiobacillus ferrooxidans. BMC Genomics 10, 394. doi: 10.1186/1471-2164-10-394
Quatrini, R., Appia-Ayme, C., Denis, Y., Ratouchniak, J., Veloso, F., Valdes, J., Lefimil, C., Silver, S., Roberto, F., Orellana, O., Denizot, F., Jedlicki, E., Holmes, D., and Bonnefoy, V. (2006). Insights into the iron and sulfur energetic metabolism of Acidithiobacillus ferrooxidans by microarray transcriptome profiling. Hydrometallurgy 83, 263–272.
Ramirez, P., Guiliani, N., Valenzuela, L., Beard, S., and Jerez, C. A. (2004). Differential protein expression during growth of Acidithiobacillus ferrooxidans on ferrous iron, sulfur compounds, or metal sulfides. Appl. Environ. Microbiol. 70, 4491–4498.
Rawlings, D. E., and Johnson, D. B. (2007). The Microbiology of biomining: development and optimization of mineral-oxidizing microbial consortia. Microbiology 153, 315–324.
Rohwerder, T., and Sand, W. (2003). The sulfane sulfur of persulfides is the actual substrate of the sulfur-oxidizing enzymes from Acidithiobacillus and Acidiphilium spp. Microbiology 149, 1699–1709.
Rohwerder, T., and Sand, W. (2007). Oxidation of inorganic sulfur compounds in acidophilic prokaryotes. Engineering in Life Sciences 7, 301-309.
Rzhepishevska, O. I., Valdés, J., Marcinkeviciene, L., Algora Gallardo, C., Meskys, R., Bonnefoy, V., Holmes, D. S., and Dopson, M. (2007). Regulation of a novel Acidithiobacillus caldus gene cluster involved in reduced inorganic sulfur compound metabolism. Appl. Environ. Microbiol. 73, 7367–7372.
Sakurai, H., Ogawa, T., Shiga, M., and Inoue, K. (2010). Inorganic sulfur oxidizing system in green sulfur bacteria. Photosyn. Res. 104, 163–176.
Sauer, K., and Camper, A. K. (2001). Characterization of phenotypic changes in Pseudomonas putida in response to surface-associated growth. J. Bacteriol. 183, 6579–6589.
Sauer, K., Camper, A. K., Ehrlich, G. D., Costerton, J. W., and Davies, D. G. (2002). Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J. Bacteriol. 184, 1140–1154.
Schippers, A., and Sand, W. (1999). Bacterial leaching of metal sulfides proceeds by two indirect mechanisms via thiosulfate or via polysulfides and sulfur. Appl. Environ. Microbiol. 65, 319–321.
Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858.
Suzuki, I. (1999). Oxidation of inorganic sulfur compounds: chemical and enzymatic reactions. Can. J. Microbiol. 45, 97–105.
Takle, G. W., Toth, I. K., and Brurberg, M. B. (2007). Evaluation of reference genes for real-time RT-PCR expression studies in the plant pathogen Pectobacterium atrosepticum. BMC Plant Biol. 7, 50. doi: 10.1186/1471-2229-7-50
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., Krylov, D. M., Mazumder, R., Mekhedov, S. L., Nikolskaya, A. N., Rao, B. S., Smirnov, S., Sverdlov, A. V., Vasudevan, S., Wolf, Y. I., Yin, J. J., and Natale, D. A. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41. doi: 10.1186/1471-2105-4-41
Urich, T., Bandeiras, T. M., Leal, S. S., Rachel, R., Albrecht, T., Zimmermann, P., Scholz, C., Teixeira, M., Gomes, C. M., and Kletzin, A. (2004). The sulphur oxygenase reductase from Acidianus ambivalens is a multimeric protein containing a low-potential mononuclear non-haem iron centre. Biochem. J. 381, 137–146.
Urich, T., Gomes, C. M., Kletzin, A., and Frazao, C. (2006). X-ray Structure of a self-compartmentalizing sulfur cycle metalloenzyme. Science 311, 996–1000.
Wakai, S., Tsujita, M., Kikumoto, M., Manchur, M. A., Kanao, T., and Kamimura, K. (2007). Purification and characterization of sulfide:quinone oxidoreductase from an acidophilic iron-oxidizing bacterium, Acidithiobacillus ferrooxidans. Biosci. Biotechnol. Biochem. 71, 2735–2742.
Valdes, J., Pedroso, I., Quatrini, R., Dodson, R. J., Tettelin, H., Blake, R., Eisen, J. A., and Holmes, D. S. (2008). Acidithiobacillus ferrooxidans metabolism: from genome sequence to industrial applications. BMC Genomics 9, 597. doi: 10.1186/1471-2164-9-597
Valdes, J., Quatrini, R., Hallberg, K., Dopson, M., Valenzuela, P. D., and Holmes, D. S. (2009). Draft genome sequence of the extremely acidophilic bacterium Acidithiobacillus caldus ATCC 51756 reveals metabolic versatility in the genus Acidithiobacillus. J. Bacteriol. 191, 5877–5878.
Valenzuela, L., Chi, A., Beard, S., Shabanowitz, J., Hunt, D. F., and Jerez, C. A. (2008). “Differential-expression proteomics for the study of sulfur metabolism in the chemolithoautotrophic Acidithiobacillus ferrooxidans,” in Microbial Sulfur Metabolism, eds C. Dahl and C. G. Friedrich (Berlin: Springer), 77–86.
Vilain, S., Cosette, P., Hubert, M., Lange, C., Junter, G. A., and Jouenne, T. (2004a). Comparative proteomic analysis of planktonic and immobilized Pseudomonas aeruginosa cells: a multivariate statistical approach. Anal. Biochem. 329, 120–130.
Vilain, S., Cosette, P., Zimmerlin, I., Dupont, J. P., Junter, G. A., and Jouenne, T. (2004b). Biofilm proteome: homogeneity or versatility? J. Proteome Res. 3, 132–136.
Keywords: Acidithiobacillus caldus, elemental sulfur, inorganic sulfur compounds, metabolism, attachment, proteomics
Citation: Mangold S, Valdés J, Holmes DS and Dopson M (2011) Sulfur metabolism in the extreme acidophile Acidithiobacillus caldus. Front. Microbio. 2:17. doi: 10.3389/fmicb.2011.00017
Received: 02 November 2010; Accepted: 25 January 2011; Published online: 10 February 2011.
Edited by:
Thomas E. Hanson, University of Delaware, USAReviewed by:
Kathleen Scott, University of South Florida, USADimitry Y. Sorokin, Delft University of Technology, Netherlands
Copyright: © 2011 Mangold, Valdés, Holmes and Dopson. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Stefanie Mangold, Department of Molecular Biology, Umeå University, SE-901 87 Umeå, Sweden. e-mail:c3RlZmFuaWUubWFuZ29sZEBtaW9sYmlvbC51bXUuc2U=