


- Veterinary Pathobiology and Faculty of Genetics, Texas A&M University and Texas AgriLife Research, College Station, TX, USA
Lipopolysaccharide-deficient mutants of smooth Brucella species (rough mutants) have been shown to arise spontaneously in culture. However, in situ analysis of Brucella infected macrophages using antibody directed against O-polysaccharide suggested a loss of reactivity of Brucella consistent with the appearance of rough organisms, and a potential contribution to infection. The experiments reported describe the direct recovery of Brucella from macrophages infected in vitro and from the spleens of infected mice at a frequency similar to that described in vitro, suggesting that Brucella dissociation is not simply an in vitro artifact. The frequency of appearance of spontaneous rough organisms deficient in O-polysaccharide expression measured in vitro is approximately 2–3 logs higher than the appearance of mutation to antibiotic resistance, purine auxotrophy, or reversion of erythritol sensitive ΔeryC mutants to tolerance. Genetic trans-complementation using a plasmid-based expression of Brucella manBA successfully restored O-polysaccharide expression in only one-third of O-polysaccharide deficient spontaneous mutants. Suggesting that the appearance of rough mutants is caused by mutation at more than one locus. In addition, Sanger sequencing of the manBA structural genes detected multiple sequence changes that may explain the observed phenotypic differences. The presence of O-polysaccharide resulted in macrophage and neutrophil infiltration into the peritoneal cavity and systemic distribution of the organism. In contrast, rough organisms are controlled by resident macrophages or by extracellular killing mechanisms and rapidly cleared from this compartment consistent with the inability to cause disease. Loss of O-polysaccharide expression appears to be stochastic giving rise to organisms with biological properties distinct from the parental smooth organism during the course of infection.
Introduction
Brucellosis continues to be a major worldwide zoonosis, despite eradication programs. Members of the genus Brucellae are small, non-motile, Gram-negative, non-encapsulated, intracellular zoonotic pathogens. The genus is divided into 10 species, including the 6 classical species Brucella abortus, B. suis, B. melitensis, B. neotomae, B. canis, and B. ovis and newly recognized species B. ceti, B. microti, B. pinnipedialis and B. inopinata. Studies of antigenicity have largely focused on outer membrane proteins and expression of lipopolysaccharide (LPS; Palmer and Douglas, 1989; Ficht et al., 1990, 1996; Rojas et al., 1994; Cloeckaert et al., 1996, 1999; Cassataro et al., 2005; Jimenez de Bagues et al., 2005; Connolly et al., 2006; Barquero-Calvo et al., 2007; Pasquevich et al., 2010). On the basis of LPS expression, the classical species can be broken into two broad categories: smooth (S-) or rough (R-). The classification between S- and R-strains is based on the observation of Brucella colonies using the oblique light method (Henry, 1933). In this paper, we are focused on the transition of B. abortus or B. melitensis from smooth LPS expression to rough LPS expression. Events leading to the transition of smooth LPS expression to rough LPS expression are poorly characterized and just beginning to be explored (Mancilla et al., 2010).
The O-polysaccharide of Brucella LPS is critical for virulence of the classical Brucella species (B. melitensis, B. abortus, B. suis). Mutants lacking O-polysaccharide are rapidly cleared from animal models of infection. In the host, this attenuated survival has been explained by increased susceptibility to complement-mediated lysis and/or professional phagocyte cell killing mechanisms (Allen et al., 1998; Gonzalez et al., 2008). Within the B. melitensis and B. abortus species, differences in survival and replication appear to be based on the nature of the genetic defect (Porte et al., 2003; Pei and Ficht, 2004; Chen and He, 2009) leading to loss of LPS expression. Mutants defective in core biosynthesis are more sensitive to macrophage killing while mutants defective in expression of O-polysaccharide alone have been shown to replicate to varying degrees (Gonzalez et al., 2008). Presence of Brucella O-polysaccharide may restrict macrophage activation and promote productive infection by actively inhibiting or preventing activation of macrophage function (Kreutzer et al., 1979a,b; Forestier et al., 1999a,b, 2000; Rittig et al., 2003). This has lead to the conclusion that the O-polysaccharide portion of the LPS is critical to persistence and stealth behavior of smooth organisms (Barquero-Calvo et al., 2007; de Jong et al., 2010). The lipid A portion of Brucella LPS is also distinct in its structure containing longer chain fatty acids (C28:C30) than enterobacterial lipid A and is the basis for it is reduced toxicity (Viadas et al., 2010).
Spontaneous rough variants of the classical Brucella species exhibit enhanced activation of macrophages in culture, in contrast to the parental smooth strains (Fernandez-Prada et al., 2003; Rittig et al., 2003). Differences in uptake result in increased intracellular numbers and enhanced macrophage activation that has been shown to result in necrotic and oncotic cell death (Fernandez-Prada et al., 2003; Pei and Ficht, 2004). Enhanced intracellular killing of rough organisms has been documented in macrophage ex vivo, as has intracellular replication (Porte et al., 2003). However, the sensitivity of rough Brucella to complement-mediated lysis is still thought to be responsible for extracellular killing (Fernandez-Prada et al., 2001). Release of a mixture of smooth and rough organisms from the host cell would result in extracellular killing of rough organisms, but may also provide a route of escape for the more resistant smooth organisms (Hybiske and Stephens, 2008).
The first step to exploring a role for the spontaneous appearance of rough derivatives is characterizing the frequency and rate with which rough organisms appear. Recently, this topic was explored, by Mancilla et al. (2010) who characterized phage integrase mediated recombination, resulting in deletion of two O-polysaccharide glycosyltransferases within the main LPS locus. In addition to genes in the main LPS locus, genetic mutation at either B. melitensis or B. abortus manBA (phosphomannomutase/phosphomannoisomerase; BMEII0899 and BMEII0900 or BAB2_0855 and BAB2_0856 respectively) is cause for loss of LPS expression and putative core defect (Allen et al., 1998; Gonzalez et al., 2008). Previous work in our lab characterizing rough mutants made manBA an ideal starting place to assess the biology of spontaneous manBA rough mutants (Allen et al., 1998; Pei and Ficht, 2004; Pei et al., 2006, 2008; Wu et al., 2006). The inability to block the generation of rough variants prevents addressing the role rough mutants play in infection directly. However, properties of spontaneous rough mutants should be considered in the context of infection, where they arise; the properties of a clonal culture of rough organism may be artifactual and may not represent the biological role these organisms play in the host. The first step in this process is to determine whether rough organisms arise in situ as has been reported in culture (Henry, 1933) and to determine whether the rate of appearance differs from the background mutation rate. The next step is to characterize a genetic basis for the spontaneous appearance of rough mutants.
In order to quantify the rate of smooth to rough transition, a fluctuation test using bacteriophage Bk2 was used to identify the frequency of mutations in bacterial populations and is directly analogous to the use of T1-phage by Luria and Delbrück (1943). Trans-complementation with the B. melitensis manBA locus restored O-polysaccharide expression and DNA sequence analysis of the involved loci was performed in an effort to characterize the mutation frequency and rate. Use of the fluctuation test allows characterization of a rate of appearance, as opposed to the frequency of appearance, establishing that the observed genetic mutations occur, regardless of selection pressure (Luria and Delbrück, 1943).
The results shown demonstrate the frequency and rate of appearance of rough variants in vivo and ex vivo and a potential genetic locus involved in their appearance. It may be surmised that the appearance of rough organisms enhances survival and perhaps virulence. The demonstration that clonal cultures of rough organisms are rapidly cleared during infection provides further evidence that the recovered organisms are appearing in situ as does the results from macrophage culture (Allen et al., 1998; Elzer et al., 1998).
Demonstration of recovery from cultured macrophages and mouse tissue from the latter stages of infection were used to confirm in situ appearance. The inability of clonal rough variants to persist under either condition is accepted as evidence of de novo appearance. Unfortunately, it will become obvious that characterization of a specific role for rough mutants cannot be performed, since their appearance involves genetic variation at multiple loci and possibly by more than one mechanism and as such cannot be readily prevented.
Materials and Methods
Bacteria, Growth Conditions, and Strain Typing
All bacteria and plasmids used during the course of this investigation are described in Table 1. E. coli cultures were routinely incubated on Luria–Bertani agar (LB, Difco Laboratories) plates for 18 h with appropriate antibiotics. Virulent B. abortus S2308 (B. Deyoe, NADC) and B. melitensis 16M (ATCC), were re-isolated from aborted fetuses of cattle or goats, respectively. These strains were used for all challenge organisms and generation of deletion mutants. Brucella strains used in this study or their derivatives were routinely grown as previously described (Kahl-McDonagh and Ficht, 2006) or on Brucella minimal medium (BMM), prepared as described by Gerhardt (1958). Brucella cultures were inactivated by autoclaving or heat killing in phenol-saline [0.05% (v/v) phenol/0.85% (w/v) NaCl] at 65°C for a minimum of 2 h. When appropriate, antibiotics were added to media with the following final concentrations: kanamycin, 100 μg/mL; chloramphenicol, 30 μg/mL; and ampicillin, 100 μg/mL. All bacterial strains were stored frozen at −80°C in medium supplemented with 50% (v/v) glycerol.
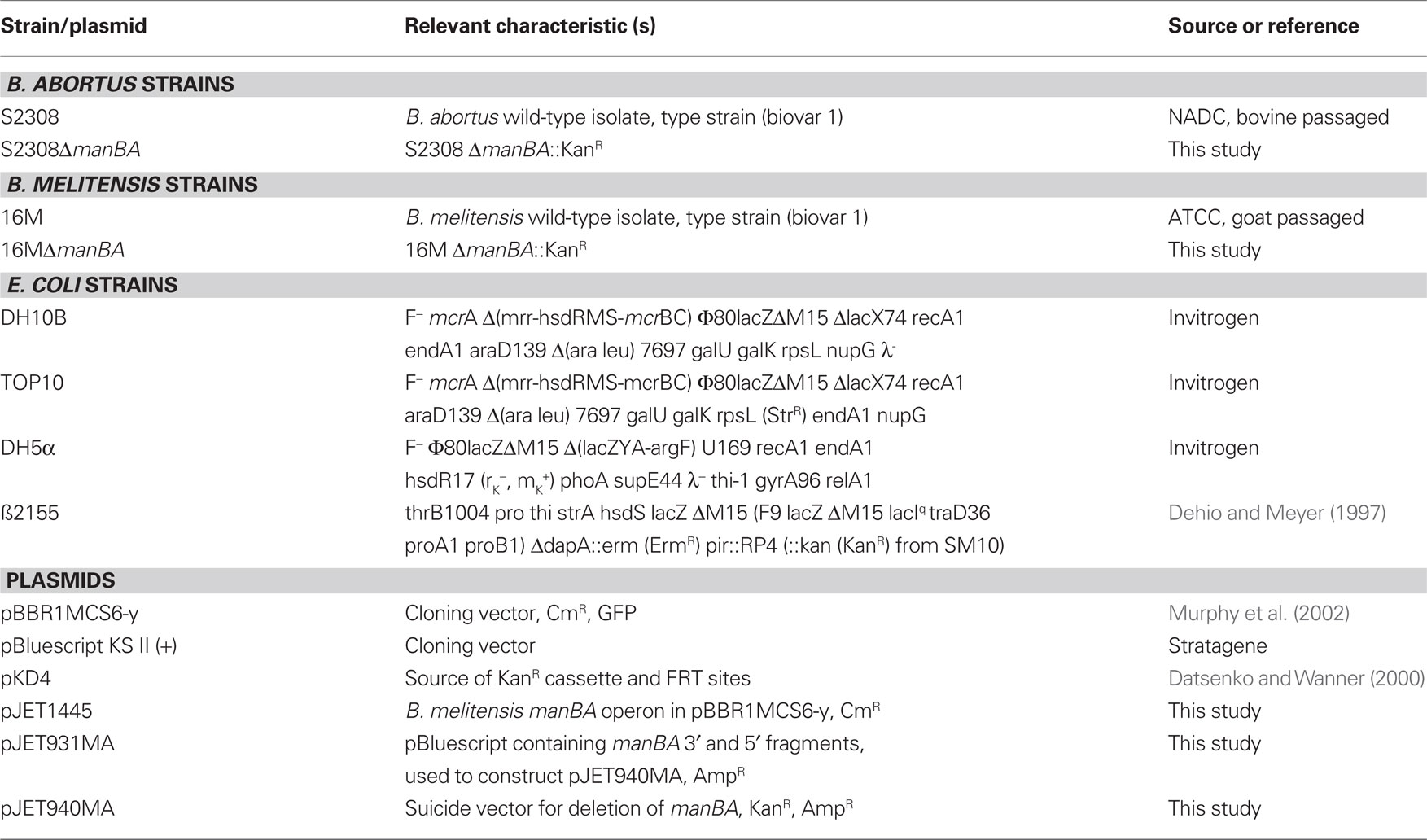
Table 1. List of strains and plasmids.
Dilutions of bacterial cultures were performed in either peptone saline [1% (w/v) Bacto peptone, 0.5% (w/v) NaCl], or phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4). Unless otherwise noted, all bacterial strains were grown at 37°C, in an atmosphere containing 5% (v/v) CO2. The phenotype of rough Brucella mutants in suspension was confirmed by acriflavine agglutination (Alton et al., 1975). Alternatively, crystal violet [2%(w/v) crystal violet dye, 20% (v/v) ethanol, 0.8% (w/v) ammonium oxalate] staining was used to test the presence of rough colonies on solid media (Alton et al., 1975). Crystal violet was used to flood a Petri plate containing the bacteria; the dye is excluded by smooth colonies while colonies formed from rough mutants absorb the dye readily and turn a dark purple. Individual colonies were resuspended in 200 μL peptone saline [1% (w/v) Bacto peptone and 0.85% (w/v) NaCl] diluted 1000-fold and then further diluted 10-fold into 200 μL fresh tryptic soy broth (TSB; 1.5% trypticase peptone, 0.5% phytone peptones, 0.5% NaCl) in the wells of a microtiter dish. The resulting cultures were removed after 48 h incubation. Acriflavine agglutination was used to confirm the rough phenotype of the cultures unless otherwise specified (Alton et al., 1975).
Determination of the Mutation Rate and Frequency in Brucella
Independent Brucella colonies were resuspended in 200 μL peptone saline from culture on tryptic soy agar (TSA; 1.5% trypticase peptone, 0.5% phytone peptones, 0.5% NaCl, 1.5% agar). This suspension was then diluted 1000-fold in fresh peptone saline. A final 10-fold dilution was made into 200 μL aliquots of TSB in the wells of a 96-well microtiter plate. The resulting cultures, each inoculated with approximately 1000 cfu, were incubated for 2 days, to an approximate cell density of 5 × 107 cfu/mL as determined by plating a subset of these cultures on non-selective media. To measure background mutation rate, 70-μL portions of each culture were plated on selective medium, BMM containing 40 μg/mL 5-fluoroorotic acid (FOA), and 50 μg/mL uracil. Uracil auxotrophs, FOA-resistant colonies, were analyzed after 8–10 days of incubation. As a positive control, the E. coli, uracil auxotroph strain, AT2538 (E. coli Genetic Stock Center, New Haven, CT, USA) was carried through all steps of the experiment. For growth comparison between rough and smooth strains, genetically engineered rough B. melitensis were mixed with wild-type B. melitensis at approximately 1:1 in 5 mL of TSB. After 48 h, 50 μL was removed and passaged to a fresh 5 mL aliquot of TSB for further culture. Passage was repeated three times and rough content from each passage was ascertained by differential plating (TSA versus TSA with kanamycin).
The method of the median is generally accepted as an unbiased method to determine the mutation rate in bacterial populations (Koch, 1982; Rosche and Foster, 2000). Mutant frequency is the ratio of mutants detected divided by the total number of bacteria in the population. The mutation rate is the number of mutations per cell division. The most probable number of mutational events per culture for the set, m, was determined by using the method of the median of Lea and Coulson, introduced in 1949 (Luria and Delbrück, 1943; Lea and Coulson, 1949). The method consists of calculating the median number of mutants and using that number to find the theoretical number of mutants per culture from the tables developed by Lea and Coulson (1949). Calculation of the mutation rate from m required determining the average number of cells per culture. Viable titers were determined by serial dilution and plating on non-selective medium. Values were averaged, deriving the average number of bacteria in a given culture (Nav). The Luria–Delbrück mutation rate per cell generation, micron (μ), was calculated by using the relationship μ = (ln2)(m/Nav).
Plasmids and Molecular Biology Methods
Genetic manipulations were carried out using standard techniques (Maniatis et al., 1982). Primers used in this study are described in Table 2. All primer design was performed using MacVector™. All of the restriction endonucleases used in this study were purchased from Roche Biochemicals, with the exception of AscI, which was purchased from New England Biolabs. FastStart Taq was purchased from Roche Biochemicals. AccuTaq LA was purchased from Sigma Chemicals. T4 DNA Ligase was purchased from Promega. DNA manipulations including restriction digestion, ligation, and PCR amplification were performed according to the manufacturers instructions. PCR reactions were typically performed using 50–200 ng target DNA, 0.5 μM of each primer, 200 μM of each deoxyribonucleoside triphosphate, 2 U of FastStart Taq polymerase, 1× PCR buffer, 2 mM MgCl2. As necessary, PCR products and digested vectors were gel purified and extracted using the Qiagen QIAEXII gel purification kit. Hybrid PCR products were produced in order to construct an allelic exchange knockout vector as previously described (Kahl-McDonagh and Ficht, 2006). Southern blotting is a routine technique and was used for examining the presence or absence of genes in total genomic DNA (Ficht et al., 1990). The presence of manBA was determined for all spontaneous rough isolates by PCR amplification. Samples were applied to an FTA CloneSaver™ card, which inactivates the bacteria (Whatman). Two-millimeter punches were taken from each sample and used in a subsequent PCR. Complementation tests with the expression vector pJET1445 were performed for 120 recovered rough colonies (Table 3). Brucella mutants were constructed using recombinant techniques as described elsewhere (Kahl-McDonagh and Ficht, 2006). Conjugation was performed as described previously and smooth phenotype was confirmed by acriflavine agglutination (Wu et al., 2006).

Table 2. Primers used in this study.
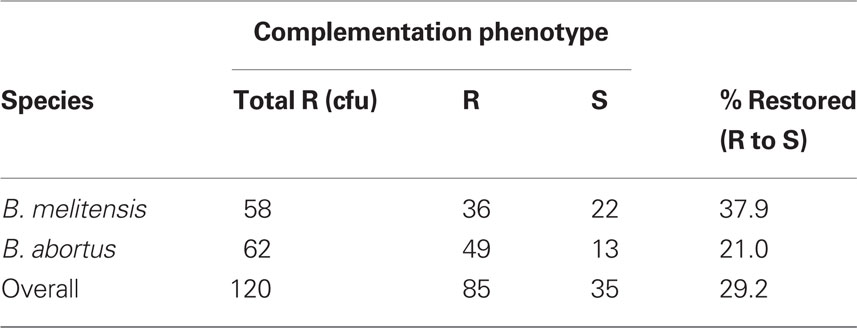
Table 3. Complementation test with spontaneous rough isolates and plasmid pJET1445.
Bacteriophage Bk2 Selection of Rough Brucella
Brucella phage, Bk2, was obtained from Douglas and Elberg (1976, 1978). Propagation of Bk2 phage was carried out in tryptic soy broth inoculated with a suspension from a 24-h culture of B. abortus S2308, so that the final bacterial concentration was approximately 5 × 108 cfu/mL. Phage was added at a final multiplicity of infection (MOI) of 0.1 and the culture was incubated at 37°C, with agitation, for 18 h. The lysed bacterial suspension was clarified by centrifugation at 10,000 × g to remove cellular debris and filtered through a 0.2-μm polyethersulfone (PES; Nalgene, Rochester, NY, USA) membrane syringe filter. B. abortus S2308 and B. melitensis 16M isolates were grown on TSA and five independent colonies selected from each plate were suspended in 1 ml of peptone saline, serially diluted, and plated on TSA in the presence or absence of Bk2 phage. The plates were overlaid with TSA–soft agar [SA; TSB supplemented with 7% (w/v) Bacto agar] containing at least 600,000 pfu of Bk2 phage. After 72 h, the colony forming units (cfu) present on either plate were enumerated and confirmed to be rough using acriflavine agglutination as described earlier.
In vitro Tissue Culture
Murine macrophage-like J774.A1 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% (v/v) fetal bovine serum, 1 mM l-glutamine, and 1 mM non-essential amino acids. To assess invasion and replication, 2.5 × 105 cells were seeded into each well of a 24-well plate. Cell cultures were placed in a 37°C incubator in the presence of 5% CO2 for 20 min. Bacteria were added to macrophages at MOI of 100. The infected cell culture plate was centrifuged (room temperature, 200 × g for 5 min) to associate the bacteria with the macrophage monolayer. The infected monolayers were then washed three times with peptone saline. Tissue culture media containing gentamicin was added back to monolayer, killing bacteria external to the macrophage. Invasion was determined after 1 h by lysing the macrophage monolayer and plating the lysate on TSA with or without appropriate antibiotics. To measure uptake, the recovered bacterial cfu is compared with the infectious dose. Survival was similarly determined at later time points.
In vitro loss of Brucella LPS was determined using similar culture conditions. J774.A1 cells were seeded in four-well Lab-Tek chambered slides (Nalge-Nunc, Rochester, NY, USA) with 2.3 × 105 cells/chamber 1 day before infection. The cells were infected at an MOI of 20 using GFP expressing B. melitensis 16M. At 1, 4, 12, 24, 48 h chambers were removed, and the cells were fixed with 3.7% (v/v) formaldehyde in PBS (pH 7.4) for 30 min at room temperature. Cells were washed three times with PBS, before antibody staining. Cells were incubated 30 min with a primary mouse anti-S-Brucella LPS monoclonal antibody 39 diluted 1:500 in 0.1% saponin/PBS before being stained with Alexa Fluor 568 anti-mouse IgG (1:1000) in 0.1% saponin/PBS. Treatment of GFP expressing Brucella with red anti-Brucella LPS antibody meant that bacteria either expressing S-LPS or R-LPS could be distinguished from one another. Colocalization of green and red fluorescence results in a yellow, colocalized signal representing S-LPS. Green fluorescent signal alone indicates presence of rough LPS. Observations were made using an Olympus IX70 microscope. Results were quantified using ImageJ (http://rsbweb.nih.gov/ij/). To rule out the potential for physical loss of O-polysaccharide under low pH conditions in the Brucella some, B. melitensis was resuspended in 25 mM homopiperazine buffer, pH 4.2 or PBS, pH 7.5, and monitored for loss of LPS over a 48-h time period (data not shown; Kohler et al., 2002a,b, 2003).
Mouse Model of Infection
The mouse model was used to evaluate the in vivo clearance kinetics and spontaneous appearance of rough Brucellae. Experiments to determine clearance kinetics used groups of three to five 6- to 8-week-old female BALB/c mice infected with 108 wild-type B. melitensis 16M, B. melitensis 16MΔmanBA, or sham infected with PBS. Mice from each treatment group were euthanized at 1, 4, 8, 24, 72, and 168 h post-infection. Peritoneal lavage with 4 mL of ice-cold PBS was used to harvest samples from the infected animals. Bacterial load was assessed in the spleen and liver, and in the ascites samples were plated with or without the addition of 50 μg/mL gentamicin to distinguish intracellular from extracellular organisms. The recovery of rough isolates from infected mice was performed using groups of 6- to 8-week-old female BALB/c mice, injected intraperitoneally with 5 × 105 cfu of inoculant composed of one of the following: (i) B. melitensis 16M, (ii) a 1:1 mixture of B. melitensis 16M and 16MΔmanBA, (iii) B. melitensis 16MΔmanBA, (iv) B. abortus S2308, (v) a 1:1 mixture of B. abortus S2308 and B. abortus S2308ΔmanBA, or (vi) B. abortus S2308ΔmanBA. Mice were euthanized at 1 and 8 weeks post-inoculation by CO2 asphyxiation. Spleens were collected and weighed prior to homogenization (Omni 2000; Omni International, Inc.). Bacterial recovery was determined by plating portions of serial diluted homogenates on TSA plates with and without kanamycin. In addition, a portion of each homogenate was plated on Bk2 phage plates as previously described, for enumeration of total rough organism. Data are the average log10 value of cfu per spleen over five mice.
Immunological Methods
Characterization of Brucella LPS was performed following growth and recovery of the organism as described previously (Palmer and Douglas, 1989). Fluorescent detection of LPS was performed directly in polyacrylamide gels, utilizing the fluorescent hydrazide, Pro-Q Emerald 300 dye (Steinberg et al., 2001), and standard gel documentation setup with UV transilluminator, as described by the manufacturer, Molecular Probes. Alternatively, Brucella LPS was visualized by Western blotting utilizing sensitized anti-B. melitensis goat sera or anti-B. abortus LPS mouse monoclonal antibody 39 (Holman et al., 1983) and a secondary antibody, either rabbit anti-goat or goat anti-mouse, conjugated to alkaline phosphatase and visualized according to manufacturer’s instructions (Roche Biochemicals). Fresh sera were obtained from goat, cow, and human donors for use in these experiments and complement-mediated killing was measured as described elsewhere (Corbeil et al., 1988). A rough strain of E. coli JM101 with no LPS side chain was included as a positive control in the bactericidal assay since it is serum susceptible and sensitive to complement-mediated killing from all sera tested.
Statistical Analysis
Statistical significance was determined using Student’s t-test. A p-value of <0.05 was considered significant.
Ethics Statement
Animals were handled in strict accordance with good animal practice as defined by the Texas A&M University Institutional Animal Care and Use Committee (IACUC) and meet all federal requirements, as defined in the Animal Welfare Act (AWA) and the Public Health Service (PHS) Policy Humane Care and Use of Laboratory Animals.
Results
Determination of the Spontaneous Mutation Rate in B. abortus s2308 and B. melitensis 16m
Conversion of wild-type Brucella to pyrimidine auxotrophy provided an estimate of background genetic mutation via selection of spontaneous mutants on 5-FOA. This approach selects for mutants deficient in either of two UMP biosynthetic enzymes: orotate phosphoribosyl transferase (OPRTase) or orotidine 5′-monophosphate decarboxylase, encoded by the pyrE and pyrF genes, respectively (Grogan et al., 2001). FOA is not toxic outright, but the 5-fluorouridine 5′-monophosphate synthesized via the sequential action of the PyrE and PyrF kills bacterial cells. Loss of either enzyme thus spares the mutant in the presence of FOA, but also renders it dependent on exogenous uracil for growth. The pyrE/pyrF aggregate mutation rate was calculated from a total of 94 independent B. abortus and 94 independent B. melitensis colonies. Excluding jackpot cultures (>3000 colonies/plate), 261 mutant colonies were collected from B. abortus and 930 mutant colonies were collected from B. melitensis. Median numbers of mutants for each culture were 2 and 18.5, respectively. The most probable number of mutational events per culture for the set, m, was determined by using the method of the median of Lea and Coulson (Luria and Delbrück, 1943; Lea and Coulson, 1949). This leads to an estimation of 1.32 and 6.07 mutational events per culture for B. abortus and B. melitensis, respectively. To calculate the average number of cfu per culture, the viable titers were determined by serial dilution and plating on non-selective medium to derive the average number in a given culture (Nav). The Luria–Delbrück mutation frequency per cell generation, micron (μ), was calculated by using the relationship μ = (ln2)(m/Nav). For B. abortus this value was 1.63 × 10−8, and for B. melitensis 7.48 × 10−8 mutations per genome per generation. The results suggest that the selected strains have extremely stable genomes, at least with regard to pyrimidine auxotrophy.
Rate of Appearance of Rough Mutants in B. abortus s2308 and B. melitensis 16m
Similar experiments were carried out to estimate the mutation rate for manBA, using the phenotypic marker of loss, or dissociation, of LPS expression. The rate of dissociation was determined for 60 and 59 individual smooth colonies of B. melitensis and B. abortus, respectively. Each colony was serially diluted in peptone saline and plated onto TSA media with and without Bk2 phage. The dissociation rate is equal to the number of rough cfu, enumerated via growth on bacteriophage Bk2 saturated plates, divided by the number of smooth cfu from each colony, as observed by crystal violet staining. The rate of dissociation for B. melitensis is 1.15 × 10−4, while the frequency of dissociation of B. abortus rough variants is 1.27 × 10−5. Using the median of all rough colonies (B. melitensis or B. abortus) to find m, the most probable number of mutational events per culture, led to m = 293 mutations per culture, for B. melitensis and m = 312 mutations per culture for B. abortus. To assess contribution of genetic events at the manBA locus (Figure 1A) a complementation test was performed for each mutant (Table 3). Plasmid pJET1445 was introduced into each rough isolate to determine if a defect in manBA was the reason these strains had lost O-polysaccharide expression. The results indicated that 22 B. melitensis, 12 B. abortus – 34 overall rough mutants were trans complemented by manBA. The ability of this expression vector to restore LPS expression in manBA deficient strains was confirmed by both Western blotting (not shown) and staining with Pro-Q Emerald 300, a glycoprotein stain (Figure 1C). Using the median of only those colonies complemented by manBA in trans, m = 197 mutations per culture for B. melitensis and 331 mutations per culture for B. abortus. Luria–Delbrück mutation frequencies, micron were calculated. The frequency of dissociation for B. melitensis is 1.15 × 10−4, while the frequency of dissociation of B. abortus rough variants is 1.27 × 10−5. In general the rate of mutation is about 100–1000 times greater than that observed for the spontaneous appearance of Brucella pyrimidine auxotrophs. These rates and frequencies, summarized in Table 3, are consistent with a mutator phenotype (Le Chat et al., 2006).
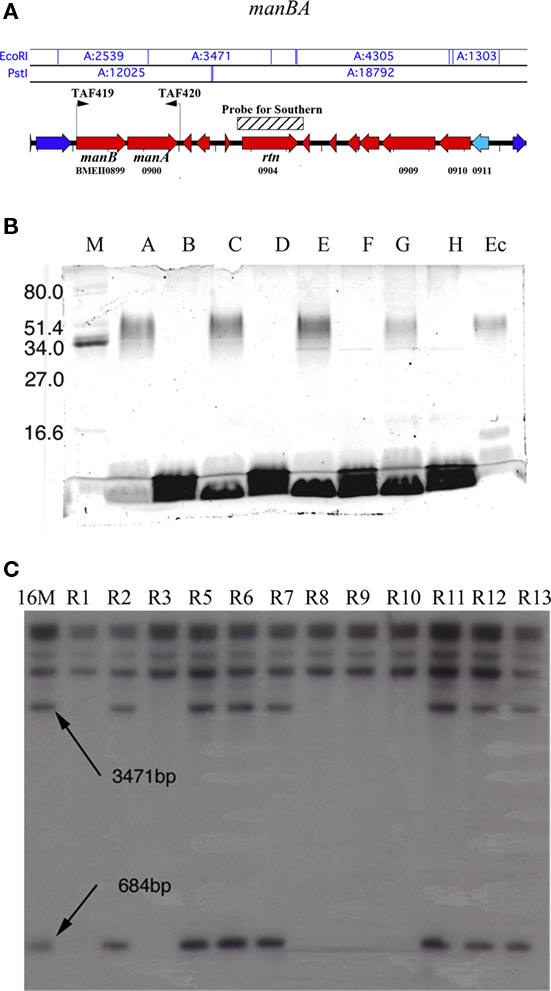
Figure 1. Genomic variation among isolates of B. melitensis 16M in stock culture originally obtained from ATCC. A preliminary genetic screen of Brucella rough variants revealed large genomic rearrangement. (A) Schematic of the manBA locus, showing the full extent of the deletion as characterized by primer walking. Primers for amplification of manBA (TAF419 and TAF420) are indicated. Unaffected genes (dark blue), truncated genes (light blue) and completely deleted genes (red). (B) Representative figure demonstrating Pro-Q Emerald 300 glycoprotein staining of Brucella LPS separated on SDS-PAGE using manBA knockout strains developed for this work and the ability to complement the deletion with the pJET1445 expression construct (C); marker (lane M), B. melitensis 16M (lane A), B. melitensis ΔmanBA (lane B), B. melitensis 16MΔmanBA/pJET1445 (lane C), B. melitensis 16MΔmanBA/pBBR1MCS6-y (lane D), B. abortus S2308 (lane E), B. abortus ΔmanBA (lane F), B. abortus 16MΔmanBA/pJET1445 (lane G), B. abortus 16MΔmanBA/pBBR1MCS6-y (lane H), E. coli O55:B5 expressing smooth LPS (lane Ec), (rtn, BMEII0904), demonstrating naturally occurring deletion of manBA. The arrows indicate two digested fragments of the rtn locus (3471 and 684 bp, respectively) that are lost with the deletion of manBA. R1–R13 are different spontaneous rough isolates, representing two broad classes of mutants. (C) Southern blot of PstI/EcoRI-digested genomic DNA isolated from B. melitensis rough isolates probed with a randomly 32P-labeled probe to phosphodiesterase.
Sequence Changes at the manBA Locus of Rough Variants
The ability to complement only one-third of spontaneously arising rough variants suggested that mutations occur at more than one locus. However, rather than characterizing all of these defects we decided to focus on the nature and extent of variation at the manBA locus (BMEII0899-0900). Initial efforts using Southern blotting of the manBA locus in B. melitensis revealed the potential for spontaneous genomic rearrangements as a cause of the loss of function of manBA in natural rough variants (Figure 1B). Genomic DNA was digested with restriction enzymes EcoRI and PstI. Using randomly labeled 32P-labeled probe to RTN protein (rtn, BMEII0904), large deletion events were identified in approximately one-half of the rough variants detected in the original stock culture received from the ATCC (Figure 1B, lanes R1, R3, R8, R9, R10). Complementation with the manBA expression plasmid, pJET1445, restored smooth LPS production in the mutants. Primer walking indicated that the deletion was up to 12 kbp in extent. A second class of rough mutants lacking any significant genomic rearrangements was also identified (Figure 1B, lanes R2, R4, R5, R6, R7, R11, R12, R13). Identification of the first group of mutants suggested that changes at the manBA locus could account for the loss of O-polysaccharide expression in spontaneously arising mutants. To increase the number of rough mutants being characterized, the Brucella phage Bk2 was used to select rough mutants. Gross deletions were not observed in spontaneously arising rough variants characterized utilizing bacteriophage Bk2 selection. In sequenced roughs, two transitions, T135C and G335A were found. These transitions cause a coding shift from serine to leucine and glutamic acid to glycine, respectively. Both transitions were found within the putative phosphoglucomutase/phosphomannomutase, alpha/beta/alpha domain I of phosphomannomutase (manB, BMEII0899). The majority of the characterized sequence changes in the manBA were similar polymorphisms, with the exception of an insertion 934TCGGGCA935 within phosphomannomutase. The accumulation of these mutations leading to a rough phenotype, in the culture, may be explained by their enhanced growth rate demonstrated in vitro culture (data not shown).
Detection of Rough Variants in the Mouse and Macrophage Models of Infection
In order to determine whether rough organisms appear under all growth conditions, mice were inoculated with smooth parental strain, a ΔmanBA (kanR) derivative or a 1:1 mixture of parental smooth and ΔmanBA (kanR) rough mutant. Survival was compared following organism recovery from the spleen over an 8-week-period post-inoculation. Mice infected with the rough mutant alone eliminated the bacteria rapidly, and both B. melitensis and B. abortus rough derivatives were undetectable within 1 week post-inoculation. At 1 week, rough mutants were recovered from mice infected with smooth B. melitensis at a frequency of 5.94 × 10−3 and mice infected with B. abortus at a frequency of 9.95 × 10−3. After 8 weeks, the frequencies changed slightly to 1.42 × 10−2 and 6.44 × 10−4, respectively. In mice infected with 1:1 ratio of smooth and rough bacteria, rough strains were recovered out to 8 weeks, at similar frequencies to animals infected with clonal populations of smooth strains. However, the rough organisms recovered from these animals were not kanamycin resistant suggesting that the rough mutants recovered represent newly derived variants arising in vivo from the smooth organisms present in the inoculum (Figure 2).
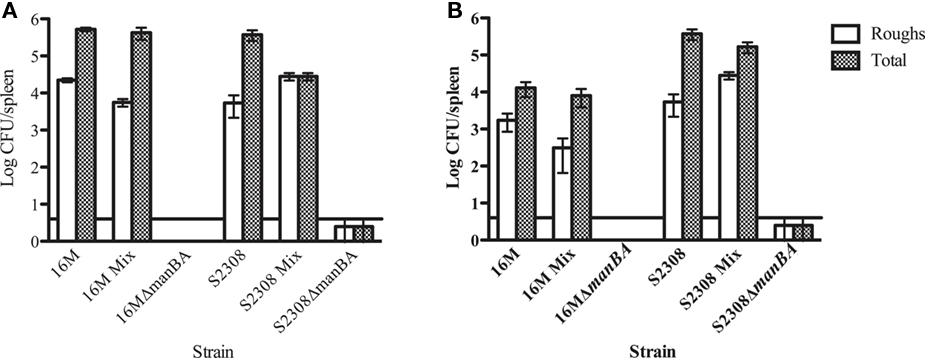
Figure 2. Recovery of rough Brucella from the spleens of infected mice. Mice were infected with clonal populations of smooth or rough B. melitensis, or a mixture of smooth and rough B. melitensis. Bacterial colonization was evaluated at 1-week (A) and 8-weeks (B) post-infection. Mice infected with a clonal population of rough organisms cleared the infection as early as 1 week post-infection. In contrast, spontaneous rough mutants are recovered from mice infected with smooth strains (either clonal smooth strains or a mixture of smooth and rough strains) at both 1 and 8 weeks post-infection. The horizontal line above the x-axis represents the limit of detection. Differences between rough and smooth organisms are significant at p < 0.05.
To determine if the appearance of rough mutants in vitro was consistent with in vivo conditions, recovery of rough derivatives from the macrophage model of infection was evaluated. Macrophages were infected with smooth Brucella melitensis and the inoculum and surviving bacteria were plated on the surface of plates saturated with the Brucella phage Bk2. The ratio of rough to smooth Brucella from macrophages is 9.12 × 10−4, indicating conservation of smooth to rough transition in macrophages. Visualization of these events was performed using macrophages infected with GFP expressing B. melitensis 16M at 1, 4, 12, 24, 48 h time points. At each time point, changes in the detection of smooth Brucella LPS was revealed by the loss of colocalization of the red and green signals from the Alexa Fluor 568 and GFP respectively (Figure 3). These results were quantified revealing that upward of 60% of bacteria within macrophages, in the fields examined, had lost reactivity for smooth LPS antibody (Figure 3, inset). Loss of reactivity to monoclonal anti-Brucella smooth LPS is consistent with a change in the nature or presence of Brucella LPS during the course of infection. Experiments using rough-specific anti-sera received from NVSL indicated the increased number of rough organisms present in the macrophage. Western blot analysis with the monoclonal anti-Brucella smooth LPS of recovered mutants confirmed the absence of O-polysaccharide (data not shown). Brucella, outside the macrophage, incubated in low pH conditions show no apparent change in LPS expression.
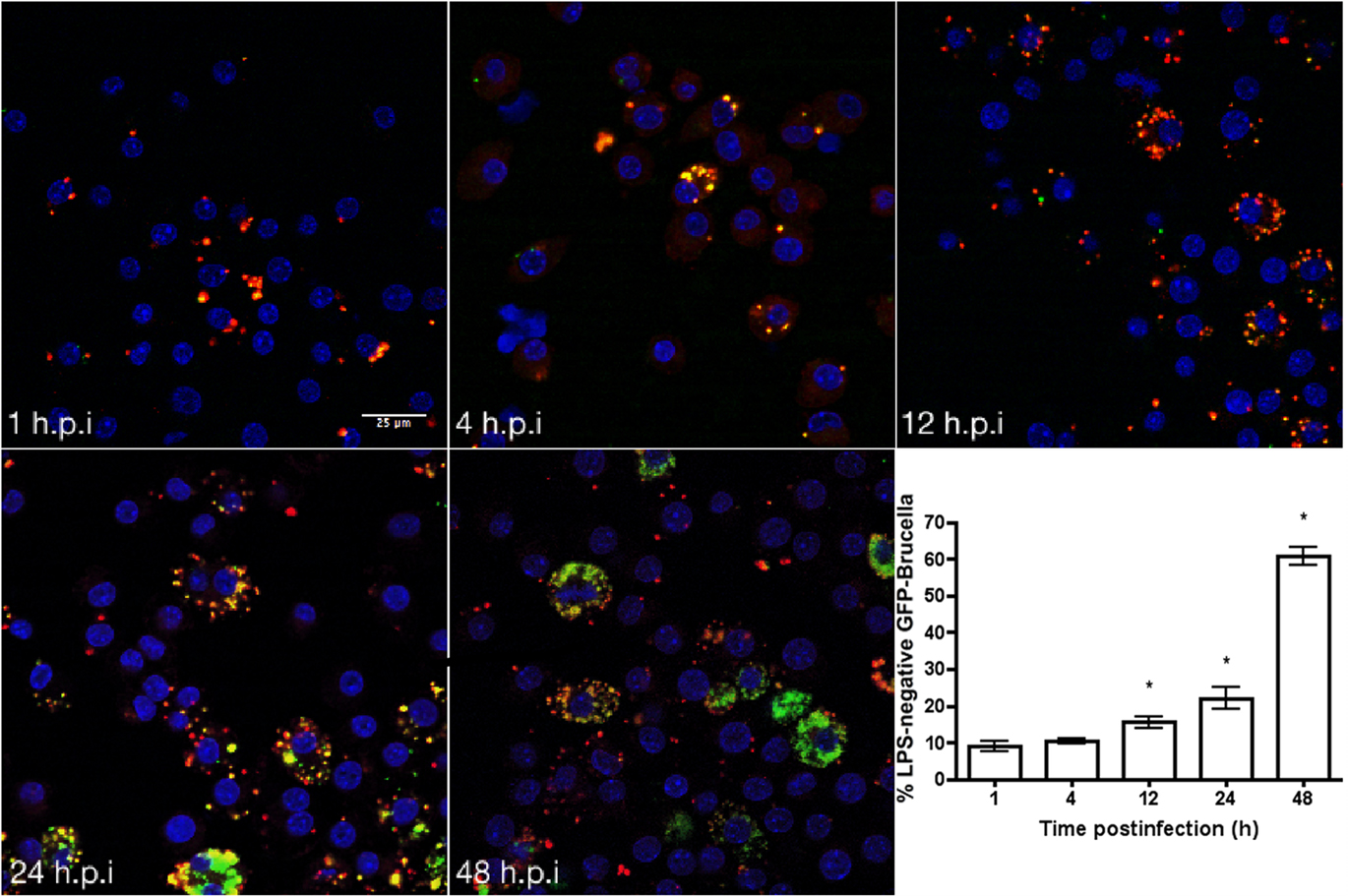
Figure 3. Spontaneous loss of O-polysaccharide reactivity among Brucella organism growing in macrophages ex vivo. Macrophages were infected with smooth B. melitensis 16M and in vitro loss of Brucella LPS was monitored using primary mouse anti-Brucella over a 48-h time period. Presence of bacteria either expressing smooth LPS (yellow, colocalized signal) or rough LPS (red) at each time point was observed using an Olympus IX70 microscope. Results were quantified using ImageJ (http://rsbweb.nih.gov/ij/). Ten fields per time point were quantified using ImageJ, the average suggests up to of 60% of bacteria within macrophages may lose reactivity with anti-LPS antibody. Results with an asterisk (*) are significant with p < 0.05. The bar shown is 25 μm.
Comparison of Brucella Survival and Host Cellular Activity in Response to Infection
The inability of rough Brucella to cause disease in the mouse model is consistent with a critical role for O-polysaccharide in the early stages of infection. In experiments following the fate of rough Brucella shortly after intraperitoneal inoculation, rough organisms became resistant to gentamicin within 1 h of inoculation (Figure 4A) while smooth Brucella remained sensitive to gentamicin treatment up to 24 h post-infection (Figure 4B). This is consistent with in vitro results demonstrating enhanced uptake of rough Brucella (Rittig et al., 2003; Pei and Ficht, 2004). During this time frame the number of rough organisms present in the ascites declined nearly four logs while the number of smooth organisms in the ascites declined by little more than one log. This may well explain the attenuated phenotype of rough Brucella mutants and is presumably the result of increased sensitivity of rough organisms to either extracellular or intracellular killing mechanisms or both. The rapid decline of rough organisms in the peritoneum may, in part, explain the lower numbers of rough organisms present in the spleen (Figure 4C) and liver (Figure 4D). An evaluation of the sensitivity of the B. melitensis rough mutant 16MΔmanBA and a natural rough isolate to serum complement was not enhanced by the loss of O-polysaccharide in contrast with the results reported for other rough Brucella spp. and confirmed by B. abortus strain RB51 (Figure 4E).
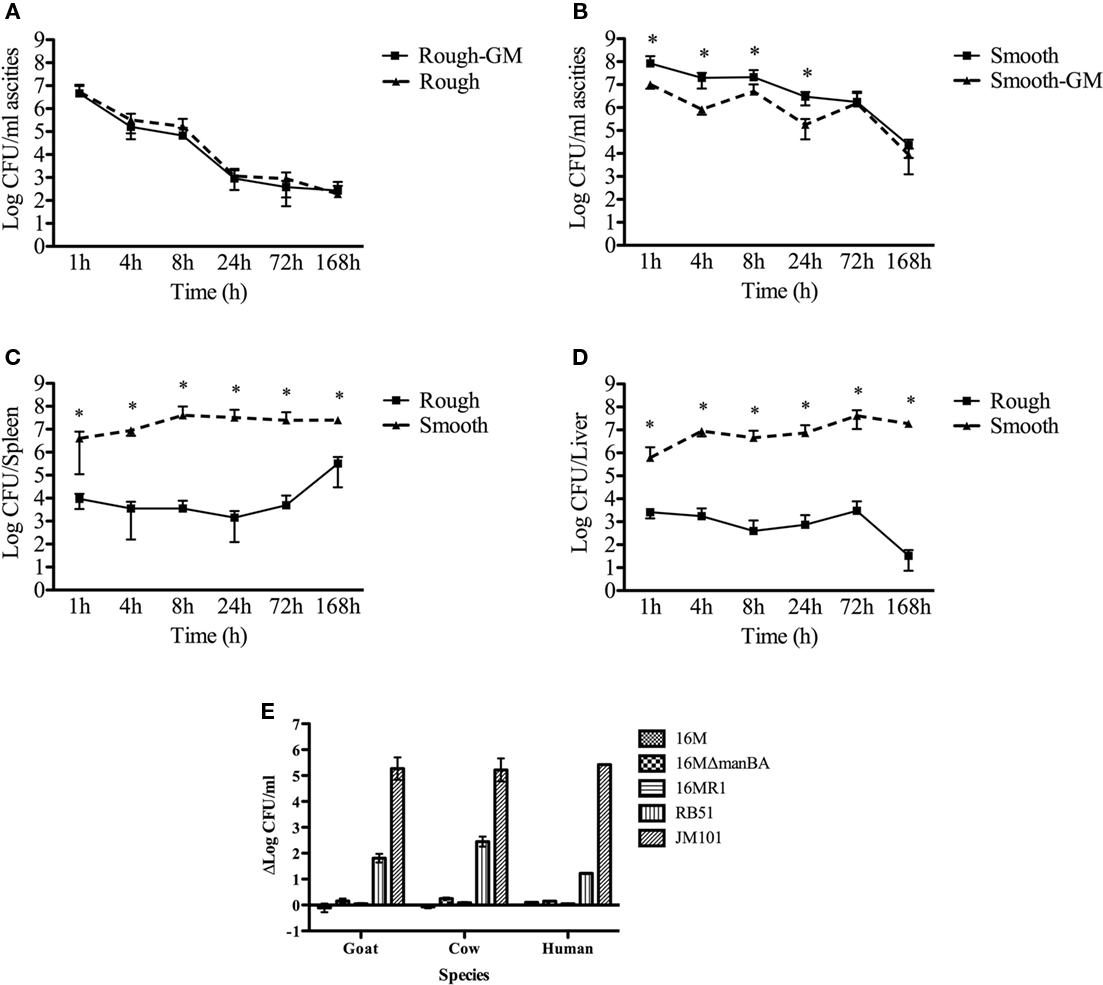
Figure 4. Clearance kinetics of smooth and rough Brucella from the murine peritoneal cavity and in vitro susceptibility to complement. Mice were infected with 108 cfu of either smooth B. melitensis 16M or rough B. melitensis 16MΔmanBA. Bacterial recovery from the ascites over a 7-day period revealed a lower percentage of smooth organisms (A) than rough organisms (B) internalized. Systemic infection was determined by recovery of smooth and rough organisms from secondary lymphoid organs, spleen (C), and liver (D). Significant differences (p ≤ 0.05) are represented with asterisks (*). (E) Shows the differences in susceptibility/resistance to complement-mediated lysis of smooth (B. melitensis 16M) and rough organisms (B. melitensis 16M R1, B. abortus RB51, and E. coli JM101) using sera from several host species. ΔLogcfu represents the difference in recovered colony forming units (cfu) following incubation in the presence or absence of naïve sera. Resistant organisms will exhibit very little difference in survival (values close to zero) while sensitive organisms will exhibit a difference in recovery. The inoculum reveals the cfu per milliliter used in these experiments. The error bars reveal SD of across five mice.
Discussion
The loss of surface O-polysaccharide from classical Brucella spp (melitensis, abortus, suis) is directly associated with a loss of virulence, yet there is evidence that rough variants persist and even replicate within professional phagocytic cells (Porte et al., 2003; Barquero-Calvo et al., 2007). In addition, there are two naturally occurring rough Brucella spp (canis, ovis) that retain virulence despite the loss of detectable O-polysaccharide. Outside the Brucella genus, a similar phenomenon has been found to confer an advantage to Vibrio cholera in the environment, and the hypermutable phenotype of Pseudomonas aeruginosa provides a changing pattern of phenotypes and virulence characteristics in infected individuals (Rice et al., 1992; Oliver et al., 2000; Rashid et al., 2003). Because of discrepancies associated with in vitro characterization of rough Brucella organisms, it was hypothesized that evaluation in vivo would provide improved insight concerning the fate and role of organisms lacking O-polysaccharide. Although rough organism exhibits an immediate reduction in number and effective internalization by resident cells, there appears to be an absence of associated inflammatory response and influx of cells (data not shown) to a lesser degree than reported for the parental smooth strains (Barquero-Calvo et al., 2007). Internalization of smooth organisms appears to be less efficient ≤10%, and this difference has been documented in ex vivo internalization kinetics (Pei et al., 2008). To explain the rapid decline in the viability of rough organisms consideration should be given to the differences in resident versus recruited inflammatory cells (Rabinowitz and Gordon, 1989). Enhanced uptake and intracellular killing by resident macrophages may explain the differences in the ability of smooth and rough organisms to cause disease. Many studies have been conducted on rough derivatives of Brucella due to interest in the use of rough strains as vaccines. As a result, research has focused on quantifying the immunological responses to and differences between rough and smooth strains. The recovery of rough variants in both mice and macrophages demonstrates the presence of organisms with an alternate LPS conformation is a normal state during infection. At this point the recovery of rough variants either reflects the replication of incoming rough variants or their spontaneous appearance during the course of infection. However, the rapid clearance of inoculated rough organisms suggests the continued appearance of rough variants during the course of infection is the most likely scenario leading to the hypothesis that the appearance of rough organisms may be part of the natural course of infection.
The results obtained are consistent with smooth organisms recruiting cells to the peritoneal cavity that enhance systemic spread of the organism. In contrast, rough organisms are inefficient at recruiting professional phagocytes to the site of infection in the peritoneal cavity, and are reduced in number by extracellular killing mechanisms and/or sensitivity to intracellular killing by resident cells. Rough variants appear later, most likely within macrophages as suggested by the loss of reactivity with monoclonal antibody to O-polysaccharide (Figure 2). Although, intracellular replication of rough Brucella variants has been previously documented, the lack of interest can be attributed to the inability of rough variants to cause disease (Porte et al., 2003). An association between rough variants and cytotoxic activity suggests that the appearance of rough organisms contributes to the spread of the agent by lysing the host macrophages and permitting subsequent infection of neighboring cells where the cycle is repeated (Pei and Ficht, 2004).The property of Brucella organisms to induce necrotic cell death is also observed in trophoblasts lining the fetus in pregnant animals (Anderson et al., 1986a,b). Although not specifically evaluated in this study placental necrosis is consistent with the appearance of rough organisms which could play a role in this acute form of the disease. Existence of two virulent rough Brucella species, B. ovis and B. canis, lacking O-polysaccharide suggests alternative receptor–ligand interactions may permit the bacterium to participate in successful interaction with the host (Jones et al., 1976; Martin-Martin et al., 2008).
Initially, Southern blotting of the manBA locus in Brucella abortus revealed that spontaneous genomic rearrangements may be a cause of the loss of function of manBA in natural rough derivatives. The observed rearrangement constitutes a 12-kb deletion, removing manBA, a putative lysR transcriptional regulator, an rtn gene encoding a histone-like protein, several glutamate dehydrogenase genes and a gamma-amino butyric acid antiporter gene. Based on this evidence we placed rough mutants into two broad classes: mutants with a spontaneous deletion encompassing manBA, and mutants with an intact, but non-functional manBA locus. Though this assay was used to identify an initial target, additional large-scale deletions were not observed. Instead point mutations and insertions were observed in sequenced, manBA defective strains. This may reflect that the strain used in the initial experiment had an unknown history and may have been subjected to serial passage. The deletion event may represent an endpoint of gene inactivation, although there are several additional functions present, none may be necessary during growth in culture. Current views from studies of homologous recombination, adapted from studies in eukaryotic organisms, are that recombination between DNA strands may involve an exploratory homology search through the tips of single-stranded DNA from intact DNA duplexes initiating recombination (Lao and Forsdyke, 2000). The inserted sequence, CGGGCA, repeats five times within the native phosphomannomutase, between the positive and negative strands. In contrast, this same motif occurs only once in the manA gene. Duplication is probably a result of run-on during replication. This motif is common throughout the Brucella genome and may represent a recombination hot spot, similar to the E. coli crossover hot spot instigator (chi), which is distributed throughout the E. coli genome (Tracy et al., 1997).
Luria–Delbrück fluctuation calculations were applied to multiple independent cultures to estimate the rates of pyrE versus manBA mutation. Often, experiments examining spontaneous mutation report a “mutant frequency.” The mutant frequency is simply the ratio of mutants divided by the total number of bacteria in the population. The mutant frequency is easy to measure, but is less reliable than the mutation rate because it may show large fluctuations depending upon when the first mutation appeared in the population (Rosche and Foster, 2000). It is important to distinguish between m, the mean number of mutations that occur during the growth of a culture, and μ = the mutation rate, which is the mean number of mutations that occur during the lifetime of a cell. By exploiting spontaneous auxotrophy, background mutation rates for both Brucella abortus and Brucella melitensis were well within the range of rates observed for other prokaryotic organisms (Drake, 1991, 1999; Grogan et al., 2001).
Appearance of rough variants appears to be the result of a natural process during growth of the organism. Efforts to demonstrate the role of rough organism in the ultimate success of the infection, is impeded by the fact that variants contain mutation at one of several loci, and thus are difficult, if not impossible, to prevent. Although the results presented here indicate the complementation of one-third of rough mutants by introduction of a plasmid containing an unaltered version of manBA, these complemented strains were also shown to produce rough variants either as a result of mutation at other O-polysaccharide related loci or mutation within the plasmid copy of manBA. Recently, Mancilla et al. (2010) demonstrated that genomic island-2, containing the classical LPS genes, is an unstable genetic element. Loss of this element, through activation of an integrase, contributes to loss of LPS expression, at frequencies similar to those observed by us for manBA derived spontaneous rough mutants.
The loss of O-polysaccharide expression, by the bacterium, to result from mutations in the primary sequence, perhaps conferring a survival advantage to the bacterium. However, the preliminary nature of these results cannot rule out additional possibilities including epigenetic changes. At this point we cannot determine whether the mutator phenotype is the result of cis-acting or trans-acting elements or both. All of which suggest the need for deep sequencing, including an analysis of DNA methylation patterns. Alternatively, conditional mutants capable of regulated O-polysaccharide expression may offer an alternative approach to confirm the significance associated with loss of O-polysaccharide expression.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge James Douglas for the gift of the Bk2 phage, Jean Celli and members of the Ficht lab for their helpful discussions. Portions of this work were supported by grants from USDA/CSREES (99-35024-7550) and NIH (A0048496-01A1) and training grant USDA/NNTG (98-38420-5806 HEP).
References
Allen, C., Adams, L., and Ficht, T. (1998). Transposon-derived Brucella abortus rough mutants are attenuated and exhibit reduced intracellular survival. Infect. Immun. 66, 1008–1016.
Alton, G. G., Jones, L. M., and Pietz, D. E. (1975). Laboratory techniques in brucellosis. Monogr. Ser. World Health Organ. 1–163.
Anderson, T. D., Cheville, N. F., and Meador, V. P. (1986a). Pathogenesis of placentitis in the goat inoculated with Brucella abortus. II. Ultrastructural studies. Vet. Pathol. 23, 227–239.
Anderson, T. D., Meador, V. P., and Cheville, N. F. (1986b). Pathogenesis of placentitis in the goat inoculated with Brucella abortus. I. Gross and histologic lesions. Vet. Pathol. 23, 219–226.
Barquero-Calvo, E., Chaves-Olarte, E., Weiss, D. S., Guzman-Verri, C., Chacon-Diaz, C., Rucavado, A., Moriyon, I., and Moreno, E. (2007). Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS ONE 2, e631. doi: 10.1371/journal.pone.0000631
Cassataro, J., Estein, S. M., Pasquevich, K. A., Velikovsky, C. A., de la Barrera, S., Bowden, R., Fossati, C. A., and Giambartolomei, G. H. (2005). Vaccination with the recombinant Brucella outer membrane protein 31 or a derived 27-amino-acid synthetic peptide elicits a CD4+ T helper 1 response that protects against Brucella melitensis infection. Infect. Immun. 73, 8079–8088.
Chen, F., and He, Y. (2009). Caspase-2 mediated apoptotic and necrotic murine macrophage cell death induced by rough Brucella abortus. PLoS ONE 4, e6830. doi: 10.1371/journal.pone.0006830
Cloeckaert, A., Tibor, A., and Zygmunt, M. S. (1999). Brucella outer membrane lipoproteins share antigenic determinants with bacteria of the family Rhizobiaceae. Clin. Diagn. Lab. Immunol. 6, 627–629.
Cloeckaert, A., Verger, J. M., Grayon, M., and Vizcaino, N. (1996). Molecular and immunological characterization of the major outer membrane proteins of Brucella. FEMS Microbiol. Lett. 145, 1–8.
Connolly, J. P., Comerci, D., Alefantis, T. G., Walz, A., Quan, M., Chafin, R., Grewal, P., Mujer, C. V., Ugalde, R. A., and Delvecchio, V. G. (2006). Proteomic analysis of Brucella abortus cell envelope and identification of immunogenic candidate proteins for vaccine development. Proteomics 6, 3767–3780.
Corbeil, L., Blau, K., Inzana, T., Nielsen, K., Jacobson, R., Corbeil, R., and Winter, A. (1988). Killing of Brucella abortus by bovine serum. Infect. Immun. 56, 3251–3261.
Datsenko, K., and Wanner, B. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645.
de Jong, M. F., Rolan, H. G., and Tsolis, R. M. (2010). Innate immune encounters of the (type) 4th kind: Brucella. Cell. Microbiol. 12, 1195–1202.
Dehio, C., and Meyer, M. (1997). Maintenance of broad-host-range incompatibility group P and group Q plasmids and transposition of Tn5 in Bartonella henselae following conjugal plasmid transfer from Escherichia coli. J. Bacteriol. 179, 538–540.
Douglas, J., and Elberg, S. (1976). Isolation of Brucella melitensis phage of broad biotype and species specificity. Infect. Immun. 14, 306–308.
Douglas, J., and Elberg, S. (1978). Properties of the Berkeley phage lytic for “Brucella melitensis” and other species. Ann. Sclavo. 20, 681–691.
Drake, J. (1991). A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U.S.A. 88, 7160–7164.
Drake, J. (1999). The distribution of rates of spontaneous mutation over viruses, prokaryotes, and eukaryotes. Ann. N. Y. Acad. Sci. 870, 100–107.
Elzer, P., Enright, F., Colby, L., Hagius, S., Walker, J., Fatemi, M., Kopec, J., Beal, V. Jr., and Schurig, G. (1998). Protection against infection and abortion induced by virulent challenge exposure after oral vaccination of cattle with Brucella abortus strain RB51. Am. J. Vet. Res. 59, 1575–1578.
Fernandez-Prada, C., Nikolich, M., Vemulapalli, R., Sriranganathan, N., Boyle, S., Schurig, G., Hadfield, T., and Hoover, D. (2001). Deletion of wboA enhances activation of the lectin pathway of complement in Brucella abortus and Brucella melitensis. Infect. Immun. 69, 4407–4416.
Fernandez-Prada, C., Zelazowska, E., Nikolich, M., Hadfield, T., Roop, R. n., Robertson, G., and Hoover, D. (2003). Interactions between Brucella melitensis and human phagocytes: bacterial surface o-polysaccharide inhibits phagocytosis, bacterial killing, and subsequent host cell apoptosis. Infect. Immun. 71, 2110–2119.
Ficht, T., Husseinen, H., Derr, J., and Bearden, S. (1996). Species-specific sequences at the omp2 locus of Brucella type strains. Int. J. Syst. Bacteriol. 46, 329–331.
Ficht, T. A., Bearden, S. W., Sowa, B. A., and Marquis, H. (1990). Genetic variation at the omp2 porin locus of the Brucellae: species-specific markers. Mol. Microbiol. 4, 1135–1142.
Forestier, C., Deleuil, F., Lapaque, N., Moreno, E., and Gorvel, J. P. (2000). Brucella abortus lipopolysaccharide in murine peritoneal macrophages acts as a down-regulator of T cell activation. J. Immunol. 165, 5202–5210.
Forestier, C., Moreno, E., Meresse, S., Phalipon, A., Olive, D., Sansonetti, P., and Gorvel, J. P. (1999a). Interaction of Brucella abortus lipopolysaccharide with major histocompatibility complex class II molecules in B lymphocytes. Infect. Immun. 67, 4048–4054.
Forestier, C., Moreno, E., Pizarro-Cerda, J., and Gorvel, J. P. (1999b). Lysosomal accumulation and recycling of lipopolysaccharide to the cell surface of murine macrophages, an in vitro and in vivo study. J. Immunol.162, 6784–6791.
Gonzalez, D., Grillo, M. J., De Miguel, M. J., Ali, T., Arce-Gorvel, V., Delrue, R. M., Conde-Alvarez, R., Munoz, P., Lopez-Goñi, I., Iriarte, M., Marin, C. M., Weintraub, A., Widmalm, G., Zygmunt, M., Letesson, J. J., Gorvel, J. P., Blasco, J. M., and Moriyon, I. (2008). Brucellosis vaccines: assessment of Brucella melitensis lipopolysaccharide rough mutants defective in core and o-polysaccharide synthesis and export. PLoS ONE 3, e2760. doi: 10.1371/journal.pone.0002760
Grogan, D., Carver, G., and Drake, J. (2001). Genetic fidelity under harsh conditions: analysis of spontaneous mutation in the thermoacidophilic archaeon Sulfolobus acidocaldarius. Proc. Natl. Acad. Sci. U.S.A. 98, 7928–7933.
Holman, P. J., Adams, L. G., Hunter, D. M., Heck, F. C., Nielsen, K. H., and Wagner, G. G. (1983). Derivation of monoclonal antibodies against Brucella abortus antigens. Vet. Immunol. Immunopathol. 4, 603–614.
Hybiske, K., and Stephens, R. S. (2008). Exit strategies of intracellular pathogens. Nat. Rev. Microbiol. 6, 99–110.
Jimenez de Bagues, M. P., Dudal, S., Dornand, J., and Gross, A. (2005). Cellular bioterrorism: how Brucella corrupts macrophage physiology to promote invasion and proliferation. Clin. Immunol. 114, 227–238.
Jones, L., Diaz, R., and Berman, D. (1976). Endotoxic activity of rough organisms of Brucella species. Infect. Immun. 13, 1638–1641.
Kahl-McDonagh, M. M., and Ficht, T. A. (2006). Evaluation of protection afforded by Brucella abortus and Brucella melitensis unmarked deletion mutants exhibiting different rates of clearance in BALB/c mice. Infect. Immun. 74, 4048–4057.
Koch, A. (1982). Mutation and growth rates from Luria–Delbrück fluctuation tests. Mutat. Res. 95, 129–143.
Kohler, S., Foulongne, V., Ouahrani-Bettache, S., Bourg, G., Teyssier, J., Ramuz, M., and Liautard, J. (2002a). The analysis of the intramacrophagic virulome of Brucella suis deciphers the environment encountered by the pathogen inside the macrophage host cell. Proc. Natl. Acad. Sci. U.S.A. 99, 15711–15716.
Kohler, S., Porte, F., Jubier-Maurin, V., Ouahrani-Bettache, S., Teyssier, J., and Liautard, J. (2002b). The intramacrophagic environment of Brucellasuis and bacterial response. Vet. Microbiol. 90, 299–309.
Kohler, S., Michaux-Charachon, S., Porte, F., Ramuz, M., and Liautard, J. (2003). What is the nature of the replicative niche of a stealthy bug named Brucella? Trends Microbiol. 11, 215–219.
Kreutzer, D., Buller, C., and Robertson, D. (1979a). Chemical characterization and biological properties of lipopolysaccharides isolated from smooth and rough strains of Brucella abortus. Infect. Immun. 23, 811–818.
Kreutzer, D., Dreyfus, L., and Robertson, D. (1979b). Interaction of polymorphonuclear leukocytes with smooth and rough strains of Brucella abortus. Infect. Immun. 23, 737–742.
Lao, P. J., and Forsdyke, D. R. (2000). Crossover hot-spot instigator (chi) sequences in Escherichia coli occupy distinct recombination/transcription islands. Gene 243, 47–57.
Le Chat, L., Fons, M., and Taddei, F. (2006). Escherichia coli mutators: selection criteria and migration effect. Microbiology 152, 67–73.
Lea, D., and Coulson, C. (1949). The distribution of the numbers of mutants in bacterial populations. J. Genet. 49, 264–285.
Luria, S., and Delbrück, M. (1943). Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28, 491–511.
Mancilla, M., Lopez-Goñi, I., Moriyon, I., and Zarraga, A. M. (2010). Genomic island 2 is an unstable genetic element contributing to Brucella lipopolysaccharide spontaneous smooth-to-rough dissociation. J. Bacteriol. 192, 6346–6351.
Maniatis, T., Sambrook, J., Fritsch, E. F. and Laboratory C. S. H. (1982). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
Martin-Martin, A. I., Caro-Hernandez, P., Orduna, A., Vizcaino, N., and Fernandez-Lago, L. (2008). Importance of the Omp25/Omp31 family in the internalization and intracellular replication of virulent B. ovis in murine macrophages and HeLa cells. Microbes Infect. 10, 706–710.
Murphy, E., Robertson, G., Parent, M., Hagius, S., Roop, R. II, Elzer, P., and Baldwin, C. (2002). Major histocompatibility complex class I and II expression on macrophages containing a virulent strain of Brucella abortus measured using green fluorescent protein-expressing Brucellae and flow cytometry. FEMS Immunol. Med. Microbiol. 33, 191–200.
Oliver, A., Canton, R., Campo, P., Baquero, F., and Blazquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1254.
Palmer, D. A., and Douglas, J. T. (1989). Analysis of Brucella lipopolysaccharide with specific and cross-reacting monoclonal antibodies. J. Clin. Microbiol. 27, 2331–2337.
Pasquevich, K. A., Garcia Samartino, C., Coria, L. M., Estein, S. M., Zwerdling, A., Ibanez, A. E., Barrionuevo, P., Oliveira, F. S., Carvalho, N. B., Borkowski, J., Oliveira, S. C., Warzecha, H., Giambartolomei, G. H., and Cassataro, J. (2010). The protein moiety of Brucella abortus outer membrane protein 16 is a new bacterial pathogen-associated molecular pattern that activates dendritic cells in vivo, induces a Th1 immune response, and is a promising self-adjuvanting vaccine against systemic and oral acquired brucellosis. J. Immunol. 184, 5200–5212.
Pei, J., and Ficht, T. (2004). Brucella abortus rough mutants are cytopathic for macrophages in culture. Infect. Immun. 72, 440–450.
Pei, J., Turse, J. E., and Ficht, T. A. (2008). Evidence of Brucella abortus OPS dictating uptake and restricting NF-kappaB activation in murine macrophages. Microbes Infect. 10, 582–590.
Pei, J., Turse, J. E., Wu, Q., and Ficht, T. A. (2006). Brucella abortus rough mutants induce macrophage oncosis that requires bacterial protein synthesis and direct interaction with the macrophage. Infect. Immun. 74, 2667–2675.
Porte, F., Naroeni, A., Ouahrani-Bettache, S., and Liautard, J. (2003). Role of the Brucellasuis lipopolysaccharide o antigen in phagosomal genesis and in inhibition of phagosome-lysosome fusion in murine macrophages. Infect. Immun. 71, 1481–1490.
Rabinowitz, S., and Gordon, S. (1989). Differential expression of membrane sialoglycoproteins in exudate and resident mouse peritoneal macrophages. J. Cell Sci. 93(Pt 4), 623–630.
Rashid, M., Rajanna, C., Ali, A., and Karaolis, D. (2003). Identification of genes involved in the switch between the smooth and rugose phenotypes of Vibrio cholerae. FEMS Microbiol. Lett. 227, 113–119.
Rice, E., Johnson, C., Clark, R., Fox, K., Reasoner, D., Dunnigan, M., Panigrahi, P., Johnson, J., and Morris, J. Jr. (1992). Chlorine and survival of “rugose” Vibrio cholerae. Lancet 340, 740.
Rittig, M., Kaufmann, A., Robins, A., Shaw, B., Sprenger, H., Gemsa, D., Foulongne, V., Rouot, B., and Dornand, J. (2003). Smooth and rough lipopolysaccharide phenotypes of Brucella induce different intracellular trafficking and cytokine/chemokine release in human monocytes. J. Leukoc. Biol. 74, 1045–1055.
Rojas, N., Freer, E., Weintraub, A., Ramirez, M., Lind, S., and Moreno, E. (1994). Immunochemical identification of Brucella abortus lipopolysaccharide epitopes. Clin. Diagn. Lab. Immunol. 1, 206–213.
Rosche, W., and Foster, P. (2000). Determining mutation rates in bacterial populations. Methods 20, 4–17.
Steinberg, T., Pretty on Top, K., Berggren, K., Kemper, C., Jones, L., Diwu, Z., Haugland, R., and Patton, W. (2001). Rapid and simple single nanogram detection of glycoproteins in polyacrylamide gels and on electroblots. Proteomics 1, 841–855.
Tracy, R. B., Chédin, F., and Kowalczykowski, S. C. (1997). The recombination hot spot chi is embedded within islands of preferred DNA pairing sequences in the E. coli genome. Cell 90, 205–206.
Viadas, C., Rodriguez, M. C., Sangari, F. J., Gorvel, J. P., Garcia-Lobo, J. M., and Lopez-Goñi, I. (2010). Transcriptome analysis of the Brucella abortus BvrR/BvrS two-component regulatory system. PLoS ONE 5, e10216. doi: 10.1371/journal.pone.0010216
Keywords: Brucella, rough mutant, lipopolysaccharide, O-polysaccharide
Citation: Turse JE, Pei J and Ficht TA (2011) Lipopolysaccharide-deficient Brucella variants arise spontaneously during infection. Front. Microbio. 2:54. doi: 10.3389/fmicb.2011.00054
Received: 12 October 2010;
Accepted: 10 March 2011;
Published online: 23 March 2011.
Edited by:
Adel M. Talaat, University of Wisconsin–Madison, USAReviewed by:
Jean-Jacques Letesson, University of Namur, BelgiumMartin R Roop II, East Carolina University, USA
Gary Splitter, University of Wisconsin–Madison, USA
Copyright: © 2011 Turse, Pei and Ficht. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Thomas A. Ficht, Department of Veterinary Pathobiology, Texas A&M University, MS 4467, College Station, TX 77843-4467, USA. e-mail:dGZpY2h0QGN2bS50YW11LmVkdQ==