
- Viral Immunobiology, Institute of Experimental Immunology, University of Zürich, Zürich, Switzerland
Innate immune activation is initiated by recognition of pathogen associated molecular patterns (PAMPs). Delivery of PAMPs to their respective receptors, regulation of receptor activity, and effector functions downstream from these receptors, which constitute part of the initiated innate immune control, are in part mediated via macroautophagy, an evolutionary conserved pathway for cytoplasmic constituent degradation in lysosomes. In this review these facets of the recently unveiled involvement of macroautophagy in innate immunity will be summarized, and aspects that need additional investigations will be high-lighted. The improved understanding of the capabilities of macroautophagy for immunity suggests that this pathway should be harnessed in immunotherapies against infectious diseases.
Introduction
Autophagy describes a group of at least three evolutionary conserved cellular degradation processes in eukaryotes that deliver cytoplasmic constituents for lysosomal degradation (Mizushima and Klionsky, 2007). These are macro-, micro-, and chaperone mediated autophagy. During microautophagy the lysosomal membrane invaginates and buds into the lysosomal lumen taking a portion of cytoplasm with it into lysosomal break-down. This pathway, however, has so far only been described in yeast and a clear demonstration that it also occurs in higher eukaryotes is still lacking. During chaperone mediated autophagy proteins with a KFERQ recognition sequence are transported across the lysosomal membrane for degradation (Massey et al., 2006). For this purpose cytosolic chaperones, including HSC70 members, recognize KFERQ, dock to Lamp2a in the lysosomal membrane and are then assisted by another HSC70 member in the lysosomal lumen to import substrates into lysosomes. This process is also conserved in higher eukaryotes, but it remains unclear to which extent it contributes to protein turn-over in the steady-state, since it has primarily been studied under extreme starvation conditions. Macroautophagy as the third pathway is characterized by de novo formation of a double-membrane engulfed vesicle, the autophagosome, which then fuses with late endosomes or lysosomes for the degradation of its cargo. Autophagosomes can form from membranes of multiple sources, probably depending on the location of their cargo and initiation signals for vesicle formation. These membrane sources include the rough endoplasmic reticulum (Hayashi-Nishino et al., 2009; Yla-Anttila Vihinen et al., 2009), the Golgi apparatus (Lynch-Day et al., 2010; Yen et al., 2010), the outer nuclear membrane (English et al., 2009), the outer mitochondrial membrane (He et al., 2006; Hailey et al., 2010), and the plasma membrane (Ravikumar et al., 2010). Autophagosome formation and degradation requires more than 30 gene products, so called autophagy-related (Atg) proteins. The site of autophagosome generation is marked by phosphatidylinositol 3-phosphate (PI3) in membranes, which is deposited there by type III PI3 kinase complexes, containing the PI3 kinase VPS34, VPS15, Atg14L, and Atg6/Beclin-1 (Figure 1). Extension of the autophagosomal membrane is then achieved with the help of two ubiquitin-like systems (Ohsumi, 2001). In one of them Atg12 is coupled with the help of the E1- and E2-like enzymes Atg7 and 10 to Atg5, and the complex localizes then with Atg16L1 to the outer membrane of the emerging autophagosome. There it acts as an E3-like enzyme for the conjugation of the other ubiquitin-like molecule Atg8, one homolog of which in mammalian cells is called LC3. Atg8/LC3 is prior to conjugation activated by proteolytic processing through Atg4, exposing a glycine at its C-terminus. It is then activated by the E1-like enzyme Atg7, conjugated to the E2-like enzyme Atg3 and finally ligated to phosphatidylethanolamine in the inner and outer membrane of the emerging autophagosome through the E3-like activity of Atg12–Atg5/Atg16L1. At the autophagosomal membrane it mediates recruitment of ubiquitinated substrates, like protein aggregates and cell organelles, via the ubiquitin and LC3 binding proteins p62, NBR1, and NDP52 (Bjorkoy et al., 2005; Kirkin et al., 2009; Thurston et al., 2009), and probably also membrane fusion for autophagosomal membrane elongation and vesicle completion (Nakatogawa et al., 2007). While Atg8/LC3 remains at the inner autophagosomal membrane, Atg4 cleaves it from the outer autophagosomal membrane and also the Atg12–Atg5/Atg16L1 is recycled after autophagosome completion. Fusion of the completed autophagosomes with lysosomes and late endosomes is then also mediated by Atg6/Beclin-1 containing PI3 kinase complexes with UVRAG or Rubicon replacing Atg14L (Liang et al., 2008; Matsunaga et al., 2009; Zhong et al., 2009). In addition, Rab11 is required for fusion with late endosomes (Fader et al., 2008) and Rab7 and Lamp2 for fusion with lysosomes (Tanaka et al., 2000; Jager et al., 2004). Thus the molecular machinery for macroautophagy has been explored in some depth, and the gained knowledge also favored investigations of this particular autophagy pathway during innate immune responses.
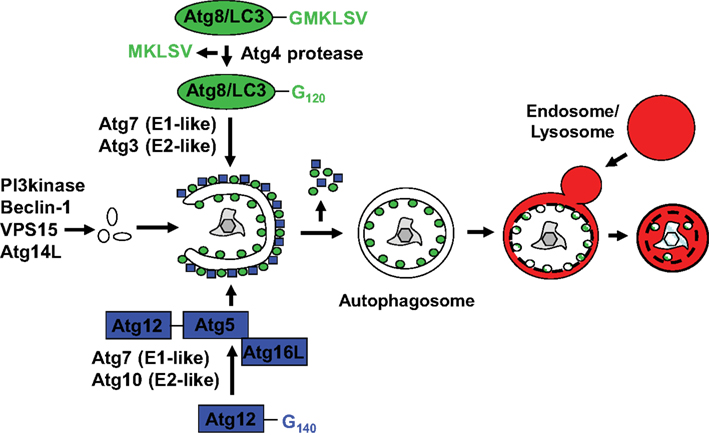
Figure 1. Macroautophagy delivers cytoplasmic constituents to lysosomal degradation via the formation of an autophagosome. While PI3 generated by Beclin-1 containing complexes (PI3 kinase, Beclin-1, VPS15, and Atg14L) marks sites of autophagosome generation, two ubiquitin-like systems are required to form autophagosomes at these sites. Atg12 is coupled to Atg5 by the indicated E1-like and E2-like enzymes Atg7 and Atg10, and associates with the outer membrane of the emerging autophagosome in complex with Atg16L. The complex guides ligation as an E3-like enzyme of Atg8, whose best studied mammalian homolog is LC3, to the outer and inner autophagosomal membrane. Prior to coupling Atg8/LC3 is processed by Atg4 and activated via the two E1- and E2-like enzymes Atg7 and Atg3. Upon completion of the autophagosomes the Atgs recycle from the outer membrane, while Atg8/LC3 is degraded with the inner autophagosomal membrane as well as the autophagosome content upon fusion to lysosomes.
Accordingly, I will primarily summarize the evidence that innate immune recognition regulates macroautophagy, how macroautophagy modulates innate immune recognition in return, which effector functions for innate immunity can be mediated by macroautophagy and how these effector functions might restrict pathogens and commensals in vivo both in animal models and even in humans.
PAMP Mediated Macroautophagy Regulation
Originally macroautophagy has been described as a response to starvation, recycling cytoplasmic constituents to generate energy and macromolecular building blocks for cell survival. The signals leading to macroautophagy up-regulation in response to amino acid deprivation and growth factor withdrawal have now in part been described (He and Klionsky, 2009). In addition, more recently it has been appreciated that pathogen detection can also up-regulate macroautophagy as an innate immune effector function (Mizushima et al., 2008). Recognition of pathogen associated molecular patterns (PAMPs) or pathogen induced changes in cell organelles stimulate macroautophagy (Figure 2). Although intuitively one would predict from the role of macroautophagy in cytosolic organelle and protein aggregate clearance that cytosolic PAMP recognition might preferentially enhance this pathway, but PAMP recognition receptors (PRRs) recognizing extra- and intra-cellular structures have been described to up-regulate macroautophagy. This already point to functions of macroautophagy beyond just engulfment of intracellular microbes for their degradation in lysosomes, and these additional effector functions of macroautophagy will be discussed in the fourth section of this review. With regard to surface and endosomal receptors that trigger macroautophagy mainly toll like receptors (TLRs) were so far evaluated for macroautophagy regulation (Sanjuan et al., 2007; Delgado et al., 2008; Shi and Kehrl, 2008). Among these TLR4, the receptor for bacterial lipopolysaccharide (LPS), and TLR7, a receptor for single stranded RNA, have been reported to induce the strongest autophagosome accumulation in the mouse macrophage cell line RAW264.7 (Xu et al., 2007; Delgado et al., 2008; Shi and Kehrl, 2008). However, up-regulation of macroautophagy by TLR4 has less consistently been observed with primary bone marrow derived mouse macrophages. While some investigators have reported such macroautophagy increase (Travassos et al., 2010), others have failed to observe this (Saitoh et al., 2008). TLR4 recruits upon LPS engagement the adaptor molecules MyD88 and Trif, which in turn bind ubiquitinated Atg6/Beclin-1 to enhance macroautophagy (Shi and Kehrl, 2008, 2010), possibly by initiating autophagosome formation at the cell membrane (Ravikumar et al., 2010). Therefore, TLR signaling seems to up-regulate macroautophagy in mouse phagocytes, but which PAMPs achieve this in human cells and which pathogens relevant to human disease are affected by it, remains to be determined.
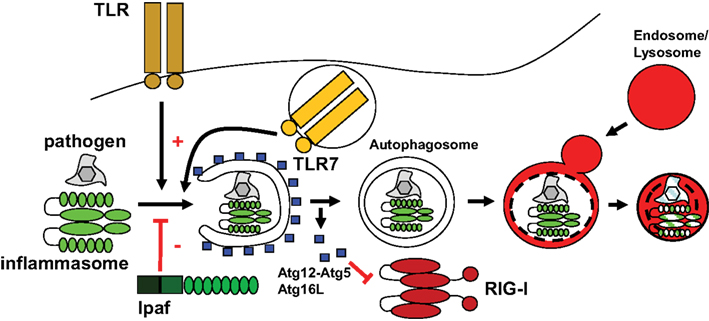
Figure 2. Some PAMP receptors stimulate macroautophagy, while others are negatively regulated by it. Toll like receptors can enhance macroautophagy, while IPAF, component of one particular inflammasome has been described to inhibit autophagosome generation. Other inflammasomes are compromised by macroautophagy, possibly by direct degradation, and RIG-I is inhibited by the Atg12–Atg5 complex.
In addition to TLR mediated up-regulation, cytosolic PRRs have also been demonstrated to up-regulate macroautophagy. These are the bacterial peptidoglycan receptors PGRP-LE in Drosophila and NOD1 and two in mammalian cells (Yano et al., 2008; Cooney et al., 2010; Travassos et al., 2010). The NOD molecules recruit Atg16L1 to the bacterial entry site for efficient degradation of invading pathogens (Travassos et al., 2010). Apart from bacterial peptidoglycan detection, double-stranded RNA (dsRNA), a PAMP associated with viral replication, stimulated the IFN-inducible eIF2 kinase PKR (dsRNA-dependent protein kinase) to induce macroautophagy in herpesvirus infected cells (Talloczy et al., 2002). Therefore, PKR and NOD molecules seem to stimulate macroautophagy upon cytosolic pathogen recognition. In contrast, NOD-like receptors (NLRs) that give rise to inflammasomes and RIG-I like receptors (RLRs) are negatively regulated by macroautophagy, as will be discussed below.
In addition to direct macroautophagy up-regulation after PAMP recognition, cytokines that are produced in response to pathogen detection, have also been identified as modulators of macroautophagy. Initially, IFN-γ was identified to mediate Mycobacterium tuberculosis clearance in part via up-regulation of macroautophagy (Gutierrez et al., 2004). However, similar to PRR mediated macroautophagy modulation, this effect was more pronounced in the mouse macrophage RAW264.7 or human macrophage THP-1 and U937 cell lines, than in primary human or mouse macrophages (Harris et al., 2007). In contrast to the cell-mediated immunity supporting cytokine IFN-γ, cytokines associated primarily with humoral immune responses, like IL-4 and IL-13, seemed to rather inhibit macroautophagy (Harris et al., 2007). Furthermore, TNF-α was reported to induce macroautophagy in cells with low NF-κB activation (Djavaheri-Mergny et al., 2006). These included Ewing sarcoma cells, in which NF-κB activation was compromised and skeletal muscle cells (Keller et al., 2011). In good agreement with TNF-α induced macroautophagy in myocytes, muscle fibers of patients with inclusion body myositis, a myopathy with chronic inflammation, harbored increased numbers of autophagosomes and were surrounded by proinflammatory infiltrates. These findings suggest that pathogen recognition can up-regulate macroautophagy directly via some PRRs and indirectly via cytokines that are produced upon pathogen triggered immune activation.
Unrelated to PRRs and cytokines, macroautophagy can also be enhanced by pathogens for their benefit. The prototypic example for this is poliovirus infection, during which the viral 2BC and 3A proteins induce the accumulation of Atg12 and Atg8/LC3 dependent double-membrane vesicles for viral replication (Dales et al., 1965; Jackson et al., 2005). Furthermore, binding of the human immunodeficiency virus (HIV) envelope protein to the chemokine receptor CXCR4 augments macroautophagy and seems to enhance bystander T cell death for immune escape (Espert et al., 2006). Finally, primarily two virus families, the herpes- and the flavi-viruses, have been described to enhance macroautophagy via the unfolded protein response (Sir et al., 2008; Lee et al., 2009; Dreux and Chisari, 2010; Lin et al., 2010), and thereby gain benefits for viral replication and latent to lytic infection transition (Dreux et al., 2009; Lee and Sugden, 2008; Lee et al., 2008). In addition to these beneficial effect of macroautophagy up-regulation for pathogens, other microbes also inhibit this pathway during infection, and these immune escape strategies from innate effector function will be discussed below.
Macroautophagy Modification of PAMP Recognition
While TLRs and NLRs enhance macroautophagy and little is know so far about C-type lectin receptor (CLR) interaction with this pathway, other cytosolic PAMP recognition pathways seem to be negatively impacted by macroautophagy (Figure 2). Along these lines, dsRNA recognition by RNA helicases of the family of RLRs is augmented in macroautophagy deficient cells (Jounai et al., 2007; Tal et al., 2009). The Atg12–Atg5 complex seems to directly associate with RIG-I and its adaptor IPS-1 to inhibit viral RNA induced type I IFN production (Jounai et al., 2007). Moreover, the accumulation of damaged mitochondria in macroautophagy deficient cells and the associated increase in reactive oxygen species (ROS) production, seem to enhance RLR activity in response to viral infections (Tal et al., 2009). Thus, RLR recognition of cytosolic dsRNA is attenuated by macroautophagy.
Parallel to decreasing cytosolic RNA recognition, macroautophagy enhances delivery of viral RNA replication intermediates to TLR containing endosomes (Lee et al., 2007). TLR7 dependent recognition of these single-stranded RNAs (ssRNAs) is essential for IFN-α production by vesicular stomatitis (VSV) and Sendai virus infected plasmacytoid dendritic cells (DCs), and depends on macroautophagy (Lee et al., 2007). Similarly, endocytosed DNA seems to be targeted to TLR9 containing endosomes via macroautophagy in B cells (Chaturvedi et al., 2008), possibly assisting autoantibody production in systemic lupus erythematosus patients. These findings indicate that macroautophagy enhances PAMP delivery to endosomal TLRs.
Among the NLRs, NOD1 and two seem to up-regulate macroautophagy in mouse phagocytes and human DCs as discussed above, whereas signaling through other members of this family that initiate inflammasome formation is inhibited by macroautophagy and deficiency in inflammasome components increases macroautophagy in Shigella flexneri infected macrophages (Suzuki et al., 2007; Saitoh et al., 2008). Indeed, loss of Atg16L1 increases IL-1β secretion by mouse macrophages, which depends on inflammasome activated caspase 1 processing. This increased IL-1β production worsens dextran sulfate sodium induced colitis in mice. The negative regulation of inflammasome dependent inflammation was suggested to be directly mediated via inflammasome degradation by macroautophagy (Harris et al., 2009), but this needs to be experimentally confirmed. In addition, it remains also unclear which inflammasomes are affected by negative regulation via macroautophagy and if the various inflammasome scaffolds are regulated differently. Thus, both RLR and inflammasome signaling in response to PAMP recognition are inhibited by macroautophagy, while the same pathway delivers RNA and DNA for endosomal PRRs.
Effector Mechanisms of Macroautophagy for Innate Pathogen Restriction
PAMP recognition receptors mediated macroautophagy up-regulation is able to exert innate restriction of intracellular pathogens, both those that escape endosomes and others that condition phagosomes to become their replication compartments (Münz, 2009). The first pathogens described in these categories were group A Streptococci and Mycobacterium tuberculosis, respectively (Gutierrez et al., 2004; Nakagawa et al., 2004). Initially it was just assumed that macroautophagy delivers these pathogens for lyosomal degradation, thereby limiting pathogen burden in infected cells (Figure 3A). However, recently it has become apparent that macroautophagy delivers in addition substrates that are then converted into bactericidal peptides by lysosomal hydrolysis (Figure 3B; Alonso et al., 2007; Ponpuak et al., 2010). In both instances ubiquitin itself or proteins containing a ubiquitin-like domain, like the ribosomal protein S30 (rpS30) precursor Fau, are transported into Mycobacterium tuberculosis containing phagosomes to be processed to bactericides. In the case of Fau, binding of its ubiquitin-like domain to p62, which in turn recruits this ribosomal protein precursor to autophagosomes by virtue of binding to Atg8/LC3, was important to deliver this source of bactericidal activity to mycobacterial phagosomes (Ponpuak et al., 2010). Thus, macroautophagy does not only deliver pathogens for lysosomal hydrolysis, but also charges pathogen containing phagosomes with proteins that give rise to bactericidal peptides after lysosomal hydrolysis.
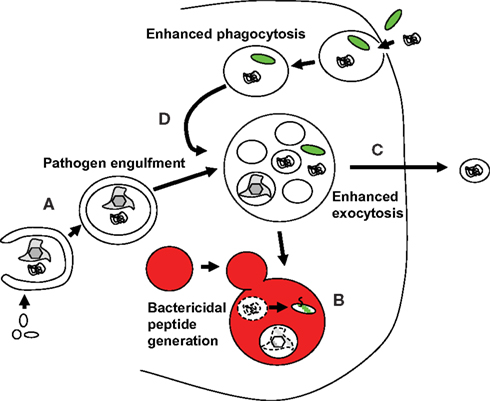
Figure 3. Macroautophagy contributes with several mechanisms to innate immune responses against pathogens. (A) Pathogens are directly taken up from the cytosol into autophagosomes and then degraded by lysosomal hydrolysis. (B) Substrates that give rise to bactericidal peptides after lysosomal degradation are transported to pathogen containing autophagosomes or phagosomes, and eliminate pathogens in these fusion vesicles. (C) Macroautophagy contributes to the release of antimicrobial granules from Paneth cells to restrict pathogens and commensals on mucosal surfaces. (D) Macroautophagy contributes to more efficient fusion of phagosomes with lysosomes for degradation of phagocytosed pathogens.
Apart from intracellular pathogen delivery to lysosomes and bactericidal source protein delivery to phagosomes, macroautophagy seems to also enhance degradation of phagocytosed pathogens (Figure 3D). A role for the molecular machinery of macroautophagy in the enhanced fusion of phagosomes with lysosomes was first identified upon TLR2 ligation in the mouse macrophage cell line RAW264.7 (Sanjuan et al., 2007). However, also in vivo processing of extracellular antigen by DCs was compromised by Atg5 deficiency in this cell subset, and this was due to diminished lysosomal hydrolase recruitment to endocytosed material (Lee et al., 2010). The enhanced phagosome maturation via Atgs seems to require either NADPH oxidase activity or diacylglycerol production at the bacterial pathogen containing endosomes in mouse myeloid cells (Huang et al., 2009; Shahnazari and Brumell, 2009). However, the nature of the quality conferred by autophagosome or just Atg recruitment to phagosomes that allows more efficient fusion with lysosomes remains to be determined.
In addition to these bactericidal effector functions in endosomes, supported by macroautophagy, the autophagic machinery seems to also support bactericidal peptide release at mucosal surfaces (Figure 3C). Paneth cells at the base of ileal crypts suffer aberrant packaging and reduced exocytosis of antimicrobial granules from the hypomorphic down-regulation of Atg16L1 expression (Cadwell et al., 2008). Moreover, this aberration was only observed upon co-infection with the murine norovirus, a positive strand RNA virus (Cadwell et al., 2010). Therefore, macroautophagy assistance for exocytosis of bactericidal granules seems to be mainly important during enteric infections.
An indication that these macroautophagic effector functions are interfering with pathogen replication is provided by the many mechanisms by which viruses and bacteria interfere with either autophagosome formation or fusion with lysosomes (Schmid and Münz, 2007). An exhaustive list of such pathogens and their immune escape strategies targeting macroautophagy is beyond the scope of this review and has been recently summarized elsewhere (Levine and Deretic, 2007). Thus, I will just high-light some general pathways of immune escape from autophagy by pathogens and discuss examples below, for which in vivo evidence for the importance of these has been gained. While DNA viruses, like Herpesviruses, seem to preferentially inhibit autophagosome formation, RNA viruses like HIV and influenza A virus seem to block autophagosome fusion with lysosomes (Gannage et al., 2009; Kyei et al., 2009; Gannage and Münz, 2010; Taylor et al., 2011). In addition, some bacterial pathogens interfere with engulfment by macroautophagy by either blocking bacterial protein recognition or covering themselves with cytosolic host cell proteins (Ogawa et al., 2005; Yoshikawa et al., 2009). Finally, membrane pore formation and/or bacterial secretion systems, which inject modulators of cellular physiology through endosomal membranes into the cytosol, seem to be required to arrest maturation of bacteria containing amphisomes, born from phagosome fusions with autophagosomes (Celli et al., 2003; Birmingham et al., 2008). Therefore, both viral and bacterial pathogens have developed strategies to evade macroautophagy, arguing for the potency of innate restriction by this pathway and of its effector functions discussed above.
In vivo Restriction of Pathogen Infection by Macroautophagy
Due to the complexity of immune control mechanisms, it is often difficult to assign in vivo phenotypes to particular immune responses without underlying effects in immune compartment development and survival. Accentuated by the well documented pro-survival role of macroautophagy (Kuma et al., 2004), this is also an issue in case of infections and their immune control in mice with deficiencies in macroautophagy. Moreover, since macroautophagy assists both innate and adaptive immunity (Münz, 2009) and macroautophagy clearly assists T cell development and T cell mediated immune responses in vivo (Pua et al., 2007; Nedjic et al., 2008; Lee et al., 2010), the contributions of innate and adaptive pathogen restriction mechanisms via macroautophagy in vertebrates still remain to be investigated in detail in in vivo infections. However, two experimental systems have been analyzed in detail with respect to innate immunity to viral infections via macroautophagy in mice. These investigated neurotropic infections with the RNA virus, Sindbis Virus (SV) and the DNA virus Herpes simplex virus (HSV; Orvedahl et al., 2007, 2010). Intracerebral inoculation with SV causes autophagosome accumulation in neurons, and mortality of infected mice is increased when macroautophagy is inhibited by either delivering a dominant negative Atg5 protein with the virus, expressing cre recombinase with recombinant SV for deletion of floxed Atg5 in infected cells or neuron specific deletion of Atg5 (Orvedahl et al., 2010). Although CNS viral titers were not increased under these conditions, infection induced protein aggregate formation probably led to increased pathology and delayed virus clearance. In addition, overexpression of Atg6/Beclin-1 in neurons protects mice from lethal intracerebral SV infection (Liang et al., 1998). In contrast to increased neuropathology of SV infection without significant elevation of viral titers in the absence of macroautophagy, HSV inhibits macroautophagy to sustain high viral titers and neurovirulence (Orvedahl et al., 2007). For this inhibition HSV encodes the ICP34.5 protein, which contains a domain that inhibits macroautophagy through binding to Atg6/Beclin-1. Deletion of this domain renders HSV neuroattenuated with decreased lethality and faster virus clearance from the brain. These data suggest that different neurotropic viruses are restricted by macroautophagy in neurons.
In addition to these mouse models, macroautophagy inhibits infection with the RNA virus VSV in flies (Shelly et al., 2009). Atg18 deficient Drosophila flies died rapidly after VSV infection. Furthermore, Drosophila uses macroautophagy to also restrict Listeria infection (Yano et al., 2008). These bacteria replicate to higher levels and kill their host flies after siRNA mediated silencing of Atg5. Moreover, host defense against Salmonella typhimurium infection in the worm Caenorhabditis elegans and the slime mold Dictyostelium discoideum depends in part on macroautophagy (Jia et al., 2009). SiRNA mediated silencing of Atg6/bec-1 and Atg8/lgg1 in worms increased their susceptibility to Salmonella infection. Similarly, deficiency in Atg1, 6, or 7 compromised survival of mold to infection by the same bacterial pathogen. In both cases diminished macroautophagy caused higher pathogen burden in the infected hosts. These invertebrate models of infectious diseases, lacking adaptive immunity, argue for a significant innate immune control function by macroautophagy in vivo.
Association between Polymorphisms in Macroautophagy Genes and Human Disease
Although animal models provide strong evidence for the importance of immune mechanisms in vivo, only clinical benefit upon manipulation of these pathways or genetic susceptibilities for diseases located in these pathways provide the final proof that some immune function is crucial for resistance against human pathogens. Along these lines, mutations in the essential macroautophagy gene Atg16L1, and macroautophagy stimulating genes, like immunity-related GTPase family M (IRGM) and NOD2 (Singh et al., 2006; Cooney et al., 2010; Travassos et al., 2010), have been described to be associated with inflammatory bowel disease in Crohn’s disease patients (Hugot et al., 2001; Ogura et al., 2001; Hampe et al., 2007; Parkes et al., 2007; Rioux et al., 2007; McCarroll et al., 2008). The modulation of macroautophagy by the respective susceptibility alleles could facilitate the development of Crohn’s disease by many mechanisms, which have been more comprehensively summarized elsewhere (Meixlsperger and Münz, 2009; Stappenbeck et al., 2011). However, with respect to the influence of macroautophagy on innate immune control of commensals in the digestive tract, whose uncontrolled invasion could trigger inflammatory bowel disease, three main mechanisms might be operational. Firstly, macroautophagy might simply restrict commensal bacteria in myeloid cells by directly targeting intracellular bacteria or assisting phagosome maturation of endocytosed extracellular commensals (Sanjuan et al., 2007; Kuballa et al., 2008; Lapaquette et al., 2010). Secondly, deficiencies in Paneth cells, caused by Atg16L1, which have been observed in Crohn’s disease patients could impair innate restriction of commensals at mucosal surfaces (Cadwell et al., 2008, 2010). Thirdly, altered macroautophagy could disturb proinflammatory cytokine production in mucosal tissues by up-regulating inflammasome dependent cytokines like IL-1β and down-regulating PAMP delivery for TLR stimulation and TLR dependent cytokine production, which then in turn would limit commensal invasion (Lee et al., 2007; Saitoh et al., 2008). Although the exact mechanisms, by which the mutations in Atg16L1, IRGM, and NOD2 facilitate the development of Crohn’s disease, still need to be characterized in more detail, the association of mutations in these genes strongly argue for an immune modulating function of this ancient catabolic pathway.
Conclusion
The multitude of studies summarized in this review that implicate macroautophagy in pathogen restriction, escape of successful pathogens from this pathway, and genetic susceptibility to human disease due to mutations in it, suggests that macroautophagy plays a crucial role in both maintenance of our coexistence with gut commensals and resistance to infections. Apart from this roles, macroautophagy regulation could counteract consequences of aging (Zhang and Cuervo, 2008), ameliorate neurodegeneration (Hara et al., 2006; Komatsu et al., 2006), and fight cancer (Mathew et al., 2009). Therefore, it would be desirable to develop therapeutic approaches to modulate macroautophagy in infectious and these other diseases. However at the moment we have very few pharmacologic substances like rapamycin, most of them with significant side effects, to regulate macroautophagy. If more specific modulators could be identified this pathway could be harnessed to increase innate immunity among other benefits.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in my laboratory is in part supported by the National Cancer Institute (R01CA108609 and R01CA101741), the Foundation for the National Institutes of Health (Grand Challenges in Global Health), and the Swiss National Science Foundation (310030_126995).
References
Alonso, S., Pethe, K., Russell, D. G., and Purdy, G. E. (2007). Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. U.S.A. 104, 6031–6036.
Birmingham, C. L., Canadien, V., Kaniuk, N. A., Steinberg, B. E., Higgins, D. E., and Brumell, J. H. (2008). Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature 451, 350–354.
Bjorkoy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., Stenmark, H., and Johansen, T. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614.
Cadwell, K., Liu, J. Y., Brown, S. L., Miyoshi, H., Loh, J., Lennerz, J. K., Kishi, C., Kc, W., Carrero, J. A., Hunt, S., Stone, C. D., Brunt, E. M., Xavier, R. J., Sleckman, B. P., Li, E., Mizushima, N., Stappenbeck, T. S., and Virgin, H. W. IV. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263.
Cadwell, K., Patel, K. K., Maloney, N. S., Liu, T. C., Ng, A. C., Storer, C. E., Head, R. D., Xavier, R., Stappenbeck, T. S., and Virgin, H. W. (2010). Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145.
Celli, J., de Chastellier, C., Franchini, D. M., Pizarro-Cerda, J., Moreno, E., and Gorvel, J. P. (2003). Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 198, 545–556.
Chaturvedi, A., Dorward, D., and Pierce, S. K. (2008). The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 28, 799–809.
Cooney, R., Baker, J., Brain, O., Danis, B., Pichulik, T., Allan, P., Ferguson, D. J., Campbell, B. J., Jewell, D., and Simmons, A. (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97.
Dales, S., Eggers, H. J., Tamm, I., and Palade, G. E. (1965). Electron microscopic study of the formation of poliovirus. Virology 26, 379–389.
Delgado, M. A., Elmaoued, R. A., Davis, A. S., Kyei, G., and Deretic, V. (2008). Toll-like receptors control autophagy. EMBO J. 27, 1110–1121.
Djavaheri-Mergny, M., Amelotti, M., Mathieu, J., Besancon, F., Bauvy, C., Souquere, S., Pierron, G., and Codogno, P. (2006). NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 281, 30373–30382.
Dreux, M., Gastaminza, P., Wieland, S. F., and Chisari, F. V. (2009). The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U.S.A. 106, 14046–14051.
English, L., Chemali, M., Duron, J., Rondeau, C., Laplante, A., Gingras, D., Alexander, D., Leib, D., Norbury, C., Lippe, R., and Desjardins, M. (2009). Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487.
Espert, L., Denizot, M., Grimaldi, M., Robert-Hebmann, V., Gay, B., Varbanov, M., Codogno, P., and Biard-Piechaczyk, M. (2006). Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 116, 2161–2172.
Fader, C. M., Sanchez, D., Furlan, M., and Colombo, M. I. (2008). Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 9, 230–250.
Gannage, M., Dormann, D., Albrecht, R., Dengjel, J., Torossi, T., Ramer, P. C., Lee, M., Strowig, T., Arrey, F., Conenello, G., Pypaert, M., Andersen, J., Garcia-Sastre, A., and Münz, C. (2009). Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380.
Gannage, M., and Münz, C. (2010). MHC presentation via autophagy and how viruses escape from it. Semin. Immunopathol. 32, 373–381.
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766.
Hailey, D. W., Rambold, A. S., Satpute-Krishnan, P., Mitra, K., Sougrat, R., Kim, P. K., and Lippincott-Schwartz, J. (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667.
Hampe, J., Franke, A., Rosenstiel, P., Till, A., Teuber, M., Huse, K., Albrecht, M., Mayr, G., De La Vega, F. M., Briggs, J., Gunther, S., Prescott, N. J., Onnie, C. M., Hasler, R., Sipos, B., Folsch, U. R., Lengauer, T., Platzer, M., Mathew, C. G., Krawczak, M., and Schreiber, S. (2007). A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211.
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., Yokoyama, M., Mishima, K., Saito, I., Okano, H., and Mizushima, N. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889.
Harris, J., De Haro, S. A., Master, S. S., Keane, J., Roberts, E. A., Delgado, M., and Deretic, V. (2007). T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 27, 505–517.
Harris, J., Hope, J. C., and Lavelle, E. C. (2009). Autophagy and the immune response to TB. Transbound. Emerg. Dis. 56, 248–254.
Hayashi-Nishino, M., Fujita, N., Noda, T., Yamaguchi, A., Yoshimori, T., and Yamamoto, A. (2009). A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 11, 1433–1437.
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93.
He, C., Song, H., Yorimitsu, T., Monastyrska, I., Yen, W. L., Legakis, J. E., and Klionsky, D. J. (2006). Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J. Cell Biol. 175, 925–935.
Huang, J., Canadien, V., Lam, G. Y., Steinberg, B. E., Dinauer, M. C., Magalhaes, M. A., Glogauer, M., Grinstein, S., and Brumell, J. H. (2009). Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. U.S.A. 106, 6226–6231.
Hugot, J. P., Chamaillard, M., Zouali, H., Lesage, S., Cezard, J. P., Belaiche, J., Almer, S., Tysk, C., O’Morain, C. A., Gassull, M., Binder, V., Finkel, Y., Cortot, A., Modigliani, R., Laurent-Puig, P., Gower-Rousseau, C., Macry, J., Colombel, J. F., Sahbatou, M., and Thomas, G. (2001). Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603.
Jackson, W. T., Giddings, T. H. Jr., Taylor, M. P., Mulinyawe, S., Rabinovitch, M., Kopito, R. R., and Kirkegaard, K. (2005). Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3, e156. doi: 10.1371/journal.pbio.0030156
Jager, S., Bucci, C., Tanida, I., Ueno, T., Kominami, E., Saftig, P., and Eskelinen, E. L. (2004). Role for Rab7 in maturation of late autophagic vacuoles. J. Cell. Sci. 117(Pt 20), 4837–4848.
Jia, K., Thomas, C., Akbar, M., Sun, Q., Adams-Huet, B., Gilpin, C., and Levine, B. (2009). Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc. Natl. Acad. Sci. U.S.A. 106, 14564–14569.
Jounai, N., Takeshita, F., Kobiyama, K., Sawano, A., Miyawaki, A., Xin, K. Q., Ishii, K. J., Kawai, T., Akira, S., Suzuki, K., and Okuda, K. (2007). The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A. 104, 14050–14055.
Keller, C. W., Fokken, C., Turville, S. G., Lünemann, A., Schmidt, J., Münz, C., and Lünemann, J. D. (2011). TNF-alpha induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J. Biol. Chem. 286, 3970–3980.
Kirkin, V., Lamark, T., Sou, Y. S., Bjorkoy, G., Nunn, J. L., Bruun, J. A., Shvets, E., McEwan, D. G., Clausen, T. H., Wild, P., Bilusic, I., Theurillat, J. P., Overvatn, A., Ishii, T., Elazar, Z., Komatsu, M., Dikic, I., and Johansen, T. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516.
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., Ueno, T., Koike, M., Uchiyama, Y., Kominami, E., and Tanaka, K. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884.
Kuballa, P., Huett, A., Rioux, J. D., Daly, M. J., and Xavier, R. J. (2008). Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS ONE 3, e3391. doi: 10.1371/journal. pone.0003391
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., Ohsumi, Y., Tokuhisa, T., and Mizushima, N. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036.
Kyei, G. B., Dinkins, C., Davis, A. S., Roberts, E., Singh, S. B., Dong, C., Wu, L., Kominami, E., Ueno, T., Yamamoto, A., Federico, M., Panganiban, A., Vergne, I., and Deretic, V. (2009). Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186, 255–268.
Lapaquette, P., Glasser, A. L., Huett, A., Xavier, R. J., and Darfeuille-Michaud, A. (2010). Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 12, 99–113.
Lee, D. Y., Lee, J., and Sugden, B. (2009). The unfolded protein response and autophagy: herpesviruses rule! J. Virol. 83, 1168–1172.
Lee, D. Y., and Sugden, B. (2008). The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 27, 2833–2842.
Lee, H. K., Lund, J. M., Ramanathan, B., Mizushima, N., and Iwasaki, A. (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401.
Lee, H. K., Mattei, L. M., Steinberg, B. E., Alberts, P., Lee, Y. H., Chervonsky, A., Mizushima, N., Grinstein, S., and Iwasaki, A. (2010). In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32, 227–239.
Lee, Y. R., Lei, H. Y., Liu, M. T., Wang, J. R., Chen, S. H., Jiang-Shieh, Y. F., Lin, Y. S., Yeh, T. M., Liu, C. C., and Liu, H. S. (2008). Autophagic machinery activated by dengue virus enhances virus replication. Virology 374, 240–248.
Levine, B., and Deretic, V. (2007). Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7, 767–777.
Liang, C., Lee, J. S., Inn, K. S., Gack, M. U., Li, Q., Roberts, E. A., Vergne, I., Deretic, V., Feng, P., Akazawa, C., and Jung, J. U. (2008). Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 10, 776–787.
Liang, X. H., Kleeman, L. K., Jiang, H. H., Gordon, G., Goldman, J. E., Berry, G., Herman, B., and Levine, B. (1998). Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72, 8586–8596.
Lin, L. T., Dawson, P. W., and Richardson, C. D. (2010). Viral interactions with macroautophagy: a double-edged sword. Virology 402, 1–10.
Lynch-Day, M. A., Bhandari, D., Menon, S., Huang, J., Cai, H., Bartholomew, C. R., Brumell, J. H., Ferro-Novick, S., and Klionsky, D. J. (2010). Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 7811–7816.
Massey, A. C., Zhang, C., and Cuervo, A. M. (2006). Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 73, 205–235.
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H. Y., Bray, K., Reddy, A., Bhanot, G., Gelinas, C., Dipaola, R. S., Karantza-Wadsworth, V., and White, E. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075.
Matsunaga, K., Saitoh, T., Tabata, K., Omori, H., Satoh, T., Kurotori, N., Maejima, I., Shirahama-Noda, K., Ichimura, T., Isobe, T., Akira, S., Noda, T., and Yoshimori, T. (2009). Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396.
McCarroll, S. A., Huett, A., Kuballa, P., Chilewski, S. D., Landry, A., Goyette, P., Zody, M. C., Hall, J. L., Brant, S. R., Cho, J. H., Duerr, R. H., Silverberg, M. S., Taylor, K. D., Rioux, J. D., Altshuler, D., Daly, M. J., and Xavier, R. J. (2008). Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 40, 1107–1112.
Meixlsperger, S., and Münz, C. (2009). Morbus Crohn – a disease of failing macroautophagy in the immune system? Int. Immunol. 21, 1205–1211.
Mizushima, N., and Klionsky, D. J. (2007). Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19–40.
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075.
Nakagawa, I., Amano, A., Mizushima, N., Yamamoto, A., Yamaguchi, H., Kamimoto, T., Nara, A., Funao, J., Nakata, M., Tsuda, K., Hamada, S., and Yoshimori, T. (2004). Autophagy defends cells against invading group A Streptococcus. Science 306, 1037–1040.
Nakatogawa, H., Ichimura, Y., and Ohsumi, Y. (2007). Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178.
Nedjic, J., Aichinger, M., Emmerich, J., Mizushima, N., and Klein, L. (2008). Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455, 396–400.
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–731.
Ogura, Y., Bonen, D. K., Inohara, N., Nicolae, D. L., Chen, F. F., Ramos, R., Britton, H., Moran, T., Karaliuskas, R., Duerr, R. H., Achkar, J. P., Brant, S. R., Bayless, T. M., Kirschner, B. S., Hanauer, S. B., Nunez, G., and Cho, J. H. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606.
Ohsumi, Y. (2001). Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2, 211–216.
Orvedahl, A., Alexander, D., Talloczy, Z., Sun, Q., Wei, Y., Zhang, W., Burns, D., Leib, D., and Levine, B. (2007). HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1, 23–35.
Orvedahl, A., MacPherson, S., Sumpter, R. Jr., Talloczy, Z., Zou, Z., and Levine, B. (2010). Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127.
Parkes, M., Barrett, J. C., Prescott, N. J., Tremelling, M., Anderson, C. A., Fisher, S. A., Roberts, R. G., Nimmo, E. R., Cummings, F. R., Soars, D., Drummond, H., Lees, C. W., Khawaja, S. A., Bagnall, R., Burke, D. A., Todhunter, C. E., Ahmad, T., Onnie, C. M., McArdle, W., Strachan, D., Bethel, G., Bryan, C., Lewis, C. M., Deloukas, P., Forbes, A., Sanderson, J., Jewell, D. P., Satsangi, J., Mansfield, J. C., Cardon, L., and Mathew, C. G. (2007). Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 39, 830–832.
Ponpuak, M., Davis, A. S., Roberts, E. A., Delgado, M. A., Dinkins, C., Zhao, Z., Virgin, H. W. t., Kyei, G. B., Johansen, T., Vergne, I., and Deretic, V. (2010). Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32, 329–341.
Pua, H. H., Dzhagalov, I., Chuck, M., Mizushima, N., and He, Y. W. (2007). A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 204, 25–31.
Ravikumar, B., Moreau, K., Jahreiss, L., Puri, C., and Rubinsztein, D. C. (2010). Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 12, 747–757.
Rioux, J. D., Xavier, R. J., Taylor, K. D., Silverberg, M. S., Goyette, P., Huett, A., Green, T., Kuballa, P., Barmada, M. M., Datta, L. W., Shugart, Y. Y., Griffiths, A. M., Targan, S. R., Ippoliti, A. F., Bernard, E. J., Mei, L., Nicolae, D. L., Regueiro, M., Schumm, L. P., Steinhart, A. H., Rotter, J. I., Duerr, R. H., Cho, J. H., Daly, M. J., and Brant, S. R. (2007). Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604.
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., Omori, H., Noda, T., Yamamoto, N., Komatsu, M., Tanaka, K., Kawai, T., Tsujimura, T., Takeuchi, O., Yoshimori, T., and Akira, S. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268.
Sanjuan, M. A., Dillon, C. P., Tait, S. W., Moshiach, S., Dorsey, F., Connell, S., Komatsu, M., Tanaka, K., Cleveland, J. L., Withoff, S., and Green, D. R. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257.
Schmid, D., and Münz, C. (2007). Innate and adaptive immunity through autophagy. Immunity 26, 11–21.
Shahnazari, S., and Brumell, J. H. (2009). Eating twice for the sake of immunity: a phagocytic receptor that activates autophagy. Cell Host Microbe 6, 297–298.
Shelly, S., Lukinova, N., Bambina, S., Berman, A., and Cherry, S. (2009). Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30, 588–598.
Shi, C. S., and Kehrl, J. H. (2008). MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 283, 33175–33182.
Shi, C. S., and Kehrl, J. H. (2010). TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 3, ra42.
Singh, S. B., Davis, A. S., Taylor, G. A., and Deretic, V. (2006). Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313, 1438–1441.
Sir, D., Chen, W. L., Choi, J., Wakita, T., Yen, T. S., and Ou, J. H. (2008). Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48, 1054–1061.
Stappenbeck, T. S., Rioux, J. D., Mizoguchi, A., Saitoh, T., Huett, A., Darfeuille-Michaud, A., Wileman, T., Mizushima, N., Carding, S., Akira, S., Parkes, M., and Xavier, R. J. (2011). Crohn disease: a current perspective on genetics, autophagy and immunity. Autophagy 7, 355–374.
Suzuki, T., Franchi, L., Toma, C., Ashida, H., Ogawa, M., Yoshikawa, Y., Mimuro, H., Inohara, N., Sasakawa, C., and Nunez, G. (2007). Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3, e111. doi: 10.1371/journal. ppat.0030111
Tal, M. C., Sasai, M., Lee, H. K., Yordy, B., Shadel, G. S., and Iwasaki, A. (2009). Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 2770–2775.
Talloczy, Z., Jiang, W., Virgin, H. W., IV Leib, D. A., Scheuner, D., Kaufman, R. J., Eskelinen, E. L., and Levine, B. (2002). Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 190–195.
Tanaka, Y., Guhde, G., Suter, A., Eskelinen, E. L., Hartmann, D., Lullmann-Rauch, R., Janssen, P. M., Blanz, J., von Figura, K., and Saftig, P. (2000). Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906.
Taylor, G. S., Mautner, J., and Münz, C. (2011). Autophagy in herpesvirus immune control and immune escape. Herpesviridae 2, 2.
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221.
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhaes, J. G., Yuan, L., Soares, F., Chea, E., Le Bourhis, L., Boneca, I. G., Allaoui, A., Jones, N. L., Nunez, G., Girardin, S. E., and Philpott, D. J. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62.
Xu, Y., Jagannath, C., Liu, X. D., Sharafkhaneh, A., Kolodziejska, K. E., and Eissa, N. T. (2007). Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27, 135–144.
Yano, T., Mita, S., Ohmori, H., Oshima, Y., Fujimoto, Y., Ueda, R., Takada, H., Goldman, W. E., Fukase, K., Silverman, N., Yoshimori, T., and Kurata, S. (2008). Autophagic control of listeria through intracellular innate immune recognition in Drosophila. Nat. Immunol. 9, 908–916.
Yen, W. L., Shintani, T., Nair, U., Cao, Y., Richardson, B. C., Li, Z., Hughson, F. M., Baba, M., and Klionsky, D. J. (2010). The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 188, 101–114.
Yla-Anttila, P., Vihinen, H., Jokitalo, E., and Eskelinen, E. L. (2009). 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 5, 1180–1185.
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., Mimuro, H., Nakagawa, I., Yanagawa, T., Ishii, T., Kakizuka, A., Sztul, E., Chakraborty, T., and Sasakawa, C. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240.
Zhang, C., and Cuervo, A. M. (2008). Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 14, 959–965.
Keywords: Atg, TLR, NOD, inflammasome, RIG-I
Citation: Münz C (2011) Macroautophagy during innate immune activation. Front. Microbio. 2:72. doi:10.3389/fmicb.2011.00072
Received: 22 October 2010;
Paper pending published: 16 November 2010;
Accepted: 28 March 2011;
Published online: 12 April 2011.
Edited by:
Amal Amer, The Ohio State University, USAReviewed by:
Jean Celli, National Institute of Allergy and Infectious Diseases, National Institutes of Health, USAAmal Amer, The Ohio State University, USA
Maria Isabel Colombo, Laboratorio de Biología Celular y Molecular Universidad Nacional de Cuyo-CONICET, Argentina
Copyright: © 2011 Münz. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Christian Münz, Viral Immunobiology, Institute of Experimental Immunology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. e-mail:Y2hyaXN0aWFuLm11ZW56QHVzei5jaA==