 Edward A. Miao1,2* and Jayant V. Rajan2,3
Edward A. Miao1,2* and Jayant V. Rajan2,3
- 1 Department of Microbiology and Immunology, University of North Carolina, Chapel Hill, NC, USA
- 2 Institute for Systems Biology, Seattle, WA, USA
- 3 Department of Medicine, University of Washington, Seattle, WA, USA
Salmonellae are intracellular pathogens that replicate within epithelial cells and macrophages, and are a significant public health threat in both developed and developing countries. The innate immune system detects microbes through pattern recognition receptors, which are compartmentalized on the subcellular level to detect either extracellular (e.g., TLRs) or cytosolic (e.g., NLRs) perturbations. Salmonella infection is detected by the NLRC4 and NLRP3 inflammasomes, which activate Caspase-1, resulting in reduced bacterial burdens during infection. NLRC4 responds to the SPI1 type III secretion system via detection of inadvertently translocated flagellin and rod protein. The signals for NLRP3 detection during Salmonella infection remain undefined. Salmonella have evolved evasion strategies to attenuate Caspase-1 responses. We review recent findings describing the interplay between detection and evasion of S. typhimurium infection by the inflammasome. We discuss how the interplay between detection and evasion affects Caspase-1 effector functions mediated by IL-1β secretion, IL-18 secretion, and pyroptosis.
Introduction
Salmonella typhi and S. paratyphi cause Typhoid fever, a continuing source of morbidity and mortality in developing countries. With 21 million cases and 200–600,000 deaths annually, antibiotic resistance is a growing concern (Parry et al., 2002; Bhutta and Threlfall, 2009). S. typhimurium and other non-typhoidal serotypes are food borne pathogens that cause self-limited gastroenteritis with periodic outbreaks from contaminated centralized food processing sites. The estimated burden of non-typhoidal Salmonellosis in the United States is 1.4 million cases annually (Voetsch et al., 2004). Invasive infections with non-typhoidal Salmonella have begun to emerge as the leading cause of community acquired bacteremia in sub-Saharan Africa and are associated with 21–47% mortality despite appropriate antibiotic therapy (Gordon et al., 2008; Kingsley et al., 2009). These isolates are predominantly S. typhimurium strains and are typically resistant to multiple antibiotics.
Type III Secretion
Salmonellae manipulate host cellular physiology during infections by using type III secretion systems (T3SS). T3SS are very common virulence factors used by a wide array of Gram-negative pathogens, which function by facilitating the translocation of effector proteins into the cytosol of host cells (Hueck, 1998). Changing the complement of effectors that are translocated allows different bacteria to exert control over distinct host signaling pathways. For example, Salmonellae encode two virulence-associated T3SS within two distinct Salmonella pathogenicity islands (SPI), each resulting in the transfer of a distinct set of effectors (Ibarra and Steele-Mortimer, 2009). The SPI1 T3SS is expressed primarily in the gut lumen, and promotes epithelial cell invasion. It is critical for the induction of gastroenteritis. The SPI2 T3SS is expressed by bacteria within the vacuolar compartment of macrophages and epithelial cells. It is important for intracellular replication and causing systemic disease.
Innate Immune Sensors
Innate immune system sensors can be classified by the cellular site of detection. Extracellular detectors, such as the Toll-like receptors (TLR) and C type Lectin receptors (CLR), respond to microbe associated molecular patterns (MAMPs) in the extracellular or vacuolar space. They tend to respond to molecules that are broadly conserved, such as LPS (TLR4) or flagellin (TLR5) and induce pro-inflammatory gene expression. Cytosolic detectors such as the Nod-like receptors (NLR), Rig-I-like receptors (RLR), and Aim2-like receptors (ALR) detect MAMPs in the cytosolic compartment or cytosolic perturbations caused by extracellular agonists. They induce either transcriptional or post-translational responses. Some of these sensors can respond to virulence properties of pathogens either directly or indirectly. For example, NLRC4 detects T3SS activity by sensing the inadvertent translocation of both flagellin and the T3SS rod protein into the cytosol of host cells (Miao et al., 2007; Miao and Warren, 2010). Interestingly, while flagellin in the extracellular space is a marker for any flagellated bacterium present within host tissues, in the cytosol it is instead a marker of a flagellated bacterium that possesses virulence factors that allow access to the cytosolic compartment. From the host’s perspective, the threat posed by these two bacteria is quite different.
Toll-like receptors induce the expression of multiple cytokines and chemokines. While most of these factors are secreted upon stimulation, pro-IL-1β and pro-IL-18 are held in reserve, awaiting a second signal. Some cytosolic receptors in the NLR and ALR families form inflammasomes, multi-protein complexes which serve as platforms for Caspase-1 activation. Activated Caspase-1 cleaves pro-IL-1β and pro-IL-18 to their mature secreted forms. Caspase-1 activation also induces a form of programmed cell death called pyroptosis (Bergsbaken et al., 2009).
Innate Immune Sensors Detecting S. typhimurium
Several sensors can detect S. typhimurium infection. Both TLR4 and TLR5 play a role in the host response to S. typhimurium (Figure 1). Tlr4−/− mice show increased susceptibility to S. typhimurium infection, and Tlr4-Tlr5DKO mice show an even more severe phenotype (Feuillet et al., 2006). S. typhimurium is also detected by NLRs, specifically NLRC4 (previously called Ipaf) and NLRP3 (previously called Nalp3 or cryopyrin), which also contribute to a reduced bacterial burden in the mouse model (Broz et al., 2010).
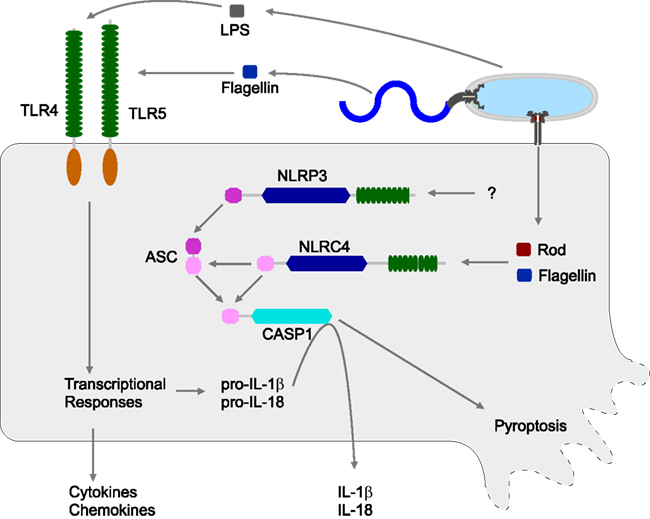
Figure 1. Innate immune detection of S. typhimurium. S. typhimurium can be detected by both extracellular (TLR) and cytosolic (NLR) sensors. TLR4 responds to lipopolysaccharide (LPS), while TLR5 responds to extracellular flagellin monomers. TLRs induce transcriptional responses, including the synthesis and secretion of multiple cytokines and chemokines. TLRs also induce the synthesis of pro-IL-1β and pro-IL-18, but these are reserved in the cytosol awaiting processing by Caspase-1. Caspase-1 activity is controlled by inflammasomes. S. typhimurium is detected by two inflammasomes, NLRC4 and NLRP3. NLRC4 responds to cytosolic flagellin monomers or T3SS rod protein monomers, both delivered by the activity of a virulence-associated T3SS. NLRP3 detects S. typhimurium via unknown mechanisms.
NLRC4 and NLRP3 both form inflammasomes and activate Caspase-1. Mice deficient in Caspase-1 have increased bacterial loads and succumb to S. typhimurium infection earlier than WT mice (Lara-Tejero et al., 2006; Raupach et al., 2006). Nlrp3-Nlrc4DKO mice have a phenotype similar to Casp1−/− mice (Broz et al., 2010; Figure 1).
Caspase-1 reduces bacterial loads after S. typhimurium infection largely via the activities of IL-18. Il18−/− and Casp1−/− mice show similar susceptibilities to S. typhimurium infection while IL-1β−/− mice have a much more subtle phenotype (Raupach et al., 2006). IL-18 has numerous effects on the immune response, including the transcriptional induction of pro-inflammatory genes and the potentiation of IFN-γ secretion by T-cells and NK cells, both of which play significant roles in host defense against S. typhimurium (Eckmann and Kagnoff, 2001).
NLRP3 Detection
NLRP3 detects S. typhimurium within macrophages at late time-points. It responds to a diverse set of agonists, including ATP, pore forming toxins, crystals, viruses, bacteria, and fungi (Schroder and Tschopp, 2010; Rajan et al., 2011). These agonists are thought to trigger convergent signaling events that involve lysosomal destabilization, electrolyte imbalances, or mitochondrial dysfunction via ROS production (Hornung and Latz, 2010; Schroder and Tschopp, 2010; Zhou et al., 2011). Whether these pathways converge on a single terminal signal or not is not known. The kinetics of detection of different NLRP3 agonists is variable: ATP and nigericin are detected within 1 h, crystals typically within 4 h, while bacteria, viruses, and fungi are detected only after 6 or more hours. These differences likely reflect the length of time required to trigger a cytosolic event that is sensed by NLRP3.
Several bacteria can trigger NLRP3 activation in vitro, including Listeria monocytogenes (Mariathasan et al., 2006; Warren et al., 2008; Kim et al., 2010; Wu et al., 2010), Staphylococcus aureus (Mariathasan et al., 2006; Craven et al., 2009; Munoz-Planillo et al., 2009; Shimada et al., 2010), Klebsiella pneumophila (Willingham et al., 2009), Porphyromonas gingivalis (Huang et al., 2009), Shigella flexneri (Willingham et al., 2007), Chlamydophila pneumoniae (He et al., 2010), Neisseria gonorrhoeae (Duncan et al., 2009), Mycobacterium tuberculosis (Koo et al., 2008), Yersinia pseudotuberculosis (Brodsky et al., 2010), and S. typhimurium (Broz et al., 2010). Although the terminal signaling events triggered by these pathogens are likely the same as those triggered by other NLRP3 agonists, the initial mechanisms are almost certainly divergent. For example Y. pseudotuberculosis triggers NLRP3 via the activity of T3SS effectors (Brodsky et al., 2010), but detection of S. typhimurium by NLRP3 does not require SPI1 or SPI2 T3SS (Broz et al., 2010). In contrast L. monocytogenes detection by NLRP3 requires lysis of the phagosome by listeriolysin O (Warren et al., 2008). S. aureus is also detected by NLRP3. Toxin-producing strains of S aureus are detected via the pore forming activities of their toxins (Craven et al., 2009; Munoz-Planillo et al., 2009) while non-toxin-producing strains are not detected, apparently evading NLRP3 detection by producing degradation resistant peptidoglycan structures. S. aureus oatA mutants that are defective in these cell wall modifications are detected via NLRP3 by a mechanism that involves peptidoglycan degradation in the phagosome (Shimada et al., 2010). Thus, even within a bacterial species different strains may be detected or may evade NLRP3.
NLRC4 Detection
NLRC4 detects the activity of T3SS and T4SS. It does so in part by responding to flagellin that is inadvertently translocated into the cytosolic compartment by T3/4SS (Figure 2; Molofsky et al., 2005, 2006; Amer et al., 2006; Franchi et al., 2006; Miao et al., 2006; Ren et al., 2006). T3SS efficiently select translocation substrates from the bacterial cytosol for injection into host cells. Because flagellin is targeted to the flagellar T3SS apparatus for secretion to form the flagellar filament (Minamino et al., 2008), it has features that likely predispose mistargeting to the virulence-associated T3SS. Specifically, it binds a secretion chaperone and has the physical properties required for export in an unfolded state through a T3SS secretion apparatus. The chaperone is required for efficient delivery and subsequent detection by NLRC4 (unpublished data). The mechanisms underlying accidental transfer by T4SS are less clear. As a cytosolic flagellin sensor, NLRC4 also detects flagellated bacteria that escape from the vacuolar compartment into the cytosol, such as Listeria monocytogenes (Warren et al., 2008).
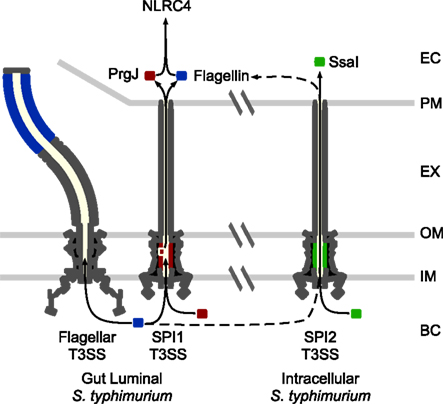
Figure 2. NLRC4 detection of S. typhimurium. There are two major phases of S. typhimurium growth, extracellular in the gut lumen (modeled on the left), and intracellular within macrophages and epithelial cells (modeled on the right). While SPI1 and flagella are expressed in the gut lumen, SPI2 is expressed in the intracellular compartment. Inadvertent translocation of flagellin (blue) or the SPI1 T3SS rod protein, PrgJ (red), is detected by NLRC4, while the SPI2 rod, SsaI (green), is not detected. NLRC4 can detect intracellular adapted S. typhimurium via flagellin transfer through the SPI2 T3SS, albeit much less efficiently than gut adapted bacteria. EC: eukaryotic cytosol. PM: eukaryotic cell plasma membrane. EX: extracellular space. OM: bacterial outer membrane. IM: bacterial inner membranel. BC: bacterial cytosol.
NLRC4 can also detect T3SS activity in non-flagellated bacteria (Miao et al., 2006; Sutterwala et al., 2007; Suzuki et al., 2007), responding to inadvertent translocation of the T3SS rod protein (Miao and Warren, 2010; Miao et al., 2010b; Figure 2). The rod component of the T3SS apparatus forms the inner channel that spans the bacterial periplasm. Similar to flagellin, it is believed to polymerize into a hollow tube that permits the passage of secreted proteins (Marlovits et al., 2004), and there is sequence conservation at the carboxy-terminus of flagellin and the rod protein (Miao et al., 2010b). If rod proteins are inadvertently translocated into the macrophage cytosol, they are detected by NLRC4 (Miao et al., 2010b). This may occur if the T3SS exports excess rod monomers from the bacterial cytosol, or if rod monomers slough into the interior channel of the needle apparatus, resulting in translocation. In their correct location within the T3SS apparatus, rod proteins are not detected.
Macrophages detect S. typhimurium SPI1 T3SS via inadvertent translocation of flagellin and the SPI1 rod protein PrgJ. Because flagellin is one of the most highly produced proteins in the bacteria during growth in LB, which is thought to mimic the gut lumen, it is the dominant NLRC4 signal in vitro. Both flagellin and SPI1 are highly induced in the late logarithmic phase of growth in LB, and these bacteria are readily detected within 1 h by macrophages in vitro. However, if bacteria are grown to stationary phase, they no longer express SPI1 and NLRC4 does not detect S. typhimurium within the first several hours (Miao et al., 2010b). Thus the transcriptional profile of the bacterium is critical for detection by NLRC4.
Evasion of NLRC4
Salmonella typhimurium have developed evasion strategies to prevent NLRC4 detection during intracellular replication in macrophages. Flagellin is repressed in the intracellular environment when SPI2 T3SS is active. In addition, the SPI2 T3SS rod protein, SsaI, has amino acid changes that prevent its detection by NLRC4 (Miao et al., 2010b; Figure 2). Elimination of these two evasion strategies by expressing PrgJ or flagellin from a SPI2 co-regulated promoter results in strong and persistent detection via NLRC4 and complete clearance of the bacteria in vivo, demonstrating that evasion of NLRC4 is essential for S. typhimurium virulence (Miao et al., 2010a,b).
These evasion strategies appear to be efficient, but not perfect. NLRC4 can detect S. typhimurium that are grown under SPI1/flagellin non-expressing conditions (stationary phase LB), but only at very late times post-infection (Broz et al., 2010). After infection of macrophages in vitro, NLRC4 does not detect the bacteria during the early hours after infection. However, between 8- and 17-h post-infection, NLRC4 does detect S. typhimurium apparently via SPI2 T3SS translocation of flagellin (Broz et al., 2010; we have verified these results). Whether the detected flagellin arises from transcriptional re-induction at late time-points after infection of macrophages, or is due to residual protein left over from the prior growth in LB remains to be determined. In either case, this detection is sufficient to provide a benefit to the host in vivo, as NLRC4 dependent Caspase-1 activation results in reduced bacterial burden in infected mice, and a delay in the time of death (Broz et al., 2010). SPI2 detection is significantly delayed compared to SPI1 detection, and the amount of IL-1β secreted is considerably lower.
Thus, there is a fine interplay between detection and evasion of NLRC4 during S. typhimurium infection. NLRC4 does detect S. typhimurium in vivo, providing a benefit to the host. S. typhimurium expresses flagellin in the gut lumen, and represses it in the intracellular environment. We propose that NLRC4 only detects S. typhimurium which have recently emigrated from the gut to deeper tissues because these bacteria have residual flagellin present within their cytosol. Indeed, GFP expressed under the control of the fliC promoter is detectable in bacteria recovered from the Peyer’s patches, but not from the draining lymph nodes or spleen (Cummings et al., 2006). Once the bacteria reprogram their gene expression for the intracellular environment, perhaps after one or more rounds of replication within host cells, we hypothesize that they subsequently evade NLRC4 completely by degrading residual flagellin in the bacterial cytosol and repressing its transcription.
Pyroptosis is an Effective Innate Immune Effector Mechanism in vivo
Caspase-1 activation results in both cytokine processing, and a form of cell death called pyroptosis (Bergsbaken et al., 2009), which is associated with stronger Caspase-1 activation than is required for cytokine processing. Like apoptosis, pyroptosis is a form of programmed cell death (Labbe and Saleh, 2008; Duprez et al., 2009). Both pyroptosis and apoptosis require the activation of specific signaling pathways triggered downstream of different caspase family proteases. This requirement contrasts with necrotic cell death (also called oncosis), which is an accidental form of cell death that does not have an absolute requirement for cellular signaling pathways. Pyroptosis, like necrosis, results in the lysis of the affected cell, releasing cytosolic contents after disruption of the plasma membrane, an inherently inflammatory event. In contrast, apoptotic blebs are cleared in an orderly fashion and are non-inflammatory manner (Labbe and Saleh, 2008; Duprez et al., 2009). Pyroptosis occurs rapidly, within 30–60 min in vitro and to date has only been characterized in macrophages and dendritic cells.
Pyroptosis was first observed in 1992 in vitro (Zychlinsky et al., 1992). Although there was some evidence that it occurred in vivo (Bergsbaken et al., 2009), the relevance of pyroptosis in vivo was largely undefined until recently. As mentioned above, we generated a S. typhimurium strain that persistently expresses flagellin (FliCON; Miao et al., 2010a). This strain was effectively detected and cleared by NLRC4 dependent Caspase-1 activation. Given that IL-18 secretion is the effector mechanism by which Caspase-1 clears WT S. typhimurium (Raupach et al., 2006), we expected that IL-18 would be required to clear FliCONS. typhimurium. However, mice deficient for both IL-1β and IL-18 retained their ability to clear these bacteria. Instead, pyroptosis released FliCONS. typhimurium into the extracellular space, where they are phagocytosed and killed by neutrophils (Miao et al., 2010a). We obtained similar results for L. monocytogenes strains engineered to persistently express and secrete flagellin or the S. typhimurium PrgJ rod protein; clearance of these strains could not be attributed to IL-1β and IL-18 secretion (unpublished data). Furthermore, after IP infection with Legionella pneumophila or Burkholderia thailandensis, Casp1−/− mice had increased bacterial burdens in the draining lymph nodes that could not be completely attributed to IL-1β or IL-18 secretion (Miao et al., 2010a). These results provide strong evidence that pyroptosis is an important innate immune effector mechanism against intracellular bacteria, including FliCONS. typhimurium.
Salmonella typhimurium Effectively Evade Pyroptosis, but Fail to Evade IL-18
Our work presents an apparent conundrum: both WT S. typhimurium and FliCONS. typhimurium are detected by NLRC4, but the former is affected by IL-18 while the latter is cleared through pyroptosis. We propose that this difference is attributable to two variables between these strains: the time to detection in a single infected cell, and the persistence of detection over the course of days.
At the single cell level under conditions that mimic systemic infection S. typhimurium does not trigger NLRC4 and NLRP3 until 17-h post-infection (under SPI1 repressing growth conditions; for example stationary phase LB cultures; Broz et al., 2010). In contrast, if FliCONS. typhimurium trigger NLRC4 within 6 h (Miao et al., 2010a). This is a significant difference. Under both conditions, Caspase-1 mediates cytokine secretion and pyroptosis. In the case of FliCONS. typhimurium, relatively early pyroptosis occurs before the bacteria can replicate in the macrophage, essentially short-circuiting the replicative cycle. Thus, FliCON are released and exposed to neutrophil killing before replication occurs. Some FliCON bacteria will also be taken up by macrophages, where they are again predicted to induce early pyroptosis before replication, exposing them to neutrophils before they can undergo a replicative cycle within the macrophage. This mechanism of clearance is so effective that any contribution of IL-1β and IL-18 is obscured in the mouse model. In contrast, WT S. typhimurium do not trigger NLRC4/NLRP3 until late times after infection of a single cell. At this time, pyroptosis should still occur, but only after the bacteria have replicated. Released bacteria are predicted to be exposed to neutrophils, but many will also infect new macrophages and continue the replicative cycle. Thus, we propose that WT S. typhimurium do not prevent pyroptosis from occurring, rather they void its effectiveness by delaying its onset.
Since S. typhimurium prevents the effectiveness of pyroptosis, one might be tempted to conclude that the role of pyroptosis in host defense against real world pathogens is negligible. However, we have evidence that during systemic infection with Legionella pneumophila or Burkholderia thailandensis, Caspase-1 reduces bacterial burdens independent of IL-1β and IL-18 (Miao et al., 2010a). These models require further research to accurately define the role of pyroptosis. Nevertheless, the fact that S. typhimurium actively constrains the effectiveness of pyroptosis attests to its potency in clearing intracellular pathogens that target macrophages or dendritic cells.
Unlike pyroptosis the effectiveness of IL-18 secretion is not avoided by WT S. typhimurium. In part, this may relate to the fact that pyroptosis is a cell intrinsic effector mechanism while IL-18 exerts its effects in a paracrine manner, inducing IFN-γ expression in NK cells and T cells. Therefore, although the effects of IL-18 can be delayed for several hours by delaying NLRC4/NLRP3 detection, it is likely that they cannot be circumvented, as is the case with pyroptosis.
Complex Interplay of Detection and Evasion
WT C57BL/6 mice succumb to S. typhimurium infection despite detection by TLR4, TLR5, NLRC4, and NLRP3, raising the question of the utility of these sensors to the host. However, it is crucial to note that this strain of mice is highly susceptible to S. typhimurium infection, at least in part due to a defect in Nramp1 (also called Slc11A1), a metal ion transporter that depletes the phagosome of Fe2+ and Mn2+ (Gruenheid et al., 1997; Cellier et al., 2007). Even so, infection in C57BL/6 mice remains a powerful model to study innate immunity and bacterial pathogenesis because of the availability of knockout mice. The extension of results in this mouse strain to human disease should be interpreted appropriately. In humans, it is likely that detection by TLR4, TLR5, NLRC4, and NLRP3 will result in reduced bacterial burdens during both gastroenteritis and systemic disease, and we would predict that this will result in decreased symptom severity and earlier resolution of the infection. Further, IL-1β and IL-18 are expected to promote protective adaptive responses to reinfection (Sims and Smith, 2010).
Future Directions
Salmonella typhimurium is an important pathogen, and understanding how it interacts with the immune system during infection has implications for human disease caused by it and many other pathogens. In addition to the importance of studying S. typhimurium in its own right, it remains a particularly useful tool for dissecting the innate immune system because of the powerful genetic tools available. Studies using S. typhimurium have revealed the mechanisms of NLRC4 activation and the utility of pyroptosis. One of the crucial paradigms to emerge from the study of S. typhimurium detection is the complex interplay between detection and evasion, wherein innate immune detectors are able to respond to pathogens, but pathogens have evolved strategies that prevent maximal responses. Future studies examining the intricacies of detection and evasion of NLRC4 and NLRP3 will undoubtedly lead to novel insights into the mechanisms by which these sensors respond to S. typhimurium as well as many other bacterial pathogens.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Amer, A., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Ozoren, N., Brady, G., Meshinchi, S., Jagirdar, R., Gewirtz, A., Akira, S., and Nunez, G. (2006). Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223.
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109.
Bhutta, Z. A., and Threlfall, J. (2009). Addressing the global disease burden of typhoid fever. JAMA 302, 898–899.
Brodsky, I. E., Palm, N. W., Sadanand, S., Ryndak, M. B., Sutterwala, F. S., Flavell, R. A., Bliska, J. B., and Medzhitov, R. (2010). A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387.
Broz, P., Newton, K., Lamkanfi, M., Mariathasan, S., Dixit, V. M., and Monack, D. M. (2010). Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755.
Cellier, M. F., Courville, P., and Campion, C. (2007). Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect. 9, 1662–1670.
Craven, R. R., Gao, X., Allen, I. C., Gris, D., Bubeck Wardenburg, J., McElvania-Tekippe, E., Ting, J. P., and Duncan, J. A. (2009). Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 4, e7446. doi: 10.1371/journal.pone.0007446
Cummings, L. A., Wilkerson, W. D., Bergsbaken, T., and Cookson, B. T. (2006). In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol. Microbiol. 61, 795–809.
Duncan, J. A., Gao, X., Huang, M. T., O’Connor, B. P., Thomas, C. E., Willingham, S. B., Bergstralh, D. T., Jarvis, G. A., Sparling, P. F., and Ting, J. P. (2009). Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol. 182, 6460–6469.
Duprez, L., Wirawan, E., Vanden, T., Berghe, and Vandenabeele, P. (2009). Major cell death pathways at a glance. Microbes Infect. 11, 1050–1062.
Eckmann, L., and Kagnoff, M. F. (2001). Cytokines in host defense against Salmonella. Microbes Infect. 3, 1191–1200.
Feuillet, V., Medjane, S., Mondor, I., Demaria, O., Pagni, P. P., Galan, J. E., Flavell, R. A., and Alexopoulou, L. (2006). Involvement of toll-like receptor 5 in the recognition of flagellated bacteria. Proc. Natl. Acad. Sci. U.S.A. 103, 12487–12492.
Franchi, L., Amer, A., Body-Malapel, M., Kanneganti, T. D., Ozoren, N., Jagirdar, R., Inohara, N., Vandenabeele, P., Bertin, J., Coyle, A., Grant, E. P., and Nunez, G. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582.
Gordon, M. A., Graham, S. M., Walsh, A. L., Wilson, L., Phiri, A., Molyneux, E., Zijlstra, E. E., Heyderman, R. S., Hart, C. A., and Molyneux, M. E. (2008). Epidemics of invasive Salmonella enterica serovar Enteritidis and S. enterica serovar Typhimurium infection associated with multidrug resistance among adults and children in Malawi. Clin. Infect. Dis. 46, 963–969.
Gruenheid, S., Pinner, E., Desjardins, M., and Gros, P. (1997). Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J. Exp. Med. 185, 717–730.
He, X., Mekasha, S., Mavrogiorgos, N., Fitzgerald, K. A., Lien, E., and Ingalls, R. R. (2010). Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. J. Immunol. 184, 5743–5754.
Hornung, V., and Latz, E. (2010). Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 40, 620–623.
Huang, M. T., Taxman, D. J., Holley-Guthrie, E. A., Moore, C. B., Willingham, S. B., Madden, V., Parsons, R. K., Featherstone, G. L., Arnold, R. R., O’Connor, B. P., and Ting, J. P. (2009). Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J. Immunol. 182, 2395–2404.
Hueck, C. J. (1998). Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62, 379–433.
Ibarra, J. A., and Steele-Mortimer, O. (2009). Salmonella – the ultimate insider. Salmonella virulence factors that modulate intracellular survival. Cell. Microbiol. 11, 1579–1586.
Kim, S., Bauernfeind, F., Ablasser, A., Hartmann, G., Fitzgerald, K. A., Latz, E., and Hornung, V. (2010). Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 40, 1545–1551.
Kingsley, R. A., Msefula, C. L., Thomson, N. R., Kariuki, S., Holt, K. E., Gordon, M. A., Harris, D., Clarke, L., Whitehead, S., Sangal, V., Marsh, K., Achtman, M., Molyneux, M. E., Cormican, M., Parkhill, J., MacLennan, C. A., Heyderman, R. S., and Dougan, G. (2009). Epidemic multiple drug resistant Salmonella typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 19, 2279–2287.
Koo, I. C., Wang, C., Raghavan, S., Morisaki, J. H., Cox, J. S., and Brown, E. J. (2008). ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell. Microbiol. 10, 1866–1878.
Labbe, K., and Saleh, M. (2008). Cell death in the host response to infection. Cell Death Differ. 15, 1339–1349.
Lara-Tejero, M., Sutterwala, F. S., Ogura, Y., Grant, E. P., Bertin, J., Coyle, A. J., Flavell, R. A., and Galan, J. E. (2006). Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 203, 1407–1412.
Mariathasan, S., Weiss, D. S., Newton, K., McBride, J., O’Rourke, K., Roose-Girma, M., Lee, W. P., Weinrauch, Y., Monack, D. M., and Dixit, V. M. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232.
Marlovits, T. C., Kubori, T., Sukhan, A., Thomas, D. R., Galan, J. E., and Unger, V. M. (2004). Structural insights into the assembly of the type III secretion needle complex. Science 306, 1040–1042.
Miao, E. A., Alpuche-Aranda, C. M., Dors, M., Clark, A. E., Bader, M. W., Miller, S. I., and Aderem, A. (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575.
Miao, E. A., Andersen-Nissen, E., Warren, S. E., and Aderem, A. (2007). TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin. Immunopathol. 29, 275–288.
Miao, E. A., Leaf, I. A., Treuting, P. M., Mao, D. P., Dors, M., Sarkar, A., Warren, S. E., Wewers, M. D., and Aderem, A. (2010a). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142.
Miao, E. A., Mao, D. P., Yudkovsky, N., Bonneau, R., Lorang, C. G., Warren, S. E., Leaf, I. A., and Aderem, A. (2010b). Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 107, 3076–3080.
Miao, E. A., and Warren, S. E. (2010). Innate immune detection of bacterial virulence factors via the NLRC4 inflammasome. J. Clin. Immunol. 30, 502–506.
Minamino, T., Imada, K., and Namba, K. (2008). Mechanisms of type III protein export for bacterial flagellar assembly. Mol. Biosyst. 4, 1105–1115.
Molofsky, A. B., Byrne, B. G., Whitfield, N. N., Madigan, C. A., Fuse, E. T., Tateda, K., and Swanson, M. S. (2006). Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203, 1093–1104.
Molofsky, A. B., Shetron-Rama, L. M., and Swanson, M. S. (2005). Components of the Legionella pneumophila flagellar regulon contribute to multiple virulence traits, including lysosome avoidance and macrophage death. Infect. Immun. 73, 5720–5734.
Munoz-Planillo, R., Franchi, L., Miller, L. S., and Nunez, G. (2009). A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J. Immunol. 183, 3942–3948.
Parry, C. M., Hien, T. T., Dougan, G., White, N. J., and Farrar, J. J. (2002). Typhoid fever. N. Engl. J. Med. 347, 1770–1782.
Rajan, J. V., Rodriguez, D., Miao, E. A., and Aderem, A. (2011). The NLRP3 inflammasome detects EMCV and VSV infection. J. Virol. 85, 4167–4172.
Raupach, B., Peuschel, S. K., Monack, D. M., and Zychlinsky, A. (2006). Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 74, 4922–4926.
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F., and Vance, R. E. (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. doi: 10.1371/journal.ppat.0020018
Shimada, T., Park, B. G., Wolf, A. J., Brikos, C., Goodridge, H. S., Becker, C. A., Reyes, C. N., Miao, E. A., Aderem, A., Gotz, F., Liu, G. Y., and Underhill, D. M. (2010). Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 7, 38–49.
Sims, J. E., and Smith, D. E. (2010). The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10, 89–102.
Sutterwala, F. S., Mijares, L. A., Li, L., Ogura, Y., Kazmierczak, B. I., and Flavell, R. A. (2007). Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245.
Suzuki, T., Franchi, L., Toma, C., Ashida, H., Ogawa, M., Yoshikawa, Y., Mimuro, H., Inohara, N., Sasakawa, C., and Nunez, G. (2007). Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3, e111. doi: 10.1371/journal.ppat.0030111
Voetsch, A. C., Van, T. J., Gilder, Angulo, F. J., Farley, M. M., Shallow, S., Marcus, R., Cieslak, P. R., Deneen, V. C., and Tauxe, R. V. (2004). FoodNet estimate of the burden of illness caused by nontyphoidal Salmonella infections in the United States. Clin. Infect. Dis. 38(Suppl. 3), S127–S134.
Warren, S. E., Mao, D. P., Rodriguez, A. E., Miao, E. A., and Aderem, A. (2008). Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 180, 7558–7564.
Willingham, S. B., Allen, I. C., Bergstralh, D. T., Brickey, W. J., Huang, M. T., Taxman, D. J., Duncan, J. A., and Ting, J. P. (2009). NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J. Immunol. 183, 2008–2015.
Willingham, S. B., Bergstralh, D. T., O’Connor, W., Morrison, A. C., Taxman, D. J., Duncan, J. A., Barnoy, S., Venkatesan, M. M., Flavell, R. A., Deshmukh, M., Hoffman, H. M., and Ting, J. P. (2007). Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2, 147–159.
Wu, J., Fernandes-Alnemri, T., and Alnemri, E. S. (2010). Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 30, 693–702.
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225.
Keywords: Salmonella, Caspase-1, inflammasome, IL-1β, pyroptosis
Citation: Miao EA and Rajan JV (2011) Salmonella and caspase-1: a complex interplay of detection and evasion. Front. Microbio. 2:85. doi: 10.3389/fmicb.2011.00085
Received: 01 March 2011;
Accepted: 11 April 2011;
Published online: 25 April 2011.
Edited by:
John S. Gunn, The Ohio State University, USAReviewed by:
Maya Saleh, McGill University, CanadaRenee M. Tsolis, University of California-Davis, USA
Copyright: © 2011 Miao and Rajan. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Edward A. Miao,University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA. e-mail:ZWRtaWFvMUBnbWFpbC5jb20=