
 Takane Kikuchi1
Takane Kikuchi1
- 1 Department of Veterinary Biosciences, The Ohio State University, Columbus, OH, USA
- 2 Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, WA, USA
Anaplasma phagocytophilum and Ehrlichia chaffeensis are obligatory intracellular α-proteobacteria that infect human leukocytes and cause potentially fatal emerging zoonoses. In the present study, we determined global protein expression profiles of these bacteria cultured in the human promyelocytic leukemia cell line, HL-60. Mass spectrometric (MS) analyses identified a total of 1,212 A. phagocytophilum and 1,021 E. chaffeensis proteins, representing 89.3 and 92.3% of the predicted bacterial proteomes, respectively. Nearly all bacterial proteins (≥99%) with known functions were expressed, whereas only approximately 80% of “hypothetical” proteins were detected in infected human cells. Quantitative MS/MS analyses indicated that highly expressed proteins in both bacteria included chaperones, enzymes involved in biosynthesis and metabolism, and outer membrane proteins, such as A. phagocytophilum P44 and E. chaffeensis P28/OMP-1. Among 113 A. phagocytophilum p44 paralogous genes, 110 of them were expressed and 88 of them were encoded by pseudogenes. In addition, bacterial infection of HL-60 cells up-regulated the expression of human proteins involved mostly in cytoskeleton components, vesicular trafficking, cell signaling, and energy metabolism, but down-regulated some pattern recognition receptors involved in innate immunity. Our proteomics data represent a comprehensive analysis of A. phagocytophilum and E. chaffeensis proteomes, and provide a quantitative view of human host protein expression profiles regulated by bacterial infection. The availability of these proteomic data will provide new insights into biology and pathogenesis of these obligatory intracellular pathogens.
Introduction
Anaplasma phagocytophilum and Ehrlichia chaffeensis are small (ca. 0.4 by 1.5 μm), pleomorphic gram-negative bacteria that belong to the family Anaplasmataceae in the order Rickettsiales, the class α-proteobacteria (Dumler et al., 2001; Rikihisa, 2010b). The infection of humans by A. phagocytophilum and E. chaffeensis causes human granulocytic anaplasmosis [HGA, first reported in 1994, formerly known as human granulocytic ehrlichiosis (HGE)] and human monocytic ehrlichiosis (HME, first reported in 1987), respectively (Maeda et al., 1987; Chen et al., 1994). HGA and HME are similar systemic febrile diseases characterized by fever, headache, myalgia, anorexia, and chills, and are frequently accompanied by leukopenia, thrombocytopenia, anemia, and elevations in serum hepatic aminotransferases (Paddock and Childs, 2003; Bakken and Dumler, 2008; Thomas et al., 2009). Neurological signs are more frequently reported with HME than HGA (Paddock and Childs, 2003). Although doxycycline is generally effective in treating human ehrlichioses, delayed therapy, the presence of underlying allergies or poor health, and immuno-suppression often lead to severe complications or death. As important life-threatening tick-borne emerging zoonoses, HGA and HME were designated as nationally notifiable diseases by US Centers for Disease Control and Prevention in 1998 (Gardner et al., 2003). Since them, reported cases have increased every year. During 2008, cases attributed to A. phagocytophilum and E. chaffeensis increased by 21 and 16% from 2007, respectively (Hall-Baker et al., 2010).
Anaplasma phagocytophilum and E. chaffeensis are obligatory intracellular bacteria with a life cycle that requires repeated transmission between mammalian hosts and tick vectors (Rikihisa, 1991, 2010b; Dumler et al., 2001). Once transmitted to mammals, these bacteria replicate in membrane-bound compartments inside the primary host immune defensive cells: granulocytes (A. phagocytophilum) or monocytes/macrophages (E. chaffeensis). Since culture isolation of these organisms in the 1990s (Dawson et al., 1991; Goodman et al., 1996), unique strategies employed by A. phagocytophilum and E. chaffeensis for their survival in hostile environment have begun to be unraveled, including hijacking host cell signaling pathways, altering vesicular trafficking, usurping nutritional and cytoskeletal components, and subverting several host innate immune responses (Carlyon and Fikrig, 2003, 2006; Carlyon et al., 2004; Sukumaran et al., 2005; Huang et al., 2010a; Rikihisa, 2010a,b; Sultana et al., 2010; Wakeel et al., 2010). The complete genome sequences of A. phagocytophilum (1,471,282 base pairs) and E. chaffeensis (1,175,764 bp), and detailed analyses of their protein-coding genes have proven a great resource for studying these bacteria and the diseases they cause (Dunning Hotopp et al., 2006). These two species share approximately 500 genes; most of them encode proteins with homologies to those with known functions. However, approximately 470–580 genes are unique to each species (Dunning Hotopp et al., 2006), and approximately 45% of predicted open reading frames (ORFs) in the two genomes were annotated as uncharacterized “hypothetical proteins” or proteins without any functional assignment (Table 1). However, whether they really encode proteins and are actually expressed in living organisms remains largely unknown.
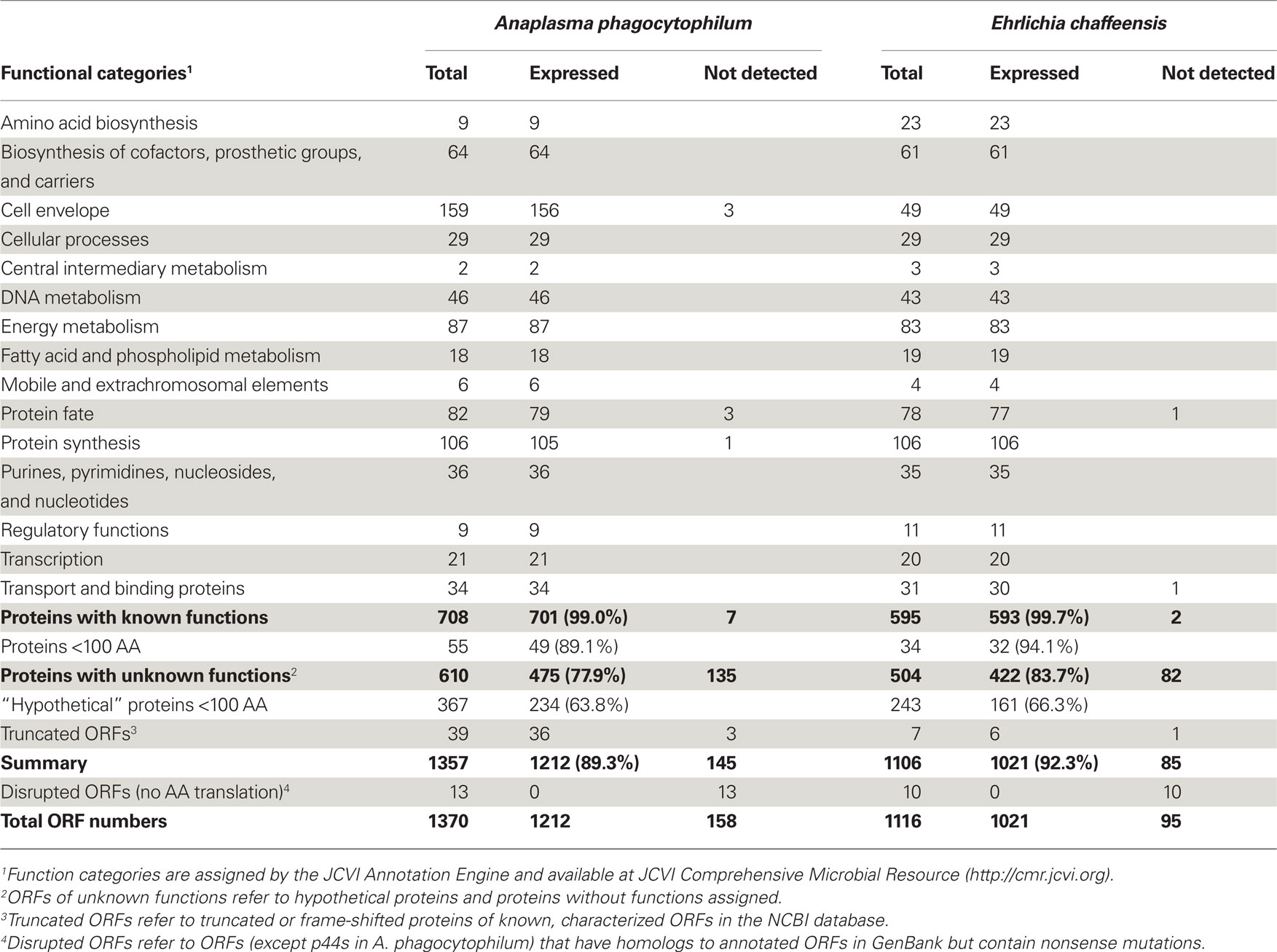
Table 1. Numbers of protein expression classified by functional categories.
Owing to the recent technical advance in transcriptome and proteome analyses, a holistic view of the numerous expressed genes and proteins of an organism has become available. Whole genome transcriptome analysis of A. phagocytophilum in human HL-60 cells showed the expression of approximately 70% of the bacterial gene transcripts (Nelson et al., 2008). Proteomics studies based on 1-D and 2-D gel analyses of E. chaffeensis identified one-fourth of the total ORFs from human and tick cell-derived bacterial cultures (Singu et al., 2005; Seo et al., 2008). However, there are major difficulties in proteomic studies of obligatory intracellular bacteria; because a high-purity bacterial sample is not easily obtainable, and the presence of a large amount of host proteins reduces the sensitivity and lowers the identification scores of bacterial proteins (Li and Lostumbo, 2010). The development of more sensitive nano-liquid chromatography combined with tandem MS/MS (nano-LC–MS/MS)-based proteomic approach improves global protein analysis of obligatory intracellular bacteria, as low levels of proteins can be identified in samples mixed with a large amount of host proteins (Zimmer et al., 2006). Furthermore, label-free protein quantitation based on LC–MS peptide peak intensity information becomes possible due to the reproducibility and sensitivity of intensity data measurements, and multiple samples from different conditions can be compared directly without stable isotope labeling (Old et al., 2005; Zimmer et al., 2006; Shi et al., 2009).
Here, we present the first comprehensive proteomes of two human pathogens A. phagocytophilum and E. chaffeensis, their relative protein expression abundances, and the influence of infection with these two pathogens on human host protein expression using multidimensional nano-LC–MS/MS approaches developed at Pacific Northwest National Laboratory1 (Zimmer et al., 2006; Mottaz-Brewer et al., 2008). The determination of protein expression profiles of A. phagocytophilum and E. chaffeensis in human leukocytes will help advance understanding cell biology, physiology of these bacteria, and complex interplay between bacteria and their host, and enhance the opportunities for investigation of novel targets for antimicrobial therapy or blocking of pathogenic pathways.
Materials and Methods
Bacteria Culture and Purification
Anaplasma phagocytophilum HZ (type strain; Rikihisa et al., 1997) and E. chaffeensis Arkansas (type strain; Dawson et al., 1991) were cultured in HL-60 cells, which are undifferentiated human promyelocytic leukemia cells from ATCC (#CCL-240, Manassas, VA, USA). Cells were maintained in RPMI 1640 medium supplemented with 5% fetal bovine serum and 2 mM l-glutamine, and incubated at 37°C in a humidified 5% CO2–95% air atmosphere. No antibiotic was used throughout the study. When infectivity reached greater than 95% as assessed by Diff-Quik staining of cytocentrifuged preparations (Baxter Scientific Products, Obetz, OH, USA), infected cells were harvested, extensively washed to remove serum proteins, and host cell-free bacteria were released by sonication for 10 s at an output setting of 2 with an ultrasonic processor W-380 (Heat Systems, Farmington, NY, USA). After low-speed centrifugation at 700×g to remove nuclei and unbroken cells, the supernatant was filtered through a 5-μm then 0.8-μm filter (Millipore, Billerica, MA, USA) to remove cellular debris. The filtrate was then centrifuged at 10,000 × g for 10 min, and the pellet enriched with host cell-free bacteria was collected.
Sample Preparation for Proteomics Analysis: Protein Partitioning, Digestion, and Clean-Up
To obtain comprehensive coverage of protein expression profiles, including both hydrophilic and hydrophobic proteins, proteins with very high or low pIs, and proteins with different cellular distributions, three optimized protein extraction protocols, including global, soluble, and insoluble protein extracts, were applied to purified host cell-free bacteria and uninfected or infected HL-60 cells as described previously (Mottaz-Brewer et al., 2008). For tryptic digestion with global protein extracts, pellets containing purified bacteria or host cells were suspended in 100 mM NH4HCO3 buffer (pH 8.4). The resulting suspension was transferred to a 2.0-mL cryovial tube with O-ring in cap, and lysed by beating with 0.1-mm zirconia/silica disruption beads (BioSpec Products, Bartlesville, OK, USA). Protein samples were denatured and reduced by adding urea, thiourea, and dithiothreitol (DTT) at final concentrations of 7 M, 2 M, and 5 mM, respectively. Following incubation at 60°C for 30 min, the samples were diluted 10-fold with NH4HCO3 buffer. Global digest was performed by adding trypsin at 1:50 (w:w) enzyme:protein ratio, and CaCl2 at a final concentration of 1 mM. The samples were incubated at 37°C for 3 h, snap frozen in liquid N2 to stop the digestion, and stored at −80°C until further analysis. Clean-up was performed using a Discovery C-18 solid phase extraction (SPE) column (Supelco, Bellefonte, PA, USA) to prepare the samples for MS analysis. Peptides were then concentrated by a Savant SpeedVac manifold (Thermo Fisher, Milford, MA, USA), and a BCA protein assay (Thermo Fisher/Pierce, Rockford, IL, USA) was performed to determine the final sample concentration.
For digestion with soluble and insoluble protein extracts, purified bacterial or host cell pellets were resuspended in 50 mM NH4HCO3 buffer and centrifuged at 355,000×g at 4°C for 10 min to separate the protein lysates into two parts: soluble and insoluble protein fractions. The supernatant was tryptically digested and cleaned up in the same fashion as in the global digest method and designated as soluble digest samples. The pellet after ultracentrifugation, containing the insoluble protein fraction, was washed and resuspended in a denaturing solution (7 M urea, 2 M thiourea, 1% CHAPS, 10 mM DTT, 50 mM NH4HCO3, pH 7.8). Insoluble protein samples were digested as described above. Removal of salts and detergent was performed using a Discovery strong cation exchange (SCX) SPE column (Supelco). Peptides were concentrated, and the concentration measured as described above. All trypsin-digested peptides were snap frozen in liquid N2 and stored at −80°C until proteomic analysis.
Mass Spectrometry and Data Analysis
In order to enhance proteome coverage, all peptide samples were further separated by SCX chromatography coupled off-line with nano-LC−MS/MS analyses. Peptide mixtures from each proteome sample were fractionated into 35–70 fractions as previously described (Qian et al., 2005). A description of the instrumentation and specifics of the high-performance liquid chromatography (HPLC–MS/MS) and HPLC–MS instrumental arrangements and associated methods for each biological system have been described previously and are consistent for all experiments (Manes et al., 2008; Mottaz-Brewer et al., 2008). In brief, samples were loaded onto an in-house developed chromatography system that uses a 20-cm × 75-μm C18 reverse-phase column and ionized as they eluted from the column into a mass spectrometer using electrospray ionization. The liquid chromatography gradient was generated linearly from aqueous to organic over 100 min in acidic conditions. Typically, MS was performed in a linear trap quadrupole (LTQ; Thermo Fisher Scientific) ion trap mass spectrometer. Tandem MS (MS/MS) were collected using data-dependent settings on the top 10 ions from the precursor scan.
Tandem MS spectra (MS/MS) were matched to protein sequence files using the SEQUEST program and filtered with a combination of scores provided in the output files (Eng et al., 1994), which included the minimum threshold filter scores defined by Washburn et al. (2001), and an additional minimum discriminant score of 0.5 to reduce the false-positive identifications (Strittmatter et al., 2004). Only peptides passing these filters were populated into the initial accurate mass and time (AMT) tag database. The searches were performed using the annotated protein databases of A. phagocytophilum HZ (1,357 protein entries, GenBank Accession Number NC_007797) including newly annotated 113 A. phagocytophilum P44 proteins, E. chaffeensis Arkansas (1,106 protein entries, GenBank Accession Number NC_007799), and Homo sapiens IPI protein database (61,225 protein entries, IPI 2006, v3.36). Each bacterial and human peptide from infected host cells was identified and populated into the same AMT tag database.
Quantitative Mass Spectrometric Analysis
Before running on the mass spectrometer, the total peptide mass was measured, and the sample was diluted to 1 μg/μL for injection. After the building of the initial AMT tag database, all samples were analyzed with a 9.4-T Fourier transform ion cyclotron resonance (FTICR) mass spectrometer (Bruker Daltonic, Billerica, MA, USA) following the separation of peptides by reverse-phase capillary HPLC under identical conditions as described (Shi et al., 2006, 2009). Standard proteins were added before digestion and used to track the performance of the instruments. Five technical replicates for each sample were injected into the FTICR mass spectrometer. Relevant information, such as the elution time from the capillary LC column, the abundance of the signal (integrated area under the elution profile), and the monoisotopic mass (determined from charge state and the high accuracy m/z measurement) of each feature observed in the FTICR, was used to match the peptide identifications contained within the initial AMT tag database. These peptides, now identified and quantified, were used to infer the protein composition of the samples. Only peptides observed in at least three out of five of these technical replicates were used in data analysis, and all proteins were required to have at least three observed peptides to be included in the confident results. In addition, the number of peptides observed for each protein in a biological sample was divided by the total number of peptides determined from the same sample to give an estimate of relative abundance of each identified protein in the sample. The abundances for each peptide were then averaged across these runs before the ratio calculations.
Data Analysis and Proteomic Database Online Access
Bacterial proteins were classified based on the functional role categories using JCVI Annotation Engine and Comprehensive Microbial Resource2 as described previously (Dunning Hotopp et al., 2006; Lin et al., 2009). Human proteins were classified based on gene ontology (GO) as annotated by the GO Consortium3. All peptides and proteins identified from A. phagocytophilum, E. chaffeensis, and human HL-60 cells, together with the detailed analyses of protein expression profiles and quantitation results can be accessed and downloaded from the website4.
Results and Discussion
Overview of A. Phagocytophilum and E. Chaffeensis Proteins Identified by Proteomics
In order to identify and quantitate the comprehensive protein expression profiles of A. phagocytophilum and E. chaffeensis, 18 A. phagocytophilum protein samples and 14 E. chaffeensis samples were prepared from purified host cell-free bacteria and infected HL-60 cells using three different protein extraction protocols as described; each contained approximately 1 mg of peptides after tryptic digestion and column clean-up. Approximately 250 MS runs for each bacterium from these samples were performed until no new peptides were detected (Figure A1 in Appendix). Positively identified peptides were populated into the AMT tag database, and two databases were constructed. The A. phagocytophilum database contained search results from 60 datasets using purified bacteria from infected cells and 189 datasets from A. phagocytophilum-infected cells, respectively. The database for E. chaffeensis contained 49 datasets from purified bacteria and 192 datasets associated with E. chaffeensis-infected host cells.
In protein samples from both A. phagocytophilum and E. chaffeensis-infected HL-60 cells, greater than 126,000 peptides were identified (Figure A1 in Appendix). Among these peptides, 44,080 matched to 1,212 A. phagocytophilum proteins, and 40,004 matched to 1,021 E. chaffeensis proteins, representing 89.3 and 92.3% of the predicted bacterial proteomes, respectively (Table 1). Among these detected proteins, greater than 96% have more than one peptide match. For proteins with known functional categories assigned, nearly all of these proteins (99.0% from A. phagocytophilum and 99.7% from E. chaffeensis) were expressed in HL-60 cells, including enzymes required for metabolisms and proteins involved in pathogenesis and regulatory functions, such as outer membrane proteins, the type IV secretion system (T4SS), and two-component regulatory systems. Therefore, nearly all proteins with known functions are likely essential for the replication and survival of these two pathogens inside human host cells. These expression profiles in mammalian host also suggest that, although gene loss occurred in the family Anaplasmataceae as a result of reductive genome evolution (Blanc et al., 2007; Darby et al., 2007), these genes cannot be sacrificed from their genomes.
Among these currently identified bacterial proteins, some mRNAs or proteins have been reported previously, including 70% of A. phagocytophilum genes in HL-60 cells by whole genome transcriptome analysis (Nelson et al., 2008) and near one-fourth of E. chaffeensis proteins by 1-D and 2-D gel based proteomics studies (Singu et al., 2005; Seo et al., 2008). Dozens of surface-exposed proteins in both bacteria using NHS-SS-biotin-labeling of host cell-free, intact bacteria cultured in human host cells (Ge and Rikihisa, 2007a,b). All of these identified proteins were detected in the current proteomic analyses. In addition, several other bacterial proteins reported previously were also detected in this study. These proteins include outer membrane proteins P44 and P28/Omp-1 (Ohashi et al., 1998b; Unver et al., 2002; Zhi et al., 2002; Zhang et al., 2004; Huang et al., 2007; Kumagai et al., 2008); transcriptional factors Tr1, ApxR, and EcxR (Wang et al., 2007; Cheng et al., 2008); three pairs of two-component regulatory system proteins in both bacteria (Cheng et al., 2006; Kumagai et al., 2006); the VirB/D4 T4S apparatus and substrates like AnkA and Ats-1 (Caturegli et al., 2000; Ohashi et al., 2002; IJdo et al., 2007; Lin et al., 2007; Bao et al., 2009; Niu et al., 2010); A. phagocytophilum toxin A (AptA) protein and morulae proteins APH_0032/APH_1387 (Huang et al., 2010b,c; Sukumaran et al., 2011); and E. chaffeensis Ank200 and tandem repeat proteins Trp32/Trp47/Trp120 (Wakeel et al., 2009, 2010; Zhu et al., 2009; Luo et al., 2010).
Expression of A. Phagocytophilum and E. Chaffeensis Proteins in Biosynthesis Pathways and Phage Components
Anaplasma phagocytophilum and E. chaffeensis have significantly higher percentages of their genomes involved in nucleotide biosynthesis, cofactor and vitamin biosynthesis, and protein synthesis than their closely related free-living α-proteobacterium Caulobacter crescentus (Dunning Hotopp et al., 2006). Expression of enzymes involved in nucleotide, vitamin, and cofactor biosynthetic pathway in A. phagocytophilum and E. chaffeensis, suggests that they do not need to compete with human leukocytes for, and may even supply host cells with, essential vitamins and nucleotides. This has been proposed to occur between the obligatory intracellular bacterium Wigglesworthia glossinidia and its insect host, tsetse fly (Zientz et al., 2004). The protein synthesis category includes many essential genes, such as those encoding ribosomal proteins, tRNA synthetases, RNA modification enzymes, and translation factors. Almost all of these proteins were expressed in mammalian hosts, except for A. phagocytophilum ribosomal protein L36 (Table S1 in Supplementary Material). Previous studies have shown that ribosomal protein L36 is dispensable for Escherichia coli growth and protein synthesis (Ikegami et al., 2005), and the gene encoding ribosomal protein L36 was not identified in the closely related Neorickettsia spp (Lin et al., 2009), suggesting that L36 might not be necessary for members in the family Anaplasmataceae.
Anaplasma phagocytophilum and E. chaffeensis have a lower coding percentage for transporters compared to the free-living C. crescentus (Dunning Hotopp et al., 2006). Although nearly 100% of the proteins with known functions were expressed in HL-60 cells, few proteins involved in transport functions, like twin-arginine translocation protein TatA/E of A. phagocytophilum, and monovalent cation/proton antiporter MnhG/PhaG subunit family protein of E. chaffeensis were not detected in bacteria cultured in HL-60 cells (Tables S1 and S2 in Supplementary Material). Interestingly, although A. phagocytophilum and E. chaffeensis do not encode for intact prophage or transposable/mobile elements, a few phage core components (HK97-like portal, major capsid, and prohead protease) were identified scattered throughout their genomes, and their protein expressions were also confirmed by proteomics. The functions of these remnant phage components on bacterial infection of human hosts are unclear; however, some literature has suggested that they might be involved in lateral gene transfer, bacterial chromosome inversion, evolution, and virulence factors expression (Canchaya et al., 2003; Brussow et al., 2004).
Expression Profiling of A. Phagocytophilum and E. Chaffeensis “Hypothetical” Proteins
Since approximately 45% of the predicted ORFs in the genomes encode conserved or uncharacterized “hypothetical” proteins (Table 1; Dunning Hotopp et al., 2006), whether they really encode proteins and whether these proteins are expressed in living organisms are largely unknown. Analysis of the expression profiles of these hypothetical proteins or proteins without known functions assigned showed that only 77.9 and 83.7% of them were expressed in A. phagocytophilum and E. chaffeensis, respectively (Table 1). The much lower expression ratio of these “hypothetical” genes compared to those of proteins with known functions assigned (near 100%), suggests that the expression of certain “hypothetical” proteins might be regulated in different host environments, like the arthropod vectors, and play critical roles in responses to host adaptation.
As suggested by Ochman (2002) and Skovgaard et al. (2001), a substantial fraction of hypothetical ORFs in bacterial genomes are short (under 300 nucleotides in length); therefore, many of them might be random stretches of DNA and do not actually encode proteins. Analysis of these expressed proteins with unknown functions showed that 50.7% of them in A. phagocytophilum and 61.8% in E. chaffeensis were greater than 100 amino acids (AA) in protein length (Tables S3 and S4 in Supplementary Material). However, for “hypothetical” proteins undetectable by proteomic analysis, 97.8% of them in A. phagocytophilum and 100% in E. chaffeensis were fewer than 100 AA (Table 2). As functional assignment to an ORF during genome annotation process is based on the homology or domain structure matches to known proteins or domains, proteins with known functions assigned are most likely biased toward long proteins (Skovgaard et al., 2001). This statement is probably true since among the “hypothetical” proteins, 60.2% in A. phagocytophilum and 48.2% in E. chaffeensis are fewer than 100 AA, whereas less than 8% of proteins with known functions are fewer than 100 AA (Table 1). The shorter protein length also reduces its possibility of being detected by proteomic analysis due to the smaller number of peptides after trypsin-treatment. However, our study showed that greater than 63% of the “hypothetical” proteins with fewer than 100 AA could be detected in both bacteria (Table 1). Therefore, further bioinformatics analyses of these expressed genes, combined with comprehensive protein expression profiles under different culturing or host environmental conditions, would help in the prediction of true “hypothetical” proteins.
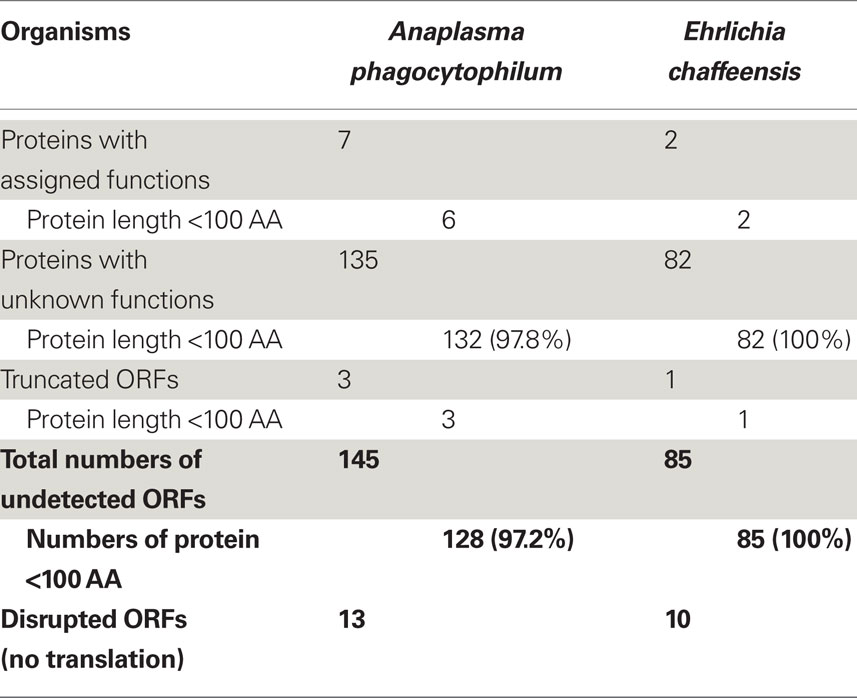
Table 2. Classification of undetected proteins by functional categories and protein lengths.
Expression of Overlapping ORFs in A. Phagocytophilum and E. Chaffeensis
Overlapping genes are detected primarily in parasitic or symbiotic bacteria and are believed to be a consequence of the reduction of originally larger genomes (Fukuda et al., 2003; Blanc et al., 2007). Analyses of the A. phagocytophilum and E. chaffeensis genomes identified overlaps among protein-coding ORFs and between RNA- and protein-coding ORFs, which occurred either at different reading frames of the same strand or on the complementary strands. Proteomic data showed that many overlapping genes were indeed expressed by A. phagocytophilum and E. chaffeensis in infected human host cells (Figures A2 and A3 in Appendix). These ORFs include one pair each of completely overlapping protein-coding ORFs in A. phagocytophilum (APH_0143/APH_0144) and E. chaffeensis (ECH_0506/ECH_0507), one pair of overlapping ORFs between protein (ECH_0472) and 6SRNA1 genes (ECH_1158), and 10 out of 21 (A. phagocytophilum) or 4 out of 26 (E. chaffeensis) partial overlapping protein-coding ORFs (Figures A2 and A3 in Appendix). These data suggest that overlapping ORFs can actually be transcribed and translated into proteins in these organisms with reduced genome contents in order to increase their coding capacities.
Expression of P44/MSP2 and Omp-1 Protein Superfamily in A. Phagocytophilum and E. Chaffeensis
Despite the reduction in their genome sizes and significantly lower coding capacity for metabolism, transport, and regulatory functions, A. phagocytophilum and E. chaffeensis not only retained but expanded a pool of genes encoding outer membrane proteins (Dunning Hotopp et al., 2006). Most of these outer membrane proteins are members of Pfam PF01617 and constitute the OMP-1/MSP2/P44 family (Dunning Hotopp et al., 2006; Finn et al., 2010). Since A. phagocytophilum and E. chaffeensis cannot be transovarially transmitted in their arthropod vectors, and ticks must acquire these organisms by feeding on an infected vertebrate reservoir animal, it was proposed that the expansion of this gene family might allow persistence in the vertebrate reservoir by providing antigenic variation, thus allowing for effective transmission from mammals to ticks (Rikihisa, 2010a).
The A. phagocytophilum genome has the largest expansion of the genes belonging to OMP-1/MSP2/P44 family among members of the family Anaplasmataceae, most of them encoding P44 outer membrane proteins (Dunning Hotopp et al., 2006). A total of 113 annotated p44 loci longer than 60 bp in gene length and some smaller DNA fragments homologous to p44 gene family can be identified throughout the genome, which consists of greater than 5% of the total genome contents (Dunning Hotopp et al., 2006). The full-length p44s consist of a central hypervariable region of approximately 280 bp encoding a signature of four conserved AA regions (C, C, WP, A) and conserved flanking sequences (Table 3; Lin and Rikihisa, 2005; Dunning Hotopp et al., 2006). By comparing the length and domains of the identified p44s to the full-length p44s, all p44 genes were annotated and classified as full-length, truncated, fragmented, or degenerated genes, as defined in Table 3 (Dunning Hotopp et al., 2006). Due to the lack of start/stop codons, silent p44 gene fragments are unlikely to be expressed at their own genomic loci and have to be recombined into and expressed from the expression locus APH_1221 (p44-18ES) by a RecF-dependent recombination, as suggested by Lin et al. (2006). To assist proteomic detection of all possible P44 peptides, in-frame AA sequences were deduced from all p44 genes (including silent pseudogenes without start/stop codons and degenerated p44 fragments containing nonsense mutations) and used in the SEQUEST search. The expression of P44-59 in the A. phagocytophilum outer membrane from these pseudogenes had been confirmed previously (Ge and Rikihisa, 2007a). Results showed that in addition to 22 full-length P44s, peptides identified by proteomic analysis were matched to protein sequences deduced from nearly all p44 genes (97.3%), including 86 silent p44 gene fragments and 2 degenerated p44 genes (Table 3). Since the N- and C-regions flanking hypervariable domains are highly conserved among P44 proteins, one peptide identified by proteomic analysis might actually match to several P44s. Therefore, we further analyzed all peptide matches to P44 proteins and confirmed that 84 P44 proteins (74.3%) were expressed with at least one unique peptide match (Table 3; Table S5 in Supplementary Material). These results showed that silent p44 gene reserves distributed throughout the A. phagocytophilum genome can actually be recombined and expressed from the p44-18ES expression locus (Figure 1). In addition, the region near this expression locus showed greater numbers of identified peptides matched to P44 proteins encoded by either full-length p44 genes that can be expressed at their own loci or silent p44 genes that have to be recombined into the p44-18ES locus for protein expression (Figure 1), which could be due to higher transcription activities in this region and/or higher recombination activities with the p44-18ES locus. The expression of the entire P44 repertoire by populations of this bacterium in human leukocytes would ensure their rapid adaptation to changing host environments and successful parasitism in new host cells, as well as escaping host immune surveillance. These results confirm our previous findings from mRNA data that diverse P44s can be expressed at the p44-expression locus by gene conversion from over 100 p44 donor loci (Lin et al., 2003, 2004; Wang et al., 2004; Lin and Rikihisa, 2005).
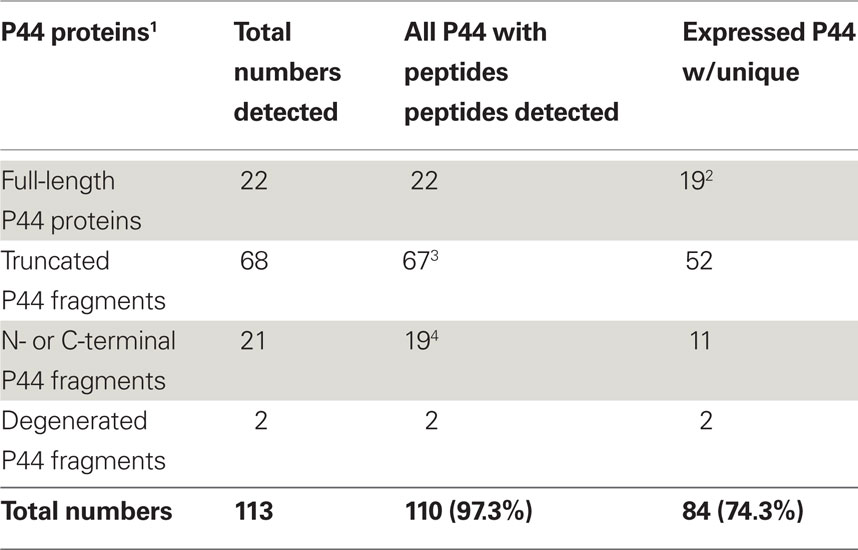
Table 3. Expression profile of Anaplasma phagocytophilum P44 outer membrane proteins.
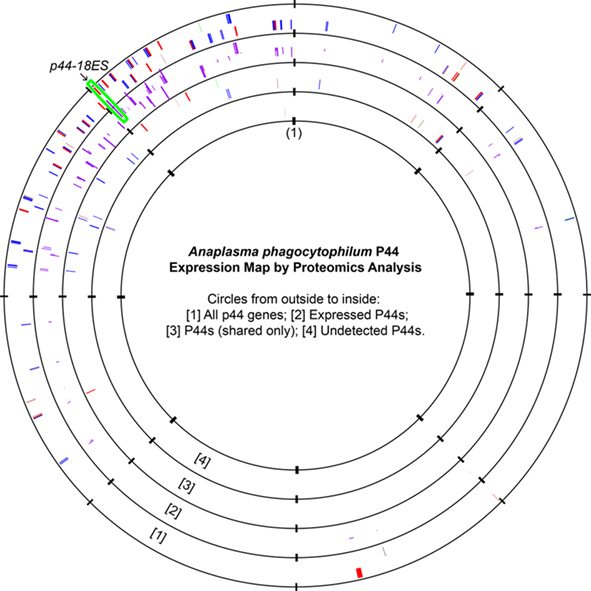
Figure 1. Anaplasma phagocytophilum P44 expression maps as detected by proteomic analysis. All genes encoding P44 outer membrane proteins were plotted on the first circle. The bar heights on the second circle represented the number of P44-matching peptides detected, with higher bars indicating greater numbers of matching unique peptides. The third circle showed P44 proteins that had matched peptides but no unique peptide matches, and the fourth circle showed P44 proteins that had no peptide matches by proteomic analysis (APH_1122/P44-75, APH_1124/P44-C, and APH_1399/P44-C). The origin of the A. phagocytophilum genome was marked as (1), and the expression locus p44-18ES was highlighted by the green box. Color codes in circles 1, 2, and 4: Red, full-length p44s; Blue, truncated p44s; Green: N-terminal p44 fragments; Brown: C-terminal p44 fragments; Gray: Degenerated P44 fragments.
Ehrlichia chaffeensis has 22 paralogous tandemly arranged p28/omp-1 genes encoding immunodominant major outer membrane proteins (Ohashi et al., 1998a,b; Dunning Hotopp et al., 2006). Proteomics analyses showed that all these proteins and 27 other cell envelope proteins are expressed by E. chaffeensis in HL-60 cells (Table 1). Nineteen out of 22 P28/OMP-1 proteins have also been confirmed by proteomic identification of surface-exposed proteins of E. chaffeensis cultured in the human acute leukemia cell line THP-1 (Ge and Rikihisa, 2007b). Temporal transcript analyses showed that mRNA expression of 16 out of 22 p28/omp-1 genes was detected in the blood from acute to chronically infected dogs (over 56 days of infection; Unver et al., 2002). Using 22 synthetic antigenic peptides unique to each of the P28/OMP-1 proteins, sera from persistently infected dogs were reacted with all P28/OMP-1 family proteins (Zhang et al., 2004). These data suggest that P28/OMP-1 family proteins are not involved in immune evasion at the population level (Unver et al., 2002; Zhang et al., 2004).
Surface expression of porins that function as passive diffusion channels is required for small hydrophilic compounds to pass through the outer membranes of gram-negative bacteria (Nikaido and Vaara, 1985; Nikaido, 2003). Our previous studies have shown that both P44 and P28/Omp-1 have porin activities as measured by liposome swelling assay, allowing the diffusion of l-glutamine, monosaccharides arabinose and glucose, disaccharide sucrose, and even tetrasaccharide stachyose (Huang et al., 2007; Kumagai et al., 2008). Since the tricarboxylic acid (TCA) cycle in A. phagocytophilum and E. chaffeensis is incomplete (Dunning Hotopp et al., 2006), porin activity of P44 and P28/Omp-1 likely feeds the TCA cycle, and the differential expression of P44 or P28/OMP-1s might influence individual bacterial physiological activity (Huang et al., 2007; Kumagai et al., 2008).
Quantitative Analyses of Protein Expression Profiles in A. Phagocytophilum and E. Chaffeensis
Following the determination of global expression profiling of these intracellular bacteria, we further determined the relative abundance of A. phagocytophilum and E. chaffeensis proteins expressed in human host cells. Quantitative analyses of protein expression were determined by averaging individual peptide abundances for the matching protein in the entire pool of peptides identified. Although different proteins do not contain the same peptides and protein abundances are not directly comparable, the relative correlation to the total abundance still exists, especially with at least threefold difference between proteins (Old et al., 2005). Quantitative analyses identified 130 proteins from A. phagocytophilum and 116 from E. chaffeensis as having relative abundances greater than 1 (Table 4; Tables S6–S8 in Supplementary Material). Among them, the most abundant proteins detected are outer membrane proteins like A. phagocytophilum P44s and E. chaffeensis P28/Omp-1, RecF of A. phagocytophilum, chaperones like GroESL and DnaK involved in protein folding/stabilization, T4S apparatus, and enzymes involved in energy metabolism, transcription, protein synthesis, and biosynthesis of cofactors, nucleotides, and phospholipids (Tables S6–S8 in Supplementary Material). Classification by functional role categories showed that A. phagocytophilum and E. chaffeensis have similar numbers of abundant proteins in all but three functional categories (Table 4, in bold font). Due to the expansion in P44 outer membrane family proteins, more proteins are expressed abundantly by A. phagocytophilum in the “Cell envelope” category. On the other hand, E. chaffeensis abundantly expresses more proteins involved in the categories including “Protein synthesis,” like ribosomal proteins and “Energy metabolism,” like electron transport chain proteins, probably because E. chaffeensis has additional ability to synthesize arginine and lysine but A. phagocytophilum does not (Dunning Hotopp et al., 2006). Interestingly, greater than 12% of these abundantly expressed proteins are hypothetical proteins or proteins with unknown functions (Table 4), suggesting that these proteins might be required for infecting human host cells and could be novel targets for the study of pathogenic mechanisms in human infection.
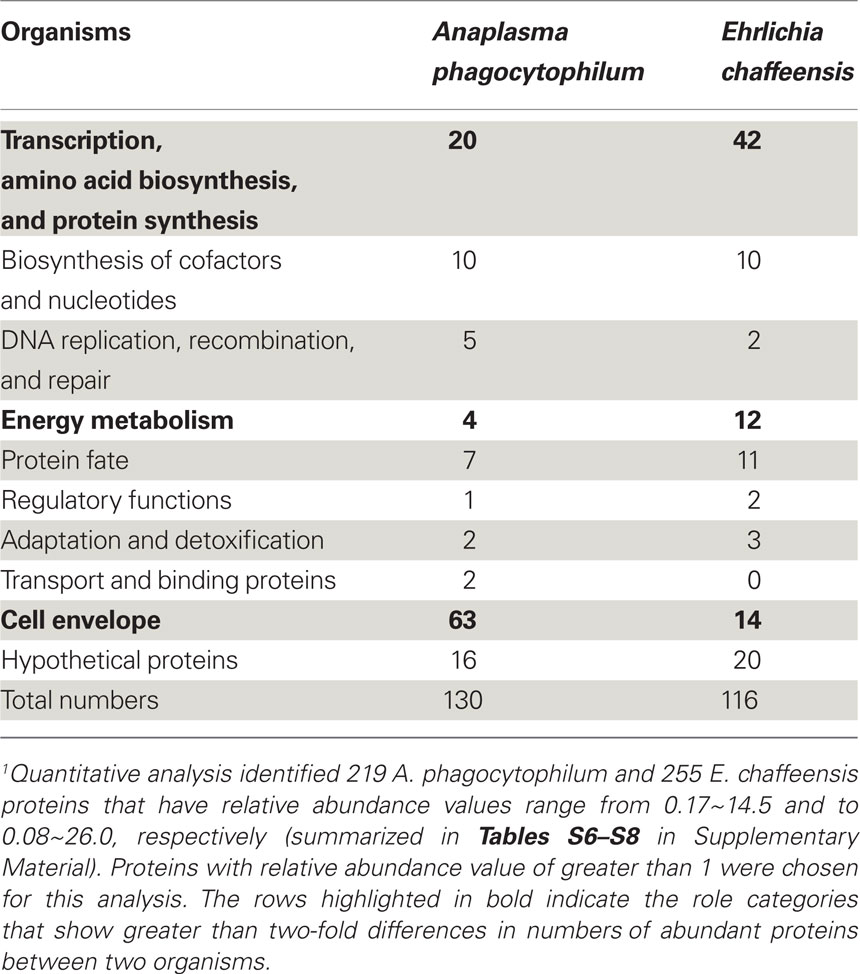
Table 4. Quantitation analysis of proteins in high abundance classified by functional categories1.
Quantitative Analyses of up- or Down-Regulated Human Proteins in A. Phagocytophilum and E. Chaffeensis-Infected HL-60 cells vs. Uninfected cells
As obligatory intracellular bacteria, the life cycles of A. phagocytophilum and E. chaffeensis are dependent on their mammalian hosts and are known to regulate or hijack host components for their survival (Rikihisa, 2010a,b). We, therefore determined the relative abundance of human proteins by comparing the LC–MS peptide peak intensity information of the same peptides from infected HL-60 cells to that from uninfected cells. A total of 48,054 human proteins were identified from HL-60 cells (Table S9 in Supplementary Material). Quantitative analyses of human proteins in infected vs. uninfected HL-60 cells showed that infection by A. phagocytophilum and E. chaffeensis up-regulated the expression of proteins involved mostly in vesicular trafficking and cytoskeleton components, protein tyrosine kinases, pro-survival proteins, and enzymes involved in metabolism and oxidative respiration (Table 5; Tables S10 and S11 in Supplementary Material). However, some proteins involved in host immune responses were down-regulated, including pattern recognition receptors like TLR1 and mannose receptor 2 (Table 6; Tables S12 and S13 in Supplementary Material).
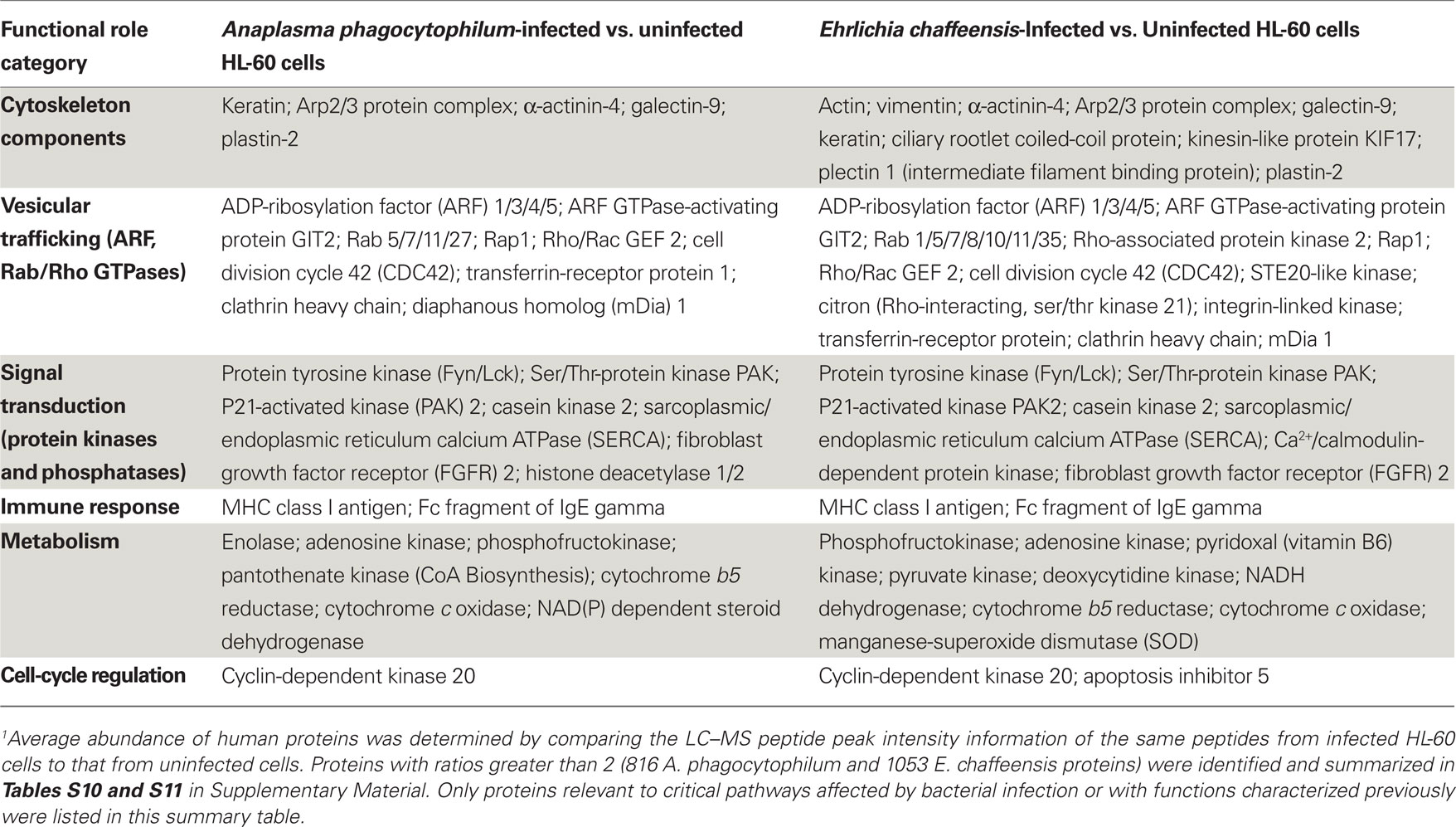
Table 5. Up-regulated human proteins in infected vs. uninfected HL-60 cells by quantitative proteomics analysis1.

Table 6. Down-regulated human proteins in infected vs. uninfected HL-60 cells by quantitative proteomics analysis1.
Several human genes that were up- or down-regulated by infection with A. phagocytophilum or E. chaffeensis have been reported previously. Up-regulated genes in human neutrophils at early stage of A. phagocytophilum infection included those that promote actin polymerization (Sukumaran et al., 2005). Up-regulation of genes involved in iron metabolism like transferrin-receptor was detected in A. phagocytophilum-infected NB4 cell, a human promyelocytic leukemia cell line (Pedra et al., 2005), and E. chaffeensis-infected THP-1 cell, a human monocytic leukemia cell line (Barnewall et al., 1999). The expression of histone deacetylase (HDAC) 1/2 was increased in A. phagocytophilum-infected THP-1 cells (Garcia-Garcia et al., 2009). Down-regulation of TLR2/4 mRNA and protein expression was reported in E. chaffeensis-infected human monocytes (Lin and Rikihisa, 2004). In addition, several reports have demonstrated the interactions between these up-regulated human proteins and bacterial proteins or activation of human proteins by bacterial infection. For example, the protein tyrosine kinase Fyn was shown to interact with E. chaffeensis TRP47 protein in THP-1 cells (Wakeel et al., 2009), whereas A. phagocytophilum induced actin phosphorylation by p21-activated kinase (PAK1) in Ixodes ticks (Sultana et al., 2010). A. phagocytophilum-containing morulae were colocalized with several Rab GTPases, including Rab11 (Huang et al., 2010a), and E. chaffeensis-containing morulae were colocalized with Rab5 (Mott et al., 1999). Both A. phagocytophilum- and E. chaffeensis-containing morulae were colocalized with major histocompatibility complex (MHC) class I and II antigens (Mott et al., 1999). Several isoforms of sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) were up-regulated in A. phagocytophilum- and E. chaffeensis-infected HL-60 cells, suggesting proteins involved in the intracellular Ca2+ regulation like phospholipase C and transglutaminase shown in previous studies are critical in bacterial infection (Lin et al., 2002; de la Fuente et al., 2005).
There are several studies using microarray analyses to identify genes differentially regulated in response to A. phagocytophilum infection in human neutrophils and the promyelocytic leukemia cell lines NB4 and HL-60 cells at different infection stages (Borjesson et al., 2005; de la Fuente et al., 2005; Pedra et al., 2005; Sukumaran et al., 2005; Lee and Goodman, 2006; Galindo et al., 2008; Lee et al., 2008). These studies identified similar sets of differentially regulated genes involved in vesicular transport, cytoskeletal remodeling, signaling and communication events, cell-cycle and apoptosis regulation, and innate immunity. However, due to the differences in host cell types, efficiency of infection, post-infection time points, experimental designs, array platforms, databases used, and statistical analyses, a large portion of the genes are difficult to compare among these studies (Pedra et al., 2005; Lee et al., 2008). Since most cell functions are carried out by proteins, the comparison of proteomic data would reflect a more accurate state of cellular physiology and pathology. Nevertheless, combining these microarray and quantitative proteomic data would allow more comprehensive understanding of host cellular changes induced by infection with these pathogens. Our proteomic analyses reveal that infection with A. phagocytophilum or E. chaffeensis could modulate human host cell machinery to produce more energy, enhance vesicular transport, and activate cell signaling events involved in bacterial entry and proliferation. Further analyses of these up- and down-regulated human proteins will provide more information about the global regulation of host cells by infection with these intracellular pathogens.
Conclusion
The determination of bacterial proteomes is an important step in converting genetic information to protein function and cell biology. This study provides the first comprehensive proteomes of obligatory intracellular pathogens. A total of 1,212 A. phagocytophilum and 1,021 E. chaffeensis proteins are identified, representing 89.3 and 92.3% of the predicted bacterial proteomes, respectively. Nearly all proteins that have functions assigned are expressed in infected human hosts, including those involved in metabolism, pathogenesis, and regulation. Bacterial infection up-regulated the expression of human proteins involved mostly in cytoskeleton components, vesicular trafficking, cell signaling, and energy metabolism, but down-regulated some pattern recognition receptors involved in innate immunity. The availability of these proteomic data will provide a wealth of information on the molecular mechanisms of bacterial pathogenesis and therefore will greatly facilitate the understanding of the biology of these ehrlichiosis agents and the signaling events between intracellular bacteria and their host cells.
Acknowledgments
Proteomics analysis was performed in the Environmental Molecular Science Laboratory, a U.S. Department of Energy (DOE) national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, Washington. PNNL is operated for the DOE by Battelle Memorial Institute under contract DE-AC05-76RLO-1830. This research was funded by the National Institute of Allergy and Infectious Diseases (NIH/DHHS) through interagency agreement Y1-AI-4894-01 to PNNL, and synergized with NIH grants R01 AI030010 and R01 AI047885 to Yasuko Rikihisa.
Supplementary Material
Tables S1–S14 can be found online at http://www.frontiersin.org/Cellular_and_Infection_Microbiology/10.3389/fmicb.2011.00024/
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Bakken, J. S., and Dumler, S. (2008). Human granulocytic anaplasmosis. Infect. Dis. Clin. North Am. 22, 433–448.
Bao, W., Kumagai, Y., Niu, H., Yamaguchi, M., Miura, K., and Rikihisa, Y. (2009). Four VirB6 paralogs and VirB9 are expressed and interact in Ehrlichia chaffeensis-containing vacuoles. J. Bacteriol. 191, 278–286.
Barnewall, R. E., Ohashi, N., and Rikihisa, Y. (1999). Ehrlichia chaffeensis and E. sennetsu, but not the human granulocytic ehrlichiosis agent, colocalize with transferrin receptor and up-regulate transferrin receptor mRNA by activating iron-responsive protein 1. Infect. Immun. 67, 2258–2265.
Blanc, G., Ogata, H., Robert, C., Audic, S., Suhre, K., Vestris, G., Claverie, J. M., and Raoult, D. (2007). Reductive genome evolution from the mother of Rickettsia. PLoS Genet. 3, e14. doi: 10.1371/journal.pgen.0030014
Borjesson, D. L., Kobayashi, S. D., Whitney, A. R., Voyich, J. M., Argue, C. M., and Deleo, F. R. (2005). Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J. Immunol. 174, 6364–6372.
Brussow, H., Canchaya, C., and Hardt, W. D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602.
Canchaya, C., Fournous, G., Chibani-Chennoufi, S., Dillmann, M. L., and Brussow, H. (2003). Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 6, 417–424.
Carlyon, J. A., Abdel-Latif, D., Pypaert, M., Lacy, P., and Fikrig, E. (2004). Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect. Immun. 72, 4772–4783.
Carlyon, J. A., and Fikrig, E. (2003). Invasion and survival strategies of Anaplasma phagocytophilum. Cell. Microbiol. 5, 743–754.
Carlyon, J. A., and Fikrig, E. (2006). Mechanisms of evasion of neutrophil killing by Anaplasma phagocytophilum. Curr. Opin. Hematol. 13, 28–33.
Caturegli, P., Asanovich, K. M., Walls, J. J., Bakken, J. S., Madigan, J. E., Popov, V. L., and Dumler, J. S. (2000). ankA: an Ehrlichia phagocytophila group gene encoding a cytoplasmic protein antigen with ankyrin repeats. Infect. Immun. 68, 5277–5283.
Chen, S. M., Dumler, J. S., Bakken, J. S., and Walker, D. H. (1994). Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. J. Clin. Microbiol. 32, 589–595.
Cheng, Z., Kumagai, Y., Lin, M., Zhang, C., and Rikihisa, Y. (2006). Intra-leukocyte expression of two-component systems in Ehrlichia chaffeensis and Anaplasma phagocytophilum and effects of the histidine kinase inhibitor closantel. Cell. Microbiol. 8, 1241–1252.
Cheng, Z., Wang, X., and Rikihisa, Y. (2008). Regulation of type IV secretion apparatus genes during Ehrlichia chaffeensis intracellular development by a previously unidentified protein. J. Bacteriol. 190, 2096–2105.
Darby, A. C., Cho, N. H., Fuxelius, H. H., Westberg, J., and Andersson, S. G. (2007). Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 23, 511–520.
Dawson, J. E., Anderson, B. E., Fishbein, D. B., Sanchez, J. L., Goldsmith, C. S., Wilson, K. H., and Duntley, C. W. (1991). Isolation and characterization of an Ehrlichia sp. from a patient diagnosed with human ehrlichiosis. J. Clin. Microbiol. 29, 2741–2745.
de la Fuente, J., Ayoubi, P., Blouin, E. F., Almazan, C., Naranjo, V., and Kocan, K. M. (2005). Gene expression profiling of human promyelocytic cells in response to infection with Anaplasma phagocytophilum. Cell. Microbiol. 7, 549–559.
Dumler, J. S., Barbet, A. F., Bekker, C. P., Dasch, G. A., Palmer, G. H., Ray, S. C., Rikihisa, Y., and Rurangirwa, F. R. (2001). Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and “HGE agent” as subjective synonyms of Ehrlichia phagocytophila. Int. J. Syst. Evol. Microbiol. 51, 2145–2165.
Dunning Hotopp, J. C., Lin, M., Madupu, R., Crabtree, J., Angiuoli, S. V., Eisen, J., Seshadri, R., Ren, Q., Wu, M., Utterback, T. R., Smith, S., Lewis, M., Khouri, H., Zhang, C., Hua, N., Lin, Q., Ohashi, N., Zhi, N., Nelson, W., Brinkac, L. M., Dodson, R. J., Rosovitz, M. J., Sundaram, J., Daugherty, S. C., Davidsen, T., Durkin, A., Gwinn, M., Haft, D. H., Selengut, J. D., Sullivan, S. A., Zafar, N., Zhou, L., Benahmed, F., Forberger, H., Halpin, R., Mulligan, S., Robinson, J., White, O., Rikihisa, Y., and Tettelin, H. (2006). Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2, e21. doi: 10.1371/journal.pgen.0020021
Eng, J. K., Mccormack, A. L., and Yates, J. R. (1994). An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989.
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., Gavin, O.L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L., Eddy, S. R., and Bateman, A. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222.
Fukuda, Y., Nakayama, Y., and Tomita, M. (2003). On dynamics of overlapping genes in bacterial genomes. Gene 323, 181–187.
Galindo, R. C., Ayoubi, P., Garcia-Perez, A. L., Naranjo, V., Kocan, K. M., Gortazar, C., and de la Fuente, J. (2008). Differential expression of inflammatory and immune response genes in sheep infected with Anaplasma phagocytophilum. Vet. Immunol. Immunopathol. 126, 27–34.
Garcia-Garcia, J. C., Barat, N. C., Trembley, S. J., and Dumler, J. S. (2009). Epigenetic silencing of host cell defense genes enhances intracellular survival of the rickettsial pathogen Anaplasma phagocytophilum. PLoS Pathog. 5, e1000488. doi: 10.1371/journal.ppat.1000488
Gardner, S. L., Holman, R. C., Krebs, J. W., Berkelman, R., and Childs, J. E. (2003). National surveillance for the human ehrlichioses in the United States, 1997–2001, and proposed methods for evaluation of data quality. Ann. N. Y. Acad. Sci. 990, 80–89.
Ge, Y., and Rikihisa, Y. (2007a). Identification of novel surface proteins of Anaplasma phagocytophilum by affinity purification and proteomics. J. Bacteriol. 189, 7819–7828.
Ge, Y., and Rikihisa, Y. (2007b). Surface-exposed proteins of Ehrlichia chaffeensis. Infect. Immun. 75, 3833–3841.
Goodman, J. L., Nelson, C., Vitale, B., Madigan, J. E., Dumler, J. S., Kurtti, T. J., and Munderloh, U. G. (1996). Direct cultivation of the causative agent of human granulocytic ehrlichiosis. N. Engl. J. Med. 334, 209–215.
Hall-Baker, P. A., Enrique Nieves, J., Jajosky, R. A., Adams, D. A., Sharp, P., Anderson, W. J., Aponte, J. J., Aranas, A. E., Katz, S. B., Mayes, M., Wodajo, M. S., Onweh, D. H., Baillie, J., and Park, M. (2010). Summary of notifiable diseases – United States, 2008. MMWR Morb. Mortal. Wkly. Rep. 57, 1–98.
Huang, B., Hubber, A., McDonough, J. A., Roy, C. R., Scidmore, M. A., and Carlyon, J. A. (2010a). The Anaplasma phagocytophilum-occupied vacuole selectively recruits Rab-GTPases that are predominantly associated with recycling endosomes. Cell. Microbiol. 12, 1292–1307.
Huang, B., Troese, M. J., Howe, D., Ye, S., Sims, J. T., Heinzen, R. A., Borjesson, D. L., and Carlyon, J. A. (2010b). Anaplasma phagocytophilum APH_0032 is expressed late during infection and localizes to the pathogen-occupied vacuolar membrane. Microb. Pathog. 49, 273–284.
Huang, B., Troese, M. J., Ye, S., Sims, J. T., Galloway, N. L., Borjesson, D. L., and Carlyon, J. A. (2010c). Anaplasma phagocytophilum APH_1387 is expressed throughout bacterial intracellular development and localizes to the pathogen-occupied vacuolar membrane. Infect. Immun. 78, 1864–1873.
Huang, H., Wang, X., Kikuchi, T., Kumagai, Y., and Rikihisa, Y. (2007). Porin activity of Anaplasma phagocytophilum outer membrane fraction and purified P44. J. Bacteriol. 189, 1998–2006.
IJdo, J. W., Carlson, A. C., and Kennedy, E. L. (2007). Anaplasma phagocytophilum AnkA is tyrosine phosphorylated at EPIYA motifs and recruits SHP-1 during early infection. Cell. Microbiol. 9, 1284–1296.
Ikegami, A., Nishiyama, K., Matsuyama, S., and Tokuda, H. (2005). Disruption of rpmJ encoding ribosomal protein L36 decreases the expression of secY upstream of the spc operon and inhibits protein translocation in Escherichia coli. Biosci. Biotechnol. Biochem. 69, 1595–1602.
Kumagai, Y., Cheng, Z., Lin, M., and Rikihisa, Y. (2006). Biochemical activities of three pairs of Ehrlichia chaffeensis two-component regulatory system proteins involved in inhibition of lysosomal fusion. Infect. Immun. 74, 5014–5022.
Kumagai, Y., Huang, H., and Rikihisa, Y. (2008). Expression and porin activity of P28 and OMP-1F during intracellular Ehrlichia chaffeensis development. J. Bacteriol. 190, 3597–3605.
Lee, H. C., and Goodman, J. L. (2006). Anaplasma phagocytophilum causes global induction of antiapoptosis in human neutrophils. Genomics 88, 496–503.
Lee, H. C., Kioi, M., Han, J., Puri, R. K., and Goodman, J. L. (2008). Anaplasma phagocytophilum-induced gene expression in both human neutrophils and HL-60 cells. Genomics 92, 144–151.
Li, Q., and Lostumbo, G. (2010). Proteomic analyses of a variety of intracellular bacterial species infecting different host cell lines. Curr. Proteomics 7, 222–232.
Lin, M., den Dulk-Ras, A., Hooykaas, P. J., and Rikihisa, Y. (2007). Anaplasma phagocytophilum AnkA secreted by type IV secretion system is tyrosine phosphorylated by Abl-1 to facilitate infection. Cell. Microbiol. 9, 2644–2657.
Lin, M., and Rikihisa, Y. (2004). Ehrlichia chaffeensis downregulates surface Toll-like receptors 2/4, CD14 and transcription factors PU.1 and inhibits lipopolysaccharide activation of NF-kB, ERK 1/2 and p38 MAPK in host monocytes. Cell. Microbiol. 6, 175–186.
Lin, M., Zhang, C., Gibson, K., and Rikihisa, Y. (2009). Analysis of complete genome sequence of Neorickettsia risticii: causative agent of Potomac horse fever. Nucleic Acids Res. 37, 6076–6091.
Lin, M., Zhu, M. X., and Rikihisa, Y. (2002). Rapid activation of protein tyrosine kinase and phospholipase C-gamma2 and increase in cytosolic free calcium are required by Ehrlichia chaffeensis for internalization and growth in THP-1 cells. Infect. Immun. 70, 889–898.
Lin, Q., and Rikihisa, Y. (2005). Establishment of cloned Anaplasma phagocytophilum and analysis of p44 gene conversion within an infected horse and infected SCID mice. Infect. Immun. 73, 5106–5114.
Lin, Q., Rikihisa, Y., Massung, R. F., Woldehiwet, Z., and Falco, R. C. (2004). Polymorphism and transcription at the p44-1/p44-18 genomic locus in Anaplasma phagocytophilum strains from diverse geographic regions. Infect. Immun. 72, 5574–5581.
Lin, Q., Rikihisa, Y., Ohashi, N., and Zhi, N. (2003). Mechanisms of variable p44 expression by Anaplasma phagocytophilum. Infect. Immun. 71, 5650–5661.
Lin, Q., Zhang, C., and Rikihisa, Y. (2006). Analysis of involvement of the RecF pathway in p44 recombination in Anaplasma phagocytophilum and in Escherichia coli by using a plasmid carrying the p44 expression and p44 donor loci. Infect. Immun. 74, 2052–2062.
Luo, T., Zhang, X., Nicholson, W. L., Zhu, B., and McBride, J. W. (2010). Molecular characterization of antibody epitopes of Ehrlichia chaffeensis ankyrin protein 200 and tandem repeat protein 47 and evaluation of synthetic immunodeterminants for serodiagnosis of human monocytotropic ehrlichiosis. Clin. Vaccine Immunol. 17, 87–97.
Maeda, K., Markowitz, N., Hawley, R. C., Ristic, M., Cox, D., and McDade, J. E. (1987). Human infection with Ehrlichia canis, a leukocytic Rickettsia. N. Engl. J. Med. 316, 853–856.
Manes, N. P., Estep, R. D., Mottaz, H. M., Moore, R. J., Clauss, T. R. W., Monroe, M. E., Du, X., Adkins, J. N., Wong, S. W., and Smith, R. D. (2008). Comparative proteomics of human monkeypox and vaccinia intracellular mature and extracellular enveloped virions. J. Proteome Res. 7, 960–968.
Mott, J., Barnewall, R. E., and Rikihisa, Y. (1999). Human granulocytic ehrlichiosis agent and Ehrlichia chaffeensis reside in different cytoplasmic compartments in HL-60 cells. Infect. Immun. 67, 1368–1378.
Mottaz-Brewer, H. M., Norbeck, A. D., Adkins, J. N., Manes, N. P., Ansong, C., Shi, L., Rikihisa, Y., Kikuchi, T., Wong, S. W., Estep, R. D., Heffron, F., Pasa-Tolic, L., and Smith, R. D. (2008). Optimization of proteomic sample preparation procedures for comprehensive protein characterization of pathogenic systems. J. Biomol. Tech. 19, 285–295.
Nelson, C. M., Herron, M. J., Felsheim, R. F., Schloeder, B. R., Grindle, S. M., Chavez, A. O., Kurtti, T. J., and Munderloh, U. G. (2008). Whole genome transcription profiling of Anaplasma phagocytophilum in human and tick host cells by tiling array analysis. BMC Genomics 9, 364. doi: 10.1186/1471-2164-9-364
Nikaido, H. (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656.
Nikaido, H., and Vaara, M. (1985). Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 49, 1–32.
Niu, H., Kozjak-Pavlovic, V., Rudel, T., and Rikihisa, Y. (2010). Anaplasma phagocytophilum Ats-1 is imported into host cell mitochondria and interferes with apoptosis induction. PLoS Pathog. 6, e1000774. doi: 10.1371/journal.ppat.1000774
Ochman, H. (2002). Distinguishing the ORFs from the ELFs: short bacterial genes and the annotation of genomes. Trends Genet. 18, 335–337.
Ohashi, N., Unver, A., Zhi, N., and Rikihisa, Y. (1998a). Cloning and characterization of multigenes encoding the immunodominant 30-kilodalton major outer membrane proteins of Ehrlichia canis and application of the recombinant protein for serodiagnosis. J. Clin. Microbiol. 36, 2671–2680.
Ohashi, N., Zhi, N., Zhang, Y., and Rikihisa, Y. (1998b). Immunodominant major outer membrane proteins of Ehrlichia chaffeensis are encoded by a polymorphic multigene family. Infect. Immun. 66, 132–139.
Ohashi, N., Zhi, N., Lin, Q., and Rikihisa, Y. (2002). Characterization and transcriptional analysis of gene clusters for a type IV secretion machinery in human granulocytic and monocytic ehrlichiosis agents. Infect. Immun. 70, 2128–2138.
Old, W. M., Meyer-Arendt, K., Aveline-Wolf, L., Pierce, K. G., Mendoza, A., Sevinsky, J. R., Resing, K. A., and Ahn, N. G. (2005). Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502.
Paddock, C. D., and Childs, J. E. (2003). Ehrlichia chaffeensis: a prototypical emerging pathogen. Clin. Microbiol. Rev. 16, 37–64.
Pedra, J. H., Sukumaran, B., Carlyon, J. A., Berliner, N., and Fikrig, E. (2005). Modulation of NB4 promyelocytic leukemic cell machinery by Anaplasma phagocytophilum. Genomics 86, 365–377.
Qian, W. J., Liu, T., Monroe, M. E., Strittmatter, E. F., Jacobs, J. M., Kangas, L. J., Petritis, K., Camp, D. G. II, and Smith, R. D. (2005). Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: the human proteome. J. Proteome Res. 4, 53–62.
Rikihisa, Y. (1991). The tribe Ehrlichieae and ehrlichial diseases. Clin. Microbiol. Rev. 4, 286–308.
Rikihisa, Y. (2010a). Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nat. Rev. Microbiol. 8, 328–339.
Rikihisa, Y. (2010b). Molecular events involved in cellular invasion by Ehrlichia chaffeensis and Anaplasma phagocytophilum. Vet. Parasitol. 167, 155–166.
Rikihisa, Y., Zhi, N., Wormser, G. P., Wen, B., Horowitz, H. W., and Hechemy, K. E. (1997). Ultrastructural and antigenic characterization of a granulocytic ehrlichiosis agent directly isolated and stably cultivated from a patient in New York state. J. Infect. Dis. 175, 210–213.
Seo, G. M., Cheng, C., Tomich, J., and Ganta, R. R. (2008). Total, membrane, and immunogenic proteomes of macrophage- and tick cell-derived Ehrlichia chaffeensis evaluated by liquid chromatography-tandem mass spectrometry and MALDI-TOF methods. Infect. Immun. 76, 4823–4832.
Shi, L., Adkins, J. N., Coleman, J. R., Schepmoes, A. A., Dohnkova, A., Mottaz, H. M., Norbeck, A.D., Purvine, S.O., Manes, N.P., Smallwood, H.S., Wang, H., Forbes, J., Gros, P., Uzzau, S., Rodland, K. D., Heffron, F., Smith, R. D., and Squier, T. C. (2006). Proteomic analysis of Salmonella enterica serovar typhimurium isolated from RAW 264.7 macrophages: identification of a novel protein that contributes to the replication of serovar typhimurium inside macrophages. J. Biol. Chem. 281, 29131–29140.
Shi, L., Chowdhury, S. M., Smallwood, H. S., Yoon, H., Mottaz-Brewer, H. M., Norbeck, A. D., Mcdermott, J. E., Clauss, T. R., Heffron, F., Smith, R. D., and Adkins, J. N. (2009). Proteomic investigation of the time course responses of RAW 264.7 macrophages to infection with Salmonella enterica. Infect. Immun. 77, 3227–3233.
Singu, V., Liu, H., Cheng, C., and Ganta, R. R. (2005). Ehrlichia chaffeensis expresses macrophage- and tick cell-specific 28-kilodalton outer membrane proteins. Infect. Immun. 73, 79–87.
Skovgaard, M., Jensen, L. J., Brunak, S., Ussery, D., and Krogh, A. (2001). On the total number of genes and their length distribution in complete microbial genomes. Trends Genet. 17, 425–428.
Strittmatter, E. F., Kangas, L. J., Petritis, K., Mottaz, H. M., Anderson, G. A., Shen, Y., Jacobs, J. M., Camp, D. G. II, and Smith, R. D. (2004). Application of peptide LC retention time information in a discriminant function for peptide identification by tandem mass spectrometry. J. Proteome Res. 3, 760–769.
Sukumaran, B., Carlyon, J. A., Cai, J. L., Berliner, N., and Fikrig, E. (2005). Early transcriptional response of human neutrophils to Anaplasma phagocytophilum infection. Infect. Immun. 73, 8089–8099.
Sukumaran, B., Mastronunzio, J. E., Narasimhan, S., Fankhauser, S., Uchil, P. D., Levy, R., Graham, M., Colpitts, T. M., Lesser, C. F., and Fikrig, E. (2011). Anaplasma phagocytophilum AptA modulates Erk1/2 signalling. Cell. Microbiol. 13, 47–61.
Sultana, H., Neelakanta, G., Kantor, F. S., Malawista, S. E., Fish, D., Montgomery, R. R., and Fikrig, E. (2010). Anaplasma phagocytophilum induces actin phosphorylation to selectively regulate gene transcription in Ixodes scapularis ticks. J. Exp. Med. 207, 1727–1743.
Thomas, R. J., Dumler, J. S., and Carlyon, J. A. (2009). Current management of human granulocytic anaplasmosis, human monocytic ehrlichiosis and Ehrlichia ewingii ehrlichiosis. Expert Rev. Anti. Infect. Ther. 7, 709–722.
Unver, A., Rikihisa, Y., Stich, R. W., Ohashi, N., and Felek, S. (2002). The omp-1 major outer membrane multigene family of Ehrlichia chaffeensis is differentially expressed in canine and tick hosts. Infect. Immun. 70, 4701–4704.
Wakeel, A., Kuriakose, J. A., and McBride, J. W. (2009). An Ehrlichia chaffeensis tandem repeat protein interacts with multiple host targets involved in cell signaling, transcriptional regulation, and vesicle trafficking. Infect. Immun. 77, 1734–1745.
Wakeel, A., Zhang, X., and McBride, J. W. (2010). Mass spectrometric analysis of Ehrlichia chaffeensis tandem repeat proteins reveals evidence of phosphorylation and absence of glycosylation. PLoS ONE 5, e9552. doi: 10.1371/journal.pone.0009552
Wang, X., Cheng, Z., Zhang, C., Kikuchi, T., and Rikihisa, Y. (2007). Anaplasma phagocytophilum p44 mRNA expression is differentially regulated in mammalian and tick host cells: involvement of the DNA binding protein ApxR. J. Bacteriol. 189, 8651–8659.
Wang, X., Rikihisa, Y., Lai, T. H., Kumagai, Y., Zhi, N., and Reed, S. M. (2004). Rapid sequential changeover of expressed p44 genes during the acute phase of Anaplasma phagocytophilum infection in horses. Infect. Immun. 72, 6852–6859.
Washburn, M. P., Wolters, D., and Yates, J. R. III. (2001). Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247.
Zhang, J. Z., Guo, H., Winslow, G. M., and Yu, X. J. (2004). Expression of members of the 28-kilodalton major outer membrane protein family of Ehrlichia chaffeensis during persistent infection. Infect. Immun. 72, 4336–4343.
Zhi, N., Ohashi, N., Tajima, T., Mott, J., Stich, R. W., Grover, D., Telford, S. R. III, Lin, Q., and Rikihisa, Y. (2002). Transcript heterogeneity of the p44 multigene family in a human granulocytic ehrlichiosis agent transmitted by ticks. Infect. Immun. 70, 1175–1184.
Zhu, B., Nethery, K. A., Kuriakose, J. A., Wakeel, A., Zhang, X., and McBride, J. W. (2009). Nuclear translocated Ehrlichia chaffeensis ankyrin protein interacts with the mid a-stretch of host promoter and intronic Alu elements. Infect. Immun. 77, 4243–4255.
Zientz, E., Dandekar, T., and Gross, R. (2004). Metabolic interdependence of obligate intracellular bacteria and their insect hosts. Microbiol. Mol. Biol. Rev. 68, 745–770.
Keywords: Anaplasma phagocytophilum, Ehrlichia chaffeensis, proteomic analysis, human granulocytic anaplasmosis, human monocytic ehrlichiosis, human leukocytes
Citation: Lin M, Kikuchi T, Brewer HM, Norbeck AD and Rikihisa Y (2011) Global proteomic analysis of two tick-borne emerging zoonotic agents: Anaplasma phagocytophilum and Ehrlichia chaffeensis. Front. Microbio. 2:24. doi: 10.3389/fmicb.2011.00024
Received: 19 December 2010;
Accepted: 31 January 2011;
Published online: 17 February 2011.
Edited by:
Robert Heinzen, NIH/NIAID-RML, USAReviewed by:
Michael F. Minnick, University of Montana, USAJere W. McBride, University of Texas Medical Branch, USA
Sanjeev K. Sahni, University of Rochester School of Medicine and Dentistry, USA
Copyright: © 2011 Lin, Kikuchi, Brewer, Norbeck and Rikihisa. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Yasuko Rikihisa, Department of Veterinary Biosciences, The Ohio State University, 1925 Coffey Road, Columbus, OH 43210, USA. e-mail:cmlraWhpc2EuMUBvc3UuZWR1