
 Brandon Feinen
Brandon Feinen
- Department of Microbiology and Immunology, Witebsky Center for Microbial Pathogenesis and Immunology, University at Buffalo, Buffalo, NY, USA
It is well-known that gonorrhea can be acquired repeatedly with no apparent development of protective immunity arising from previous episodes of infection. Symptomatic infection is characterized by a purulent exudate, but the host response mechanisms are poorly understood. While the remarkable antigenic variability displayed by Neisseria gonorrhoeae and its capacity to inhibit complement activation allow it to evade destruction by the host’s immune defenses, we propose that it also has the capacity to avoid inducing specific immune responses. In a mouse model of vaginal gonococcal infection, N. gonorrhoeae elicits Th17-driven inflammatory–immune responses, which recruit innate defense mechanisms including an influx of neutrophils. Concomitantly, N. gonorrhoeae suppresses Th1- and Th2-dependent adaptive immunity, including specific antibody responses, through a mechanism involving TGF-β and regulatory T cells. Blockade of TGF-β alleviates the suppression of specific anti-gonococcal responses and allows Th1 and Th2 responses to emerge with the generation of immune memory and protective immunity. Genital tract tissues are naturally rich in TGF-β, which fosters an immunosuppressive environment that is important in reproduction. In exploiting this niche, N. gonorrhoeae exemplifies a well-adapted pathogen that proactively elicits from its host innate responses that it can survive and concomitantly suppresses adaptive immunity. Comprehension of these mechanisms of gonococcal pathogenesis should allow the development of novel approaches to therapy and facilitate the development of an effective vaccine.
Introduction
It is now well recognized that microbial pathogenesis involves a two-way interaction with the host immune system. Numerous examples are known of the ways in which bacteria resist or evade host defense mechanisms, and in which host responses attempt to counteract the efforts of pathogens to overcome them. This concept of the dynamic response–reaction paradigm of infectious disease has been held to underpin the emergence of the discipline of Cellular Microbiology. It is conventionally imagined that bacteria invade and attack the host, the host then mounts an immune response, the bacteria counterattack or deploy evasive strategies to which the host responds further, and so forth until one gains the upper hand such that either the pathogen is eliminated and the host is cured of the infection, or conversely that the host succumbs and ultimately dies. A third outcome can be a form of stalemate, in which both pathogen and host survive, in a form of chronic infection with ongoing pathology, or even an asymptomatic carrier state in which the host appears to suffer no ill effects. In the first instance it is supposed that the host response proves superior, in the second it is inadequate, while in the third some degree of balance is struck. In all three scenarios, vaccination aims to enhance the host immune response particularly by exploiting the phenomenon of immune memory in order to curtail the infection at an early point or shorten its course and mitigate disease severity. Numerous vaccines have been very successfully developed and deployed on the basis of the paradigm that vaccination mimics the natural infection without actually causing disease and thereby stimulates the host to mount adaptive immune responses generally in the form of specific antibodies against the pathogen.
Several aspects of gonorrhea, however, suggest that this is an oversimplistic view of infectious pathogenesis. In the first place, it has proven very difficult, despite efforts spanning a century, to develop an effective vaccine against Neisseria gonorrhoeae (Russell and Hook, 2009). Furthermore, it is clear that one of the essential preconditions for the standard vaccine paradigm is not met, namely that recovery from the disease confers a state of immunity against future infection. It is well-known that gonorrhea can be acquired repeatedly without any apparent diminution in probability of infection from exposure, or the severity or duration of the disease (Noble et al., 1977). Gonorrhea is not unique in this, as there are many examples of infectious diseases that can be acquired repeatedly including influenza, rubella, and even the common cold. The generally accepted explanation for this situation is the diversity of pathogens capable of causing the syndrome, or the antigenic variation of particular pathogens. This is the conventional view of the immune response to gonorrhea (Table 1).

Table 1. Conventional view of immunity to gonorrhea*.
Our recent findings, however, support an alternative or additional hypothesis. In the first place, quantitative studies of circulating and local mucosal antibody responses to naturally acquired uncomplicated gonorrhea (i.e., gonococcal cervicitis in women, and urethritis in men, both confirmed by microbiological testing) led to the conclusion that the human adaptive immune response to the infection is minimal (Hedges et al., 1998, 1999). A modest increase in antibodies measured against the homologous clinical isolate was seen in some individuals, but responses were of short duration, and showed no relation to documented previous infections (Hedges et al., 1999). Furthermore, there was no substantially increased response in subjects who had rectal involvement, in a site where mucosal immune inductive tissues (lymphoid follicles) are abundant, in contrast to the genital tract. Yet some women, especially those having co-infection with Chlamydia trachomatis or Trichomonas vaginalis, showed considerably elevated inflammatory cytokine responses (IL-1, IL-6, IL-8, and IL-10), testifying to an acute inflammatory response (Hedges et al., 1998). Overall these findings led us to hypothesize that “gonococci avoid inducing humoral immune responses during uncomplicated natural infections … [and that] gonococci use an as yet undefined mechanism of protection which may subvert the natural immune response.” (Hedges et al., 1999).
The discovery of the novel subset of helper T cells designated Th17, on account of their ability to produce the inflammatory cytokine IL-17 (Harrington et al., 2005; Park et al., 2005), provided the basis for a new concept in pathogenesis particularly for extracellular and mucosal bacterial pathogens such as N. gonorrhoeae (Curtis and Way, 2009; Khader et al., 2009). When IL-17 activates cells that carry its receptor, IL-17R, these cells (typically endothelial and stromal cells) secrete other inflammatory cytokines including TNF-α, as well as granulocyte colony-stimulating factor (GCSF), granulocyte-monocyte colony-stimulating factor (GMCSF), and CXC chemokines (IL-8 in humans, KC, LIX, and MIP-2α in mice) which mobilize neutrophils from bone marrow and recruit them to the inflammatory focus (Kolls and Linden, 2004; Kolls et al., 2008). In addition, epithelial cells at mucosal surfaces respond to stimulation with IL-17 and IL-22 (another cytokine typically produced by Th17 cells) by upregulating the secretion of antimicrobial defense peptides including defensins, S100 proteins, and lipocalin-2 (Ouyang and Valdez, 2008). Thus Th17 cells stand at the interface of the adaptive and innate immune systems, and activate potent innate defense mechanisms, both cellular (especially neutrophils) and molecular. IL-17 is also highly inflammatory in its mode of action, and has been heavily implicated in chronic inflammatory and autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease. Thus, like many potent immune mechanisms, Th17 cell activation has beneficial as well as harmful consequences, depending on the duration of responses, and the effectiveness of their control mechanisms (Onishi and Gaffen, 2010). Nevertheless, it is clear that Th17 responses play a major role in defense against bacterial infections. The evidence for this role in gonococcal infection is summarized below.
Th17 Response to N. gonorrhoeae in the Mouse Model
We were initially inspired to investigate Th17 responses to gonococcal infection following findings that mice show an IL-17-dependent neutrophil response to the periodontal pathogen, Porphyromonas gingivalis (Yu et al., 2007). In these studies, neutrophils were recruited in response to CXC chemokines generated by cells responding to stimulation with IL-17. Mice lacking the principal receptor for IL-17 (IL-17RA-knockout or IL-17RA-ko mice) were more susceptible to oral infection with P. gingivalis and had increased periodontal bone loss as a result. Furthermore they showed reduced levels of CXC chemokines KC and LIX in response to P. gingivalis and had diminished infiltration of neutrophils into the gingival tissue. Given that a neutrophil-rich discharge is a classic diagnostic criterion of gonorrhea, we were impelled to determine whether IL-17 had a similar role in the response to gonococcal infection.
In order first to establish whether N. gonorrhoeae is capable of inducing cytokines characteristic of a Th17 response, in vitro cell culture studies were conducted in which mouse spleen mononuclear cells were incubated with N. gonorrhoeae. These showed that IL-17A, IL-22, and other cytokines typical of an innate and inflammatory response (IL-1β, IL-6, TNF-α) were released into the supernatant in a time- and dose-dependent manner (Feinen et al., 2010). Notably, there was a lack of cytokines typical of Th1 or Th2 responses, i.e., IFN-γ, IL-12, IL-4, and the little IFN-γ that was secreted came not from CD4+ T cells but from innate NK cells. The ability of N. gonorrhoeae to induce secretion of IL-17, IL-22, and IL-6, but not IFN-γ in mouse spleen cell cultures, suggests that it is capable of eliciting Th17 responses. There were in fact two sources of IL-17: both CD4+ T cells, i.e., Th17 cells, and T cells bearing the alternative γδ T cell receptor which belong to the innate immune system and are abundant at mucosal surfaces, including the genital tract. The responses were not confined to one strain of N. gonorrhoeae and were not dependent on live gonococci, as similar results were seen with strains FA1090, MS11, and PID-2, and could be replicated with outer membrane vesicle (OMV) preparations which contain most of the surface components of the intact gonococci and are naturally shed by the live organisms (Feinen et al., 2010).
This raises the issue of which gonococcal surface components are responsible for eliciting the IL-17 response, and indeed whether it is “specific” to N. gonorrhoeae. With regard to the latter question, there are now numerous species of bacteria, fungi, protozoa, and even viruses that have been reported to induce Th17 responses in animal models and humans. Bacterial species include the afore-mentioned P. gingivalis, Klebsiella pneumoniae, Escherichia coli, Citrobacter rodentium, Bordetella pertussis, Mycobacterium tuberculosis, Helicobacter pylori, Salmonella enterica, and many others, as well as the yeast Candida albicans and the protozoan Toxoplasma gondii (Higgins et al., 2006; Shibata et al., 2007; Yu et al., 2007; Caruso et al., 2008; Conti et al., 2009; Godinez et al., 2009; Khader et al., 2009). Thus it is not a unique response specific to N. gonorrhoeae, although the consequences for different infections and the implications in particular for human disease have not been fully explored. While Th17 cells may be induced by nominal antigens, the key factors in Th17 differentiation are cytokines, especially TGF-β and IL-6 as discussed below. Among gonococcal surface components, we found that the effect of whole gonococci (or OMV) can be to a large extent replicated by gonococcal lipo-oligosaccharide (LOS), which induces a similar level of IL-17 production by murine spleen cells in vitro (Feinen et al., 2010). Signaling through TLR4 is important, since TLR4-deficient mouse cells did not respond to gonococcal LOS and showed diminished responses to gonococci, whereas TLR2-knockout mouse cells responded similarly to wild-type mouse cells. Interestingly, gonococcal LOS having different terminal glycan residues have been reported to induce Th1, Th2, or Th17 responses according to their interactions with different lectin receptors on dendritic cells (van Vliet et al., 2009). Heat-treatment of OMV did not affect their ability to elicit IL-17 responses in vitro, suggesting that proteins might not be integral to this response but consistent with the role of LOS. An Opa protein deficient mutant in which all opa genes have been eliminated (Jerse et al., 1994) was still capable of inducing IL-17 responses (Feinen et al., 2010).
As a first step in determining whether findings based on mouse spleen cells in vitro are applicable to the genital tract, we made use of genital tract tissue explants which can be cultured for up to 5 days ex vivo with N. gonorrhoeae (Feinen et al., 2010). These cultures showed that N. gonorrhoeae could induce IL-17 and IL-22, as well as other inflammatory cytokines such as IL-6, but not those typical of Th1 or Th2 responses (IFN-γ and IL-4, respectively), in genital tract tissue. Furthermore, the CXC chemokines KC (CXCL1), LIX (CXCL5), and MIP-2α (CXCL2) were also secreted in response to N. gonorrhoeae. These are the murine equivalent of IL-8 (CXCL8) which does not occur in mice, and are chemoattractant for neutrophils.
In order to determine whether a Th17 response occurs in response to gonococcal infection, the murine model (Jerse, 1999) was employed to examine the immune response to N. gonorrhoeae in vivo. In this model, gonococcal infection of the genital tract persists for 1–2 weeks, and it elicits a local influx of neutrophils and the production of cytokines (Jerse, 1999; Song et al., 2008; Packiam et al., 2010). In addition, the availability of IL-17RA-ko mice (Ye et al., 2001) allowed us to determine the role of IL-17 in the course of infection and the neutrophil influx. Whereas wild-type control mice started to eliminate N. gonorrhoeae from day 4 onwards and cleared the infection by about day 7, in IL-17RA-ko mice reduction of the gonococcal load was delayed until day 9 and it took about 12 days to clear the infection (Feinen et al., 2010). The neutrophil influx, which began on day 3 in control mice, was almost completely abrogated in IL-17RA-ko mice. These results indicate that IL-17RA is important in neutrophil recruitment and in clearance of N. gonorrhoeae in this mouse model. Similar findings were obtained when mice were treated with blocking antibody to IL-17A during gonococcal infection, although the neutrophil influx was diminished but not abrogated in these experiments (Feinen et al., 2010).
The local generation of IL-17 in the genital tract was first revealed by culturing cells from the draining iliac lymph nodes taken on 1–5 days after infection; the cultures released IL-17 into the supernatants (Feinen et al., 2010). Further studies of cells isolated from the genital tracts of infected mice showed that γδ T cells were abundant and accounted for a large proportion of the IL-17 production, in addition to CD4+ Th17 cells (Liu, Y. and Russell, M.W., in preparation). Gene-expression profiling of mouse genital tract tissue revealed upregulation of genes associated with Th17 and innate immunity in response to N. gonorrhoeae, but not those associated with Th1- or Th2-driven adaptive immunity (Liu, Y. and Russell, M.W., in preparation). Thus the local response of the genital tract to infection with N. gonorrhoeae parallels the response of spleen cells, and is dominated by IL-17-driven innate inflammatory responses, with consequent recruitment of innate defense proteins and phagocytic cells, predominantly neutrophils. These findings are in accord with other studies of the mouse genital tract infection model showing that neutrophils are the predominant infiltrating cell type, with smaller numbers of macrophages, and little or no induction of a specific antibody response (Song et al., 2008). They are also reminiscent of the response in humans where inflammatory cytokines such as IL-1, IL-6, TNF-α (Hedges et al., 1998) but little or no IL-12, IFN-γ, IL-4, or IL-5 have been reported (Ramsey et al., 1995; Naumann et al., 1997; Simpson et al., 1999; Fichorova et al., 2001; Makepeace et al., 2001). Moreover, elevation of IL-17 and IL-23, which is important for the functional development of Th17 cells (Weaver et al., 2007), has now been reported in humans with gonorrhea (Gagliardi et al., 2011).
It is important to note that, while the neutrophil influx in the murine gonococcal infection model is abrogated in IL-17RA-ko mice with concomitant prolongation of the infection, it is unlikely that clearance is dependent on the neutrophils: the infection is still cleared in IL-17RA-ko mice (Feinen et al., 2010). Moreover, the extent of neutrophil influx varies considerably between mouse strains and bears no relation to susceptibility to gonococcal infection (Packiam et al., 2010). Numerous factors undoubtedly contribute to the inability of N. gonorrhoeae to persist in mice, in contrast to humans to which it has become specifically adapted.
Role of TGF-β in Suppressing Th1/Th2 Responses to N. gonorrhoeae
CD4+ T cells differentiate along at least four different pathways or lineages depending upon the cytokine milieu in which they are stimulated (Weaver et al., 2007). In the presence of IL-12, they differentiate as Th1 cells, whereas in the presence of IL-4 they become Th2 cells. Both of these are “classical” T-helper cells that are critical for adaptive immune responses whether cell-mediated (e.g., by CD8+ cytotoxic T cells) or antibody production by plasma cells. Th1 and Th2 cells are cross-regulatory, as IFN-γ produced by Th1 cells suppresses Th2 development, while IL-4 from Th2 cells suppresses Th1 development. Th17 cells differentiate in the presence of TGF-β and an inflammatory cytokine such as IL-6 or IL-1; IL-23 is further required for the maintenance and functional differentiation of Th17 cells. Both IFN-γ and IL-4 suppress Th17 development but there is no confirmed evidence for the suppression of Th1 or Th2 cells by products of Th17 cells. However, when precursor T cells are stimulated in the presence of TGF-β alone they become induced T regulatory (Treg) cells which regulate Th1 and Th2 cells (Bettelli et al., 2006). Thus TGF-β is critical for the development of both Th17 and Treg cells, the essential difference being whether or not inflammatory cytokines are also present. TGF-β is also well-known as a regulatory cytokine that is directly inhibitory for Th1 and Th2 cells. Furthermore, female genital tract tissues are known to be rich in TGF-β (Wira and Rossoll, 2003; Shen et al., 2007) which is involved in maintaining an immunosuppressive environment that is important for reproductive physiology, in particular, the admission of allogeneic sperm and the implantation of a semi-allogeneic fetus (Russell and Mestecky, 2010; Wira et al., 2010).
We therefore hypothesized that N. gonorrhoeae is capable of inducing the production of TGF-β, which in turn promotes the development of both Th17 and Treg cells. Indeed, the development of Treg cells in the mouse model of genital gonococcal infection has already been reported (Imarai et al., 2008). Studies of the interaction of N. gonorrhoeae with mouse spleen cells in vitro show that TGF-β is among the cytokines induced, and that it is produced by several different cell types, especially B cells, but also by T cells, macrophages, and dendritic cells (Liu et al., submitted). Further studies with genital tract tissue cultured in vitro, and in the mouse genital tract in vivo, showed that N. gonorrhoeae further elevated the generation of TGF-β in the genital tract. Moreover, N. gonorrhoeae inhibited the in vitro proliferation and differentiation of Th1 and Th2 cells with their characteristic cytokine production by a TGF-β-dependent mechanism. This effect was reversed in the presence of blocking antibodies to TGF-β, and under these conditions, Th1 and Th2 cells developed and secreted IFN-γ and IL-4, respectively.
The effect of TGF-β-blocking antibody could also be observed in vivo in the genital tract infection model. When mice were treated with anti-TGF-β antibody during infection with N. gonorrhoeae, the duration of infection was significantly shortened by about 4 days, and both Th1 and Th2 responses were generated in the genital tract or its draining lymph nodes (Liu, Y. and Russell, M.W., in preparation). Normally, when mice that have recovered from primary infection are later challenged with a secondary infection of N. gonorrhoeae, the duration of the infection follows the same kinetics as primary infection, and there is no significant antibody response resulting from either primary or secondary infection (Song et al., 2008). This implies that in mice, as in humans, there is no effective generation of immune memory to gonococcal infection, and consequently no protective immunity develops against genital tract infection. However, if mice that have been treated with anti-TGF-β antibody during primary infection are then re-challenged (without further treatment with anti-TGF-β antibody), the secondary infection is resisted and is cleared more rapidly than in control mice that have not received anti-TGF-β treatment during primary infection. Analysis of the immune responses developed in these mice showed that the anti-TGF-β-treated animals develop both Th1 and Th2 cell responses, as well as anti-gonococcal antibodies in the circulation (IgG) and vaginal fluid (IgG and IgA). Thus the alleviation of N. gonorrhoeae-induced immunosuppression by means of anti-TGF-β antibody treatment results in the generation of adaptive anti-gonococcal immune responses and protective immunity to N. gonorrhoeae (Liu, Y. and Russell, M.W., in preparation).
A New Hypothesis for Gonococcal–Host Interactions
Neisseria gonorrhoeae has probably been associated with humans for several millennia, and through evolution has become extremely well-adapted to the human immune system. Thus it has evolved the capacity to cope with human innate defense mechanisms such as defensins and other secreted anti-microbial proteins as well as non-opsonic phagocytosis by neutrophils. There is evidence that multiple drug exporter mechanisms enable gonococci to resist defensins (Shafer et al., 1998). Gonococci are able to survive at least partially within neutrophils (Casey et al., 1979); indeed they utilize pathogen-directed endocytosis to invade neutrophils and other cells (Jerse and Rest, 1997). Recent findings indicate that N. gonorrhoeae can survive oxygen-dependent and non-oxidative intracellular killing mechanisms within neutrophils (Criss and Seifert, 2008; Criss et al., 2009). Notably in this context, N. gonorrhoeae has multiple mechanisms that inhibit the activation of human complement, especially by the alternate pathway, and that prevent lysis by the membrane-attack complex of complement (Ram et al., 1998, 2001; Lewis et al., 2010). This not only allows it to escape from direct complement-mediated bacteriolysis, but also helps it to evade C3b-mediated opsonization and phagocytosis by neutrophils. An old body of literature indicates that antibody- and complement-mediated opsonophagocytosis is more effective in killing ingested microbes than non-opsonic phagocytosis.
Neisseria gonorrhoeae has evolved potent mechanisms for avoiding destruction by adaptive immune responses, especially specific antibodies. An obvious immune evasion strategy is its extraordinary capacity for antigenic variation, in which most of its major surface components are subject to variable expression through multiple mechanisms. These include genetic polymorphism (porins, Opa proteins, transferrin-binding proteins), phase-variable on–off switching of gene-expression either directly (Opa proteins, pili) or indirectly (LOS, through expression of enzymes involved in biosynthesis of the glycan chains), recombination of gene segments (pilin structural proteins), as well as natural competence for DNA uptake and DNA secretion that facilitate frequent horizontal gene exchange (Yang and Gotschlich, 1996; Dehio et al., 1998; Massari et al., 2003; Cornelissen, 2008; Maiden, 2008; Hill and Davies, 2009; Virji, 2009). Thus it can be argued that if the host mounts antibody responses to these components, their constantly shifting expression and antigenicity prevent recognition and binding of antibodies to the gonococcal surface. Some evidence in favor of this scenario can be seen in studies on highly exposed sex-workers in Nairobi, Kenya. Partial serovar (porin)-specific immunity to re-infection with the prevalent serovar, and reduced susceptibility to salpingitis associated with anti-Opa antibodies have been reported (Plummer et al., 1989, 1994). However, these findings were not replicated elsewhere (Fox et al., 1999), possibly because of a lower frequency of infection. Antibodies to porin or Opa proteins can mediate complement- or phagocyte-dependent killing of gonococci that express the homologous (or cross-reactive) antigens (Virji and Heckels, 1985; Heckels et al., 1989), but additional mechanisms of resistance are known. These include antibodies to the reduction-modifiable protein (RMP) which is closely associated with porin; these antibodies block bacteriolysis mediated by anti-porin antibody (Rice et al., 1986; Virji and Heckels, 1988). Sialylation of LOS also inhibits complement-mediated lysis (Wetzler et al., 1992a; Smith et al., 1995). Nevertheless, purified porin formed the basis for the development of a potential serovar-specific vaccine (Wetzler et al., 1992b). The outcome of the pilus vaccine effort also supports the concept of immune evasion by antigenic variation. This vaccine, based on the pilus structural protein (pilin) was successful in generating antibodies against homologous pilus protein, and antibodies to pilin could inhibit attachment of gonococci to epithelial cells (Tramont et al., 1981). However, the vaccine proved completely ineffective in a field trial because of the very high antigenic variability of pilin protein among naturally occurring gonococcal strains (Boslego et al., 1991).
As effective as antigenic variation may be in evading the consequences of adaptive immune responses, we further propose that N. gonorrhoeae avoids the generation of specific antibodies in the first place (Table 2). This was initially based on our observations of minimal antibody responses to uncomplicated gonorrhea in humans, despite symptomatic inflammatory disease and in some cases strong cytokine responses (Hedges et al., 1998, 1999). The human response appears to be quite well replicated in the mouse model of vaginal gonococcal infection, in that there is a neutrophil-dominated cellular infiltrate, inflammatory cytokines are induced through TLR4-dependent mechanisms, no antibody responses are detectable in the serum or genital secretions and tissues, and no protective immunity is generated against secondary infection with the same strain (Song et al., 2008; Packiam et al., 2010). We find that the LOS-TLR4-dependent inflammatory response involves IL-17 and Th17-driven innate responses, and that interference with these leads to diminished neutrophil infiltration and prolongation of the infection in mice (Feinen et al., 2010).
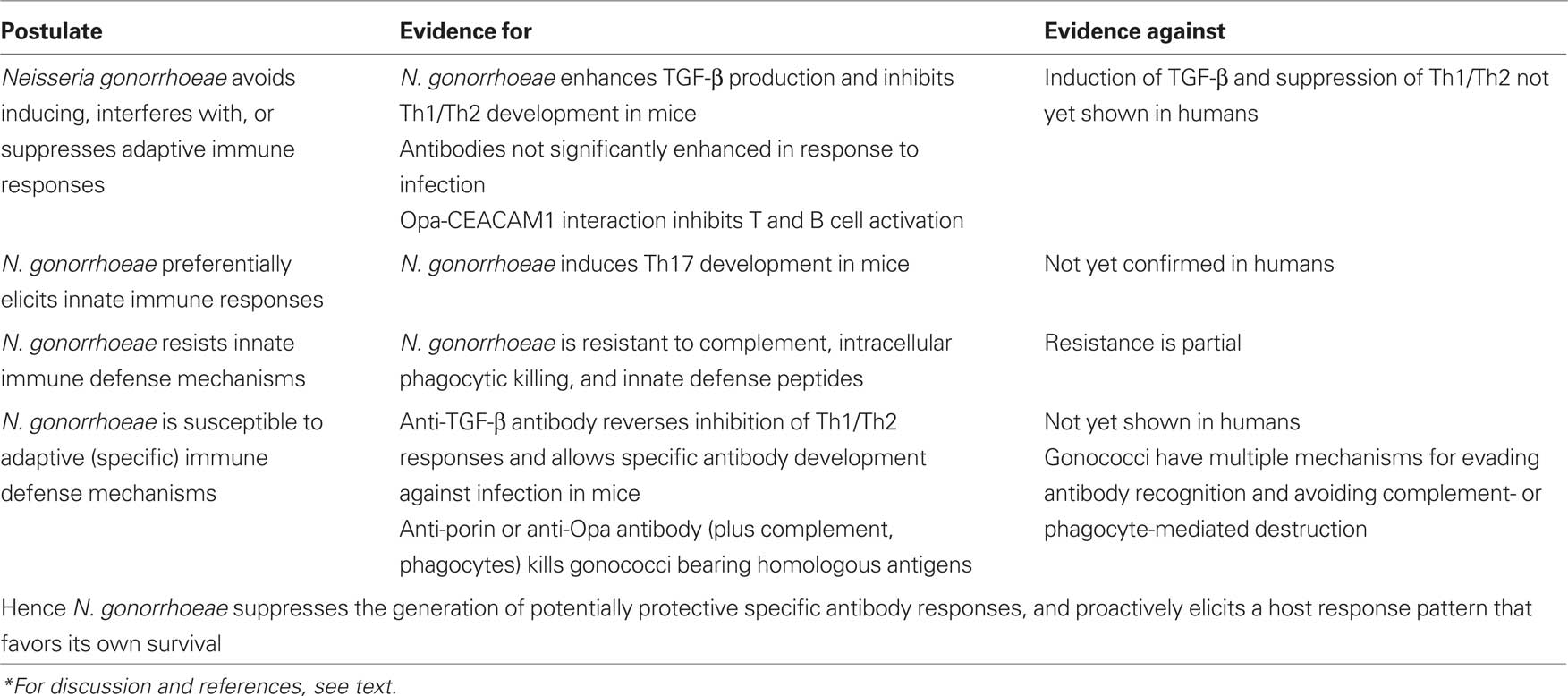
Table 2. New hypothesis concerning immunity to gonorrhea*.
Initial support for the concept of gonococcal suppression of host immune responses was provided by the findings that CEACAM1-binding Opa proteins inhibit human T cell activation and B cell differentiation (Boulton and Gray-Owen, 2002; Pantelic et al., 2005), although this was not confirmed in another study (Youssef et al., 2009). The mouse homolog of human CEACAM1, however, does not possess the residues found to be critical for Opa recognition (Virji et al., 1999). Our own studies on mouse immune cells in vitro, and on genital tract tissues from infected mice, show that N. gonorrhoeae can exploit TGF-β to suppress adaptive immune responses driven by Th1 and Th2 cells. There is preliminary evidence that this involves Treg cells which have been reported to be induced in the mouse model of gonococcal infection (Imarai et al., 2008), but this mechanism remains to be fully investigated. Blockade of TGF-β both in vitro and in vivo alleviates immunosuppression elicited by N. gonorrhoeae and permits the generation of Th1- and Th2-governed responses, with the development of immune memory and anti-gonococcal antibodies, and protection against re-infection. Thus we propose not only that N. gonorrhoeae elicits from its host Th17-driven innate responses that it can resist, but also that it suppresses adaptive responses that might be capable of eliminating it. In other words, N. gonorrhoeae does not merely react to the host’s immune responses, but it proactively elicits a pattern of immune responses that is favorable to its own survival, not what is desirable for the host. Some possible mechanisms by which this is accomplished are illustrated in Figure 1.
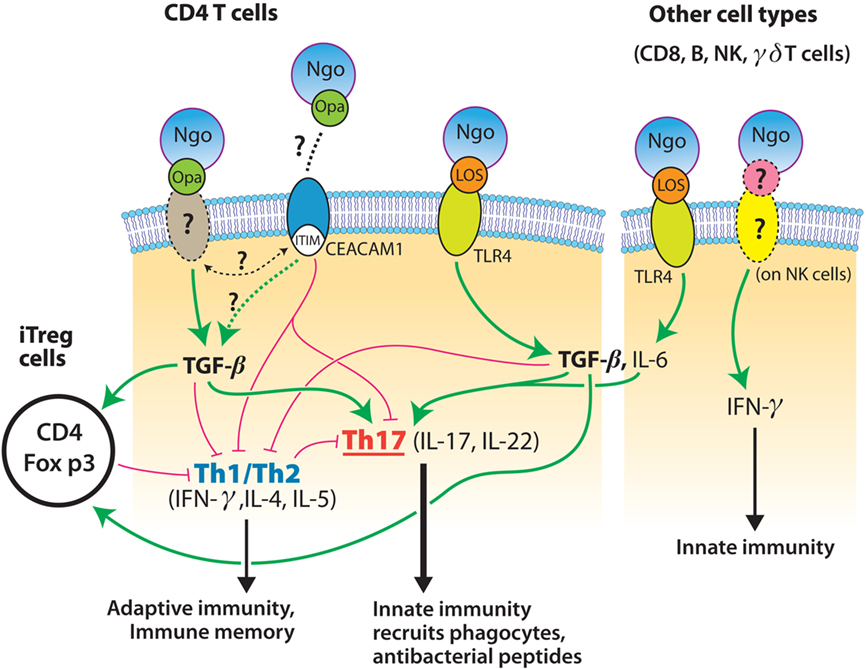
Figure 1. Model for mechanisms of interaction of N. gonorrhoeae (Ngo) with cells of the immune system. Gonococcal LOS interacts with CD4+ T cells and other cells through TLR4 to induce the production of IL-6 and TGF-β which drive the development of Th17 cells; IFN-γ is also secreted by NK cells. Gonococcal Opa proteins interact with an unknown receptor on mouse cells (or with CEACAM1 on human cells), leading to the increased production of TGF-β, which directly suppresses Th1 and Th2 cells, and also enhances the development of Treg cells which regulate Th1 and Th2 cells. Blockade of TGF-β thus relieves the suppression of Th1 and Th2 cells, which then drive adaptive immune responses, leading to memory, and antibody production.
How this relates to the human infection is unclear at present, because studies of Th17-dependent responses to gonococcal infection have not yet been conducted. We hypothesize that IL-17-driven innate responses are inadequate to eliminate gonococcal infection in humans and that potentially protective adaptive immune responses are suppressed, but this remains to be tested. However, increased serum levels of IL-17 and IL-23 have now been reported in men infected with gonorrhea (Gagliardi et al., 2011). It must be borne in mind that there are numerous differences between mouse and human systems, and that several of the known pathogenic mechanisms deployed by N. gonorrhoeae are specific to human cells or proteins. N. gonorrhoeae is not a natural pathogen of mice which do not develop signs of disease due to genital tract infection, the infection does not usually persist in mice for more than 1–2 weeks, and the mechanisms by which gonococci are eliminated from the genital tract in either mice or humans are not known. The murine studies cannot be directly replicated in humans for ethical and practical reasons, but if evidence can be obtained that human immune cells respond to N. gonorrhoeae in a similar way to murine cells, and that the natural infection of humans with gonorrhea elicits Th17 and/or TGF-β and Treg responses, then new approaches to therapy and vaccine development can be anticipated. For example, TGF-β or Treg cells might be targeted by novel therapeutics to alleviate gonococcal immunosuppression, although caution will be needed to avoid adverse consequences, such as autoimmunity, allergy, or other manifestations of undesirable immune responses that are held in check by immunoregulatory mechanisms. In vaccine development, it is clear that the standard paradigm, of seeking to mimic the natural infection without causing disease in order to elicit protective immunity, is inapplicable because the natural infection does not induce protective immunity, due to gonococcal antigenic variation and gonococcus-induced immunosuppression. Therefore, other strategies to induce effective immune responses against conserved gonococcal antigens and deliver them to the sites of infection will be needed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Studies in the authors’ laboratory were supported by USPHS grant AI074791 from the National Institute of Allergy and Infectious Diseases, and the John R. Oishei Foundation, Buffalo, NY, USA.
References
Bettelli, E., Carrier, Y., Gao, W., Korn, T., Strom, T. B., Oukka, M., Weiner, H. L., and Kuchroo, V. K. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238.
Boslego, J. W., Tramont, E. C., Chung, R. C., McChesney, D. G., Ciak, J., Sadoff, J. C., Piziak, M. V., Brown, J. D., Brinton, C. C., Wood, S. W., and Bryan, J. R. (1991). Efficacy trial of a parenteral gonococcal pilus vaccine in men. Vaccine 9, 154–162.
Boulton, I. C., and Gray-Owen, S. D. (2002). Neisserial binding to CEACAM1 arrests the activation and proliferation of CD4+ T lymphocytes. Nat. Immunol. 3, 229–236.
Caruso, R., Fina, D., Paoluzi, O. A., Blanco, G. D. V., Stolfi, C., Rizzo, A., Caprioli, F., Sarra, M., Andrei, F., Fantini, M. C., MacDonald, T. T., Pallone, F., and Monteleone, G. (2008). IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur. J. Immunol. 38, 470–478.
Casey, S. G., Veale, D. R., and Smith, H. (1979). Demonstration of intracellular growth of gonococci in human phagocytes using spectinomycin to kill extracellular organisms. J. Gen. Microbiol. 113, 395–398.
Conti, H. R., Shen, F., Nayyar, N., Stocum, E., Sun, J. N., Lindemann, M. J., Ho, A. W., Hai, J. H., Yu, J. J., Jung, J. W., Filler, S. G., Masso-Welch, P., Edgerton, M., and Gaffen, S. L. (2009). Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 206, 299–311.
Cornelissen, C. N. (2008). Identification and characterization of gonococcal iron transport systems as potential vaccine antigens. Future Microbiol. 3, 287–298.
Criss, A. K., Katz, B. Z., and Seifert, H. S. (2009). Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leucocytes. Cell. Microbiol. 11, 1074–1087.
Criss, A. K., and Seifert, H. S. (2008). Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell. Microbiol. 10, 2257–2270.
Curtis, M. M., and Way, S. S. (2009). Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 126, 177–185.
Dehio, C., Gray-Owen, S. D., and Meyer, T. F. (1998). The role of neisserial Opa proteins in interactions with host cells. Trends Microbiol. 6, 489–495.
Feinen, B., Jerse, A. E., Gaffen, S. L., and Russell, M. W. (2010). Critical role of Th17 responses in a murine model of Neisseria gonorrhoeae genital infection. Mucosal Immunol. 3, 312–321.
Fichorova, R. N., Desai, P. J., Gibson, F. C., and Genco, C. A. (2001). Distinct proinflammatory host responses to Neisseria gonorrhoeae infection in immortalized human cervical and vaginal epithelial cells. Infect. Immun. 69, 5840–5848.
Fox, K. K., Thomas, J. C., Weiner, D. H., Davis, R. H., Sparling, P. F., and Cohen, M. S. (1999). Longitudinal evaluation of serovar-specific immunity to Neisseria gonorrhoeae. Am. J. Epidemiol. 149, 353–358.
Gagliardi, M. C., Starnino, S., Teloni, R., Mariotti, S., Dal Conte, I., Di Carlo, A., and Stefanelli, P. (2011). Circulating levels of interleukin-17A and interleukin-23 are increased in patients with gonococcal infection. FEMS Immunol. Med. Microbiol. 61, 129–132.
Godinez, I., Raffatellu, M., Chu, H., Paixao, T. A., Haneda, T., Santos, R. L., Bevins, C. L., Tsolis, R. M., and Baumler, A. J. (2009). Interleukin-23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect. Immun. 77, 387–398.
Harrington, L. E., Hatton, R. D., Mangan, P. R., Turner, H., Murphy, T. L., Murphy, K. M., and Weaver, C. T. (2005). Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132.
Heckels, J. E., Fletcher, J. N., and Virji, M. (1989). The potential protective effect of immunization with outer-membrane protein I from Neisseria gonorrhoeae. J. Gen. Microbiol. 135, 2269–2276.
Hedges, S. R., Mayo, M. S., Mestecky, J., Hook, E. W., and Russell, M. W. (1999). Limited local and systemic antibody responses to Neisseria gonorrhoeae during uncomplicated genital infections. Infect. Immun. 67, 3937–3946.
Hedges, S. R., Sibley, D., Mayo, M. S., Hook, E. W., and Russell, M. W. (1998). Cytokine and antibody responses in women infected with Neisseria gonorrhoeae: effects of concomitant infections. J. Infect. Dis. 178, 742–751.
Higgins, S. C., Jarnicki, A. G., Lavelle, E. C., and Mills, K. H. G. (2006). TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T cells. J. Immunol. 177, 7980–7989.
Hill, S. A., and Davies, J. K. (2009). Pilin gene variation in Neisseria gonorrhoeae: reassessing the old paradigms. FEMS Microbiol. Rev. 33, 521–530.
Imarai, M., Candia, E., Rodriguez-Tirado, C., Tognarelli, J., Pardo, M., Perez, T., Valdes, D., Reyes-Cerpa, S., Nelson, P., Acuna-Castillo, C., and Maisey, K. (2008). Regulatory T cells are locally induced during intravaginal infection of mice with Neisseria gonorrhoeae. Infect. Immun. 76, 5456–5465.
Jerse, A. E. (1999). Experimental gonococcal genital tract infection and opacity protein expression in estradiol-treated mice. Infect. Immun. 67, 5699–5708.
Jerse, A. E., Cohen, M. S., Drown, P. M., Whicker, L. G., Isbey, S. F., Seifert, S., and Cannon, J. G. (1994). Multiple gonococcal opacity proteins are expressed during experimental urethral infection in the male. J. Exp. Med. 179, 911–920.
Jerse, A. E., and Rest, R. F. (1997). Adhesion and invasion by the pathogenic Neisseria. Trends Microbiol. 5, 217–221.
Khader, S. A., Gaffen, S. L., and Kolls, J. K. (2009). Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2, 403–411.
Kolls, J. K., and Linden, A. (2004). Interleukin-17 family members and inflammation. Immunity 21, 467–476.
Kolls, J. K., McCray, P. B., and Chan, Y. R. (2008). Cytokine-mediated regulation of antimicrobial proteins. Nat. Rev. Immunol. 8, 829–835.
Lewis, L. A., Burrowes, E., Rice, P. A., and Ram, S. (2010). “Interactions of Neisseria with complement,” in Neisseria: Molecular Mechanisms of Pathogenesis, eds. C. A. Genco and L. Wetzler (Norfolk: Caister Academic Press), 123–144.
Maiden, M. C. (2008). Population genomics: diversity and virulence in the Neisseria. Curr. Opin. Microbiol. 11, 467–471.
Makepeace, B. L., Watt, P. J., Heckels, J. E., and Christodoulides, M. (2001). Interactions of Neisseria gonorrhoeae with mature human macrophage opacity proteins influence production of proinflammatory cytokines. Infect. Immun. 69, 1909–1913.
Massari, P., Ram, S., Macleod, H., and Wetzler, L. M. (2003). The role of porins in neisserial pathogenesis and immunity. Trends Microbiol. 11, 87–93.
Naumann, M., Wessler, S., Bartsch, C., Wieland, B., and Meyer, T. F. (1997). Neisseria gonorrhoeae epithelial cell interaction leads to the activation of the transcription factors nuclear factor κB and activator protein 1 and the induction of inflammatory cytokines. J. Exp. Med. 186, 247–258.
Noble, R. C., Kirk, N. M., Slagel, W. A., Vance, B. J., and Somes, G. W. (1977). Recidivism among patients with gonococcal infection presenting to a venereal disease clinic. Sex. Transm. Dis. 4, 39–43.
Onishi, R. M., and Gaffen, S. L. (2010). Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology 129, 311–321.
Packiam, M., Veit, S. J., Anderson, D. J., Ingalls, R. R., and Jerse, A. E. (2010). Mouse strain-dependent differences in susceptibility to Neisseria gonorrhoeae infection and induction of innate immune responses. Infect. Immun. 78, 433–440.
Pantelic, M., Kim, Y.-J., Bolland, S., Chen, I., Shively, J., and Chen, T. (2005). Neisseria gonorrhoeae kills carcinoembryonic antigen-related cellular adhesion molecule 1 (CD66a)-expressing human B cells and inhibits antibody production. Infect. Immun. 73, 4171–4179.
Park, H., Li, X. R., Yang, X. O., Chang, S. H., Nurieva, R., Wang, Y.-H., Wang, Y., Hood, L., Zhu, Z., Tian, Q., and Dong, C. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6, 1133–1141.
Plummer, F. A., Chubb, H., Simonsen, J. N., Bosire, M., Slaney, L., Nagelkerke, N. J. D., Maclean, I., Ndinya-Achola, J., Waiyaki, P., and Brunham, R. C. (1994). Antibodies to opacity proteins (Opa) correlate with a reduced risk of gonococcal salpingitis. J. Clin. Invest. 93, 1748–1755.
Plummer, F. A., Simonsen, J. N., Chubb, H., Slaney, L., Kimata, J., Bosire, M., Ndinya-Achola, J. O., and Ngugi, E. N. (1989). Epidemiologic evidence for the development of serovar-specific immunity after gonococcal infection. J. Clin. Invest. 83, 1472–1476.
Ram, S., Cullinane, M., Blom, A. M., Gulati, S., McQuillen, D. P., Monks, B. G., O’Connell, C., Boden, R., Elkins, C., Pangburn, M. K., Dahlbäck, B., and Rice, P. A. (2001). Binding of C4b-binding protein: a molecular mechanism of serum resistance of Neisseria gonorrhoeae. J. Exp. Med. 193, 281–295.
Ram, S., Sharma, A. K., Simpson, S. D., Gulati, S., McQuillen, D. P., Pangburn, M. K., and Rice, P. A. (1998). A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J. Exp. Med. 187, 743–752.
Ramsey, K. H., Schneider, H., Cross, A. S., Boslego, J. W., Hoover, D. L., Staley, T. L., Kuschner, R. A., and Deal, C. D. (1995). Inflammatory cytokines produced in response to experimental human gonorrhea. J. Infect. Dis. 172, 186–191.
Rice, P. A., Vayo, H. E., Tam, M. R., and Blake, M. S. (1986). Immunoglobulin G antibodies directed against protein III block killing of serum resistant Neisseria gonorrhoeae by immune serum. J. Exp. Med. 164, 1735–1748.
Russell, M. W., and Hook, E. W. (2009). “Gonorrhea,” in Vaccines for Biodefense and Emerging and Neglected Diseases, eds. A. D. T. Barrett and L. R. Stanberry (London: Academic Press), 963–981.
Russell, M. W., and Mestecky, J. (2010). Tolerance and protection against infection in the genital tract. Immunol. Invest. 39, 500–525.
Shafer, W. M., Qu, X., Waring, A. J., and Lehrer, R. I. (1998). Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. U.S.A. 95, 1829–1833.
Shen, L., Smith, J. M., Shen, Z., Eriksson, M., Sentman, C., and Wira, C. R. (2007). Inhibition of human neutrophil degranulation by transforming growth factor-β1. Clin. Exp. Immunol. 149, 155–161.
Shibata, K., Yamada, H., Hara, H., Kishihara, K., and Yoshikai, Y. (2007). Resident Vδ1+ γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J. Immunol. 178, 4466–4472.
Simpson, S. D., Ho, Y., Rice, P. A., and Wetzler, L. M. (1999). T lymphocyte response to Neisseria gonorrhoeae porin in individuals with mucosal gonococcal infections. J. Infect. Dis. 180, 762–773.
Smith, H., Parsons, N. J., and Cole, J. A. (1995). Sialylation of neisserial lipopolysaccharide: A major influence on pathogenicity. Microb. Pathog. 19, 365–377.
Song, W., Condron, S., Mocca, B. T., Veit, S. J., Hill, D., Abbas, A., and Jerse, A. E. (2008). Local and humoral immune responses against primary and repeat Neisseria gonorrhoeae genital tract infections of 17β-estradiol-treated mice. Vaccine 26, 5741–5751.
Tramont, E. C., Sadoff, J. C., Boslego, J. W., Ciak, J., McChesney, D., Brinton, C. C., Wood, S., and Takafuji, E. (1981). Gonococcal pilus vaccine: studies of antigenicity and inhibition of attachment. J. Clin. Invest. 68, 881–888.
van Vliet, S. J., Steeghs, L., Bruijns, S. C., Vaezirad, M. M., Snijders Blok, C., Arenas Busto, J. A., Deken, M., van Putten, J. P., and van Kooyk, Y. (2009). Variation of Neisseria gonorrhoeae lipooligosaccharide directs dendritic cell-induced T helper responses. PLoS Pathog. 5, e1000625. doi: 10.1371/journal.ppat.1000625
Virji, M. (2009). Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol. 7, 274–286.
Virji, M., Evans, D., Hadfield, A., Grunert, F., Teixeira, A. M., and Watt, S. M. (1999). Critical determinants of host receptor targeting by Neisseria meningitidis and Neisseria gonorrhoeae: identification of Opa adhesiotopes on the N-domain of CD66 molecules. Mol. Microbiol. 34, 538–551.
Virji, M., and Heckels, J. E. (1985). Role of anti-pilus antibodies in host defense against gonococcal infection studied with monoclonal anti-pilus antibodies. Infect. Immun. 49, 621–628.
Virji, M., and Heckels, J. E. (1988). Nonbactericidal antibodies against Neisseria gonorrhoeae: evaluation of their blocking effect on bactericidal antibodies directed against outer membrane antigens. J. Gen. Microbiol. 134, 2703–2711.
Weaver, C. T., Hatton, R. D., Mangan, P. R., and Harrington, L. E. (2007). IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 25, 821–852.
Wetzler, L. M., Barry, K., Blake, M. S., and Gotschlich, E. C. (1992a). Gonococcal lipooligosaccharide sialylation prevents complement dependent killing by immune sera. Infect. Immun. 60, 39–43.
Wetzler, L. M., Blake, M. S., Barry, K., and Gotschlich, E. C. (1992b). Gonococcal porin vaccine evaluation: Comparison of Por proteosomes, liposomes, and blebs isolated from rmp deletion mutants. J. Infect. Dis. 166, 551–555.
Wira, C. R., Fahey, J. V., Ghosh, M., Patel, M. V., Hickey, D. K., and Ochiel, D. O. (2010). Sex hormone regulation of innate immunity in the female reproductive tract: the role of epithelial cells in balancing reproductive potential with protection against sexually transmitted pathogens. Am. J. Reprod. Immunol. 63, 544–565.
Wira, C. R., and Rossoll, R. M. (2003). Oestradiol regulation of antigen presentation by uterine stromal cells: role of transforming growth factor-β production by epithelial cells in mediating antigen-presenting cell function. Immunology 109, 398–406.
Yang, Q. L., and Gotschlich, E. C. (1996). Variation of gonococcal lipooligosaccharide structure is due to alterations in poly-G tracts in lgt genes encoding glycosyl transferases. J. Exp. Med. 183, 323–327.
Ye, P., Rodriguez, F. H., Kanaly, S., Stocking, K. L., Schurr, J., Schwarzenberger, P., Oliver, P., Huang, W., Zhang, P., Zhang, J., Shellito, J. E., Bagby, G. J., Nelson, S., Charrier, K., Peschon, J. J., and Kolls, J. K. (2001). Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194, 519–527.
Youssef, A.-R., van der Flier, M., Estevao, S., Hartwig, N. G., van der Ley, P., and Virji, M. (2009). Opa+ and Opa− isolates of Neisseria meningitidis and Neisseria gonorrhoeae induce sustained proliferative responses in human CD4+ T cells. Infect. Immun. 77, 5170–5180.
Yu, J. J., Ruddy, M. J., Wong, G. C., Sfintescu, C., Baker, P. J., Smith, J. B., Evans, R. T., and Gaffen, S. L. (2007). An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood 109, 3794–3802.
Keywords: Th17, IL-17, TGF-β, innate immunity, adaptive immunity, immunosuppression
Citation: Liu Y, Feinen B and Russell MW (2011) New concepts in immunity to Neisseria gonorrhoeae: innate responses and suppression of adaptive immunity favor the pathogen, not the host. Front. Microbio. 2:52. doi: 10.3389/fmicb.2011.00052
Received: 31 January 2011;
Paper pending published: 17 February 2011;
Accepted: 08 March 2011;
Published online: 22 March 2011.
Edited by:
Cynthia N. Cornelissen, Virginia Commonwealth University School of Medicine, USAReviewed by:
Dennis Metzger, Albany Medical College, USAJoseph Duncan, University of North Carolina at Chapel Hill, USA
Copyright: © 2011 Liu, Feinen and Russell. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Michael W. Russell, Department of Microbiology and Immunology, University at Buffalo, Farber 138, University at Buffalo, 3435 Main Street, Buffalo, NY 14214, USA. e-mail:cnVzc2VsbG1AYnVmZmFsby5lZHU=