
 Catherine A. Mueller
Catherine A. Mueller
- Department of Microbiology and Immunology, Columbia University Medical Center, New York, NY, USA
Legionella pneumophila is the causative agent of Legionnaires’ disease, an acute pulmonary infection. L. pneumophila is able to infect and multiply in both phagocytic protozoa, such as Acanthamoeba castellanii, and mammalian professional phagocytes. The best-known L. pneumophila virulence determinant is the Icm/Dot type IVB secretion system, which is used to translocate more than 150 effector proteins into host cells. While the transcriptional response of Legionella to the intracellular environment of A. castellanii has been investigated, much less is known about the Legionella transcriptional response inside human macrophages. In this study, the transcriptome of L. pneumophila was monitored during exponential and post-exponential phase in rich AYE broth as well as during infection of human cultured macrophages. This was accomplished with microarrays and an RNA amplification procedure called selective capture of transcribed sequences to detect small amounts of mRNA from low numbers of intracellular bacteria. Among the genes induced intracellularly are those involved in amino acid biosynthetic pathways leading to l-arginine, l-histidine, and l-proline as well as many transport systems involved in amino acid and iron uptake. Genes involved in catabolism of glycerol are also induced during intracellular growth, suggesting that glycerol could be used as a carbon source. The genes encoding the Icm/Dot system are not differentially expressed inside cells compared to control bacteria grown in rich broth, but the genes encoding several translocated effectors are strongly induced. Moreover, we used the transcriptome data to predict previously unrecognized Icm/Dot effector genes based on their expression pattern and confirmed translocation for three candidates. This study provides a comprehensive view of how L. pneumophila responds to the human macrophage intracellular environment.
Introduction
Legionella pneumophila is a human opportunistic pathogen and the causative agent of Legionnaires’ disease, a pulmonary infection acquired by inhaling contaminated aerosols (Fraser et al., 1977). Once in the lung, L. pneumophila infects and multiplies in alveolar macrophages. Legionellosis accounts for 2–15% of community acquired pneumonia cases that require hospitalization (Marston et al., 1994). In most cases healthy individuals clear L. pneumophila from their lungs but immune compromised patients or individuals with sustained lung damage often suffer complications. Nosocomial infection with L. pneumophila is usually more severe and the fatality rate even with effective antibiotic therapy is close to 50% (Carratala et al., 1994). In the environment, L. pneumophila is found in many natural and man made water systems where it infects amebae and other protozoa.
To successfully infect and grow inside host cells, L. pneumophila circumvents normal endocytic trafficking pathways and inhibits phagosome maturation, including acidification and fusion with lysosomes. This results in a permissive replication niche called the Legionella containing vacuole (LCV; reviewed in Franco et al., 2009). The LCV is characterized by recruitment of early secretory vesicles, mitochondria, and membrane vesicles derived from the golgi and the endoplasmic reticulum (Roy and Tilney, 2002; Molofsky and Swanson, 2004; Shin and Roy, 2008; Hubber and Roy, 2010).
The Icm/Dot type IVB secretion system (TFBSS) of L. pneumophila is essential for intracellular growth of the bacteria in all known hosts. It translocates ∼200 diverse effector proteins, which often contain eukaryotic-like domains, into host cells (Segal and Shuman, 1998; Segal et al., 1998; Vogel et al., 1998; Cazalet et al., 2004; Chien et al., 2004; de Felipe et al., 2005; Burstein et al., 2009; Huang et al., 2011). In the absence of a functional Icm/Dot TFBSS, effectors are not translocated and the bacteria are unable to avoid intracellular degradation. There appears to be a high degree of functional redundancy among the effectors, because deletion of a single effector gene, or even groups of related genes usually has no or a very limited impact on the fate of the LCV (Ninio and Roy, 2007). This apparent functional redundancy has made the characterization of the effectors difficult.
It has been proposed that L. pneumophila adopts a biphasic lifestyle consisting of the replicative phase inside the LCV and an extracellular transmissive phase (Molofsky and Swanson, 2004). The replicative phase is characterized by exponential growth, no motility, and repression of transmissive traits. At the transition to the transmissive phase the bacteria become motile and more cytotoxic. The current assumption is that the L. pneumophila replicative and transmissive phases are equivalent to exponential and post-exponential growth in broth (Molofsky and Swanson, 2004).
In this study we used microarray technology to monitor global gene expression changes among several different conditions, to gain further insight into the processes occurring during intracellular growth of L. pneumophila inside human macrophages and to directly evaluate the relationship between the patterns of gene expression during growth in axenic medium and during growth in macrophages. At early times following macrophage infection at low multiplicity of infection (MOI), extracted RNA is largely of eukaryotic origin, which interferes with, and reduces the hybridization signal on microarrays. Due to this limitation other studies of gene expression have been limited to infections at non-physiologic MOI. To circumvent this limitation, we used a previously described method called selective capture of transcribed sequences (SCOTS), to enrich bacterial transcripts and discard host cell transcripts and rRNA (Graham and Clark-Curtiss, 1999; Faucher et al., 2006).
Materials and Methods
Bacterial Strains and Growth Conditions
Legionella pneumophila strain JR32, a streptomycin-resistant, restriction-negative mutant of L. pneumophila strain Philadelphia-1 was used. Media and antibiotics were used as previously described (Chen et al., 2004). For liquid culture, AYE broth was inoculated with a culture grown overnight to a final OD600 of 0.1 and incubated at 37°C with vigorous shaking. Exponential (E) phase bacteria were harvested at an OD600 of 0.7–0.8 and post-exponential (PE) phase bacteria were harvested approximately 6 h after the cessation of growth.
Infection of Cultured Human Macrophages
The human monocyte cell line THP-1 (ATCC TIB-202) was maintained in Advanced RPMI (Invitrogen) supplemented with 10% (v/v) fetal calf serum (Invitrogen) and 2 mM L-glutamine (Invitrogen). A stock culture of the cells was maintained as monocyte-like, non-adherent cells at 37°C in an atmosphere containing 5% (v/v) CO2. For macrophage infection, cells were seeded at 2 × 107 cells in 10 cm culture dishes and were differentiated by addition of 10−7 M phorbol 12-myristate 13-acetate for 48 h (Sigma). Before infection, macrophages were treated with antibodies raised against the major outer membrane protein (MOMP) of L. pneumophila for 30 min (Charpentier et al., 2009). Bacteria were grown overnight shaking in AYE at 37°C and were then added to the cell monolayer at a MOI of 1, and centrifuged for 5 min at 800 × g to synchronize bacterial uptake. After incubation for 2 h at 37°C, the infected cells were washed three times with phosphate buffered saline (PBS) pH 7.4 and fresh complete RPMI medium containing 100 μg ml−1 gentamicin was added to each well. After incubation for 1 h at 37°C, cells were washed three times with PBS and the cells were either harvested (T0) or incubated with fresh complete RPMI medium for a further 6 h (T6) or 18 h (T18). At each time point, cells were scraped into 2 ml PBS and 100 μl were removed to determine the number of colony forming units (CFU) by plating dilutions on AYE plates. Samples were then centrifuged for 5 min at 1000 × g and the pellet was lysed in TRIzol (Invitrogen) and stored at −80°C.
RNA Extraction
RNA was isolated using TRIzol reagents as described by the manufacturer (Invitrogen). The RNA was subsequently treated with DNase I (Invitrogen) for 1 h at 37°C. The DNase was then inactivated by incubation at 75°C for 5 min and after acid phenol–chloroform (Ambion) extraction the RNA was precipitated with NaAc–ethanol. Purity and quantity of RNA was determined by spectrophotometry.
Selective Capture of Transcribed Sequences
Each RNA sample was converted to cDNA in five independent reverse-transcription reactions. Briefly, 5 μg of RNA was converted to first strand cDNA by random priming, using primer RB1-RNA (Table 1) containing a defined 5′ end and random non amer at the 3′ end, with Superscript II (Invitrogen), according to the manufacturer’s instruction. A second strand of cDNA was synthesized using Klenow fragment (New England Biolab) according to the manufacturer’s instruction. Bacterial transcripts were then separated from host cDNA by SCOTS, a selective hybridization to bacterial genomic DNA (gDNA) as described previously (Graham and Clark-Curtiss, 1999; Daigle et al., 2001; Faucher et al., 2006). Briefly, denatured, biotinylated, and sonicated L. pneumophila gDNA fragments (0.3 μg) were mixed with 5 μg of sonicated ribosomal DNA (from plasmid pSF6) to pre-block rRNA encoding regions on the gDNA. After hybridization at 60°C for 30 min, total cDNA (1 μg) was added, and hybridization was allowed to proceed for 22 h at 60°C. Bacterial cDNA that was hybridized to biotinylated gDNA was then captured by binding hybrids to streptavidin-coated magnetic beads (Promega) according to manufacturer’s instructions. Captured cDNA was eluted, precipitated, and amplified by PCR using primer RB1-PCR (Table 1). For each condition, three rounds of capture were performed.
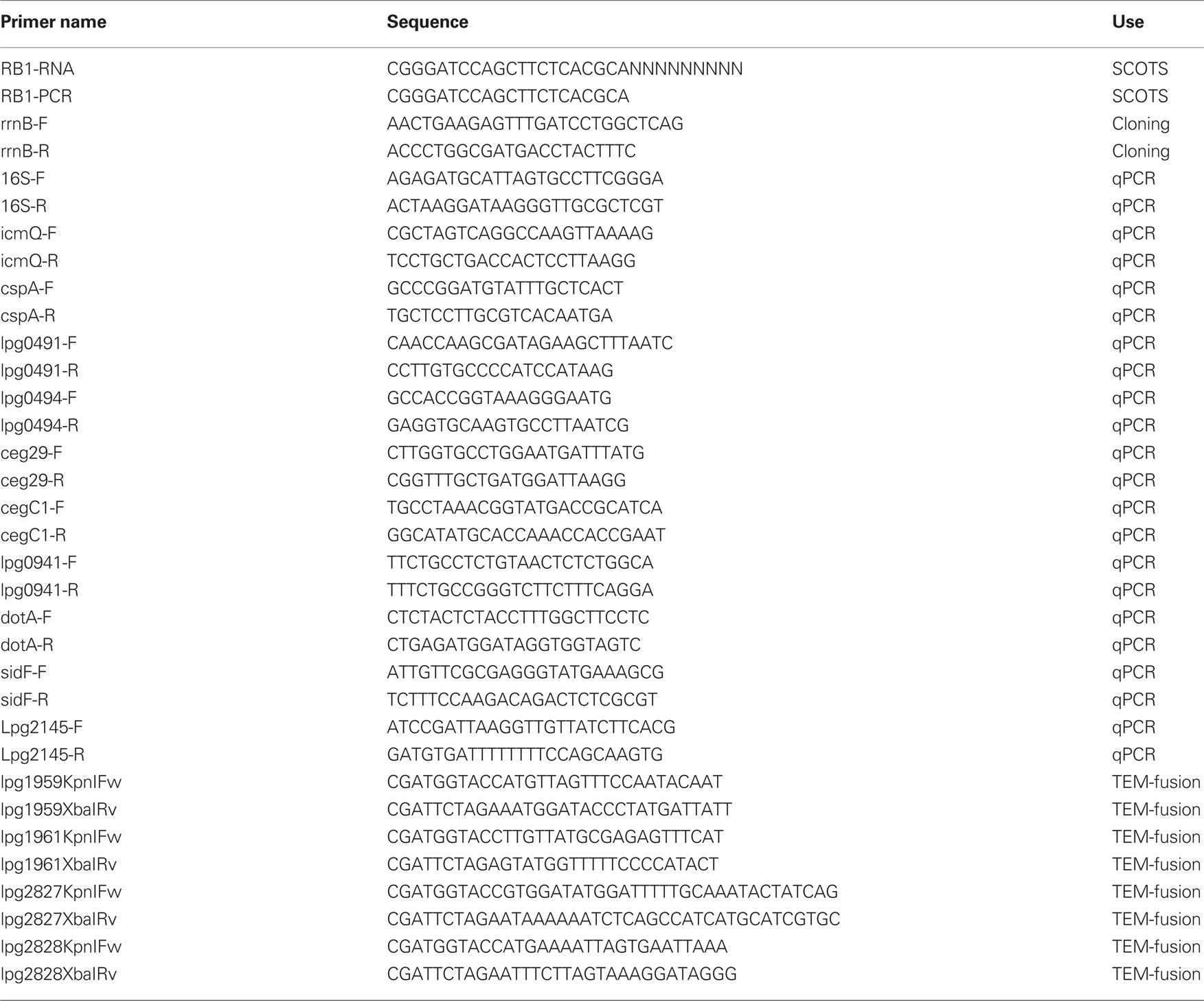
Table 1. Primers used in this study.
Microarray Procedure
The whole genome microarray of L. pneumophila has been previously described (Hovel-Miner et al., 2009). Two micrograms of cDNA from each condition/replicate was labeled independently by PCR using amino-allyl dUTP and RB1 primer. Bacterial gDNA was used as the reference channel on each slide to allow comparison of each time point and of different samples (Talaat et al., 2002). Five micrograms of gDNA was labeled with amino-allyl dUTP using Klenow fragment and random primers (Invitrogen) at 37°C for 18 h (Faucher et al., 2006). DNA was subsequently coupled to the succinimidyl ester fluorescent dye AlexaFluor 546 (for cDNA) or AlexaFluor 647 (for gDNA) (Invitrogen) following the manufacturer’s protocols. Hybridization and data acquisition were performed as previously described (Hovel-Miner et al., 2009). Local background was removed from spot signal intensity and normalization was carried out by calculating the fraction over the total signal intensity in both channels as previously described (Faucher et al., 2006). Signal levels that were lower than background in experiments and controls were filtered out. A total of 12 cDNA to reference ratios were recorded for each time point. Statistical analysis between test and control conditions was performed using an unpaired one-tailed Student’s t-test. Genes were considered differentially expressed if they demonstrated a ratio to control value of ±2-fold with a p < 0.001. The microarray data have been submitted to the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE23029 and GSE23032.
Quantitative Real-Time PCR
RNA extraction was performed as described above. cDNA was synthesized in triplicate using Superscript II (Invitrogen) with random hexamers (Invitrogen), according to the manufacturer’s instructions. For each sample, a no reverse-transcriptase reaction served as a no template control (NTC). qPCR was performed using the Applied Biosystems StepOne Plus 96 well RT-PCR system with Power Syber green PCR master mix following the manufacturer’s instructions (Applied Biosystems). Primers are described in Table 1. For each qPCR run, the calculated threshold cycle (Ct) was normalized to the Ct of the internal control 16S rRNA amplified from the corresponding samples and the fold-change was calculated as previously described (Livak and Schmittgen, 2001).
Southern Blot
The PCR DIG labeling mix (Roche Molecular Biochemicals) was used to produce Digoxigenin-labeled cDNA probes according to the manufacturer’s instructions. gDNA was extracted with the Wizard kit (Promega) and digested with HincII (NEB). The DNA was transferred to a positively charged membrane. Membranes were prehybridized and subsequently incubated at 42°C with the digoxigenin-labeled specific probe in 50% (v/v) formamide, 5× SSC; 2% (w/v) blocking reagent (Roche Molecular Biochemicals), 0.1% (w/v) N-laurylsarcosine, 0.02% (w/v) SDS. The blots were hybridized overnight and subjected to stringency washes in 0.1× SSC and 0.1% (w/v) SDS. Hybridization signals were detected with a DIG Luminescent Detection Kit (Roche Molecular Biochemicals) according to the manufacturer’s instructions.
β-Lactamase (TEM) Fusion Translocation Assay
The putative effector genes (lpg1959, lpg1961, lpg2827, lpg2828) were PCR amplified and cloned in frame with the beta-lactamase gene at the KpnI/XbaI sites of pXDC61 (de Felipe et al., 2008). The resulting plasmids were introduced into KS79 (JR32ΔcomR) or KS79 dotA::Tn903dIIlacZ by transformation. All primers, strains, and plasmids used are listed in Tables 1 and 2. The TEM-translocation assay was performed as previously described (de Felipe et al., 2008) with the difference that the MOI was lowered from 50 to 20. Images of cells after the translocation were obtained by epifluorescence on individual assay wells (data not shown). For immunoblots bacteria used for the TEM-translocation assay were resuspended in 1× laemmli buffer and boiled. Whole cell lysate corresponding to 2 × 106 bacteria was loaded per lane. Immunoblotting was carried out using rabbit polyclonal antibodies directed against β-lactamase (anti-TEM). Detection was performed with secondary antibodies conjugated to horseradish peroxidase (1:5000, Goat Anti-Rabbit-HRP conjugated, Pierce) before development with supersignal chemiluminescent substrate (Pierce).
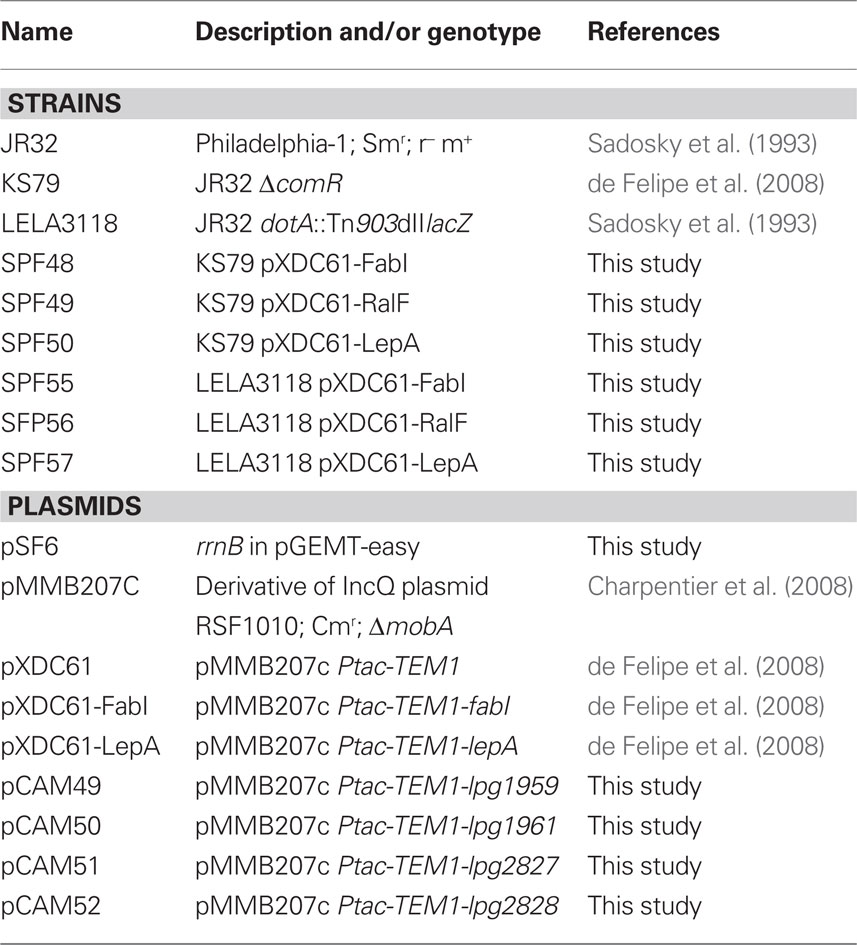
Table 2. Strains used in this study.
Results
Infection Model, Effect of Scots, and Validation of the Method
The aim of this work was to study the gene expression profile of L. pneumophila during infection of macrophages infected at a MOI of 1. SCOTS is a method that allows amplification of small amounts of bacterial RNA from infected host cells, while discarding host cell transcripts and ribosomal RNA (Faucher et al., 2006). Macrophage-like cells derived from the human THP-1 monocyte cell line were infected with L. pneumophila opsonized with antibodies raised against the L. pneumophila MOMP, which increases the efficiency of bacterial entry into host cells (Charpentier et al., 2009). After 2 h of infection, the macrophages were washed and treated with gentamicin for 1 h to kill extracellular bacteria, and cells were washed three times, and fresh medium was added. Samples for the first time point (T0) were collected after the gentamicin treatment. Samples were also collected after 6 h (T6) and 18 h (T18). Only 1 × 106 CFU were recovered at T0, indicating that approximately 1 in 20 macrophages were infected by L. pneumophila (Figure 1A). At T6, the number of bacteria increased and by T18, a 10-fold increase in the number of intracellular bacteria was observed (Figure 1B) and some of the cells were detached from the cell monolayer (data not shown). T0 can be considered an early time point of infection where the bacteria are still adapting to the intracellular environment. At T6 the bacteria are actively growing and by T18 the bacteria have reached their maximum number and start to lyse the host cells.
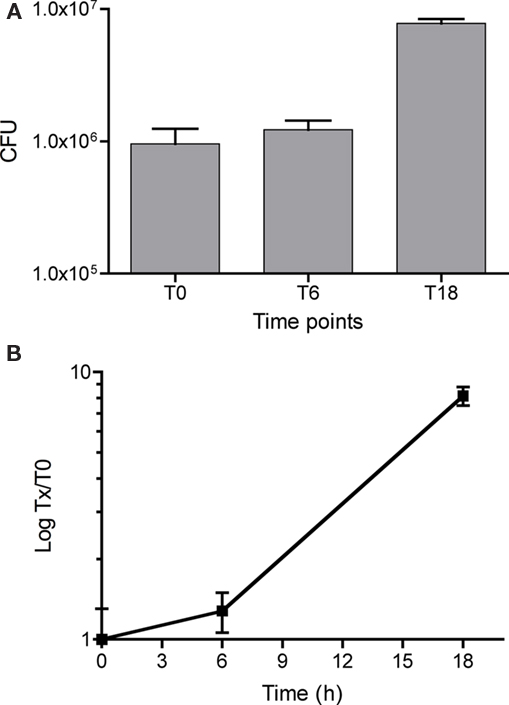
Figure 1. Kinetics of THP-1 macrophage infection by Legionella pneumophila. (A) Ten million human cultured THP-1 macrophages were infected with L. pneumophila at a MOI of 1. At each time point, cells were washed three times and a fraction was resuspended in distilled water to release intracellular bacteria. Shown is the number of colony forming units (CFU) present inside the totality of cells. (B) CFU was normalized against the number of CFU at T0 to show change in intracellular multiplication.
Following isolation of total RNA of the infected cells, the RNA was converted to cDNA with reverse-transcriptase and processed using the SCOTS method as described in the Section “Materials and Methods.” The effect of SCOTS on the cDNA pool was visualized by Southern blotting (Figure 2A). Bacterial cDNA not treated by SCOTS is almost exclusively of prokaryotic ribosomal origin (Figure 2A lane 1 and 5). The diversity of the bacterial cDNA increases with the number of SCOTS rounds performed while the amount of cDNA of ribosomal origin decreases (Figure 2A lanes 1 through 4) as previously described (Graham and Clark-Curtiss, 1999). Therefore, samples from all conditions, including growth in AYE broth to E or PE phase, were treated with three consecutive rounds of SCOTS and the resulting cDNA was labeled and used to hybridize to the microarray slides. As an internal reference, labeled L. pneumophila gDNA was also hybridized to the microarrays (Talaat et al., 2002). For each condition studied, three independent biological replicates and two technical replicates were analyzed, resulting in six replicates for each condition. The background values were subtracted, the data were normalized by calculating the contribution of each spot to the total signal intensity and the ratio to the gDNA signal was recorded. A one-tailed Student’s t-test was used for statistical analysis and the ratio between test conditions (PE, T0, T6, and T18) and the control conditions (E phase or T0) was calculated. Despite the very small number of bacteria present during infection, SCOTS allowed us to obtain easily measurable microarray signals.
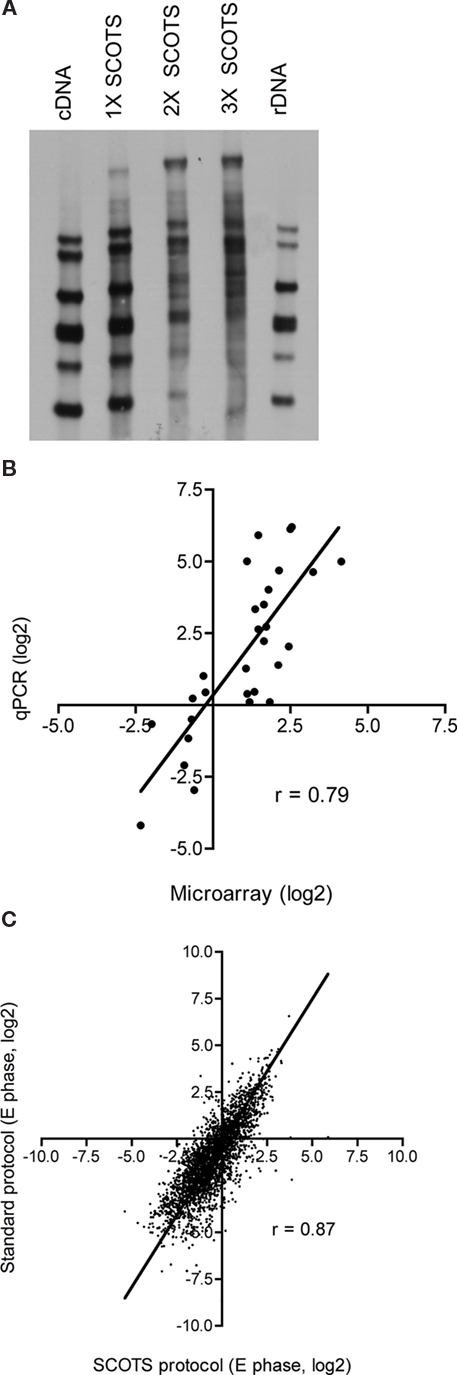
Figure 2. Effects of SCOTS on the cDNA population. (A) Southern blot of L. pneumophila gDNA digested with HincII and hybridized with labeled cDNA from the T0 time point obtained before (lane 1) and after the first (lane 2), the second (lane 3), and the third (lane 4) round of SCOTS. Lane 5 was hybridized with labeled rDNA. (B) qPCR was used to validate the expression profiles obtained by microarray for eight genes in all the conditions. (C) Comparison of microarray data obtained when using the SCOTS amplification method and when using a standard microarray protocol. Shown is the normalized signal intensity obtained from E phase growth in rich AYE broth. See text for details.
We used qPCR to validate the expression patterns of eight genes (Figure 2B) and the correlation between the microarray values and the qPCR values was 0.79 with a slope of 1.4. Next, we compared the expression patterns obtained by SCOTS to those obtained when a standard microarray protocol was used. RNA from axenically grown bacteria in E phase was treated using a standard microarray protocol where the cDNA is labeled during the reverse-transcription reaction as previously published (Faucher et al., 2010). The resulting cDNA was hybridized to the microarray slides as described above and data analysis was carried out the same way as for the SCOTS treated samples. The normalized signal intensities of the transcriptome in E phase obtained by SCOTS and by the standard microarray protocol were compared (Figure 2C). The correlation of the two datasets is 0.87, which indicates that SCOTS treatment of samples does not introduce significant bias on the gene expression data, in agreement with previous reports (Faucher et al., 2006; Poirier et al., 2008).
Comparison of Gene Expression Patterns between Growth in THP-1 Cells and Growth IN Broth
The normalized signal intensities for each gene were subjected to hierarchical clustering which revealed that there are some similarities, but also striking differences between PE phase and intracellular growth (T0, T6, T18, Figure 3A). To quantify these differences on a global level, a correlation matrix analysis of the five conditions was performed using Bioconductor (Figure 3B; Gentleman et al., 2004). The correlation between PE and any intracellular condition was approximately 0.6, indicating that PE phase is quite different than intracellular growth in human cultured macrophages. Interestingly, the correlation between E phase and in vivo time points was higher for early time points (0.77 and 0.70 for T0 and T6 respectively) than the later time point (0.64 for T18). The T0 pattern was more similar to T6 (0.86) than to T18 (0.80) and T6 was very similar to T18 (0.94). These observations suggest that the pattern of gene expression gradually changes as the infection proceeds from T0 to T18 and that T6 represents a mixture between genes differentially expressed at T0 and T18.
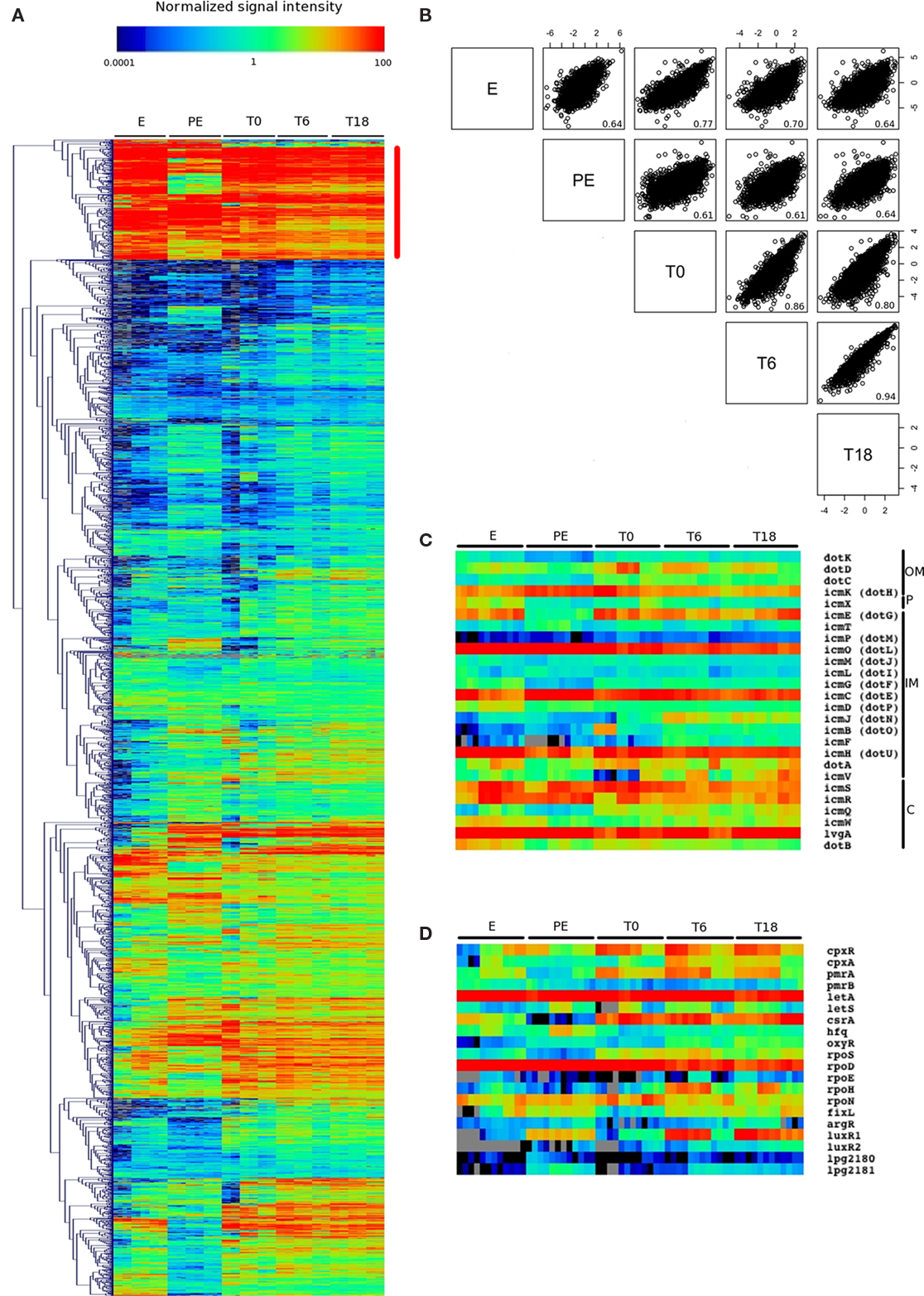
Figure 3. Analysis of the normalized signal intensities for each condition reveals similarity between in vivo conditions. (A) Hierarchical clustering of the normalized signal intensity of each replicate for each condition. The red line marks a cluster of genes highly expressed in all conditions tested. (B) The Bioconductor package was used to generate paired correlation matrix showing the degree of similarity between conditions. The correlation value is displayed in the lower right part of each graph. The x and y axis represent the median of the log2 transform of the normalized signal intensity. (C) Normalized signal intensities for genes encoding proteins of the Icm/Dot Type IVB secretion system. Putative location of each gene products is shown on the left side of the annotation: OM, outer membrane; P, periplasm; IM, inner membrane; C, cytoplasm. (D) Normalized signal intensities for genes encoding known and putative regulators.
Some Genes are Highly Expressed in Most Conditions
Hierarchical clustering of signal intensities reveals a cluster of genes that are highly expressed in most conditions tested (Figure 3A, red line). A value of 1 (blue–green) means that the normalized signal intensity of the cDNA was equal to the normalized signal intensity of the gDNA used as a reference. This cluster contains genes involved in basic cell functions such as transcription (rpoA, rpoB, rpoC, and the sigma factor rpoD), translation (ribosomal genes, tRNA genes, and elongation factors), replication (dnaB, dnaG, and topoisomerase genes such as gyrA, gyrB, parC), and cell division (minC, minD, and ftsY). In addition, some genes known to be involved in virulence, such as the macrophage infectivity potentiator (mip), the regulator letA and a number of Icm/Dot genes (icmC, icmH, icmO, icmR, and icmS) as well as some Icm/Dot translocated effectors (legA15, lem3, lem21, legA14, ceg19, and sidB) are present in this cluster of genes. The Icm/Dot secretion system is essential for virulence and almost all of the icm/dot genes show high signal intensities with low variability between all conditions (Figure 3C). Most well-known virulence regulators are also expressed at similar levels during infection (Figure 3D), including cpxR, cpxA, pmrA, pmrB, letA, and letS.
Gene Expression Patterns During Growth in Macrophages Compared to Exponential Growth in Aye Broth
The transcriptome of L. pneumophila during infection of THP-1 macrophage-like cells at T0, T6, and T18 post-infection, and during PE phase in broth were compared to that of bacteria in E phase and subjected to hierarchical clustering (Figure 4A; Table S1 in Supplementary Material). For clarity, a transcript with a higher or lower steady-state level in the test condition (PE, T0, T6, T18) compared to the control (E) is considered induced or repressed, respectively. Globally there were 1956 genes (65.6%) that show significant changes of expression (−2 > log2 > 2, P < 0.001) in at least one of the test conditions. Three hundred thirty-four genes were induced at all three time points in THP-1 cells and 110 were repressed at all three time points (Figure 4B). There were also a number of genes that were either induced or repressed in one specific condition (Figure 4B). Table 3 displays the genes with the highest level of induction or repression during intracellular growth. Interestingly, 8 of the 10 most highly induced genes have no assigned function, suggesting that novel virulence strategies could be used by L. pneumophila to infect host cells. The most highly induced gene during intracellular growth is an Icm/Dot effector of unknown function (lpg2527). Also highly induced intracellularly is a gene encoding a putative glutamine ABC transporter (lpg0491), which is encoded next to the argR gene (lpg0490). The ArgR arginine repressor is required for maximal growth of L. pneumophila in its ameba host Acanthamoeba castellanii (Hovel-Miner et al., 2009).
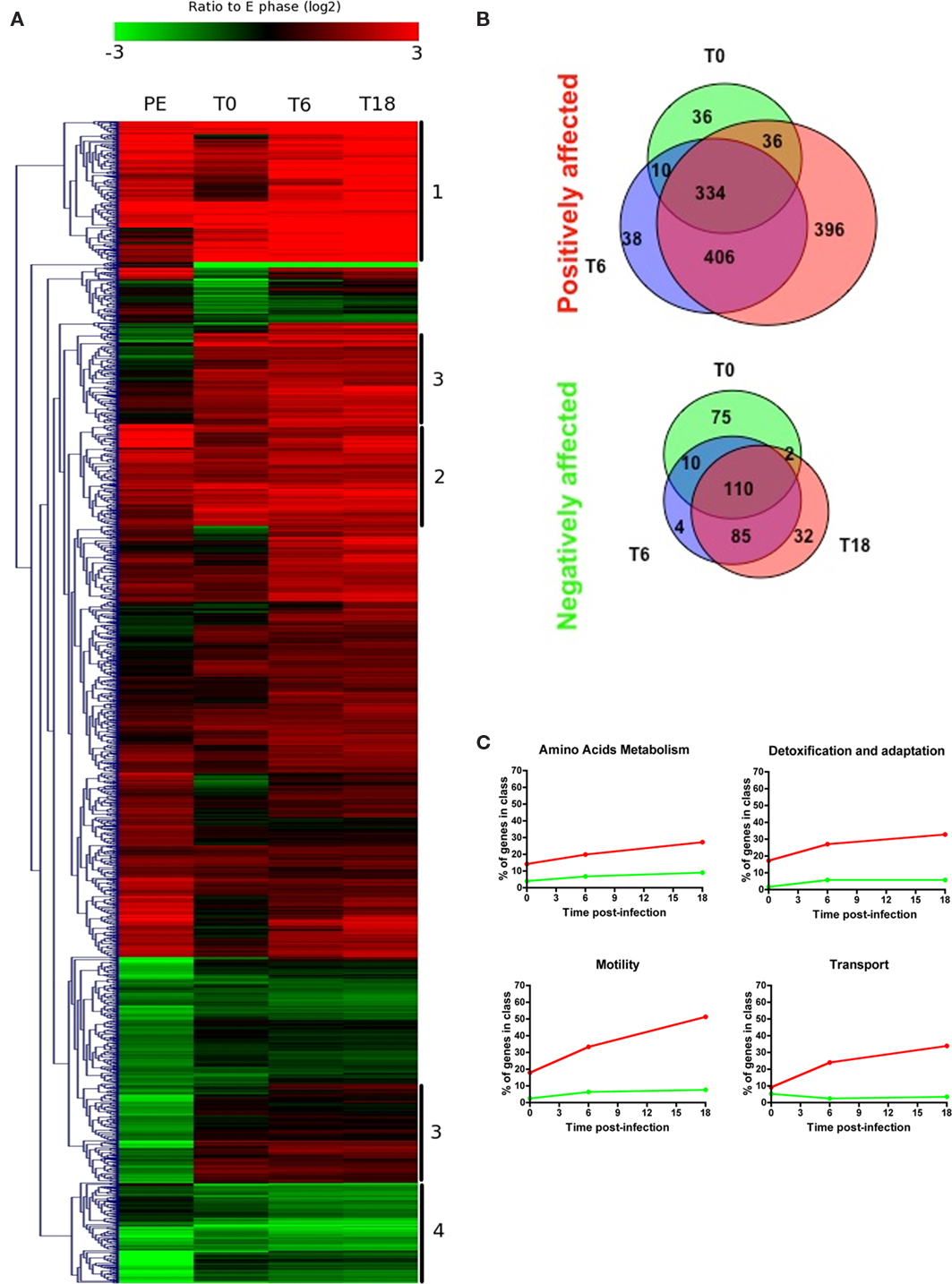
Figure 4. Genes differentially expressed compared to the E phase control. (A) Hierarchical clustering of the relative expression of L. pneumophila genes during intracellular multiplication in macrophages and during PE phase in rich AYE broth compared to exponential growth in broth. Only genes with significant change in expression (−2 > log2 > 2, P < 0.001) in at least one condition are shown. (B) The number of genes positively or negatively affected during intracellular growth compared to E phase is displayed in Venn diagrams. (C) Genes with significant changes in expression were grouped according to their published COG class. The percentage of genes that change positively (red line) or negatively (green line) during intracellular growth, compared to the E phase control, is shown for selected COG classes. Refer to Figure 5 for the results of other COG classes.
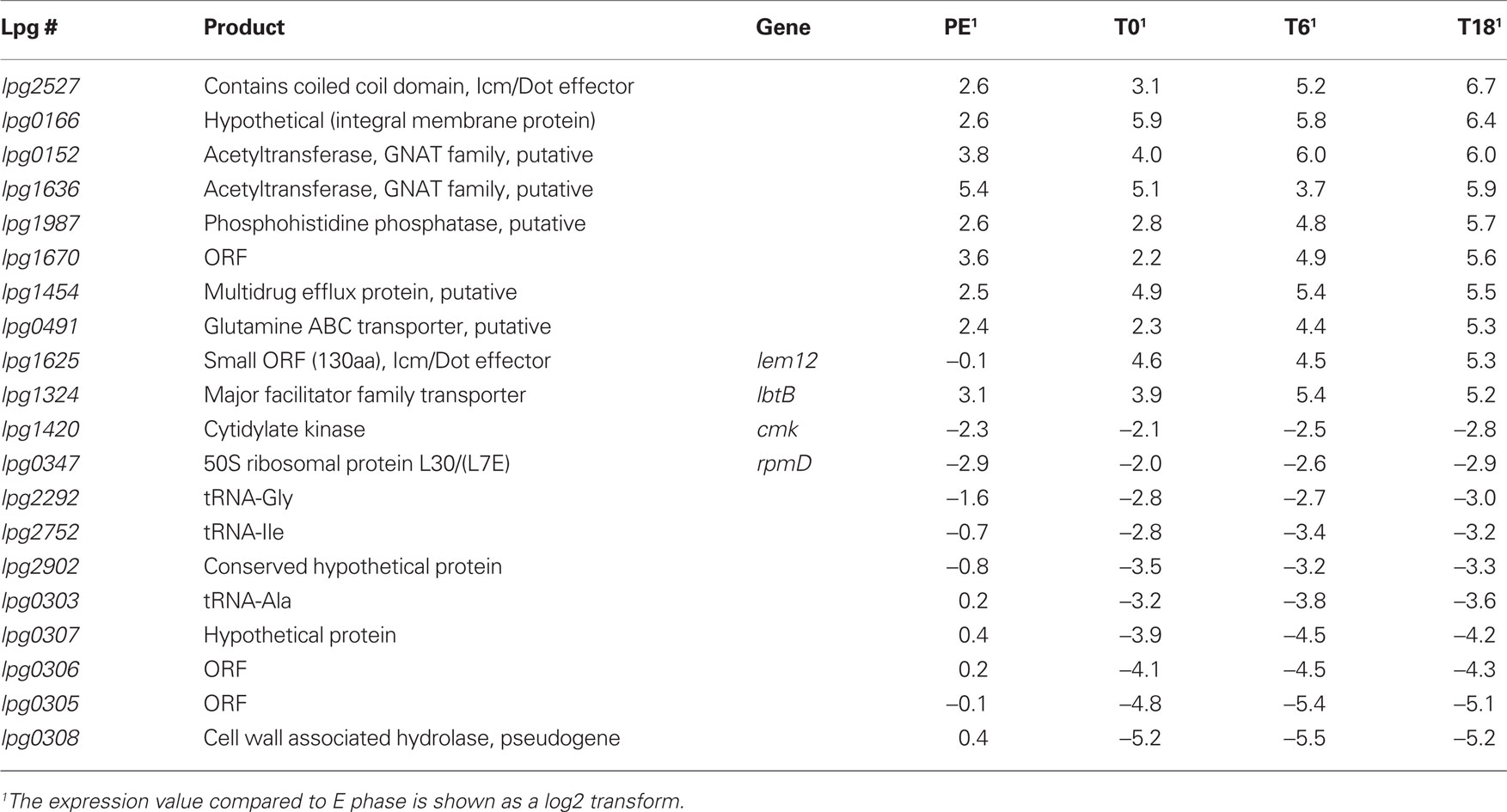
Table 3. The 10 most highly induced and repressed genes during intracellular growth, compared to E phase.
Based on the hierarchical clustering, four different groups of genes were defined (Figure 4A): (i) genes that were strongly induced inside cells (237 genes, group #1), (ii) genes induced at all three time points in THP-1 cells (171 genes, group #2), (iii) genes that were induced inside cells but repressed or unchanged in PE phase (224 genes, group #3), and (iv) genes that were repressed at all time points (169 genes, group #4). The complete list of genes present in these groups is shown in Table S3 in Supplementary Material. Group #1 and #2 contain 61 genes encoding Icm/Dot effectors, many genes involved in flagella production, a number of Lvh Type IV secretion system genes and genes of unknown or putative function. Group #3 contains 23 genes encoding Icm/Dot effectors that are only induced intracellularly, including SidF and SdhA, which are known to inhibit macrophage apoptosis following infection (Laguna et al., 2006; Banga et al., 2007). The virulence factor RtxA, which is involved in attachment of L. pneumophila to host cells (Cirillo et al., 2000, 2001) is also present in group #3 and its expression increases over the course of infection. Group #4 includes two Icm/Dot effectors (MavG and MavM) and a number of genes involved in translation, such as ribosomal subunits, tRNAs and elongation factors (EF-TU, EF-G, EF-P) and genes involved in transcription, including RNA polymerase subunits (rpoA, rpoB, and rpoC), and the σ70 gene rpoD.
To obtain a broad overview of the data, genes with significant changes in expression were clustered based on the genome annotation and their known or predicted function (Figures 4C and 5). The number of induced genes involved in motility (flagella), transport (ABC transporters, permeases, multidrug efflux pumps, Type II, and Lvh/Lvr secretion system), and detoxification/adaptation increases during the course of infection (Figure 4C). Of the 176 genes involved in amino acid metabolism 20% were induced during intracellular growth.
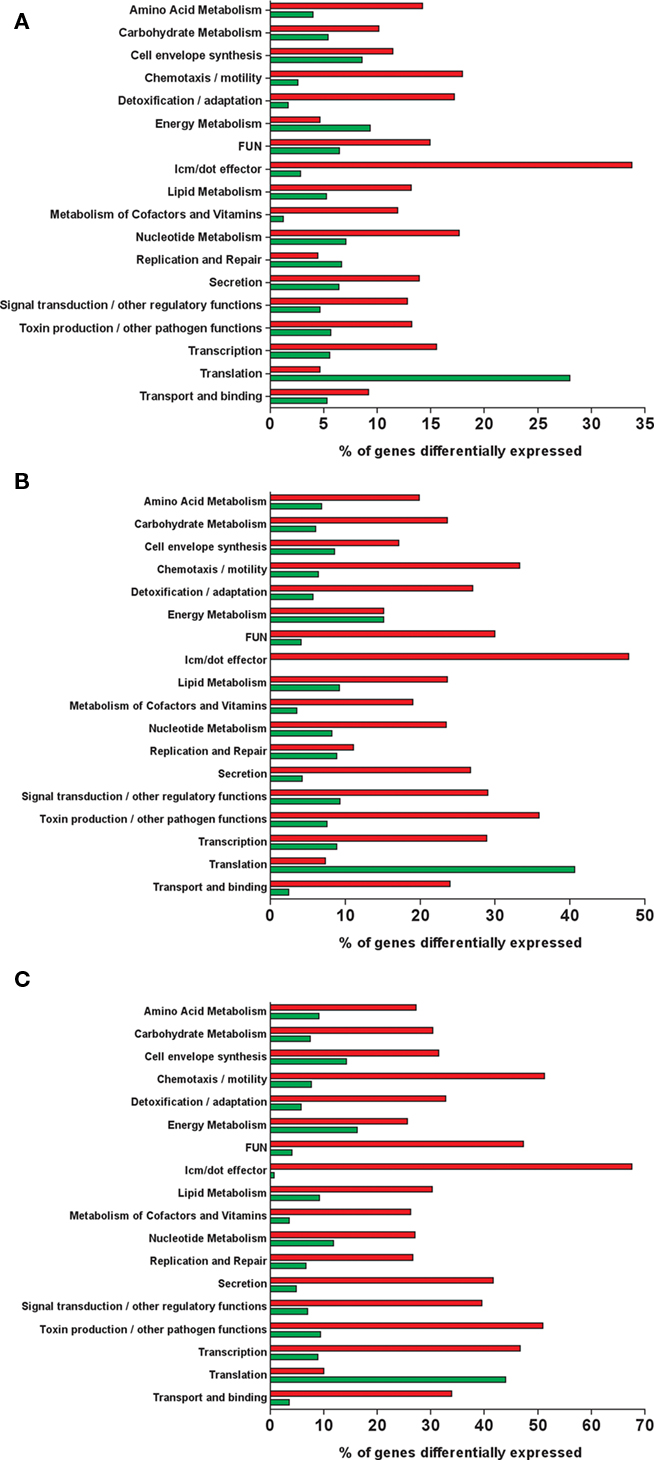
Figure 5. Fraction of genes in orthologous groups deferentially expressed during infection. Number of genes significantly induced (red bar) or repressed (green bar) intracellularly at T0 (A), T6 (B), and T18 (C) compared to the E phase control.
Nutrition of L. pneumophila during intracellular multiplication in human macrophages
Of all the genes involved in the metabolism of amino acids, lipids, carbohydrates, nucleotides, cofactors, and vitamins a larger proportion was induced than repressed inside cells, regardless of the time post-infection (Figures 4C and 5). The pathway tool Omics Viewer from the BioCyc Database was used to analyze trends in expression of genes involved in catabolism and anabolism pathways (Paley and Karp, 2006). The most highly induced pathway inside human macrophages was the one leading to thiamine synthesis (Figure 6). L. pneumophila has been shown to rely on amino acids as a carbon and nitrogen source (Tesh et al., 1983). Many pathways involved in the synthesis of amino acids (L-histidine, L-arginine, L-aspartate, L-lysine, and L-proline) are induced during growth inside macrophages (Figure 6). In addition, amino acid transporters and oligopeptide transporters are also highly induced during growth in macrophages. The degradation pathways for L-lysine, L-arginine, L-histidine, L-threonine, L-glutamine, and L-glutamate were all induced inside cells. In contrast, genes involved in translation and the tRNA-charging pathway are repressed during intracellular growth (Figure 5). Taken together these observations suggest that L. pneumophila can acquire amino acids from the host but the induction of synthesis pathways for certain amino acids and the repression of translation and tRNA-charging suggests that L. pneumophila has limited access to certain amino acids inside the host cell. However, this should be taken cautiously, since the intracellular transcriptome was compared to exponential growth in rich broth, in which all the amino acids, sugars, and cofactors, essential or not, for Legionella growth are likely supplied in unlimited quantity. Whether or not these pathways are essential for intracellular growth remains to be elucidated. However, it is known that L-arginine biosynthesis is not essential for growth inside a protozoan host (Hovel-Miner et al., 2010), but its role during infection of mammalian cells has not been investigated. Interestingly, genes required for glycerol catabolism (lpg1414 and glpD) are induced intracellularly (Figure 6). However most of the genes involved in glycolysis were not differentially regulated compared to growth in rich broth. We also noted the induction of lpg1607 and lpg0466 that are predicted to encode enzymes that would mediate oxaloacetate production, from phospho-enolpyruvate and pyruvate respectively, which can then be used in the TCA cycle or for the production of L-aspartate and L-lysine (Figure 6).
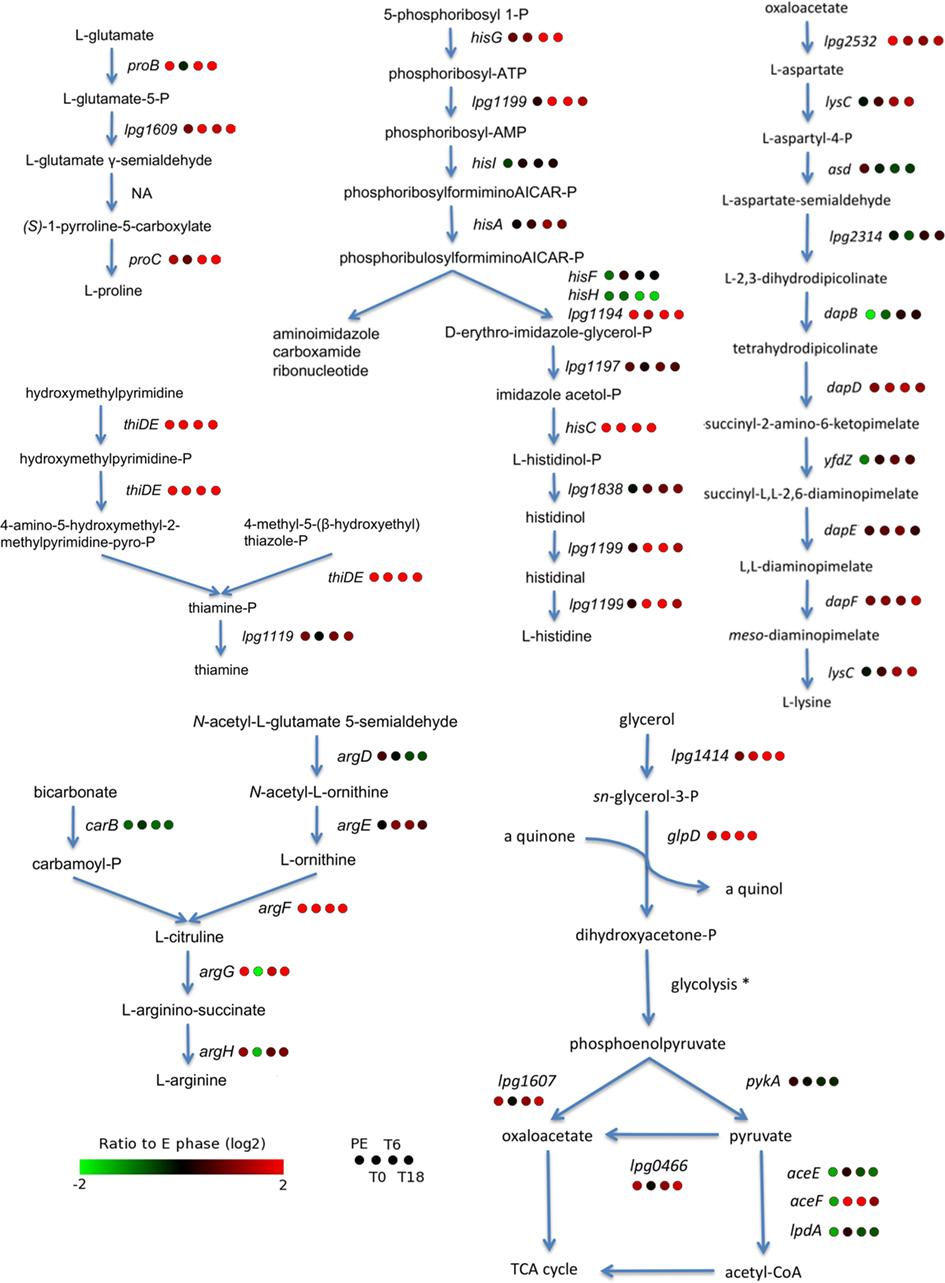
Figure 6. Some metabolic pathways are induced during intracellular growth. The synthesis pathways were drawn according to the Biocyc database (http://biocyc.org). A series of four circles next to each gene display its relative expression value compared to the E phase control. NA, There is no known or predicted gene mediating this reaction in L. pneumophila; *glycolysis genes are not differentially expressed.
Legionella pneumophila requires relatively large amounts of iron to grow in broth and inside host cells and has many systems to acquire sufficient amounts (Cianciotto, 2007). Iron transport systems were induced during intracellular growth (Figure 7A), including genes involved in legiobactin production (lbtAB), ferrous iron uptake (feoAB), and iron acquisition by an unknown mechanism (iraAB). Of note, lbtB is one of the most highly induced genes during infection (Table 3). The two genes pvcA and pvcB, which encode proteins similar to the pyoverdine synthesis genes of Pseudomonas aeruginosa, are also induced during intracellular growth even though these two proteins do not seem to be involved in iron assimilation in L. pneumophila (Allard et al., 2006). A recent study showed that pvcA and pvcB are highly induced in L. pneumophila within biofilms and the authors suggest that they might be involved in resistance to oxidative stress generated by an overload of ferrous iron (Hindre et al., 2008).
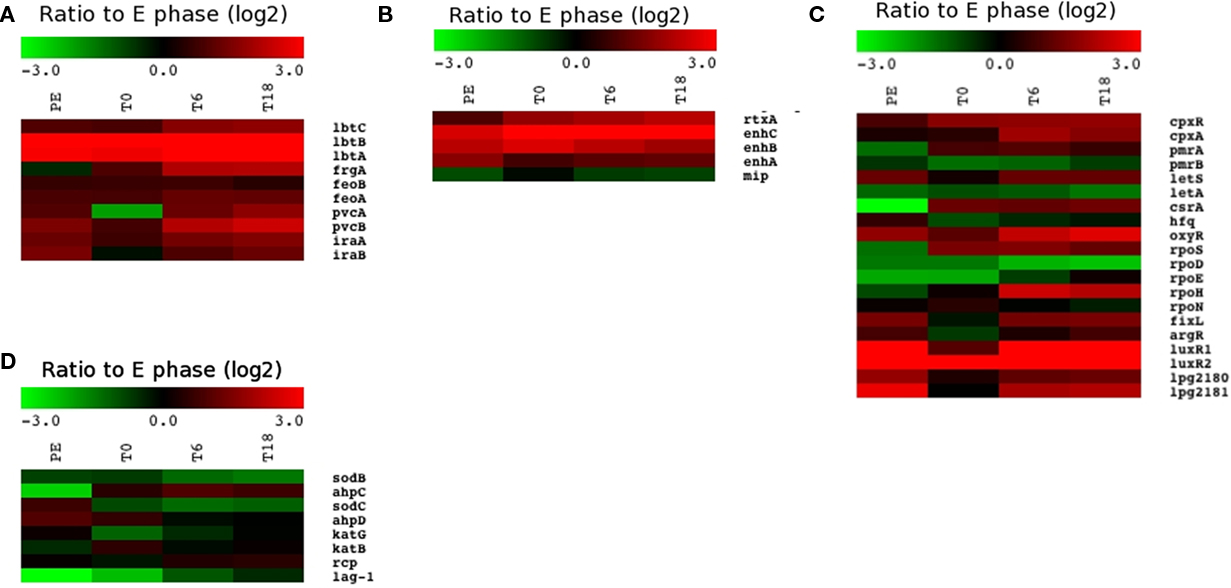
Figure 7. Relative expression of genes involved in various functions. Heat map of the expression ratio to E phase of (A) genes involved in iron acquisition, (B) known virulence factors other than the Icm/Dot system, (C) genes encoding regulators, and (D) genes encoding proteins involved in defense mechanisms against oxidative stress and antimicrobial peptides.
Expression of the Genes for the Icm/Dot Secretion System and its Substrates
Most of the 27 icm/dot genes show high signal intensities in all conditions (Figure 3C) and were not differentially expressed during intracellular growth compared to the E phase control (Table S1 in Supplementary Material). Only five icm/dot genes (icmQ, icmB, icmJ, icmF, and icmV) were somewhat induced at later time points during intracellular growth compared to the E phase control. This suggests that the secretion machinery is likely always present and ready to translocate effectors when needed. The normalized signal intensities of the genes encoding 191 Icm/Dot effectors were analyzed by hierarchical clustering (Figure 8A). Most Icm/Dot effector genes show good signal intensity (around or above 1) in all or a subset of the conditions tested. Interestingly, the sidM/drrA, vipA, legC7, lepA, and lepB genes were not differentially expressed and all show strong signal intensities in all of the conditions tested and are therefore likely produced and usable during intracellular growth in human macrophages. One hundred and three (64%) of the 191 genes encoding Icm/Dot effectors were significantly induced at T18 compared to E phase (Figure 8B), including genes encoding some well-characterized effectors (ralF, sidH) and two effector genes are among the most highly induced genes (lpg2527 and lem12, Table 3). The only known effector genes that are repressed inside host cells are lem3, mavG, mavK, mavM, and mavP. Many effector genes are expressed throughout the infection process, including ralF, lepA, lepB, sidM/drrA, vipA, and legC7, whereas others are more highly expressed during the initial phase (sidF) or at the end of infection (ceg17, legK2, lirB, lem26, lem5).
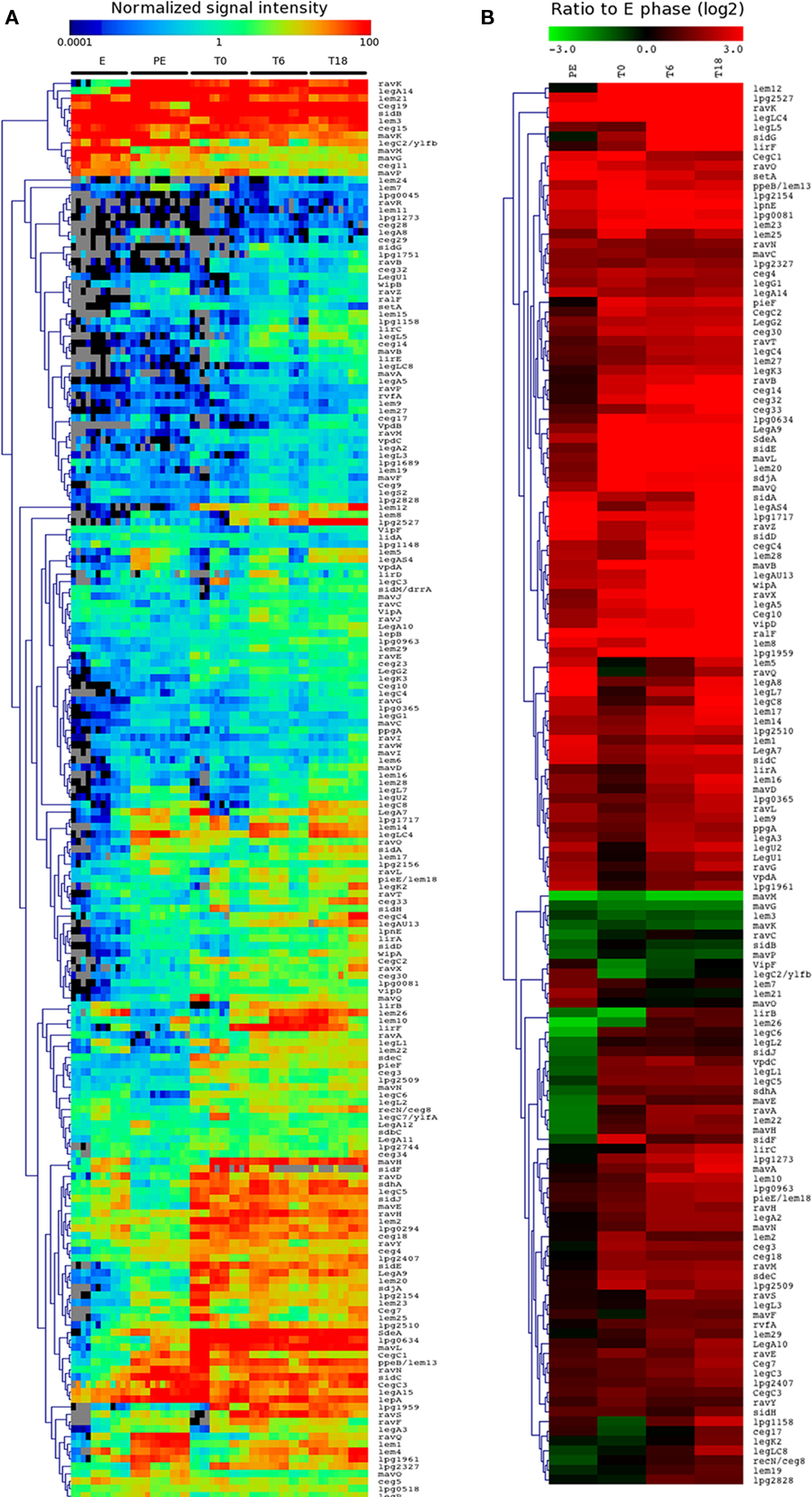
Figure 8. Icm/Dot effectors are differentially expressed inside human cells. (A) Hierarchical clustering of the normalized signal intensity of the genes encoding Icm/Dot effectors for each replicate and each condition. (B) Hierarchical clustering of the expression ratio of the genes encoding Icm/Dot effectors compared to the E phase control. Only genes with significant change in expression (−2 > log2 > 2, P < 0.001) in at least one condition are shown.
We wanted to examine the patterns of gene expression in order to look for additional genes that encode effectors. In the past, Icm/Dot effectors were identified using a variety of bioinformatic and experimental approaches. In total these approaches led to the identification of around 200 effectors. In general, the effectors are scattered across the L. pneumophila genome, but there are four areas of the genome that contain clusters of effector genes (Burstein et al., 2009). As an example, 17 effectors are grouped together in the region bordered by lpg1933 and lpg1978. In order to identify candidate effector genes based on gene expression patterns, the genome was manually scanned for regions enriched in genes encoding known Icm/Dot effectors and genes of unknown function with similar expression patterns as the neighboring effector genes. Two regions were identified, lpg1958–lpg1966 and lpg2826–lpg2831, containing four putative effectors (lpg1959, lpg1961, lpg2827, and lpg2828). In order to test the products of these genes for their ability to be translocated by the Icm/Dot TFBSS, they were fused to the TEM-1 β-lactamase and translocation of the hybrid proteins into J774 macrophages was measured as previously described (de Felipe et al., 2008). Using this approach we found two new Icm/Dot effector proteins (lpg1959, lpg1961) encoded within the previously identified cluster of effector genes (lpg1933–lpg1978) and one new effector gene (lpg2828) within the other region located between lpg2826 and lpg2831 (Figure 9A). Lpg2827 appeared not to be translocated. Expression of the four β-lactamase fusion proteins in Legionella was analyzed by western blotting using an anti-TEM polyclonal antibody (Figure 9B). Three of the four proteins (TEM-Lpg1961, TEM-Lpg2827, and TEM-Lpg2828) are stably expressed, but the third protein (TEM-Lpg1959) appears to be unstable, which may explain the low level of translocation observed. The expression pattern of these newly identified effector genes closely resembles the pattern of known effectors (Figure 8B).
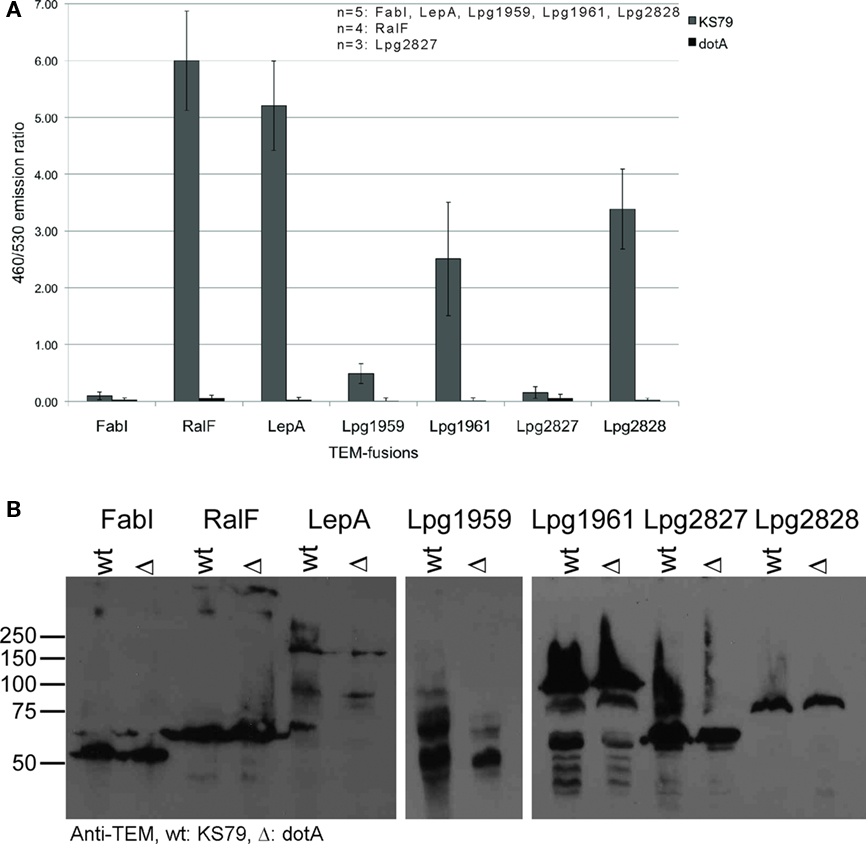
Figure 9. Identification of new Icm/Dot effectors. (A) Translocation of the TEM-effector fusions leads to the cleavage of CCF4/AM. Translocation was determined for each TEM-effector fusion by measuring the ratio of cleaved (460 nm) to uncleaved (530 nm) CCF4/AM in wild type KS79 or KS79 dotA (Type IV secretion deficient). FabI serves as negative control; RalF and LepA are known Legionella effector and serves as positive control. The number of biological replicates analyzed is shown. (B) Immunoblot on whole cell lysate using an anti-TEM rabbit polyclonal antibody showing expression of the TEM-effector fusions. Moleular weight of fusion proteins: TEM-FabI: 59.3 kDa, TEM-RalF: 73 kDa, TEM-LepA: 161.9 kDa, TEM-lpg1959: 106 kDa, TEM-lpg1961: 88 kDa, TEM-lpg2827: 66.7 kDa, TEM-lpg2828: 78.5 kDa.
Expression of Regulators
Several two-component systems, including PmrA/PmrB (Zusman et al., 2007), CpxR/CpxA (Altman and Segal, 2008), and LetA/LetS (Hammer et al., 2002), are known to be involved in the regulation of virulence factors of L. pneumophila. The cpxR and cpxA genes are significantly induced at T6 and T18 (Figure 7C). Although the pmrA gene encoding the response regulator component is not differentially expressed inside cells, the pmrB gene encoding the cognate sensor kinase is significantly repressed at early time points (T0 and T6, Figure 7C), suggesting that a negative feedback loop acts on the expression of pmrB early in infection, which is released at later time points to activate the system. In contrast, the gene encoding the LetS sensor is induced inside cells, but the gene encoding its cognate transcription activator, LetA is repressed. The LetA/LetS system controls the expression of two small RNAs (RsmY and RsmZ), which, in turn, control the activity of CsrA, a global regulator that represses the expression of post-exponential traits during exponential growth (Molofsky and Swanson, 2003; Forsbach-Birk et al., 2004; Rasis and Segal, 2009; Sahr et al., 2009). Inside host cells, CsrA was reported to control transmissive traits to allow intracellular multiplication. In accordance with this, CsrA is highly expressed at all time points inside host cells. A number of other two-component systems and regulators are strongly induced inside host cells, including oxyR, fixL, the putative two-component system lpg2180/lpg2181 and the two luxR homologs (Figure 7C).
Sigma factors also regulate gene expression in response to stress or other environmental signals. While the rpoD gene encoding the vegetative σ70 is repressed during growth inside THP-1 cells compared to the E phase control, the rpoS gene encoding σS is strongly induced (Figure 7C). RpoS (σS) has been shown to regulate a number of known virulence factors such as the Icm/Dot effectors (Hovel-Miner et al., 2009) and is required for intracellular multiplication in ameba and primary macrophages (Hales and Shuman, 1999; Abu-Zant et al., 2006). Other sigma factor genes are also induced inside human macrophages including rpoH, which is strongly induced at T6 and T18 inside human macrophages.
Legionella Response to Host Antimicrobial Systems
Phagocytes use a variety of strategies to kill bacteria, which include: (i) acidification of the phagosome, (ii) production of reactive oxygen and nitrogen species (ROS and NOS), and (iii) production of antimicrobial peptides (Flannagan et al., 2009). L. pneumophila has evolved several ways to alter host cell responses after infection, but the mechanisms remain unclear in most cases. The Icm/Dot effectors play a central role in altering host cell responses. Inhibition of acidification has recently been shown to be mediated by the SidK effector (Xu et al., 2010), which is induced at all time points inside cells (Figure 8B). It has been shown that infection of macrophages with L. pneumophila prevents the formation of ROS (Harada et al., 2007), which may explain why genes involved in oxidative stress adaptation such as sodB, sodC, katG, katB, aphC, and aphD were not induced during intracellular growth (Figure 7D). Legionella expresses a number of proteases and peptidases during intracellular growth, which could be a countermeasure against antimicrobial peptides produced by the host cell. Even though the protein Rcp has been reported to have a role in resistance against antimicrobial peptides in macrophages (Robey et al., 2001), the rcp gene was not induced inside cells compared to E phase (Figure 7D). Similarly, lag-1, which encodes an O-acetyltransferase involved in lipopolysaccharide modification (Luck et al., 2001), is not induced inside cells.
Genes Differentially Expressed During Intracellular Growth
To get a better view of the genes differentially expressed during the intracellular stages of infection, T0 was used as the control condition and compared to T6 and T18 (Figure 10A). As a result of this comparison, 667 (22%) genes with a significant change in expression at later time points compared to T0 were identified, 290 of which were induced at both T6 and T18 (Figure 10B). Hierarchical clustering of the data shows that the gene expression patterns at T6 and T18 are very similar (Figure 10A). This is not surprising since the correlation between the normalized signal intensity of T6 and T18 was 0.94 (Figure 3B). The genes can be grouped in three clusters based on their expression: (i) genes that were strongly induced (around eightfold) compared to T0 (group #1, 39 genes), (ii) genes repressed compared to T0 (group #2, 12 genes), and (iii) genes induced in vivo but repressed in PE phase (group #3, 83 genes). The complete list of genes present in these groups is shown in Table S4 in Supplementary Material. Most of the genes contained in group #1 have no known or putative function (33 out of 39 genes). The remaining six genes are the Icm/Dot effectors lirB and lpg2527, a gene involved in arginine synthesis (argG), a gene involved in proline synthesis (proB), the pyoverdine synthesis gene pvcA and a gene involved in tRNA modification (gidA). Group #2 contains two Icm/Dot effectors (cegC1 and lem25) and many genes of unknown or putative function. Group #3 contains 6 Icm/Dot effector genes (ravA, lpg1751, legLC8, sidM/drrA, lirE, and lem26) and 62 genes of unknown or putative function. In addition a number of transcriptional regulators, such as oruR and the sigma factor rpoH are found in this cluster.
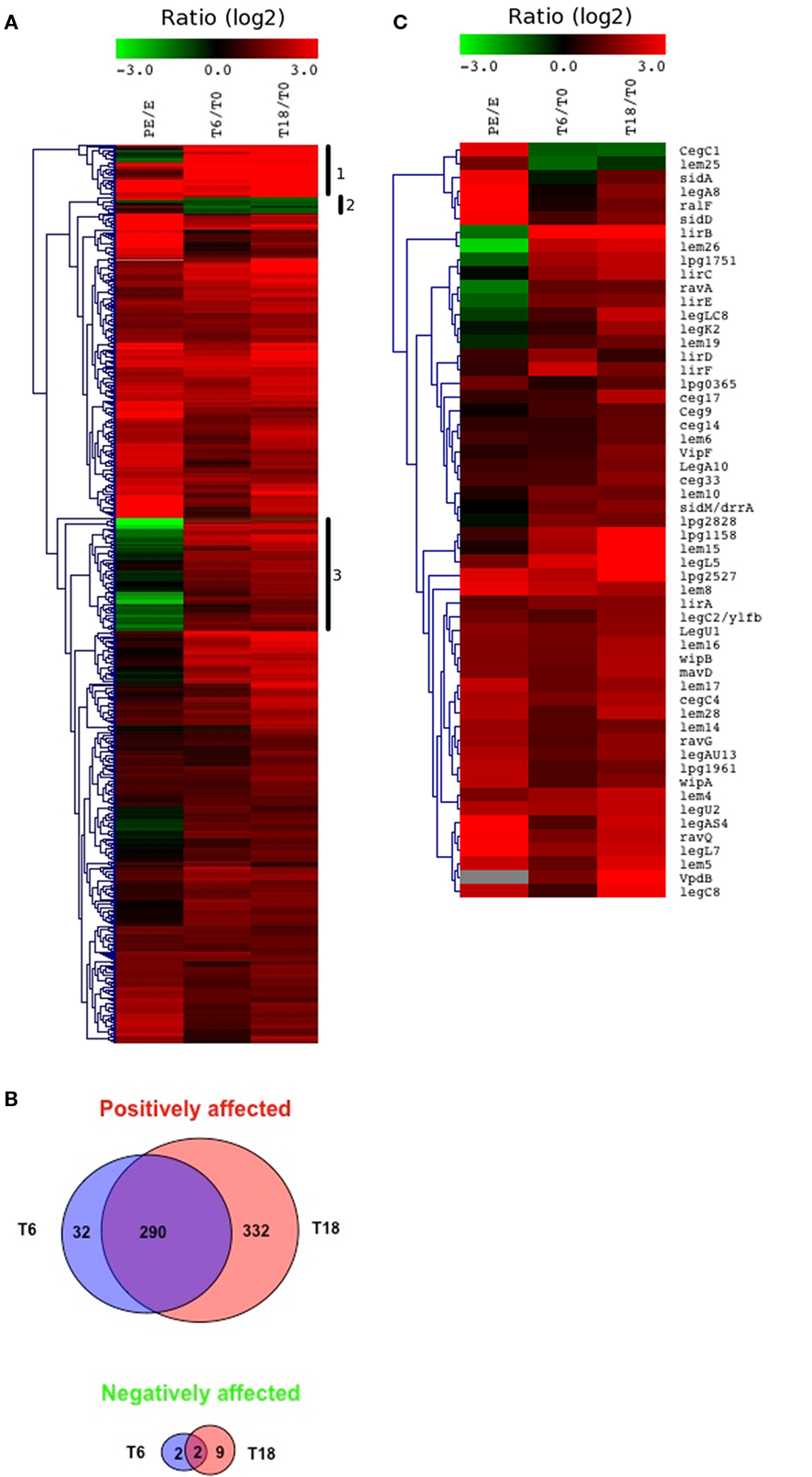
Figure 10. Identification of genes differentially expressed compared to the T0 control. (A) Hierarchical clustering of the transcriptome of L. pneumophila during intracellular multiplication in THP-1 macrophages compared to intracellular growth at T0. PE phase compared to E phase is shown as a reference. Only genes with significant change in expression (−2 > log2 > 2, P < 0.001) in at least one condition are shown. (B) The number of genes positively or negatively affected during intracellular growth compared to T0 is displayed in Venn diagrams. (C) Heat map of the Icm/Dot effectors differentially expressed compared to T0. PE phase compared to E phase is shown as a reference.
Overall 50/191 (35%) of Icm/Dot effector genes were differentially expressed at T6 or T18 compared to T0 (Figure 10C), 13 of which were not induced when compared to E phase (vipF, ceg9, wipB, lem4, lem6, vpdB, lpg1751, lem15, lirD, lirE, legC2, sidM/drrA, and lem26). Other genes induced at later time points compared to T0 include flagella biosynthesis genes (flgA, flhB, motA, and motB), amino acid and peptide transporters, arginine synthesis genes (argG and argF), lipid A modification (waaM), and genes of unknown function (298 genes). Unlike what was observed when E phase was used as the control condition, whole metabolism pathways were not induced when T6 and T18 were compared to T0. However, some genes involved in amino acid synthesis (proB, argG, and argH) were induced at T6 or T18 compared to T0 and were not induced at T0 compared to E phase. The differences in gene expression patterns that are obtained when using E phase and T0 as the control conditions are likely reflective of the vast re-organization of gene expression that occurs when the bacteria transition from growing exponentially in rich media to the intracellular environment, compared to the gradual changes in gene expression that occur as intracellular growth proceeds.
Discussion
In this study, we analyzed the gene expression profile of L. pneumophila during multiplication inside human macrophage-like cells shortly after infection is established (T0) and at 6 and 18 h later (T6 and T18) and compared this profile to the profile obtained during exponential and post-exponential growth in rich media. To circumvent the inherent problem of low levels of bacterial RNA during intracellular growth conditions at low multiplicities of infection, we used a method called SCOTS (Graham and Clark-Curtiss, 1999; Faucher et al., 2006) to successfully remove host cell transcripts and amplify bacterial transcripts. Additionally, this method discards bacterial ribosomal RNA (Figure 2A) resulting in improved hybridization signals. The data obtained from the microarrays were validated by qPCR performed on eight randomly chosen genes (Figure 2B) and by comparison of expression patterns obtained using the SCOTS protocol and a standard microarray protocol (Figure 2C) for exponentially growing bacteria. Globally, expression of 65.6% of the L. pneumophila genome is affected during intracellular growth when compared to exponential growth in rich media (Figure 4A). The number of genes induced increases as the infection proceeds, with only a few genes with decreased expression levels (Figure 4B) suggesting that additional functions may be required as intracellular growth proceeds. Some genes whose expression level increases during the latter stages of intracellular growth may be induced prematurely in preparation for stresses that appear later in infection, or in preparation for host cell lysis and escape. This phenomenon of anticipation, although not investigated in L. pneumophila has been recently observed in Escherichia coli (Mitchell et al., 2009).
Comparison of the gene expression profiles of L. pneumophila growing inside host cells to bacteria in E or PE phase in broth revealed some striking differences between the growth conditions. The current assumption is that early stages of infection and intracellular growth can be compared to E phase and later stages of infection and transmission are comparable to PE phase (Molofsky and Swanson, 2004). Our analysis shows that there are significant differences between intracellular growth and PE phase even if a late stage in infection (T18) is compared to PE phase (Figure 3B). This argues that processes that occur in PE phase are not necessarily representative of what happens inside host cells. However, the early stages of intracellular growth and exponential growth in broth are at least partially comparable (Figure 3B). Still, growth in rich media and intracellular growth have clearly distinct effects on the L. pneumophila transcriptome.
Legionella pneumophila relies on amino acids as a carbon and nitrogen source, when grown in broth and inside host cells (Tesh et al., 1983). Many genes involved in amino acid transport and degradation were induced during intracellular growth (Figure 6). Induction of amino acid transport genes was also observed during intracellular growth of Yersinia pestis, Salmonella typhimurium, Salmonella typhi, Shigella flexneri, and Bacillus anthracis (Eriksson et al., 2003; Lucchini et al., 2005; Faucher et al., 2006; Bergman et al., 2007; Fukuto et al., 2010). Moreover, many amino acid transporters were identified as essential for intracellular growth of L. monocytogenes (Schauer et al., 2010). Therefore, amino acid acquisition from the host during intracellular multiplication seems to be a general strategy and one could speculates that a fraction of the virulence strategies employed by intracellular pathogens is aimed at the modification of the host cell process in order to supply essential amino acids and other nutrients to the vacuole. The Icm/Dot mediated modification of the LCV and acquisition of vesicles coming from the endoplasmic reticulum, containing polypeptides, is a striking example of that (reviewed in Hubber and Roy, 2010).
Genes involved in the biosynthesis of thiamine, L-arginine, L-aspartate, L-lysine, and L-histidine were induced as well during Legionella intracellular growth, suggesting that the concentration of these metabolites are lower inside cells than during exponential growth in rich broth. It is unclear if the intermediates needed for the synthesis of thiamine, L-arginine, and L-histidine are provided directly by the host or if L. pneumophila encodes as yet unidentified enzymes that could provide them from other molecules. Induction of L-arginine biosynthesis genes and aspartate-family biosynthesis genes was also observed during intracellular infection of macrophages by B. anthracis and by Y. pestis, respectively (Bergman et al., 2007; Fukuto et al., 2010).
Interestingly, glycerol catabolism seems to be induced during intracellular growth and suggests that L. pneumophila could use this carbon source inside mammalian macrophages. The metabolism of Listeria monocytogenes during intracellular growth in murine cultured macrophages was previously investigated by using 13C-isotopologue profiling (Eylert et al., 2008). This study showed that L. monocytogenes acquires a significant proportion of its amino acids from the host. Moreover, it shows that a C3-metabolite, probably glycerol, serves as a carbon source during intracellular growth of L. monocytogenes and that L-aspartate is synthesized from oxaloacetate derived from the carboxylation of pyruvate. In addition, a L. monocytogenes deletion mutant of glycerol-3-phosphate dehydrogenase (glpD) shows reduced intracellular growth (Schauer et al., 2010). Therefore, one could postulate that L. pneumophila also uses glycerol as a carbon source and that carboxylation of pyruvate by Lpg0466 or phospho-enol-pyruvate by Lpg1607 provide oxaloacetate that could then be use for the biosynthesis of L-aspartate and L-lysine (Figure 6), but it will need further investigation. Interestingly, in L. monocytogenes, a deletion mutant of pyruvate carboxylase is defective for intracellular growth (Schar et al., 2010).
Based on our results (Figure 8), the effector proteins which are secreted by the Icm/Dot TFBSS can be organized into several distinct groups, based on relative expression to the E phase control: (i) effectors induced during intracellular growth (64% of all effectors), (ii) effectors repressed inside cells (2.6% of all effectors) and effectors not differentially regulated (33.4% of all effectors). Some Icm/Dot effector genes were not differentially expressed when E phase was used as the control condition, but when expression at T6 and T18 was compared to T0, 13 effector genes were differentially expressed, including legC2, sidM/drrA, and others (see above). Some of these effectors appear to be expressed during growth in broth (Figure 8A), which precludes detecting their induction during intracellular growth when the transcription levels were compared to E phase.
Comparison to T0 reveals that the expression of these effectors increases as the infection proceeds and suggests that their function is required at later stages in infection. Consistent with this observation, LegC2 was found to localize only to large structures that resemble mature LCV (de Felipe et al., 2008). Expression of some Icm/Dot effector genes decreases over time (cegC1 and lem25) suggesting that these effectors are required during the initial stages of infection (Figure 10C). Taken together, these results indicated that the effectors are regulated independently of the Icm/Dot secretion system and their expression is probably linked to the stage in infection where they are relevant.
Previously, some effectors were identified by searching for genes encoding proteins containing eukaryotic-like domains in the different Legionella genomes available (Chien et al., 2004; de Felipe et al., 2005; Bruggemann et al., 2006; Burstein et al., 2009). Here we show that effectors can also be identified based on microarray data by comparing the expression patterns of genes of unknown function to the patterns of closely linked, known effector genes. The three new effectors identified here are located in two large clusters containing several other known effectors and are regulated similarly to the other effector genes in the cluster. These results show that microarray data and the organization of effector genes can be used to predict the identity of novel effector genes.
We also compared our data to the previously published transcriptome of L. pneumophila growing inside its protozoan host A. castellanii (Bruggemann et al., 2006; Jules and Buchrieser, 2007). It is important to keep in mind that this study of Bruggemann et al. (2006) and the present study used different infection protocols (MOI, infection medium, time points). Also, in the case of the A. castellanii study, the intracellular time points (8, 11, and 14 h post-infection) were analyzed two-by-two, in order to unravel how genes expression evolved during intracellular growth, whereas we compare the intracellular transcriptome (T0, T6, and T18) to exponential growth in broth, in order to identify infection-related changes in gene expression. We also analyzed how gene expression evolved during infection but in contrast to Bruggemann et al. (2006) we used a very early time point (T0) as the control. Therefore, comparison between the data or the conclusion of both studies should be done cautiously. In other words, differences between these data sets are likely due to differences in the experimental design and these two studies should be seen as complementary. For example, glycerol catabolism, although induced inside THP-1 cells compared to exponential growth, is not differentially regulated in A. castellanii between intracellular time points. However, when the THP-1 dataset was analyzed for differential expression during intracellular multiplication (T6/T0 and T18/T0), glycerol catabolism is not differentially expressed, because it is highly expressed at T0. Table S5 in Supplementary Material contains the complete data sets of L. pneumophila growing inside A. castellanii (8 vs 14, 11 vs 14, and 8 vs 11) and the complete data sets of L. pneumophila growing inside THP-1 macrophages (T0/E, T6/E, T18/E and T6/T0, T18/T0). Nonetheless, some of the similarities and the differences will be mentioned hereafter but should be taken cautiously. Many genes involved in amino acid transport and degradation (Figure 6) as well as genes predicted to encode myo-inositol catabolism pathways were induced in both protozoan and mammalian host. In addition, protein synthesis machinery was repressed in both the protozoan and the mammalian host. Many known virulence factors are induced in both host cells, including enhA, enhB, enhC, and rtxA (Figure 7B). However, inside protozoa, L. pneumophila does not seem to induce expression of genes involved in amino acid synthesis pathways, contrary to what was observed during growth inside macrophages. The Entner–Doudoroff pathway, which was induced in A. castellanii was not induced during growth in human macrophages. Strikingly, we did not observe differential regulation of the icm/dot genes during growth in human macrophages contrary to what was observed inside A. castellanii even when we compare the late time points (T6, T18) to T0. Induction of genes encoding many Icm/Dot effectors was observed in both cases (for example RalF) but some genes were not induced in A. castellanii (for example LegC4, LegL5, and the three new effectors identified: Lpg1959, Lpg1961, and Lpg2828).
In conclusion, we have analyzed the transcriptome of L. pneumophila during infection of human tissue culture macrophages. The use of SCOTS to enrich bacterial transcript allowed us to use a low MOI and to study time points where the number of bacteria would not have yielded sufficient levels of RNA for standard microarray protocols. Acquisition of amino acids and biosynthesis of L-arginine, L-histidine, L-aspartate, and L-lysine were induced during intracellular multiplication. Interestingly, glycerol catabolism was also induced, suggesting that inside cells, Legionella not only acquires carbon from amino acids, but also from glycerol. The high proportion of Icm/Dot effectors induced during infection, together with the lack of induction of a stress response enforces the idea that during L. pneumophila infection, the Icm/Dot system is the major mediator of virulence.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank lab members for helpful discussion and useful suggestions. This work was supported by NIH grant AI064481 to Howard A. Shuman. Sébastien P. Faucher is a recipient of a postdoctoral fellowship from NSERC. Catherine A. Mueller is a recipient of fellowships from the Swiss National Science Foundation (SNSF) and the European Molecular Biology Organization (EMBO).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/cellular_and_infection_microbiology/10.3389/fmicb.2011.00060/abstract/
Reference
Abu-Zant, A., Asare, R., Graham, J. E., and Abu Kwaik, Y. (2006). Role for RpoS but not RelA of Legionella pneumophila in modulation of phagosome biogenesis and adaptation to the phagosomal microenvironment. Infect. Immun. 74, 3021–3026.
Allard, K. A., Viswanathan, V. K., and Cianciotto, N. P. (2006). lbtA and lbtB are required for production of the Legionella pneumophila siderophore legiobactin. J. Bacteriol. 188, 1351–1363.
Altman, E., and Segal, G. (2008). The response regulator CpxR directly regulates expression of several Legionella pneumophila icm/dot components as well as new translocated substrates. J. Bacteriol. 190, 1985–1996.
Banga, S., Gao, P., Shen, X., Fiscus, V., Zong, W. X., Chen, L., and Luo, Z. Q. (2007). Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc. Natl. Acad. Sci. U.S.A. 104, 5121–5126.
Bergman, N. H., Anderson, E. C., Swenson, E. E., Janes, B. K., Fisher, N., Niemeyer, M. M., Miyoshi, A. D., and Hanna, P. C. (2007). Transcriptional profiling of Bacillus anthracis during infection of host macrophages. Infect. Immun. 75, 3434–3444.
Bruggemann, H., Hagman, A., Jules, M., Sismeiro, O., Dillies, M. A., Gouyette, C., Kunst, F., Steinert, M., Heuner, K., Coppee, J. Y., and Buchrieser, C. (2006). Virulence strategies for infecting phagocytes deduced from the in vivo transcriptional program of Legionella pneumophila. Cell. Microbiol. 8, 1228–1240.
Burstein, D., Zusman, T., Degtyar, E., Viner, R., Segal, G., and Pupko, T. (2009). Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 5, e1000508. doi: 10.1371/journal.ppat.1000508
Carratala, J., Gudiol, F., Pallares, R., Dorca, J., Verdaguer, R., Ariza, J., and Manresa, F. (1994). Risk factors for nosocomial Legionella pneumophila pneumonia. Am. J. Respir. Crit. Care Med. 149, 625–629.
Cazalet, C., Rusniok, C., Bruggemann, H., Zidane, N., Magnier, A., Ma, L., Tichit, M., Jarraud, S., Bouchier, C., Vandenesch, F., Kunst, F., Etienne, J., Glaser, P., and Buchrieser, C. (2004). Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat. Genet. 36, 1165–1173.
Charpentier, X., Faucher, S. P., Kalachikov, S., and Shuman, H. A. (2008). Loss of RNase R induces competence development in Legionella pneumophila. J. Bacteriol. 190, 8126–8136.
Charpentier, X., Gabay, J. E., Reyes, M., Zhu, J. W., Weiss, A., and Shuman, H. A. (2009). Chemical genetics reveals bacterial and host cell functions critical for type IV effector translocation by Legionella pneumophila. PLoS Pathog. 5, e1000501. doi: 10.1371/journal.ppat.1000501
Chen, J., de Felipe, K. S., Clarke, M., Lu, H., Anderson, O. R., Segal, G., and Shuman, H. A. (2004). Legionella effectors that promote nonlytic release from protozoa. Science 303, 1358–1361.
Chien, M., Morozova, I., Shi, S., Sheng, H., Chen, J., Gomez, S. M., Asamani, G., Hill, K., Nuara, J., Feder, M., Rineer, J., Greenberg, J. J., Steshenko, V., Park, S. H., Zhao, B., Teplitskaya, E., Edwards, J. R., Pampou, S., Georghiou, A., Chou, I. C., Iannuccilli, W., Ulz, M. E., Kim, D. H., Geringer-Sameth, A., Goldsberry, C., Morozov, P., Fischer, S. G., Segal, G., Qu, X., Rzhetsky, A., Zhang, P., Cayanis, E., De Jong, P. J., Ju, J., Kalachikov, S., Shuman, H. A., and Russo, J. J. (2004). The genomic sequence of the accidental pathogen Legionella pneumophila. Science 305, 1966–1968.
Cirillo, S. L., Bermudez, L. E., El-Etr, S. H., Duhamel, G. E., and Cirillo, J. D. (2001). Legionella pneumophila entry gene rtxA is involved in virulence. Infect. Immun. 69, 508–517.
Cirillo, S. L., Lum, J., and Cirillo, J. D. (2000). Identification of novel loci involved in entry by Legionella pneumophila. Microbiology 146(Pt 6), 1345–1359.
Daigle, F., Graham, J. E., and Curtiss, R. III. (2001). Identification of Salmonella typhi genes expressed within macrophages by selective capture of transcribed sequences (SCOTS). Mol. Microbiol. 41, 1211–1222.
de Felipe, K. S., Glover, R. T., Charpentier, X., Anderson, O. R., Reyes, M., Pericone, C. D., and Shuman, H. A. (2008). Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 4, e1000117. doi: 10.1371/journal.ppat.1000117
de Felipe, K. S., Pampou, S., Jovanovic, O. S., Pericone, C. D., Ye, S. F., Kalachikov, S., and Shuman, H. A. (2005). Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J. Bacteriol. 187, 7716–7726.
Eriksson, S., Lucchini, S., Thompson, A., Rhen, M., and Hinton, J. C. (2003). Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47, 103–118.
Eylert, E., Schar, J., Mertins, S., Stoll, R., Bacher, A., Goebel, W., and Eisenreich, W. (2008). Carbon metabolism of Listeria monocytogenes growing inside macrophages. Mol. Microbiol. 69, 1008–1017.
Faucher, S. P., Friedlander, G., Livny, J., Margalit, H., and Shuman, H. A. (2010). Legionella pneumophila 6S RNA optimizes intracellular multiplication. Proc. Natl. Acad. Sci. U.S.A. 107, 7533–7538.
Faucher, S. P., Porwollik, S., Dozois, C. M., McClelland, M., and Daigle, F. (2006). Transcriptome of Salmonella enterica serovar Typhi within macrophages revealed through the selective capture of transcribed sequences. Proc. Natl. Acad. Sci. U.S.A. 103, 1906–1911.
Flannagan, R. S., Cosio, G., and Grinstein, S. (2009). Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7, 355–366.
Forsbach-Birk, V., McNealy, T., Shi, C., Lynch, D., and Marre, R. (2004). Reduced expression of the global regulator protein CsrA in Legionella pneumophila affects virulence-associated regulators and growth in Acanthamoeba castellanii. Int. J. Med. Microbiol. 294, 15–25.
Franco, I. S., Shuman, H. A., and Charpentier, X. (2009). The perplexing functions and surprising origins of Legionella pneumophila type IV secretion effectors. Cell. Microbiol. 11, 1435–1443.
Fraser, D. W., Tsai, T. R., Orenstein, W., Parkin, W. E., Beecham, H. J., Sharrar, R. G., Harris, J., Mallison, G. F., Martin, S. M., McDade, J. E., Shepard, C. C., and Brachman, P. S. (1977). Legionnaires’ disease: description of an epidemic of pneumonia. N. Engl. J. Med. 297, 1189–1197.
Fukuto, H. S., Svetlanov, A., Palmer, L. E., Karzai, A. W., and Bliska, J. B. (2010). Global gene expression profiling of Yersinia pestis replicating inside macrophages reveals the roles of a putative stress-induced operon in regulating type III secretion and intracellular cell division. Infect. Immun. 78, 3700–3715.
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., Ellis, B., Gautier, L., Ge, Y., Gentry, J., Hornik, K., Hothorn, T., Huber, W., Iacus, S., Irizarry, R., Leisch, F., Li, C., Maechler, M., Rossini, A. J., Sawitzki, G., Smith, C., Smyth, G., Tierney, L., Yang, J. Y., and Zhang, J. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80.
Graham, J. E., and Clark-Curtiss, J. E. (1999). Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc. Natl. Acad. Sci. U.S.A. 96, 11554–11559.
Hales, L. M., and Shuman, H. A. (1999). The Legionella pneumophila rpoS gene is required for growth within Acanthamoeba castellanii. J. Bacteriol. 181, 4879–4889.
Hammer, B. K., Tateda, E. S., and Swanson, M. S. (2002). A two-component regulator induces the transmission phenotype of stationary-phase Legionella pneumophila. Mol. Microbiol. 44, 107–118.
Harada, T., Miyake, M., and Imai, Y. (2007). Evasion of Legionella pneumophila from the bactericidal system by reactive oxygen species (ROS) in macrophages. Microbiol. Immunol. 51, 1161–1170.
Hindre, T., Bruggemann, H., Buchrieser, C., and Hechard, Y. (2008). Transcriptional profiling of Legionella pneumophila biofilm cells and the influence of iron on biofilm formation. Microbiology 154, 30–41.
Hovel-Miner, G., Faucher, S. P., Charpentier, X., and Shuman, H. A. (2010). ArgR-regulated genes are derepressed in the Legionella-containing vacuole. J. Bacteriol. 192, 4504–4516.
Hovel-Miner, G., Pampou, S., Faucher, S. P., Clarke, M., Morozova, I., Morozov, P., Russo, J. J., Shuman, H. A., and Kalachikov, S. (2009). SigmaS controls multiple pathways associated with intracellular multiplication of Legionella pneumophila. J. Bacteriol. 191, 2461–2473.
Huang, L., Boyd, D., Amyot, W. M., Hempstead, A. D., Luo, Z. Q., O’Connor, T. J., Chen, C., Machner, M., Montminy, T., and Isberg, R. R. (2011). The E Block motif is associated with Legionella pneumophila translocated substrates. Cell. Microbiol. 13, 227–245.
Hubber, A., and Roy, C. R. (2010). Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26, 261–283.
Jules, M., and Buchrieser, C. (2007). Legionella pneumophila adaptation to intracellular life and the host response: clues from genomics and transcriptomics. FEBS Lett. 581, 2829–2838.
Laguna, R. K., Creasey, E. A., Li, Z., Valtz, N., and Isberg, R. R. (2006). A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U.S.A. 103, 18745–18750.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408.
Lucchini, S., Liu, H., Jin, Q., Hinton, J. C., and Yu, J. (2005). Transcriptional adaptation of Shigella flexneri during infection of macrophages and epithelial cells: insights into the strategies of a cytosolic bacterial pathogen. Infect. Immun. 73, 88–102.
Luck, P. C., Freier, T., Steudel, C., Knirel, Y. A., Luneberg, E., Zahringer, U., and Helbig, J. H. (2001). A point mutation in the active site of Legionella pneumophila O-acetyltransferase results in modified lipopolysaccharide but does not influence virulence. Int. J. Med. Microbiol. 291, 345–352.
Marston, B. J., Lipman, H. B., and Breiman, R. F. (1994). Surveillance for Legionnaires’ disease. Risk factors for morbidity and mortality. Arch. Intern. Med. 154, 2417–2422.
Mitchell, A., Romano, G. H., Groisman, B., Yona, A., Dekel, E., Kupiec, M., Dahan, O., and Pilpel, Y. (2009). Adaptive prediction of environmental changes by microorganisms. Nature 460, 220–224.
Molofsky, A. B., and Swanson, M. S. (2003). Legionella pneumophila CsrA is a pivotal repressor of transmission traits and activator of replication. Mol. Microbiol. 50, 445–461.
Molofsky, A. B., and Swanson, M. S. (2004). Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 53, 29–40.
Ninio, S., and Roy, C. R. (2007). Effector proteins translocated by Legionella pneumophila: strength in numbers. Trends Microbiol. 15, 372–380.
Paley, S. M., and Karp, P. D. (2006). The pathway tools cellular overview diagram and Omics viewer. Nucleic Acids Res. 34, 3771–3778.
Poirier, K., Faucher, S. P., Beland, M., Brousseau, R., Gannon, V., Martin, C., Harel, J., and Daigle, F. (2008). Escherichia coli O157:H7 survives within human macrophages: global gene expression profile and involvement of the Shiga toxins. Infect. Immun. 76, 4814–4822.
Rasis, M., and Segal, G. (2009). The LetA-RsmYZ-CsrA regulatory cascade, together with RpoS and PmrA, post-transcriptionally regulates stationary phase activation of Legionella pneumophila Icm/Dot effectors. Mol. Microbiol. 72, 995–1010.
Robey, M., O’Connell, W., and Cianciotto, N. P. (2001). Identification of Legionella pneumophila rcp, a pagP-like gene that confers resistance to cationic antimicrobial peptides and promotes intracellular infection. Infect. Immun. 69, 4276–4286.
Roy, C. R., and Tilney, L. G. (2002). The road less traveled: transport of Legionella to the endoplasmic reticulum. J. Cell Biol. 158, 415–419.
Sadosky, A. B., Wiater, L. A., and Shuman, H. A. (1993). Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect. Immun. 61, 5361–5373.
Sahr, T., Bruggemann, H., Jules, M., Lomma, M., Albert-Weissenberger, C., Cazalet, C., and Buchrieser, C. (2009). Two small ncRNAs jointly govern virulence and transmission in Legionella pneumophila. Mol. Microbiol. 72, 741–762.
Schar, J., Stoll, R., Schauer, K., Loeffler, D. I., Eylert, E., Joseph, B., Eisenreich, W., Fuchs, T. M., and Goebel, W. (2010). Pyruvate carboxylase plays a crucial role in carbon metabolism of extra- and intracellularly replicating Listeria monocytogenes. J. Bacteriol. 192, 1774–1784.
Schauer, K., Geginat, G., Liang, C., Goebel, W., Dandekar, T., and Fuchs, T. M. (2010). Deciphering the intracellular metabolism of Listeria monocytogenes by mutant screening and modelling. BMC Genomics 11, 573. doi: 10.1186/1471-2164-11-573
Segal, G., Purcell, M., and Shuman, H. A. (1998). Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. U.S.A. 95, 1669–1674.
Segal, G., and Shuman, H. A. (1998). How is the intracellular fate of the Legionella pneumophila phagosome determined? Trends Microbiol. 6, 253–255.
Shin, S., and Roy, C. R. (2008). Host cell processes that influence the intracellular survival of Legionella pneumophila. Cell. Microbiol. 10, 1209–1220.
Talaat, A. M., Howard, S. T., Hale, W. IV, Lyons, R., Garner, H., and Johnston, S. A. (2002). Genomic DNA standards for gene expression profiling in Mycobacterium tuberculosis. Nucleic Acids Res. 30, e104.
Tesh, M. J., Morse, S. A., and Miller, R. D. (1983). Intermediary metabolism in Legionella pneumophila: utilization of amino acids and other compounds as energy sources. J. Bacteriol. 154, 1104–1109.
Vogel, J. P., Andrews, H. L., Wong, S. K., and Isberg, R. R. (1998). Conjugative transfer by the virulence system of Legionella pneumophila. Science 279, 873–876.
Xu, L., Shen, X., Bryan, A., Banga, S., Swanson, M. S., and Luo, Z. Q. (2010). Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6, e1000822. doi: 10.1371/journal.ppat.1000822
Keywords: THP-1, microarray, SCOTS, Icm/Dot effectors, iron
Citation: Faucher SP, Mueller CA and Shuman HA (2011) Legionella pneumophila transcriptome during intracellular multiplication in human macrophages. Front. Microbio. 2:60. doi: 10.3389/fmicb.2011.00060
Received: 19 November 2010;
Paper pending published: 17 December 2010;
Accepted: 18 March 2011;
Published online: 04 April 2011.
Edited by:
Carmen Buchrieser, Pasteur Institute, FranceReviewed by:
Alain Charbit, University Paris Descartes, FranceWerner Goebel, Max von Pettenkofer-Institute, Germany
Gil Segal, Tel-Aviv University, Israel
Copyright: © 2011 Faucher, Mueller and Shuman. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Howard A. Shuman, Department of Microbiology, Cummings Life Sciences Center, University of Chicago, 920 E. 58th Street, Chicago, IL 60637, USA. e-mail:aHNodW1hbkBic2QudWNoaWNhZ28uZWR1