 Dalia H. A. Abdelaziz1,2
Dalia H. A. Abdelaziz1,2 Mikhail A. Gavrilin1
Mikhail A. Gavrilin1 Anwari Akhter1 Kyle Caution1 Sheetal Kotrange1 Arwa Abu Khweek1 Basant A. Abdulrahman1,2 Zeinab A. Hassan2 Fathia Z. El-Sharkawi2 Simranjit S. Bedi1 Katherine Ladner3 M. Elba Gonzalez-Mejia4 Andrea I. Doseff4 Mahmoud Mostafa1
Anwari Akhter1 Kyle Caution1 Sheetal Kotrange1 Arwa Abu Khweek1 Basant A. Abdulrahman1,2 Zeinab A. Hassan2 Fathia Z. El-Sharkawi2 Simranjit S. Bedi1 Katherine Ladner3 M. Elba Gonzalez-Mejia4 Andrea I. Doseff4 Mahmoud Mostafa1 Thirumala-Devi Kanneganti5 Dennis Guttridge3 Clay B. Marsh1 Mark D. Wewers1
Thirumala-Devi Kanneganti5 Dennis Guttridge3 Clay B. Marsh1 Mark D. Wewers1 Amal O. Amer1*
Amal O. Amer1*
- 1 Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine, Center for Microbial Interface Biology and the Department of Internal Medicine, Ohio State University, Columbus, OH, USA
- 2 Faculty of Pharmacy, Department of Biochemistry and Molecular Biology, Helwan University, Helwan, Egypt
- 3 Human Cancer Genetics Program, Ohio State University, Columbus, OH, USA
- 4 Department of Molecular Genetics, Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH, USA
- 5 Department of Immunology, St Jude Children’s Research Hospital, Memphis, TN, USA
The apoptosis-associated speck-like protein containing a caspase recruitment domain (Asc) is an adaptor molecule that mediates inflammatory and apoptotic signals. Legionella pneumophila is an intracellular bacterium and the causative agent of Legionnaire’s pneumonia. L. pneumophila is able to cause pneumonia in immuno-compromised humans but not in most inbred mice. Murine macrophages that lack the ability to activate caspase-1, such as caspase-1−/− and Nlrc4−/− allow L. pneumophila infection. This permissiveness is attributed mainly to the lack of active caspase-1 and the absence of its down stream substrates such as caspase-7. However, the role of Asc in control of L. pneumophila infection in mice is unclear. Here we show that caspase-1 is moderately activated in Asc−/− macrophages and that this limited activation is required and sufficient to restrict L. pneumophila growth. Moreover, Asc-independent activation of caspase-1 requires bacterial flagellin and is mainly detected in cellular extracts but not in culture supernatants. We also demonstrate that the depletion of Asc from permissive macrophages enhances bacterial growth by promoting L. pneumophila-mediated activation of the NF-κB pathway and decreasing caspase-3 activation. Taken together, our data demonstrate that L. pneumophila infection in murine macrophages is controlled by several mechanisms: Asc-independent activation of caspase-1 and Asc-dependent regulation of NF-κB and caspase-3 activation.
Introduction
The apoptosis-associated speck-like protein containing a caspase recruitment domain (Asc encoded by the Pycard gene) is an adaptor molecule that mediates inflammatory and apoptotic signals and is predominantly expressed in monocytes and mucosal epithelial cells (Taniguchi and Sagara, 2007; Hasegawa et al., 2009). Asc contains an N-terminal pyrin/PAAD (PYD) death domain and a C-terminal CARD protein–protein interaction domain (CARD; Masumoto et al., 1999, 2001; Liepinsh et al., 2003; Stehlik et al., 2003). Both domains enable Asc to recruit other PYD and CARD-containing proteins through homotypic protein–protein interactions (Fernandes-Alnemri et al., 2007; Mariathasan, 2007). Proteins with pyrin and/or caspase recruitment domains have roles in inflammation, apoptosis, and innate immunity. Many pyrin domain proteins, such as Asc, modulate NF-κB activity. Asc also participates in the assembly of multiprotein complexes called “inflammasomes” (Srinivasula et al., 2002; Fernandes-Alnemri et al., 2007; Fernandes-Alnemri and Alnemri, 2008). Within the inflammasome, Asc is able to link caspase-1 to NOD-like receptors (NLRs) via its CARD domain, leading to the activation of caspase-1 (Srinivasula et al., 2002; Fernandes-Alnemri et al., 2007; Fernandes-Alnemri and Alnemri, 2008). NLRs act as intracellular sensors to stress, and foreign molecules like microbial components (Kanneganti et al., 2007; Martinon and Tschopp, 2007). Once activated, caspase-1 subsequently cleaves pro-IL-1β/IL-18 and accompanies the active cytokines when secreted out of the cell (Stehlik et al., 2002, 2003; Liepinsh et al., 2003; Hasegawa et al., 2005, 2007, 2009; Sarkar et al., 2006; Bedoya et al., 2007). Caspase-1 also activates caspase-7 in response to flagellin or LPS (Franchi et al., 2008; Lamkanfi et al., 2008, 2009; Akhter et al., 2009; Lamkanfi and Kanneganti, 2010; Shaw et al., 2010). Many Gram-negative bacteria, such as Salmonella typhimurium, Pseudomonas aeruginosa, Shigella flexneri, and Legionella pneumophila, are recognized in murine macrophages by the NLR Nlrc4/Ipaf leading to caspase-1 activation through the inflammasome (Abdelaziz et al., 2010; Amer, 2010).
Furthermore, when Asc is over expressed, it cooperates with Nlrp3 (cryopyrin) and Nlrp12 (Pypaf7) in order to promote NF-κB activity in an over expression system (Masumoto et al., 2003; Hasegawa et al., 2005). However, other reports show that Asc is a suppressor of NF-κB activity and that it uses its CARD interaction not only to induce caspase-1 activation, but also to down regulate NF-κB signaling (Sarkar et al., 2006; Bedoya et al., 2007). Additionally, other studies demonstrate that Asc can be either an inducer or an inhibitor of NF-κB depending on expression level and location (Stehlik et al., 2002; Yu et al., 2006). Thus, Asc regulates the inflammasome and other signaling complexes depending on the stoichiometry of its expression and also whether certain other PYD family proteins are expressed upon activation. On the other hand, Asc mediates apoptosis by serving as an adaptor molecule for Bax and regulates a p53-Bax mitochondrial pathway of apoptosis resulting in the activation of caspase-2 and -3 (Ohtsuka et al., 2004; Hasegawa et al., 2007; Fernandes-Alnemri and Alnemri, 2008; Motani et al., 2010).
Legionella pneumophila is an intracellular bacterium and the causative agent of Legionnaire’s pneumonia (Horwitz and Silverstein, 1981, 1983; Nash et al., 1984). The ability of L. pneumophila to cause pneumonia in humans is dependent on its capability to evade the immune system and multiply within human monocytes and derived macrophages (Horwitz and Silverstein, 1981, 1983; Nash et al., 1984). In murine macrophages, L. pneumophila activates the Nlrc4 inflammasome leading to the production of active caspase-1 and IL-1β (Amer et al., 2006; Ren et al., 2006; Zamboni et al., 2006; Coers et al., 2007; Lightfield et al., 2008; Akhter et al., 2009; Abdelaziz et al., 2010; Amer, 2010; Vance, 2010). Then, Naip5 mediates caspase-7 activation downstream of caspase-1 which restricts the intracellular survival of the organism (Akhter et al., 2009; Abdelaziz et al., 2010). Therefore, mice and their derived macrophages lacking Nlrc4, caspase-1, or caspase-7 allow L. pneumophila growth and are ideal models to study L. pneumophila pathogenesis (Amer et al., 2006; Ren et al., 2006; Zamboni et al., 2006; Coers et al., 2007; Lightfield et al., 2008; Akhter et al., 2009; Abdelaziz et al., 2010; Amer, 2010; Vance, 2010).
Here we demonstrate that murine macrophages control L. pneumophila infection through an Asc-dependent mechanism by decreasing NF-κB activation and an Asc-independent mechanism by modestly activating caspase-1 in the cytosol. Together, our data show that Asc controls L. pneumophila infection in the absence of caspase-1.
Results
Asc Controls l. Pneumophila Infection in Murine Macrophages Lacking Caspase-1
WT mouse macrophages effectively activate and release caspase-1 in response to L. pneumophila infection. This is accompanied by the fusion of the L. pneumophila-containing phagosome with the lysosome, bacterial degradation, and elimination (Amer et al., 2006; Akhter et al., 2009). Consistent with that, caspase-1 knockout (−/−) macrophages are permissive to L. pneumophila replication. Asc is an adaptor molecule involved in caspase-1 activation in response to a variety of agents. However, its role during L. pneumophila infection is not well established. To characterize the role of Asc during L. pneumophila infection, Asc was down regulated in both caspase-1−/− (Figure 1A) and WT macrophages (Figure 1B) using Asc specific siRNA. After transfection, Asc protein level declined in both sets of cells, while other components of the inflammasome, such as Nlrc4 and pro-caspase-1, were not affected (Figures 1A,B). After depletion of Asc, cells were infected with L. pneumophila and the bacterial replication was assessed by counting colony-forming units (CFU). Depletion of Asc in caspase-1−/− cells supported significantly more L. pneumophila replication compared to caspase-1−/− cells treated with control siRNA (Figure 1C). In WT mouse macrophages, depletion of Asc did not have an effect on the bacterial growth compared to that of cells treated with control siRNA or untreated cells (Figure 1D). Therefore, Asc controls L. pneumophila replication in murine macrophages in the absence of caspase-1.
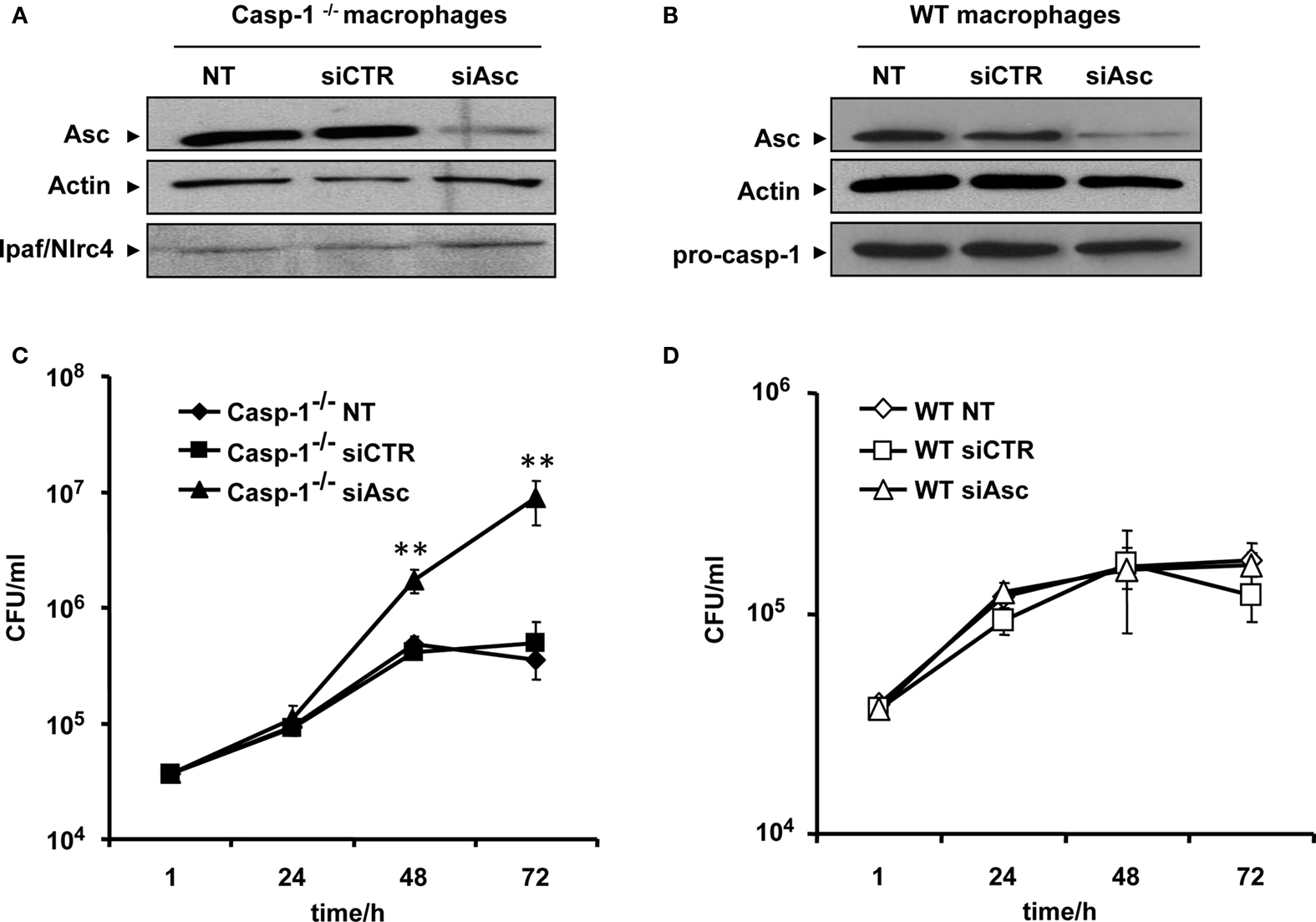
Figure 1. Asc controls Legionella pneumophila replication in caspase-1−/− macrophages. (A) Caspase-1−/− (casp-1−/−) macrophages and (B) WT macrophages were either left untreated (NT) or transfected with Asc specific siRNA (siAsc) or control siRNA (siCTR). After 48 h transfection Asc, levels were assessed using Western blotting. Actin was used as a loading control. Casp-1−/− macrophages (C) and wild type macrophages (D) were transfected or not (NT) with siAsc or siCTR and 48 h after transfection cells were infected with L. pneumophila (Leg) at MOI = 0.1 and the bacterial replication was assessed by counting the CFU after 1, 24, 48, and 72 h. Results are displayed as mean ± SD of three independent wells. **P ≤ 0.01. The data shown in (C,D) are representative of four independent experiments showing the same results.
Asc Reduces NF-κB Activation Induced by L. pneumophila Infection
It has been shown by several reports that L. pneumophila triggers NF-κB activation in TLR5 dependent and independent manner depending on the stage of the infection (Bartfeld et al., 2009; Losick et al., 2010). Nevertheless, the role of Asc in NF-κB modulation depends on its protein levels and its location within the cells (Stehlik et al., 2002; Sarkar et al., 2006; Yu et al., 2006; Bedoya et al., 2007). Our results showed that Asc controls L. pneumophila replication in a caspase-1-independent manner. To determine if the mechanism by which Asc restricts L. pneumophila (independently of caspase-1) involves the NF-κB pathway, Asc was depleted in caspase-1−/− macrophages by siRNA against Asc. Then, cells were infected with L. pneumophila and NF-κB activation was assessed in nuclear extracts by electrophoretic mobility shift assay (EMSA). We found that caspase-1−/− macrophages depleted for Asc allowed more NF-κB activation at 1, 4, and 8 h of infection compared with cells treated with control siRNA (Figure 2A). These results suggest that Asc decreased NF-κB during L. pneumophila infection. To confirm these findings, Asc−/− and WT macrophages were infected with L. pneumophila and EMSA assay was performed. Initially, WT and Asc−/− macrophages showed NF-κB activation within 1 h after infection. This activation declined in WT macrophages by 8 h infection whereas NF-κB pathway remained activated in Asc−/− 8 h after L. pneumophila infection (Figure 2B). Thus, in the context of L. pneumophila infection, Asc hinders NF-κB activation, and decreases cell survival.
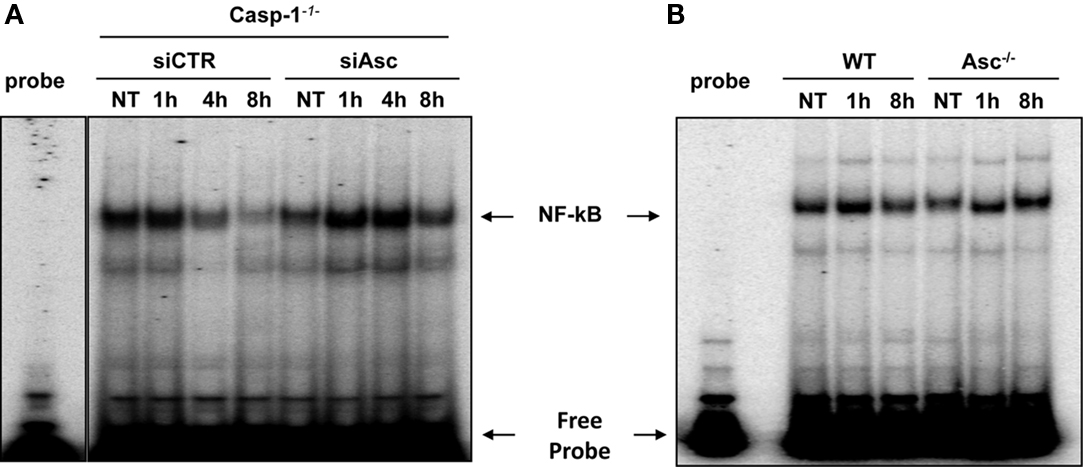
Figure 2. Asc hinders the activation of NF-κB induced by L. pneumophila. (A) Caspase-1−/− (casp-1-/-) macrophages were transfected with Asc specific siRNA (siAsc) or control siRNA (siCTR) and 48 h after transfection cells were infected or not (NT) with L. pneumophila (Leg) at MOI = 0.5 for 1, 4, and 8 h. Afterward, NF-κB activation was examined using electrophoretic mobility shift assay (EMSA). (B) WT and Asc−/− mouse macrophages were infected or not (NT) with L. pneumophila (Leg) at MOI = 0.5 for 1 and 8 h. Subsequently, NF-κB activation was examined using EMSA.
Caspase-1 is Activated in the Cytosol of Murine Macrophages Lacking Asc
The involvement of Asc in Nlrc4 inflammasome is still unclear (Zamboni et al., 2006). To discern the role of Asc in L. pneumophila-mediated caspase-1 activation, Asc−/− macrophages were left untreated or infected with L. pneumophila at a low multiplicity of infection (MOI). Then, cleaved caspase-1 was examined in cellular extracts and culture supernatants of Asc−/− macrophages and of their WT counterparts. Notably, cells lacking Asc still allowed the cleavage of caspase-1 within their cytosols when infected with L. pneumophila (Figure 3A). The amount of cleaved caspase-1 in Asc−/− cell lysates was less than that of WT macrophages (Figure 3A). Cleaved caspase-1 was detected in culture supernatants of infected WT macrophages but not that of infected Asc−/− macrophages (Figure 3B). This data indicates that in the absence of Asc, a fraction of caspase-1 is cleaved by a yet unknown mechanism.
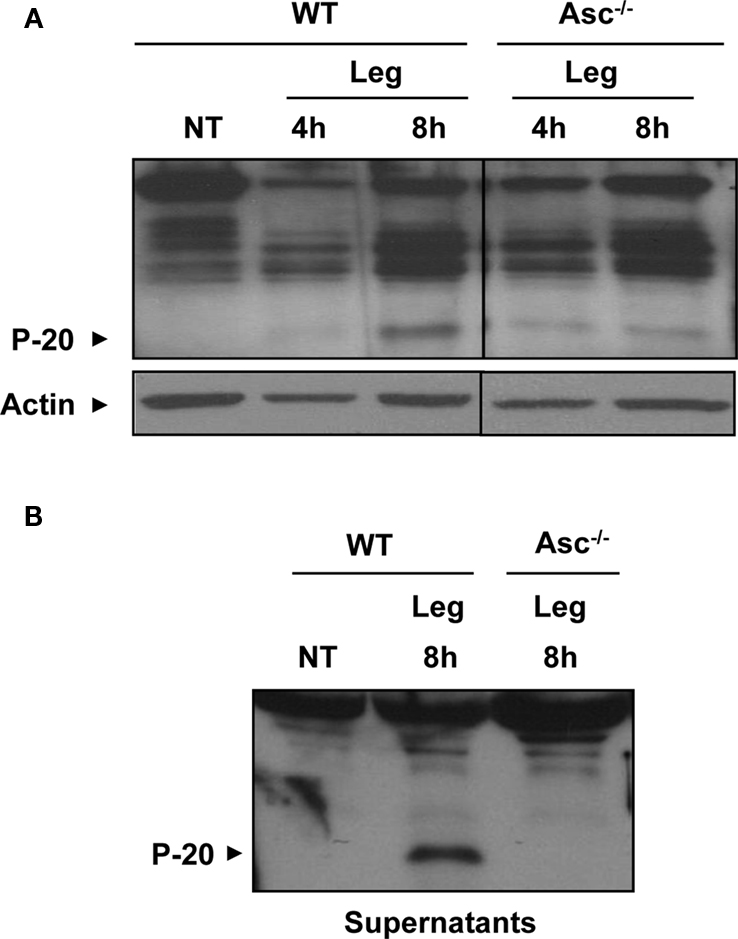
Figure 3. A fraction of caspase-1 is activated in the cytosol of murine macrophages lacking Asc. (A) WT and Asc−/− mouse macrophages were either not treated (NT) or infected with L. pneumophila (Leg) at MOI = 0.5 for 4 and 8 h. Then active caspase-1 (p-20) was detected in the cell extracts (A) and the supernatants (B) by Western blotting. Actin was used as a loading control.
To further investigate the role of Asc in caspase-1 activation during L. pneumophila of WT macrophages, Asc was depleted in WT murine macrophages, as in Figure 1B and then infected with L. pneumophila. The amount of cleaved caspase-1 detected in extracts of cells treated with Asc specific siRNA was less than that in cells treated with control siRNA (Figure 4A). Accordingly, the release of active caspase-1 from siAsc treated cells was diminished (Figure 4B). In support of this data, total cleaved caspase-1 in combined cell extracts and culture supernatants was less in Asc heterozygote macrophages than that of WT ones (Figure A1 of Appendix). Furthermore, IL-1β release from macrophages was impeded when Asc was depleted (Figure 4C). Therefore, Asc contributes to the activation of a portion of pro-caspase-1, while the rest of caspase-1 pro-form is cleaved independently of Asc.
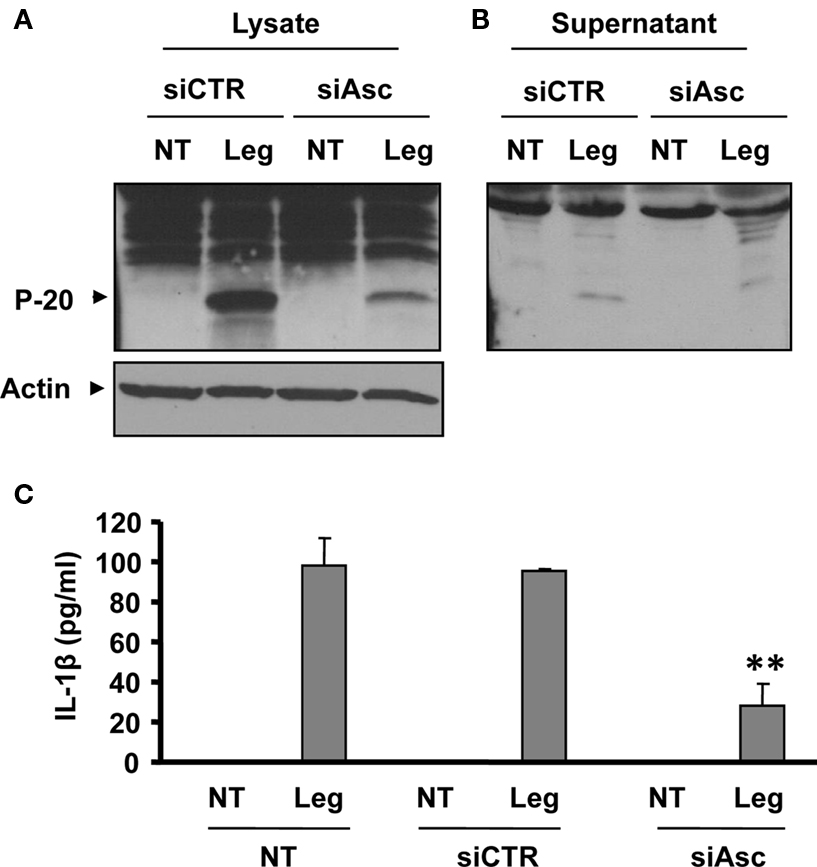
Figure 4. Depletion of Asc decreases caspase-1 activation in response to L. pneumophila infection. (A,B,C) WT mouse macrophages were transfected or not (NT) with Asc specific siRNA (siAsc) or control siRNA (siCtr) and 48 h after transfection cells were infected with L. pneumophila (Leg). Then, active caspase-1 (p-20) was detected in the cell extracts (A) and in the supernatants (B) of 8 h infected samples by Western blotting. Active IL-1β was detected in supernatants using ELISA (C). The results displayed as mean ± SD (**P ≤ 0.01) of three independent wells.
Asc-Independent Activation of Caspase-1 within Macrophage Cytosols Requires Flagellin and Contributes to the Restriction of L. pneumophila Infection
The activation of caspase-1 requires flagellin, thereby promoting the restriction of L. pneumophila infection (Amer et al., 2006; Ren et al., 2006; Zamboni et al., 2006; Lamkanfi et al., 2007; Akhter et al., 2009; Case et al., 2009; Amer, 2010; Kang et al., 2010). Thus, we next examined the growth of L. pneumophila in WT, caspase-1−/−, and Asc−/− macrophages. As previously reported by our group and by others, caspase-1−/− macrophages were permissive, whereas WT and Asc−/− macrophages restricted L. pneumophila replication (Amer et al., 2006; Ren et al., 2006; Zamboni et al., 2006; Lamkanfi et al., 2007; Akhter et al., 2009; Case et al., 2009; Amer, 2010; Kang et al., 2010). However, our data demonstrate that caspase-1 is cleaved in the cytosol of Asc−/− macrophages, therefore, we examined if this cytosolic cleavage required flagellin and if it is sufficient for restriction of L. pneumophila infection. WT macrophages were infected with the L. pneumophila mutant lacking flagellin (Fla), then, caspase-1 activation in cell extracts was examined by Western blots. Figure 5 shows that the Fla mutant did not activate caspase-1 within the cytosols of macrophages (Figure 5A). Because caspase-1 activation requires flagellin and is accompanied with restriction to L. pneumophila infection, we examined the growth of L. pneumophila mutants lacking flagellin (Fla) in macrophages lacking Asc in comparison to WT cells, since both cells lack caspase-1 activation in response to Fla (data not shown). Specifically, Figure 5B demonstrates that the growth of Fla mutants in Asc−/− macrophages exceeds that exhibited in WT cells. Yet, NF-κB activation during Fla was similar in WT and Asc−/− macrophages (Figure 5C). Therefore, there must be another pathway mediated by Asc that maintains the replication of Fla mutant under control in WT macrophages and is missing in Asc−/− macrophages. To answer this question, we next determined the levels of LDH release in culture supernatants of WT and Asc−/− macrophages infected with L. pneumophila or the Fla mutant. We found that the absence of Asc allows more host cell survival during Fla infection. To further understand the mechanism by which Asc modulates cell survival, we examined the activation of caspase-3, a caspase involved in apoptosis and know to be activated during L. pneumophila infection (Molmeret et al., 2004). First, we tested if the Fla mutant activates caspase-3 in wild type macrophages. Until 4 h post infection only WT L. pneumophila activated caspase-3 (Figure 6A). However, at 5 h post infection, the Fla mutant activated caspase-3 in WT macrophages but not in Asc−/− macrophages (Figure 6B). Taken together, these data suggest that Asc is involved in caspase-3 activation which mediates cell death in WT macrophages at later stages of Fla infection.
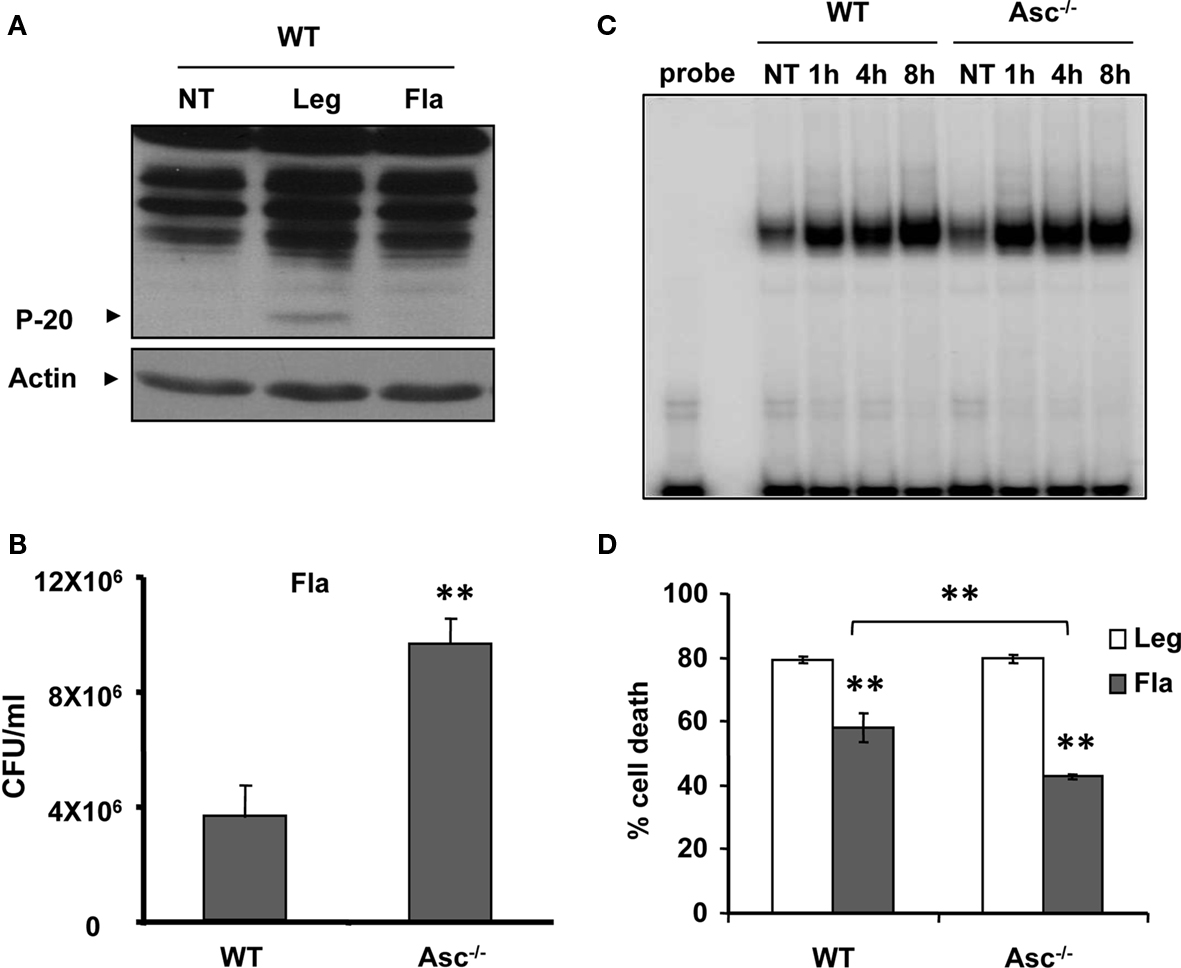
Figure 5. Asc-/- macrophages are permissive to flagellin mutant. (A) WT macrophages were infected with L. pneumophila (Leg), with corresponding flagellin mutant (Fla), or left untreated (NT). Active caspase-1 (p-20) was then detected in the cell extracts. (B) WT and Asc-/- macrophages were infected with Fla mutant and CFUs were scored 72 h after infection. (C) WT and Asc-/- macrophages were infected with Fla mutant for 1, 4, and 8 h then, nuclear extracts were processed for determination of NF-κB activation using electrophoretic mobility shift assay (EMSA). (D) WT and Asc macrophages were infected with Leg or Fla for 24 h then LDH release was determined and presented as percent cell death on the Y axis.
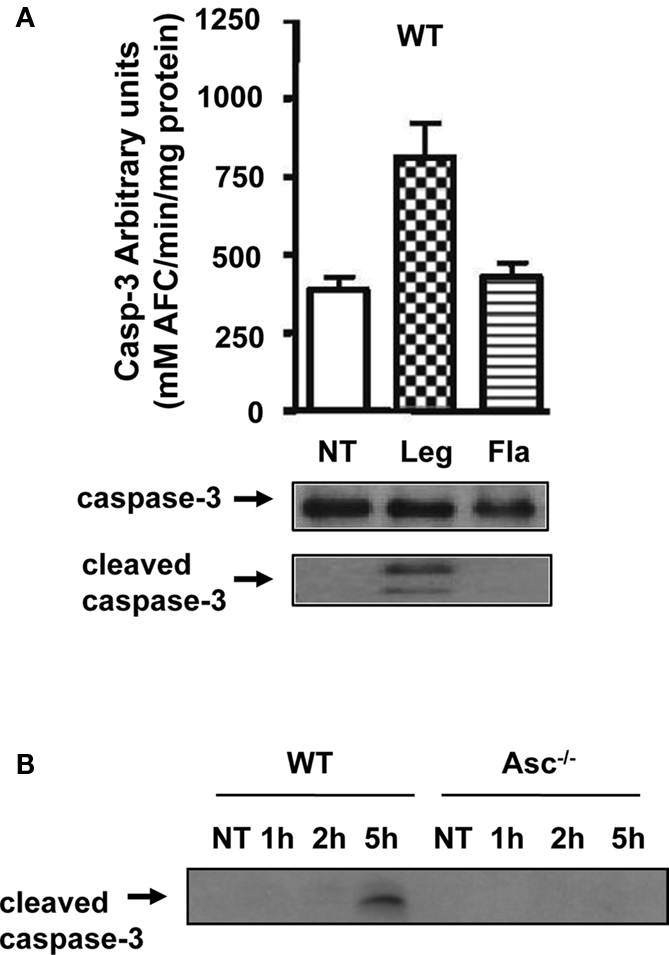
Figure 6. (A) Lysates from WT macrophages infected with L. pneumophila (Leg), the Fla mutant, or the type IV secretion mutant DotA were used to determine caspase-3 activity by the DEVD-AFC assay, or inactive full-length (FL-casp-3) and cleaved caspase-3 (cleaved casp-3) by immunoblotting. (B) WT and Asc−/− macrophages were infected with Fla mutant for 1, 2, and 5 h then cell lysates were obtained and used to determine cleaved caspase-3 by Western Blotting.
Legionella pneumophila Modulates the Expression of Nlrc4 Inflammasome Components in Wild Type Mouse Macrophages
In WT murine macrophages L. pneumophila flagellin is detected by Nlrc4 with subsequent activation of caspase-1 (Amer et al., 2006; Ren et al., 2006; Zamboni et al., 2006; Lamkanfi et al., 2007; Akhter et al., 2009; Case et al., 2009; Amer, 2010; Kang et al., 2010). This response is completely lacking in human phagocytes. In contrast to murine phagocytes, human phagocytes which are permissive to L. pneumophila do not activate caspase-1 in response to the pathogen (Abdelaziz et al., 2011). This lack of activation is due to the down regulation of ASC in human cells upon infection with L. pneumophila (Abdelaziz et al., 2011). To characterize the effect of L. pneumophila on the expression of the Nlrc4 inflammasome components in the mouse, murine WT macrophages were infected with L. pneumophila and the expression of caspase-1, IL-1β, Nlrc4, and Asc was assessed on both mRNA and protein levels. The expression of both caspase-1 and IL-1β proteins were induced significantly within 4 h after infection and remained up regulated for 24 h of infection (Figures A2A,B of Appendix). Subsequently, both caspase-1 and IL-1β were activated early upon infection (Figures A2A,B lower panel of Appendix) and their active forms were released into the supernatant (Data not shown). Remarkably, in contrast to mouse macrophages, the expression of Asc was enhanced 24 h after infection (Figure A2C). As for Nlrc4, mRNA, and protein levels were decreased later in infection (Figure A2D lower panel of Appendix). We found that this decrease in Nlrc4 protein levels is due to its release with the rest of the inflammasome components into the media (data not shown). Therefore, L. pneumophila differentially modulates the expression of Asc in murine and in human phagocytes.
Discussion
Asc is an adaptor molecule necessary for the assembly and activation of several inflammasome complexes in response to stress signals and microbial molecules (Taniguchi and Sagara, 2007; Hasegawa et al., 2009). Asc is required for caspase-1 activation downstream of Nlrc4/Ipaf during Salmonella or Shigella infections, yet its role during L. pneumophila infection has been controversial (Zamboni et al., 2006; Akhter et al., 2009; Case et al., 2009). In this study, we demonstrate that a fraction of caspase-1 is activated within the cytosol of infected macrophages independently of Asc. Data from our group and others indicate that restriction of L. pneumophila infection is mediated by several mechanisms. This can occur either via caspase-1-dependent or independent mechanisms. Moreover, caspase-1 dependent control of L. pneumophila infection can be either Asc-dependent or independent.
First, caspase-1-dependent restriction of L. pneumophila is by modulation of pyroptosis and phagosome–lysosome fusion. Several studies have demonstrated that at high MOIs with L. pneumophila, the activation of caspase-1 leads to pyroptotic cell death, which contributes to resistance to infection (Zamboni et al., 2006; Case et al., 2009). Other reports have shown that during extreme MOIs of L. pneumophila, murine caspase-1 is activated independently of Nlrc4 (Case et al., 2009). This is at odds with other data that clearly demonstrates that the activation of caspase-1 during physiological levels of infection requires Nlrc4 (Amer et al., 2006; Ren et al., 2006; Zamboni et al., 2006; Coers et al., 2007; Lightfield et al., 2008; Akhter et al., 2009; Abdelaziz et al., 2010; Amer, 2010; Vance, 2010). It seems that these discrepancies are largely due to different infection doses. Consequently, it is plausible to suggest that the activation of caspase-1 by high bacterial burdens overrides the signaling pathways controlling their activation at physiological levels of infection. Yet, our data suggest that Asc is required for the recruitment and cleavage of at least a part of pro-caspase-1 protein into its active form since the amount of cleaved p20 detected in the cell extracts of Asc−/+ macrophages was much less than that of WT macrophages.
Secondly, macrophages deploy an Asc-dependent restriction mechanism to control L. pneumophila infection by controlling NF-κB pathway. Dixit and colleagues demonstrated that macrophages from Asc−/− mice are markedly resistant to Salmonella typhimurium induced cell death (Mariathasan et al., 2004). However, in their study, cell death was mainly attributed to caspase-1 mediated pyroptosis while other pathways such as NF-κB activation were not examined. Several reports by Isberg and Abu Kwaik showed that L. pneumophila activates NF-κB in two phases to extend the host cell survival to permit intracellular growth (Losick and Isberg, 2006; Abu-Zant et al., 2007; Bartfeld et al., 2009). They clearly demonstrated that NF-κB activation is absolutely required for L. pneumophila growth within macrophages. L. pneumophila promotes two phases of NF-κB activation, a TLR5-dependent and a TLR5-independent activation at an early and later stages of infection respectively (Losick and Isberg, 2006; Bartfeld et al., 2009). The role of Asc in L. pneumophila-induced NF-κB activation was not explored previously. A number of studies suggest that Asc promotes NF-κB activation (Chamaillard et al., 2003; Masumoto et al., 2003; Hasegawa et al., 2005). Conversely, several studies by Wewers and Reed showed that THP-1 cells treated with small interfering RNA for human ASC decreased their caspase-1 activity while enhancing their NF-κB signal (Stehlik et al., 2002; Sarkar et al., 2006; Yu et al., 2006). The later studies showed that the interaction of caspase-1 and RIP2 mediates NF-κB activation which is prevented by human ASC as it hinders their interaction. Here, we report that mouse Asc deters the activation of NF-κB during L. pneumophila infection since the depletion of Asc allowed more NF-κB activation and additional L. pneumophila growth.
Importantly, murine Asc is induced in response to L. pneumophila infection (Figure A2). This is in stark contrast to human ASC, which is down regulated in human monocytes upon infection with L. pneumophila (Abdelaziz et al., 2011). The down regulation of human ASC contributed to the permissiveness to L. pneumophila growth. These findings are among the first reports clarifying the mechanism of permissiveness of human monocytes to L. pneumophila (Abdelaziz et al., 2011). Thus, regulating Asc availability in human phagocytes could be a mechanism employed by L. pneumophila to modulate caspase-1, NF-κB, and pyroptosis.
The detection of cleaved caspase-1 in supernatants of cultured cells has been widely used as the hallmark for caspase-1 activation. Few studies have shown that Nlrc4/Ipaf−/− and Asc−/− macrophages fail to produce cleaved caspase-1 in response to L. pneumophila. The absence of active caspase-1 promoted L. pneumophila growth. Yet, it is unclear as to why Asc−/− macrophages do not allow L. pneumophila replication. Here we show that in Asc−/− murine macrophages, a small portion of caspase-1 is cleaved during L. pneumophila infection but is not detected in culture supernatants. However, we cannot exclude that cleaved caspase-1 was released in amounts below our detection threshold. Our data agree with recent work by Monack group although the interpretation differs. They suggested that caspase-1 can be active without being cleaved and independently of Asc as suggested by the absence of cleaved caspase-1 in culture supernatants (Broz et al., 2010). However, it is possible that caspase-1 was modestly cleaved within the cytosol (which was not examined) and not released in supernatants. Irrespective of its release, the active cytosolic caspase-1 is required and sufficient to restrict L. pneumophila growth in Asc−/− macrophages.
On the other hand, the L. pneumophila mutant lacking flagellin did not activate cytosolic caspase-1 in macrophages and replicated efficiently. These findings support the idea that cytosolic cleavage of caspase-1 is sufficient to restrict L. pneumophila growth within macrophages and requires flagellin. The flagellin mutant replicated more effectively in Asc−/− macrophages than in WT ones although both cells lacked cytosolic caspase-1 activation and activated NF-κB at comparable levels. However, Asc−/− macrophages survive more that WT ones allowing more time for Fla replication. This was because caspase-3 was activated in WT macrophages during late stages of Fla infection but not in Asc−/− macrophages. It is possible that L. pneumophila activates caspase-3 directly or through the Bax/Bak pathway which is governed by Asc (Abu-Zant et al., 2005; Fischer et al., 2006). Taken together, these data suggest that Asc is involved in caspase-3 activation and apoptosis induction during L. pneumophila infection. Altogether, our data demonstrate that the host employs more than one mechanism to prevent L. pneumophila infection and that many of these are governed by Asc.
Materials and Methods
Mice and Macrophages
Wild type C57BL/6 (B6) and Asc−/− mice were previously described (Mariathasan et al., 2004). Caspase-1−/− mice were from Dr. Amy Hise at Case Western University. All knockout mice were in a C57BL/6 background. Bone marrow-derived macrophages (BMDMs) were isolated from femurs of 6- to 12-week-old mice and were cultured in IMDM containing 10% heat-inactivated FBS, 20% L cell-conditioned medium, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2. After 5 days of incubation, cells were collected and plated in 6-well plates or in 24-well plates in IMDM containing 10% heat-inactivated FBS (Stanley, 1997; Amer et al., 2005, 2006; Akhter et al., 2009).
Bacterial Strains
Legionella pneumophila strain Lp02, is a thymine auxotrophic derivative of Philadelphia-1 (Brenner et al., 1979; McDade and Shepard, 1979). L. pneumophila flagellin (Fla) mutant was previously described (Albert-Weissenberger et al., 2010). L. pneumophila was cultured as described previously (Sturgill-Koszycki and Swanson, 2000; Akhter et al., 2009) All experiments were performed at a low MOI of 0.5, followed by centrifugation and rinsing of the wells after 30 min except when otherwise indicated (Derre and Isberg, 2004).
Intracellular Growth of L. pneumophila
All experiments were performed at an MOI ranging 0.1–1, followed by centrifugation and rinsing of the wells after 30 min except when otherwise indicated (Derre and Isberg, 2004). All experiments were performed in the absence of ferric nitrate and L-cysteine from the monocytes or macrophage culture medium, to allow L. pneumophila multiplication only intracellularly. At designated time points, macrophages were lysed and plated on AYE plates for CFUs (Amer et al., 2006; Abdelaziz et al., 2010). The quantification of the CFU in vitro was performed more than four independent times as described (Amer et al., 2006; Abdelaziz et al., 2010).
Immunoblotting
Cell extracts of macrophages were prepared and immunoblotted with an antibody that recognizes Nlrc4, Asc (Alexis Biochemicals), caspase-1 (Santa Cruz), IL-1β (National Cancer Institute), caspase-3 (Cell Signalling), actin (Abcam), followed by appropriate secondary antibody as described (Amer et al., 2006; Abdelaziz et al., 2010).
NF-κB DNA Binding Activity Assay
Nuclear extracts of L. pneumophila treated or untreated BMDMs from WT and Asc−/− mice were prepared as previously described (Akhter et al., 2009). EMSA was used to measure NF-κB DNA binding activity as described (Sarkar et al., 2006).
Quantitative PCR
Total RNA was extracted from cells lysed in Trizol (Invitrogen Life Technologies) and 1–2 μg of the RNA was converted to cDNA by ThermoScript RNase H− Reverse Transcriptase (Invitrogen, Life Technologies). 20–60 ng of the converted cDNA was then used for quantitative PCR with SYBR Green I PCR Master Mix using the StepOne Plus Real Time PCR System (Applied Biosystems). The target gene Ct values were normalized to the Ct values of two housekeeping genes (mouse GAPDH and CAP-1) and expressed as relative copy number (RCN), as described earlier (Gavrilin et al., 2006; Zakharova et al., 2010). Primers used in RT-PCR are presented in Table A1 of Appendix. We also evaluated expression of an around 600 genes with Open Array Mouse Inflammatory Panel (BioTrove, Life Technologies). All individual Ct values were normalized to the average of 18 housekeeping genes used in this array, and also expressed as RCN.
Caspase-3 Activity Assay
Active caspase-3 was determined by the AFC assay, as previously described (Gonzalez-Mejia et al., 2010). Lysates were incubated in a cyto-buffer (10% glycerol, 50 mM Pipes, pH 7.0, 1 mM EDTA, containing 1 mM DTT) containing 20 mM of the tetrapeptide substrate DEVD-AFC. The tetrapeptide was obtained from Enzyme Systems Products (Livermore, CA, USA). Release of free AFC was determined using a Cytofluor 4000 fluorometer (Perseptive Company, Framingham, MA, USA; Filters: excitation; 400 nm, emission; 508 nm).
LDH Release Assay
LDH release into cell culture medium was used as an indicator of cell death using NAD+ reduction assay, according to the manufacturer (Roche Applied Science). Cells were plated in 12-well plate at the density 0.5 × 106, and 0.5 MOI of both wild type L. pneumophila (Leg) and its flagellin mutant (Fla) were added. Cell culture medium was collected 24 h post infection; clarified from floating bacteria by centrifugation; and used for LDH assay. To determine spontaneous cell death, referred as a negative control, we collected medium from cells incubated the same time without bacteria. To measure total LDH content in cells, referred as a positive control, cells were lysed by adding TritonX-100 (1% final concentration) to the well. Media alone was used as a blank. LDH concentration in the medium was detected at OD 490 nm. Cell death was calculated by the following formula: cytotoxicity (%) = (sample−blank/positive control−blank) × 100.
Statistical Analysis
Data are displayed as mean of three independent experiments ± SD. P value ≤0.05 was considered significant.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Studies in the Amer laboratory are supported by grants R01HL094586 and R21AI083871 from the National Institute of Health (NIH) and GRT00013604 from the American Lung Association (ALA). Grants RO1HL075040-01 and NSF-MCB-0542244 to Andrea I. Doseff. Dalia H. A. Abdelaziz and Basant A. Abdulrahman are supported by The Egyptian Cultural and Educational Bureau Fellowship.
References
Abdelaziz, D. H., Amr, K., and Amer, A. O. (2010). Nlrc4/Ipaf/CLAN/CARD12: more than a flagellin sensor. Int. J. Biochem. Cell Biol. 42, 789–791.
Abdelaziz, D. H., Gavrilin, M. A., Akhter, A., Caution, K., Kotrange, S., Khweek, A. A., Abdulrahman, B. A., Grandhi, J., Hassan, Z. A., Marsh, C., Wewers, M. D., and Amer, A. O. (2011). ASC controls Legionella pneumophila infection in human monocytes. J. Biol. Chem. 286, 3203–3208.
Abu-Zant, A., Jones, S., Asare, R., Suttles, J., Price, C., Graham, J., and Kwaik, Y. A. (2007). Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell Microbiol. 9, 246–264.
Abu-Zant, A., Santic, M., Molmeret, M., Jones, S., Helbig, J., and Abu Kwaik, Y. (2005). Incomplete activation of macrophage apoptosis during intracellular replication of Legionella pneumophila. Infect. Immun. 73, 5339–5349.
Akhter, A., Gavrilin, M. A., Frantz, L., Washington, S., Ditty, C., Limoli, D., Day, C., Sarkar, A., Newland, C., Butchar, J., Marsh, C. B., Wewers, M. D., Tridandapani, S., Kanneganti, T. D., and Amer, A. O. (2009). Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. 5, e1000361. doi: 10.1371/journal.ppat.1000361
Albert-Weissenberger, C., Sahr, T., Sismeiro, O., Hacker, J., Heuner, K., and Buchrieser, C. (2010). Control of flagellar gene regulation in Legionella pneumophila and its relation to growth phase. J. Bacteriol. 192, 446–455.
Amer, A., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Ozoren, N., Brady, G., Meshinchi, S., Jagirdar, R., Gewirtz, A., Akira, S., and Nunez, G. (2006). Regulation of Legionella phagosome maturation and infection through flagellin and host ipaf. J. Biol. Chem. 281, 35217–35223.
Amer, A. O. (2010). Modulation of caspases and their non-apoptotic functions by Legionella pneumophila. Cell. Microbiol. 12, 140–147.
Amer, A. O., Byrne, B. G., and Swanson, M. S. (2005). Macrophages rapidly transfer pathogens from lipid raft vacuoles to autophagosomes. Autophagy 1, 53–58.
Bartfeld, S., Engels, C., Bauer, B., Aurass, P., Flieger, A., Bruggemann, H., and Meyer, T. F. (2009). Temporal resolution of two-tracked NF-kappaB activation by Legionella pneumophila. Cell. Microbiol. 11, 1638–1651.
Bedoya, F., Sandler, L. L., and Harton, J. A. (2007). Pyrin-only protein 2 modulates NF-kappaB and disrupts ASC:CLR interactions. J. Immunol. 178, 3837–3845.
Brenner, D. J., Steigerwalt, A. G., and McDade, J. E. (1979). Classification of the Legionnaires’ disease bacterium: Legionella pneumophila, genus novum, species nova, of the family Legionellaceae, familia nova. Ann. Intern. Med. 90, 656–658.
Broz, P., von Moltke, J., Jones, J. W., Vance, R. E., and Monack, D. M. (2010). Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483.
Case, C. L., Shin, S., and Roy, C. R. (2009). Asc and ipaf inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect. Immun. 77, 1981–1991.
Chamaillard, M., Hashimoto, M., Horie, Y., Masumoto, J., Qiu, S., Saab, L., Ogura, Y., Kawasaki, A., Fukase, K., Kusumoto, S., Valvano, M. A., Foster, S. J., Mak, T. W., Nunez, G., and Inohara, N. (2003). An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707.
Coers, J., Vance, R. E., Fontana, M. F., and Dietrich, W. F. (2007). Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol. 9, 2344–2357.
Derre, I., and Isberg R. R. (2004). Macrophages from mice with the restrictive Lgn1 allele exhibit multifactorial resistance to Legionella pneumophila. Infect. Immun. 72, 6221–6229.
Fernandes-Alnemri, T., and Alnemri E. S. (2008). Assembly, purification, and assay of the activity of the ASC pyroptosome. Meth. Enzymol. 442, 251–270.
Fernandes-Alnemri, T., Wu, J., Yu, J. W., Datta, P., Miller, B., Jankowski, W., Rosenberg, S., Zhang, J., and Alnemri, E. S. (2007). The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604.
Fischer, S. F., Vier, J., Muller-Thomas, C., and Hacker, G. (2006). Induction of apoptosis by Legionella pneumophila in mammalian cells requires the mitochondrial pathway for caspase activation. Microbes Infect. 8, 662–669.
Franchi, L., Park, J. H., Shaw, M. H., Marina-Garcia, N., Chen, G., Kim, Y. G., and Nunez, G. (2008). Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 10, 1–8.
Gavrilin, M. A., Bouakl I. J., Knatz, N. L., Duncan, M. D., Hall, M. W., Gunn, J. S., and Wewers, M. D. (2006). Internalization and phagosome escape required for Francisella to induce human monocyte IL-1beta processing and release. Proc. Natl. Acad. Sci. U.S.A. 103, 141–146.
Gonzalez-Mejia, M. E., Voss, O. H., Murnan, E. J., and Doseff, A. I. (2010). Apigenin-induced apoptosis of leukemia cells is mediated by a bimodal and differentially regulated residue-specific phosphorylation of heat-shock protein-27. Cell Death Dis. 1, e64.
Hasegawa, M., Imamura, R., Kinoshita, T., Matsumoto, N., Masumoto, J., Inohara, N., and Suda, T. (2005). ASC-mediated NF-kappaB activation leading to interleukin-8 production requires caspase-8 and is inhibited by CLARP. J. Biol. Chem. 280, 15122–15130.
Hasegawa, M., Imamura, R., Motani, K., Nishiuchi, T., Matsumoto, N., Kinoshita, T., and Suda, T. (2009). Mechanism and repertoire of ASC-mediated gene expression. J. Immunol. 182, 7655–7662.
Hasegawa, M., Kawase, K., Inohara, N., Imamura, R., Yeh, W. C., Kinoshita, T., and Suda, T. (2007). Mechanism of ASC-mediated apoptosis: bid-dependent apoptosis in type II cells. Oncogene 26, 1748–1756.
Horwitz, M. A., and Silverstein, S. C. (1981). Interaction of the Legionnaires’ disease bacterium (Legionella pneumophila) with human phagocytes. I. L. pneumophila resists killing by polymorphonuclear leukocytes, antibody, and complement. J. Exp. Med. 153, 386–397.
Horwitz, M. A., and Silverstein, S. C. (1983). Intracellular multiplication of Legionnaires’ disease bacteria (Legionella pneumophila) in human monocytes is reversibly inhibited by erythromycin and rifampin. J. Clin. Invest. 71, 15–26.
Kang, T. J., Lee G. S., Kim, S. K., Jin, S. H., and Chae, G. T. (2010). Comparison of two mice strains, A/J and C57BL/6, in caspase-1 activity and IL-1beta secretion of macrophage to Mycobacterium leprae infection. Mediators Inflamm. 2010, 708713.
Kanneganti, T. D., Lamkanfi, M., and Nunez, G. (2007). Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559.
Lamkanfi, M., Amer, A., Kanneganti, T. D., Munoz-Planillo, R., Chen, G., Vandenabeele, P., Fortier, A., Gros, P., and Nunez, G. (2007). The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J. Immunol. 178, 8022–8027.
Lamkanfi, M., and Kanneganti, T. D. (2010). Caspase-7: a protease involved in apoptosis and inflammation. Int. J. Biochem. Cell Biol. 42, 21–24.
Lamkanfi, M., Kanneganti T. D., Van Damme, P., Vanden Berghe, T., Vanoverberghe, I., Vandekerckhove, J., Vandenabeele, P., Gevaert, K., and Nunez, G. (2008). Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell Proteomics 7, 2350–2363.
Lamkanfi, M., Moreira, L. O., Makena, P., Spierings, D. C., Boyd, K., Murray, P. J., Green, D. R., and Kanneganti, T. D. (2009). Caspase-7 deficiency protects from endotoxin-induced lymphocyte apoptosis and improves survival. Blood 113, 2742–2745.
Liepinsh, E., Barbals, R., Dahl, E., Sharipo, A., Staub, E., and Otting, G. (2003). The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J. Mol. Biol. 332, 1155–1163.
Lightfield, K. L., Persson, J., Brubaker, S. W., Witte, C. E., von Moltke, J., Dunipace, E. A., Henry, T., Sun, Y. H., Cado, D., Dietrich, W. F., Monack, D. M., Tsolis, R. M., and Vance, R. E. (2008). Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178.
Losick, V. P., Haenssler, E., Moy, M. Y., and Isberg, R. R. (2010). LnaB: a Legionella pneumophila activator of NF-kappaB. Cell. Microbiol. 12, 1083–1097.
Losick, V. P., and Isberg, R. R. (2006). NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203, 2177–2189.
Mariathasan, S. (2007). ASC, ipaf and cryopyrin/Nalp3: bona fide intracellular adapters of the caspase-1 inflammasome. Microbes Infect. 9, 664–671.
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., Roose-Girma, M., Erickson, S., and Dixit, V. M. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218.
Martinon, F., and Tschopp, J. (2007). Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 14, 10–22.
Masumoto, J., Dowds, T. A., Schaner, P., Chen, F. F., Ogura, Y., Li, M., Zhu, L., Katsuyama, T., Sagara, J., Taniguchi, S., Gumucio, D. L., Nunez, G., and Inohara, N. (2003). ASC is an activating adaptor for NF-kappa B and caspase-8-dependent apoptosis. Biochem. Biophys. Res. Commun. 303, 69–73.
Masumoto, J., Taniguchi, S., Ayukawa, K., Sarvotham, H., Kishino, T., Niikawa, N., Hidaka, E., Katsuyama, T., Higuchi, T., and Sagara, J. (1999). ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 274, 33835–33838.
Masumoto, J., Taniguchi, S., Nakayama, K., Ayukawa, K., and Sagara, J. (2001). Murine ortholog of ASC, a CARD-containing protein, self-associates and exhibits restricted distribution in developing mouse embryos. Exp. Cell Res. 262, 128–133.
McDade, J. E., and Shepard, C. C. (1979). Virulent to avirulent conversion of Legionnaires’ disease bacterium (Legionella pneumophila) – its effect on isolation techniques. J. Infect. Dis. 139, 707–711.
Molmeret, M., Zink S. D., Han, L., Abu-Zant, A., Asari, R., Bitar, D. M., and Abu Kwaik, Y. (2004). Activation of caspase-3 by the Dot/Icm virulence system is essential for arrested biogenesis of the Legionella-containing phagosome. Cell. Microbiol. 6, 33–48.
Motani, K., Kawase, K., Imamura, R., Kinoshita, T., Kushiyama, H., and Suda, T. (2010). Activation of ASC induces apoptosis or necrosis, depending on the cell type, and causes tumor eradication. Cancer Sci. 101, 1822–1827.
Nash, T. W., Libby, D. M., and Horwitz, M. A. (1984). Interaction between the legionnaires’ disease bacterium (Legionella pneumophila) and human alveolar macrophages. Influence of antibody, lymphokines, and hydrocortisone. J. Clin. Invest. 74, 771–782.
Ohtsuka, T., Ryu, H., Minamishima, Y. A., Macip, S., Sagara, J., Nakayama, K. I., Aaronson, S. A., and Lee, S. W. (2004). ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat. Cell Biol. 6, 121–128.
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F., and Vance, R. E. (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. doi: 10.1371/journal.ppat.0020018
Sarkar, A., Duncan, M., Hart, J., Hertlein, E., Guttridge, D. C., and Wewers, M. D. (2006). ASC directs NF-kappaB activation by regulating receptor interacting protein-2 (RIP2) caspase-1 interactions. J. Immunol. 176, 4979–4986.
Shaw, P. J., Lamkanfi, M., and Kanneganti, T. D. (2010). NOD-like receptor (NLR) signaling beyond the inflammasome. Eur. J. Immunol. 40, 624–627.
Srinivasula, S. M., Poyet, J. L., Razmara, M., Datta, P., Zhang, Z., and Alnemri, E. S. (2002). The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 277, 21119–21122.
Stehlik, C., Fiorentino, L., Dorfleutner, A., Bruey, J. M., Ariza, E. M., Sagara, J., and Reed, J. C. (2002). The PAAD/PYRIN-family protein ASC is a dual regulator of a conserved step in nuclear factor kappaB activation pathways. J. Exp. Med. 196, 1605–1615.
Stehlik, C., Krajewska, M., Welsh, K., Krajewski, S., Godzik, A., and Reed, J. C. (2003). The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation. Biochem. J. 373(Pt 1), 101–113.
Sturgill-Koszycki, S., and Swanson, M. S. (2000). Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J. Exp. Med. 192, 1261–1272.
Taniguchi, S., and Sagara, J. (2007). Regulatory molecules involved in inflammasome formation with special reference to a key mediator protein, ASC. Semin. Immunopathol. 29, 231–238.
Yu, J. W., Wu, J., Zhang, Z., Datta, P., Ibrahimi, I., Taniguchi, S., Sagara, J., Fernandes-Alnemri, T., and Alnemri, E. S. (2006). Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 13, 236–249.
Zakharova, E., Grandhi, J., Wewers, M. D., and Gavrilin, M. A. (2010). Mycoplasma suppression of THP-1 Cell TLR responses is corrected with antibiotics. PLoS ONE 5, e9900. doi: 10.1371/journal.pone.0009900
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., Kuida, K., Mariathasan, S., Dixit, V. M., Flavell, R. A., Dietrich, W. F., and Roy, C. R. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325.
Keywords: inflammasome, caspase-1, Legionella pneumophila, Asc
Citation: Abdelaziz DHA, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA, Abdulrahman BA, Hassan ZA, El-Sharkawi FZ, Bedi SS, Ladner K, Gonzalez-Mejia ME, Doseff AI, Mostafa M, Kanneganti T-D, Guttridge D, Marsh CB, Wewers MD and Amer AO (2011) Asc-dependent and independent mechanisms contribute to restriction of Legionella pneumophila infection in murine macrophages. Front. Microbio. 2:18. doi: 10.3389/fmicb.2011.00018
Received: 09 December 2010;
Paper pending published: 22 December 2010;
Accepted: 25 January 2011;
Published online: 14 February 2011.
Edited by:
Yousef Abu Kwaik, University of Louisville School of Medicine, USAReviewed by:
Maya Saleh, McGill University, CanadaMarina Santic’, University of Rijeka, Croatia (Hrvatska)
Copyright: © 2011 Abdelaziz, Gavrilin, Akhter, Caution, Kotrange, Khweek, Abdulrahman, Hassan, El-Sharkawi, Bedi, Ladner, Gonzalez-Mejia, Doseff, Mostafa, Kanneganti, Guttridge, Marsh, Wewers and Amer. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Amal O. Amer, Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine, Center for Microbial Interface Biology and The Department of Internal Medicine, Ohio State University, Biological Research Tower, 460W 12th Avenue, Room 1014, Columbus, OH 43210, USA.e-mail:YW1hbC5hbWVyQG9zdW1jLmVkdQ==