

- Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA, USA
Salmonella enterica serotypes are invasive enteric pathogens spread through fecal contamination of food and water sources, and represent a constant public health threat around the world. The symptoms associated with salmonellosis and typhoid disease are largely due to the host response to invading Salmonella, and to the mechanisms these bacteria employ to survive in the presence of, and invade through the intestinal mucosal epithelia. Surmounting this barrier is required for survival within the host, as well as for further dissemination throughout the body, and subsequent systemic disease. In this review, we highlight some of the major hurdles Salmonella must overcome upon encountering the intestinal mucosal epithelial barrier, and examine how these bacteria surmount and exploit host defense mechanisms.
Salmonella Biology
Salmonella enterica are Gram (−) bacteria responsible for causing typhoid disease and gastroenteritis. S. enterica serovar Typhi (S. Typhi) is the primary cause of typhoid fever, while non-typhoidal Salmonella (NTS) strains can cause Salmonella-induced food poisoning called salmonellosis. While young children, the elderly, and immuno-compromised individuals are most at risk for complications, people at any age are susceptible to the diarrhea, intestinal cramping, and intestinal epithelial erosion associated with salmonellosis. The disease is primarily spread by the contamination of water and food items with fecal matter from infected hosts and is often self-limiting, but can cause prolonged complications (Graham et al., 2000).
Salmonella possess Salmonella pathogenicity islands (SPI), or collections of pathogenesis-related genes acquired horizontally. At least 21 SPIs have been identified in S. Typhimurium and S. Typhi combined (for review of functions, see Sabbagh et al., 2010). In S. Typhimurium and in S. Typhi, SPI-1 and SPI-2 contain genes for two type-three secretion systems (T3SS). Specific to Gram (−) bacteria, the T3SS likely evolved from the flagella basal body, and is composed of a motor, needle complex, and translocon through which secreted effectors are injected into host cells (Stebbins and Galan, 2003). The effectors provide various functions, including promoting bacterial entry, controlling inflammatory responses, and regulating bacterial survival within the cell (summarized in Table 1). The SPI-1 T3SS (T3SS1) is primarily associated with invasion (Galan, 1996). Effectors secreted through the SPI-2 T3SS (T3SS2) seem to primarily promote the intracellular survival of Salmonella, although they may not be absolutely required for S. Typhi survival within human macrophages (Forest et al., 2010). However, in S. Typhi infections of humanized non-obese–diabetic mice, the loss of some SPI-2 genes caused a competitive disadvantage (Libby et al., 2010). Also, SPI-2 genes are up-regulated during S. Typhi infection of macrophages in vitro (Faucher et al., 2006). While the functions of SPI-1 and SPI-2 effectors have traditionally been considered distinct from each other, there is increasing evidence suggesting overlap in the times at which effectors from each system are required (Lawley et al., 2006; Brawn et al., 2007). These data highlight the intricate level of coordination between SPI-1 and SPI-2 involved with Salmonella pathogenesis. Nonetheless, recent data challenges the dependence of T3SS1 in Salmonella invasion (Radtke et al., 2010). SPI-1 mutant strains of Salmonella were capable of invading HT-29 3D intestinal cells to a higher degree than HT-29 monolayers, although the level of invasion was less than that of the wild-type strains. Additionally, SPI-1 was required for wild-type levels of intracellular replication over a 24-h period. The additional loss of SPI-2 may enhance these phenotypes. These data suggest that various pathways are required for wild-type levels of invasion and intracellular replication. Other types of secretion systems that may promote pathogenicity have been identified in additional SPI loci (Sabbagh et al., 2010).
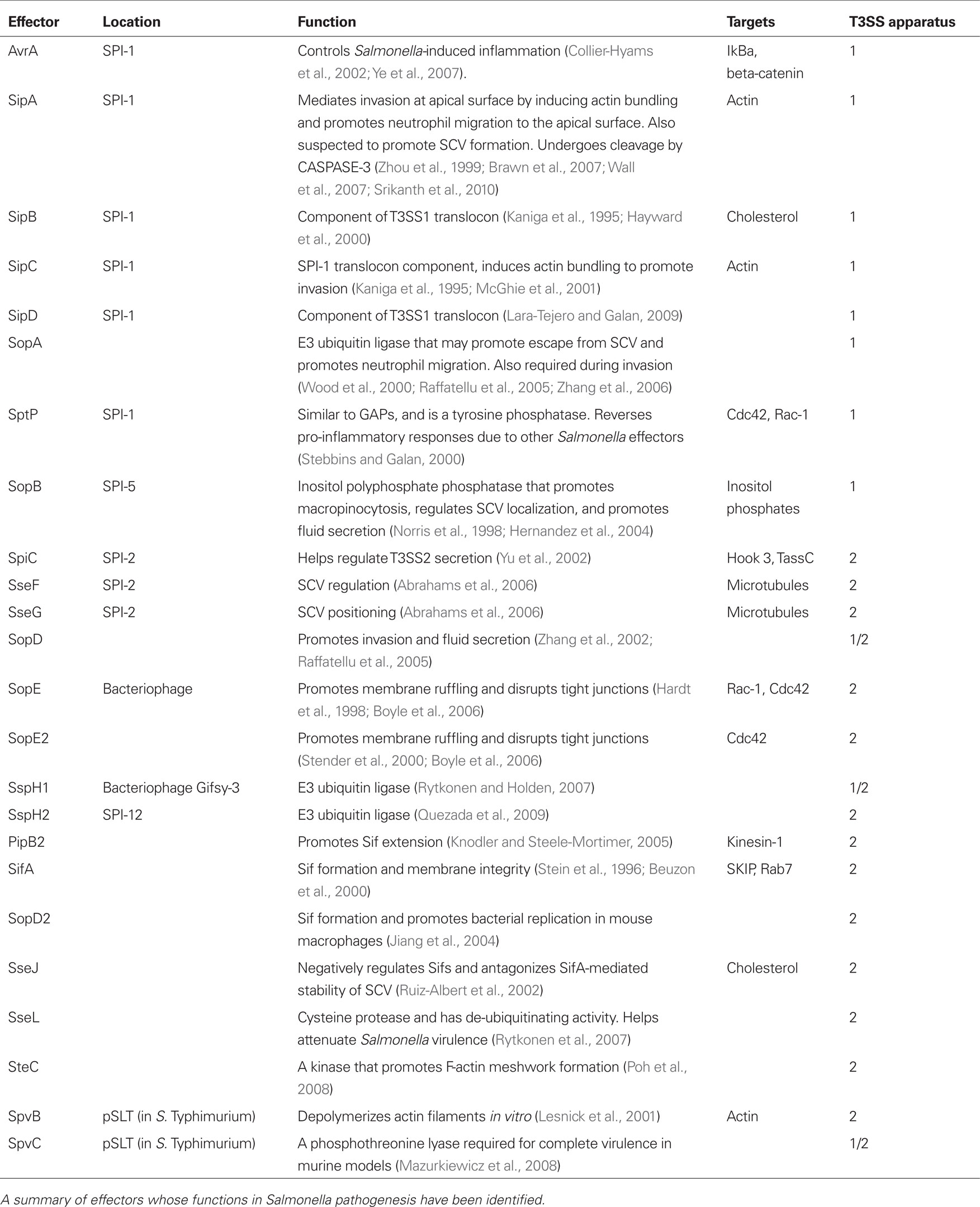
Table 1. Salmonella effectors and their roles in pathogenesis.
Salmonella Interaction with the Intestinal Mucosal Epithelia
The gastrointestinal tract is the largest mucosal surface in the human body, with the epithelial monolayer surface area alone measuring 400 m2 (MacDonald and Monteleone, 2005; Turner, 2009). Regulating the host microbiota, immune responses, and barrier functions is paramount to providing timely and controlled retaliation to pathogenic assaults, while simultaneously maintaining a healthy, balanced, intestinal environment (Figure 1).
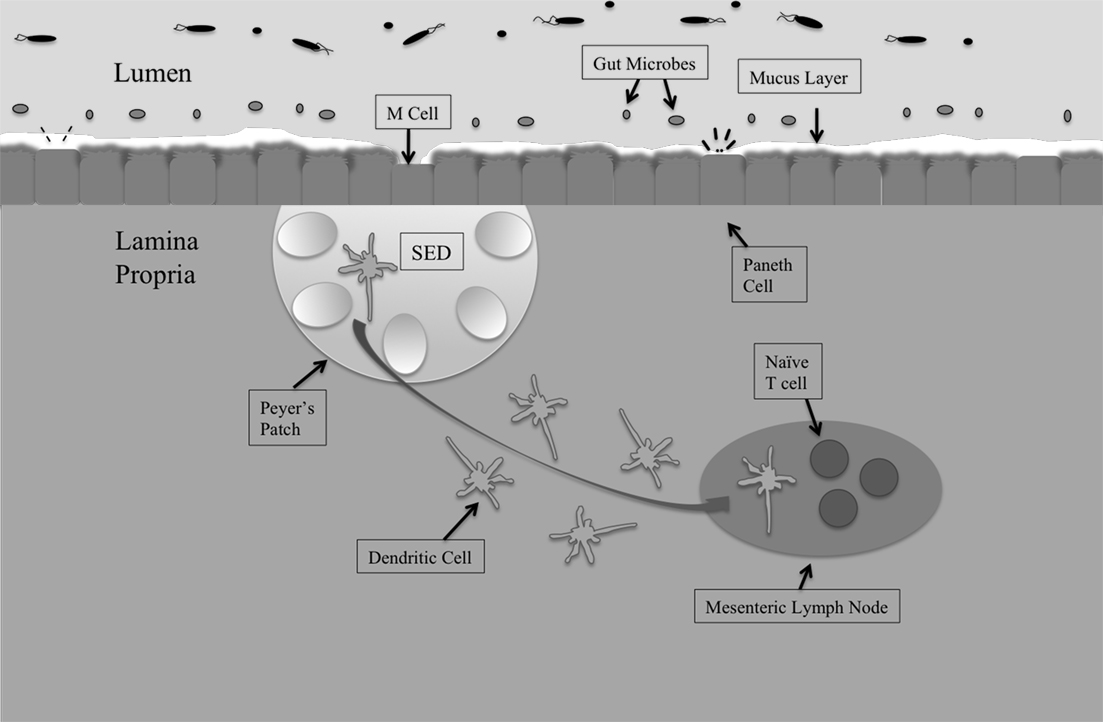
Figure 1. The intestinal mucosal epithelium is home to various interacting cell types that come together to maintain intestinal homeostasis and protect against invading pathogens. The first line of defense is the host microbiota, populations of commensal organisms that compete with invading pathogens for nutrients and space. The mucus layer protects against Salmonella invasion of epithelial cells, and the bacteria must adhere to mucus components in order to remain in the intestines. The epithelial monolayer underlying the mucus layer contains distinct cell types with different roles. M cells sample intestinal antigens and are the preferred route of entry by Salmonella. Underlying the M cells is the subepithelial dome (SED) that houses Peyer’s patches. Peyer’s patches contain germinal centers and have associated dendritic cells. Dendritic cells take whole bacteria to the mesenteric lymph node (MLN), from which Salmonella can escape to promote systemic disease.
Gut Microbiome
The human intestine is home to a plethora of mutualistic microorganisms, dominated mostly by Lactobacillus, Bacteroides, and Firmicutes (Backhed et al., 2005). Among other roles, these organisms assist immune system development and promote epithelial homeostasis (Rakoff-Nahoum et al., 2004; Mazmanian et al., 2005). Beyond these regulatory roles, the microbiome also functions as a critical barrier to invading pathogens. The population of host microbes in the gut physically blocks pathogen access to the epithelial layer, and also outcompetes pathogens for nutrients, thus reducing the survival and invasiveness of intestinal pathogens. Nevertheless, S. Typhimurium is capable of maneuvering through the microbiome to reach intestinal epithelial cells. In mouse experiments with wild-type and avirulent S. Typhimurium, Stecher et al. demonstrate that an inflammatory state in the intestines not only permits wild-type S. Typhimurium to outcompete the microbiome during colonization, but also allows a normally avirulent strain to colonize the intestines in the presence of intestinal microbiota (Stecher et al., 2007). These data unveil an intriguing concept that S. Typhimurium utilizes inflammation to outcompete host microbiota in mouse models of salmonellosis.
Host microbiota may also regulate the degree of S. Typhimurium shedding by infected mice. Following infection with 108 CFU of bacteria, mice were classified into three groups corresponding to the level of fecal shedding: supershedders (>108 CFU/g feces), moderate shedders (104–108 CFU/g feces), and low shedders (<104 CFU/g feces). Treating low shedders with streptomycin converted these mice to supershedders (Lawley et al., 2008), suggesting that the absence of host microbes may have permitted Salmonella to colonize more heavily, thus elevating the level of shed bacteria. The significance of a gut microbiota barrier to Salmonella infection was also explored by Croswell et al. (2009). The authors found that treatment of mice with streptomycin, streptomycin–bacitracin, or ampicillin–vancomycin–neomycin–metronidazole (AVNM) reduced gut microflora populations, and that each treatment was subsequently associated with an increase in Salmonella colonization and Salmonella-induced inflammation compared to untreated mice. The authors further show that allowing 3 weeks between antibiotic treatment and infection still predisposed mice to greater levels of inflammation in response to Salmonella infection than was seen in mice that were never treated with antibiotics. These results suggest that antibiotics have long-lasting effects on gut microflora populations, and that the presence and composition of gut microfloral populations may play an important role in controlling Salmonella colonization.
Additionally, Barman et al. (2008) showed that salmonellosis may alter the microfloral population in FvB mice. Infected mice showed disrupted numbers of Bacteroides, Lactobacillus/Enterococcus, and Eubacterium rectale/Clostridium coccoides groups and of the C. perfringens group compared to uninfected mice. However, Salmonella did not replace these populations, and wild-type population numbers returned after Salmonella clearance, suggesting any effect salmonellosis has on the intestinal microbiota is not permanent.
Mucosal Layer
Key to the mucosal epithelium is the formation of a mucus layer along the luminal lining of the gastrointestinal tract. Mucosal epithelial cells, specifically goblet cells, secrete glycosylated transmembrane proteins called mucins at the cell surface. These proteins generate a layer of large complexes containing thread-like structures and oligosaccharides. The resulting gelatinous layer blocks contact between the underlying epithelial monolayer and large particles, including bacteria. The mucus layer also contains trefoil factors, which are peptides produced in tissues containing mucus-producing cells, such as the intestinal epithelia. Their various proposed functions include limiting intestinal inflammation (Playford et al., 1996) and regulation of immune system responses (Baus-Loncar et al., 2005). Adhering to the mucosal layer would be necessary for Salmonella to avoid being washed out of the intestines. Indeed, there is evidence of Salmonella binding to mucus, although increased adherence does not necessarily correlate with increased mucosal penetration (Nevola et al., 1987; McCormick et al., 1988; Vimal et al., 2000).
Epithelial Monolayer: M Cells and the Associated Peyer’s Patches
The epithelial monolayer mediates interactions between triggers of immune responses, and the gut-associated lymphoid tissue (GALT), where immune responses originate (MacDonald and Monteleone, 2005; Turner, 2009). A follicle-associated epithelial (FAE) layer of columnar epithelial cells blankets lymphoid tissue within the intestinal wall. The intestinal mucosa contains a variety of cell types with unique functions including enterocytes, entero-endocrine cells, goblet cells, Paneth cells, and microfold (M) cells (Table 2). While all the unique epithelial cells found within the intestine are important for establishing the functional intestinal mucosal epithelium, in mice the M cells are a key route of invasion by Salmonella, and are thus discussed in greater detail here.
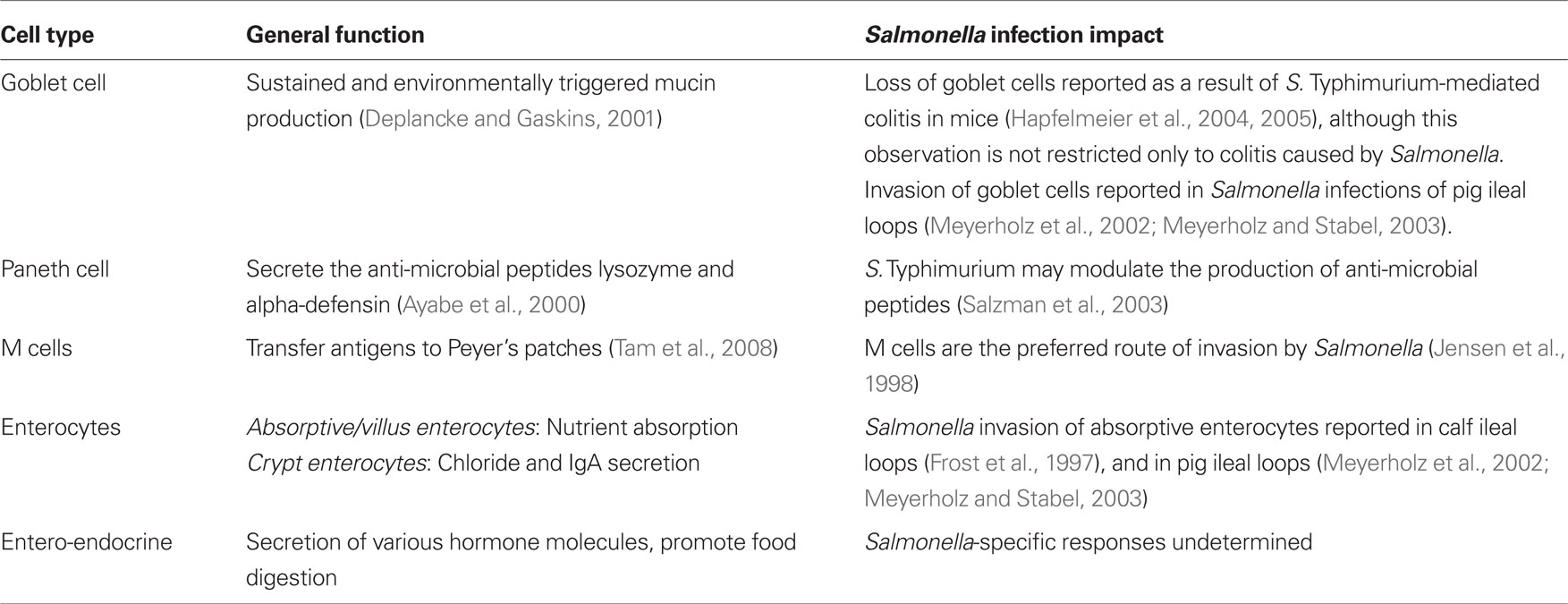
Table 2. Cells within the intestinal epithelial monolayer.
Microfold cells are specialized intestinal epithelial cells that are found in the FAE overlying mucosa-associated lymphoid tissue (MALT). Their primary role is to sample mucosal contents and transfer antigens from the lumen to underlying Peyer’s patches (Tam et al., 2008), and thus act as sentinels of the intestinal epithelium. The overall structure of M cells differs in various ways from that of enterocytes. For example, the apical (mucosal) surface of M cells is not covered by the mucus layer observed over other cells in the intestines (Frey et al., 1996). Additionally, the apical brush border characteristic of enterocytes is absent from M cells, which instead contain microfolds. These features promote the sampling role of M cells, but also inadvertently provide opportunities for enteropathogen docking and invasion.
An additional trait of M cells that promotes both their role as intestinal sentinels and promotes enteropathogen invasion is their use of a transcytotic pathway to shuttle luminal contents to lymphoid tissue (Kraehenbuhl and Neutra, 2000). Following endocytosis of extra-cellular material (including invading pathogens), M cells transfer the endosomal contents to the basolateral (serosal) surface and to the underlying MALT. This action results in appropriate immune responses to infection. Likewise, bacteria engulfed at the apical surface can escape the M cell through this same pathway and disseminate to other areas.
M cells provide a key route of invasion by S. Typhimurium (Jensen et al., 1998). SPI-1 genes help regulate Salmonella invasion through M cells, although SPI-1-independent processes are also proposed to regulate M cell invasion (Clark et al., 1996). For example, Salmonella defective for invA are less able to invade M cells (Clark et al., 1998), but SPI-1 mutants are still capable of invading M cells (Martinez-Argudo and Jepson, 2008). These results suggest that while SPI-1 genes are needed for wild-type levels of invasion, SPI-1-independent mechanisms are also important for Salmonella invasion through M cells.
The lymphoid tissue underlying M cells contains Peyer’s patches, which are large aggregates of B lymphocyte follicles that contain germinal centers, and can be found along the length of the intestine. Here, the antigens translocated by M cells are processed by local dendritic cells. Lysozyme-producing dendritic cells associated with Peyer’s patches mediate uptake of S. Typhimurium (LeLouard et al., 2010). Peyer’s patches also play a role in up-regulating intestinal IgA as a result of Salmonella infection (Hashizume et al., 2008).
Epithelial Monolayer: Structure and Salmonella Entry
The barrier function of the epithelial monolayer results from a variety of proteins that maintain close intercellular interactions. Desmosomes are composed of cadherins, which form an adhesive interface (Koch and Franke, 1994; Green and Simpson, 2007), and varying structural proteins, notably plakoglobin and desmoplakin (Hatsell and Cowin, 2001), all of which help disperse forces from physical stress. Meanwhile, the paracellular pathway, or area between adjacent epithelial cells, is kept closed by tight junctions and adherens junctions. Bacteria attempting to penetrate the epithelial monolayer must possess mechanisms for overcoming these strong interactions. As such, Salmonella possess several effectors that mediate disruption of the epithelial barrier and subsequent uptake into the non-phagocytic epithelial cells lining the intestinal lumen. These actions are carried out largely through the alteration of junctional protein localization and of Rho GTPase activity. The latter mechanism is especially important in the Salmonella-mediated modulation of tight junctions. Dis-regulating the Rho-GTPases leads to changes in organization of junctions and of actin at the cell membrane, which subsequently alters the membrane morphology in a manner referred to as “ruffling” (Finlay et al., 1991), and promotes Salmonella entry (Figure 2).
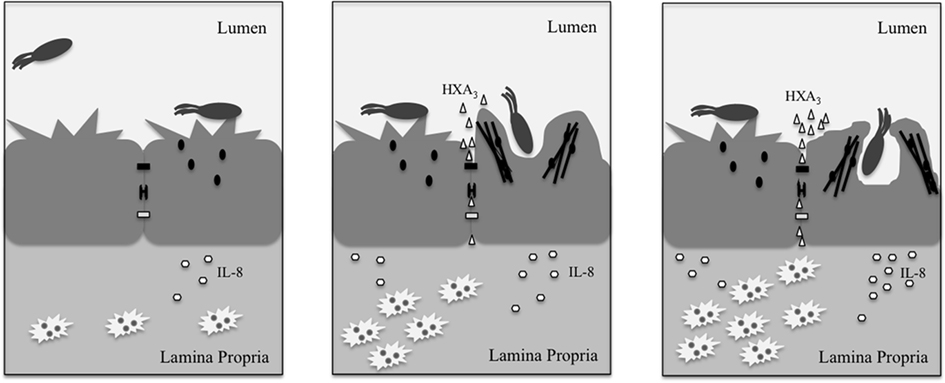
Figure 2. Upon interacting with the epithelial cell, Salmonella secrete effectors (small black ovals) that promote release of the neutrophil chemoattractant, HXA3 (small triangles), and membrane ruffling as a result of actin (black lines) re-organization. The release of HXA3 from the apical (mucosal) surface creates a concentration gradient across tight junctions (small black rectangle), desmosomes (small black bracket), and adherens junctions (small white rectangle) and through the paracellular space. Neutrophils (jagged white ovals) are recruited via basolateral secretion of IL-8, and subsequently migrate from the basolateral surface to the apical surface to the point of infection due to the HXA3 gradient. The actin re-organization alters the morphology of the cell membrane at the apical surface in a manner that promotes Salmonella uptake via macropinocytosis. Once inside the cell, Salmonella reside within a compartment termed the SCV that forms as a result of macropinocytosis.
Interactions between classical (Ca2+ dependent) cadherins and catenin family proteins are the foundation of adherens junctions. E-cadherin (epithelial cadherin) is a single pass transmembrane protein and its ability to homodimerize with E-cadherins on neighboring cells makes it a key component of adherens junctions (Takeichi, 1991). E-cadherins form small, intercellular clusters that quickly associate with actin and, overtime, expand into larger bundles that strengthen cell–cell adhesion (Adams et al., 1998). p120-catenin stabilizes E-cadherin at the cell surface, and its loss induces down-regulation of E-cadherin (Davis et al., 2003). Further, beta-catenin, a transcription factor involved in cell proliferation and differentiation, can bind to the cytoplasmic domain of E-cadherin (Hartsock and Nelson, 2008), and direct the localization of E-cadherin from the ER to the plasma membrane (Chen et al., 1999). Phosphorylation of beta-catenin promotes its ubiquitination and subsequent degradation (Aberle et al., 1997). Duan et al. (2007) demonstrated that cultured epithelial cells infected with S. Typhimurium display increased phosphorylated beta-catenin. This increase in phosphorylated beta-catenin resulting from Salmonella infection could increase the level of beta-catenin degradation, and thus could limit E-cadherin translocation to the plasma membrane. Such an action would result in weakened adherens junctions and promote Salmonella invasion through the epithelial monolayer.
The tight junction is an important regulator of epithelial monolayer permeability (Martinez-Palomo and Erlij, 1975), and of cell polarity by preventing mixing of apical (mucosal) and basolateral (serosal) components. The “leak pathway” permits passage of larger solutes, such as bacterial peptides (but not whole bacteria), while the “small pore pathway” excludes solutes larger than 4 Å, and exhibits some charge selectivity (Forster, 2008; Turner, 2009). The core of tight junction complexes is composed of occludin, ZO, and claudin proteins. Occludin binds to several crucial tight junction proteins including ZO-1 (Furuse et al., 1994), ZO-2 (Itoh et al., 1999), and ZO-3 (Haskins et al., 1998) and its activity is regulated by PKC-mediated phosphorylation (Andreeva et al., 2001). ZO-1, ZO-2, and ZO-3 are members of the membrane-associated guanylate kinase (MAGUK) family (Gonzalez-Mariscal et al., 2000). ZO-1 and ZO-2 assist in the polymerization of claudins, which permits extension of tight junctions, and also recruit ZO-3 to the tight junction (Umeda et al., 2006; Tsukita et al., 2009). A specific role for ZO-3 has yet to be determined, although it is shown to not be required for tight junction formation (Adachi et al., 2006). Claudins, which are tetraspan transmembrane proteins, are key structural elements, as they recruit occludin (Furuse et al., 1998). Another class of tight junction proteins include junction-associated adhesion molecules (JAMs), or which are integral membrane proteins that belong to the immunoglobin superfamily.
Cytoskeletal regulators of tight junctions include myosin ATPase, AMP-activated protein kinases, and especially Rho-GTPases (Turner, 2009). Rho-GTPases regulate various cell functions and are activated by guanine-nucleotide exchange factors (GEFs), which facilitate the active, GTP-bound state. GTPase activating protein (GAPs) inhibit Rho-GTPases by activating GTP hydrolysis, thereby inducing the inactive, GDP-bound state (Etienne-Manneville and Hall, 2002). Rho-GTPases are regulated by several proteins, the best-studied being Rho, Cdc42, and Rac. Rho is required for formation of focal adhesions and stress fibers, while constitutive Rac causes formation of plasma membrane extensions (Ridley and Hall, 1992; Ridley et al., 1992). Cdc42 activity promotes finger-like extrusions called filopodia (Nobes and Hall, 1995).
The tight junction is a key target of Salmonella, as infection with S. Typhimurium induces altered localization of ZO-2 and claudin-1, degradation of ZO-1, and promotes dephosphorylation of occludin in T84 cells (Kohler et al., 2007). Salmonella SPI-1 effectors SopB, SopE, SopE2, and SipA each play various roles in inducing ruffling and bacterial uptake by changing the localization and expression of ZO-1 and occludin, although SipA and SopB alone are not sufficient for this process (Boyle et al., 2006). These same effectors were also associated with altered epithelial cell polarity. Specifically, SopB (or SopB/SigD) is a phosphoinositide phosphatase that promotes formation of macropinosomes (Hernandez et al., 2004). This effector is capable of indirectly stimulating Cdc42-dependent cytoskeletal rearrangements (Zhou et al., 2001), which can lead to extension of the cell membrane around the bacterium. Similarly, SopE and SopE2 act like GEFs, and activate Cdc42 and Rac1 GTPases to induce cytoskeletal rearrangements that favor bacterial uptake (Stender et al., 2000). SipA and SipC associate with actin and promote bundling of actin filaments so as to facilitate macropinocytosis of Salmonella (McGhie et al., 2001).
As control mechanisms, Salmonella also secrete SptP and AvrA. SptP acts like a GAP, and essentially reverses the cytoskeletal rearrangements that occur during Salmonella uptake (Fu and Galan, 1999). AvrA, which stabilizes tight junctions (Liao et al., 2008) and inhibits NF-kB activation (Collier-Hyams et al., 2002), keeps the Salmonella-induced inflammation response in check.
Immune Response
PAMP Receptors
Intestinal epithelial cells can recognize invading pathogens by capturing common viral or bacterial components, known as pathogen-associated molecular patterns (PAMPs). Toll-like receptors (TLRs) are a well-studied family of PAMP receptors. Several types of intestinal epithelial cells have been shown to express TLRs (Elphick and Mahida, 2005; Tyrer et al., 2006; Palazzo et al., 2007; Gribar et al., 2008), and each type of receptor recognizes a unique PAMP. TLR4 recognizes lipopolysaccharide (LPS) from Gram (−) bacterial cell walls, TLR2 in concert with TLR1 or TLR6 recognizes triacyl or diacyl bacterial lipopeptide, respectively, and TLR5 recognizes flagellin protein from bacterial flagella. The binding of a TLR to its cognate antigen triggers activation of a signaling cascade that ultimately, via NF-kB activation, induces production of pro-inflammatory cytokines. TLR5-flagellin interaction during S. Typhimurium in mouse infections was shown to regulate the early stages of Salmonella infection and immune response. Peyer’s patches and mesenteric lymph nodes (MLN) in TLR5-deficient mice accumulate more Salmonella than wild-type littermates, and aflagellate Salmonella accumulate more in Peyer’s patches and MLN of wild-type mice than do wild-type Salmonella (Fournier et al., 2009). As Salmonella encounter Peyer’s patches after traversing the epithelial monolayer, regulation at this stage would be key to preventing further bacterial dissemination.
NLRs, or nucleotide-binding and oligomerization domain (NOD)-like receptors, are another established family of PAMP receptors that recognize components of bacterial peptidoglycan. The NLR family includes Nod1 and Nod2, both of which are expressed intracellularly by intestinal epithelial cells (MacDonald and Monteleone, 2005). Nod1 recognizes – d-glutamyl-meso-diaminopimelic (DAP) acid, expressed predominantly by Gram (−) bacteria (Chamaillard et al., 2003), while Nod2 recognizes muramyl dipeptide (MDP), which is common to Gram (−) and Gram (+) bacteria (Girardin et al., 2003). Binding between Nod1 and Nod2 and their specific PAMPs is followed by association with the adaptor protein Rip2, which initiates production of NF-kB-dependent pro-inflammatory cytokines. A recent paper suggests the role of Rip2-mediated inflammatory responses is key during Salmonella infection only when SPI-2 effectors are present (Geddes et al., 2010). As SPI-2 effectors are associated predominantly with intracellular survival, Rip2, and thus Nod1 and Nod2, may be required for dampening the ability of Salmonella to survive after invading host cells.
Neutrophil Recruitment
The release of IL-8 resulting from pathogen-induced immune signaling stimulates the recruitment of neutrophils from blood vessels to the basolateral (serosal) surface (Figure 2). Neutrophils are phagocytic white blood cells that are among the first line of the innate immune defense in response to pathogens. Recruitment of neutrophils to the apical (mucosal) surface is mediated by release of hepoxilin A3 (HXA3), a potent neutrophil chemoattractant, and a metabolite of the arachidonic acid pathway (McCormick, 2007). SipA at the apical surface induces a lipid signal cascade that includes the activation of PKC, which causes the release of arachidonic acid from the plasma membrane (Wall et al., 2007). Arachidonic acid is then converted to HXA3 by 12-lipoxygenase activity. Release of HXA3 is facilitated by the ABC transporter, MRP2 (Pazos et al., 2008), and generates a concentration gradient across the tight junction and through the paracellular space. The recruitment of neutrophils superficially seems solely an act of host defense; however the rapid migration of these cells through the epithelial monolayer may actually loosen the epithelial cell–cell interactions, and thus create space through which Salmonella can invade, further facilitating PMN infiltration (Kohler et al., 2007). Indeed, PMN transmigration has been demonstrated to reduce epithelial monolayer resistance in vitro (Nash et al., 1987).
Salmonella Reside in the Salmonella Containing Vacuole
The Salmonella containing vacuole (SCV) is an intracellular vacuole that forms via macropinocytosis of Salmonella. The SopB effector helps direct the maturation of the SCV into a compartment suitable for bacterial survival and replication (Bakowski et al., 2008), and may function to steer the compartment away from the endocytic pathway (Hernandez et al., 2004). After formation of the SCV, many T3SS2 effectors assist in formation of Salmonella-induced filaments (Sifs), localization of the SCV, and intravacuolar replication.
SifA, SseF, SseG, and SopD2 are involved with Sif formation, which results in the extension of tubules outward from the SCV after fusion with late endosomal compartments (Brumell et al., 2001). SifA is required for Sif formation (Stein et al., 1996), as its absence precludes formation of any Sif structures. SseF and SseG are also required for proper Sif formation, as mutants lacking either of these effectors only form Sif-like structures that differ in composition from fully formed Sifs (Kuhle et al., 2004). SopD2 has recently been shown to balance the effects of SifA, as its loss in SifA mutants restores SCV stability in the presence of other effectors (Schroeder et al., 2010). The role of Sifs in Salmonella survival and replication has yet to be determined. To date, these structures have not been observed in vivo. SseF and SseG also localize with, and promote bundling of, microtubules in the cytoplasm and help position the SCV near the microtubule organizing center (MTOC), which is close to the Golgi network (Kuhle et al., 2004; Ramsden et al., 2007). Proximity to the Golgi network appears necessary for efficient bacterial replication within the SCV, as disruption of the Golgi network and loss of SseG diminish bacterial growth (Salcedo and Holden, 2003).
Once Salmonella enter the host epithelial cell, they seem to form two populations with distinct doubling rates. One population, referred to as “hyper-replicating,” doubles approximately every 20 min while the population as a whole doubles approximately every 95 min (Knodler et al., 2010). Interestingly, at least a third of the hyper-replicating population was found in the cytosol and express T3SS1 genes, while the slower replicating population was found in SCVs, and was expressing T3SS2 genes. These findings suggest that while Salmonella are capable of replicating within the SCV, cytosolic replication may be more efficient.
Salmonella Induces Ileal Secretion
One of the hallmarks of salmonellosis is a severe, watery diarrhea caused by high levels of fluid secretion into the intestines. Various T3SS1 effectors are proposed to mediate the level of fluid secretion into the gut, likely due to their ability to induce inflammation, including SopA, SopB, SopD, SopE2, and SipA (Zhang et al., 2002). Additionally, the loss of some genes encoded on SPI-5, including SopB, significantly reduced the level of S. dublin-induced fluid secretion from ligated ileal loops (Wood et al., 1998). One theory is that the rapid influx of neutrophils, in response to the inflammation caused by secreted effectors, impairs the epithelial barrier to an extent that results in leakage of extravascular fluids (Zhang et al., 2003). Neutrophil migration could also lead to chloride secretion by epithelial cells (Madara et al., 1993), an event that is compensated for by the subsequent secretion of water in an attempt by the host to restore ion balance.
Mechanisms of Salmonella Escape from Epithelial Cells and Dissemination
Salmonella Typhi, using the same initial invasion mechanisms as S. Typhimurium can disseminate in humans to cause systemic disease through infection of the gallbladder, liver, spleen, and bone marrow (Gonzalez-Escobedo et al., 2011). In mice, S. Typhimurium is capable of invading and destroying M cells, allowing penetration of the intestinal epithelium (Jones et al., 1994), and subsequent phagocytosis by macrophages. S. Typhimurium survival within the macrophages is mediated by SPI-2 effectors (Cirillo et al., 1998) and is essential for dissemination.
The role of the MLN in bacterial dissemination was closely explored by Voedisch et al. Here, the authors show that dendritic cells are major suppliers of S. Typhimurium to the MLN, but they alone are not regulating dissemination. After permitting re-circulation of intestinal lymph following removal of the MLN (mesenteric adenectomy), the authors observed increased liver colonization by S. Typhimurium in wild-type mice, and increased colonization of liver and spleen in NRAMP1+ 129Sv mice. Wild-type levels of liver and spleen colonization were observed in Rag2-deficient mice, suggesting the observations in the adenectomized mice were not artifacts of a weak adaptive immune response (Voedisch et al., 2009). These results suggest that while dendritic cells supply the MLN with S. Typhimurium, the MLN is the final barrier to dissemination in this pathway, as its presence is needed to limit the circularization of infected dendritic cells.
Although most NTS infections are self-limiting, NTS strains are capable of inducing prolonged infection, particularly in those with weakened immune systems. To cause prolonged infection, Salmonella must have a mechanism in place to facilitate escape from infected host cells. A recent paper suggests Salmonella escape their intracellular niche by co-opting the epithelial cell shedding process, a host mechanism used to remove dying epithelial cells. During this process, called extrusion, cells adjacent to a dying cell contract and force the dying cell out into the lumen (Madara, 1990; Mayhew et al., 1999). The authors found that during Salmonella infection, extrusion rates were increased, and about 10% of infected cells underwent extrusion, followed by inflammatory cell death. In contrast, less than 1% of uninfected cells underwent extrusion, and those that were extruded did not exhibit inflammatory cell death (Knodler et al., 2010). Escape into the lumen can permit Salmonella to infect additional cells, or to exit the host completely as part of its transmission cycle.
Concluding Remarks
Salmonella employ various mechanisms to overcome host defense mechanisms. By evolving ways to subvert, mimic, antagonize, or exploit a defense strategy, Salmonella maintain their ability to infect vertebrate hosts. The present and future research endeavors aimed at better understanding the tools Salmonella use to invade and traverse the mucosal intestinal epithelia will provide invaluable knowledge that will help devise ways to better treat and prevent Salmonella infections.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Institutes of Health Grant DK56754 and the Crohn’s and Colitis Foundation of America to Beth A. McCormick.
References
Aberle, H., Bauer, A., Stappert, J., Kispert, A., and Kemler, R. (1997). β-catenin is a target for the ubiquitin–proteasome pathway. EMBO J. 16, 3793–3804.
Abrahams, G. L., Muller, P., and Hensel, M. (2006). Functional dissection of SseF, a Type III Effector protein involved in positioning the Salmonella-containing vacuole. Traffic 7, 950–965.
Adachi, M., Inoko, A., Hata, M., Furuse, K., Umeda, K., Itoh, M., and Tsukita, S. (2006). Normal establishment of epithelial tight junctions in mice and cultured cells lacking expression of ZO-3, a tight-junction MAGUK protein. Mol. Cell. Biol. 26, 9003–9015.
Adams, C. L., Chen, Y., Smith, S. J., and Nelson, W. J. (1998). Mechanisms of epithelial cell–cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin–green fluorescent protein. J. Cell Biol. 142, 1105–1119.
Andreeva, A. Y., Krause, E., Müller, E. C., Blasig, I. E., and Utepbergenov, D. I. (2001). Protein kinase C regulates the phosphorylation and cellular localization of occludin. J. Biol. Chem. 42, 38480–38486.
Ayabe, T., Satchell, D. P., Wilson, C. L., Parks, W. C., Selsted, M. E., and Ouellette, A. J. (2000). Secretion of microbicidal α-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 1, 113–118.
Backhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A., and Gordon, J. I. (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920.
Bakowski, M. A., Braun, V., and Brumell, J. H. (2008). Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic 9, 2022–2031.
Barman, M., Unold, D., Shifley, K., Amir, E., Hung, K., Bos, N., and Salzman, N. (2008). Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect. Immun. 76, 907–915.
Baus-Loncar, M., Schmid, J., Lalani, el-N., Rosewell, I., Goodlad, R. A., Stamp, G. W., Blin, N., and Kayademir, T. (2005). Trefoil factor 2 (Tff2) Deficiency in murine digestive track influences the immune system. Cell Physiol. Biochem. 16, 31–42.
Beuzon, C. R., Méresse, S., Unsworth, K. E. Ruíz-Albert, J., Garvis, S., Waterman, S. R. Ryder, T. A., Boucrot, E., and Holden, D. W. (2000). Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19, 3235–3249.
Boyle, E. C., Brown, N. F., and Finlay, B. B. (2006). Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell. Microbiol. 8, 1946–1957.
Brawn, L. C., Hayward, R. D., and Koronakis, V. (2007). Salmonella SPI1 effector SipA persists after entry and cooperates with a SPI2 effector to regulate phagosome maturation and intracellular replication. Cell Host Microbe 1, 63–75.
Brumell, J. H., Tang, P., Mills, S. D., and Finlay, B. B. (2001). Characterization of Salmonella-induced filaments (Sifs) reveals a delayed interaction between Salmonella-containing vacuoles and late endocytic compartments. Traffic 2, 643–653.
Chamaillard, M., Hashimoto, M., Horie, Y., Masumoto, J., Qiu, S., Saab, L., Ogura, Y., Kawasaki, A., Fukase, K., Kusumoto, S., Valvano, M. A., Foster, S. J., Mak, T. W., Nuñez, G., and Inohara, N. (2003). An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707.
Chen, Y., Stewart, D. B., and Nelson, J. W. (1999). Coupling Assembly of the E-Cadherin/β-Catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-Cadherin in polarized MDCK cells. J. Cell Biol. 144, 687–699.
Cirillo, D. M., Valdivia, R. H., Monack, D. M., and Falkow, S. (1998). Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30, 175–188.
Clark, M. A., Hirst, B. H., and Jepson, M. A. (1998). Inoculum composition and Salmonella pathogenicity island 1 regulate M-cell invasion and epithelial destruction by Salmonella typhimurium. Infect. Immun. 66, 724–731.
Clark, M. A., Reed, K. A., Lodge, J., Stephen, J., Hirst, B. H., and Jepson, M. A. (1996). Invasion of murine intestinal M cells by Salmonella typhimurium inv mutants severely deficient for invasion of cultured cells. Infect. Immun. 64, 4363–4368.
Collier-Hyams, L. S., Zeng, H., Sun, J., Tomlinson, A. D., Bao, Z. Q., Chen, H., Madara, J. L., Orth, K., and Neish, A. S. (2002). Cutting edge: Salmonella AvrA effector inhibits the key proinflammatory, anti-apoptotic NF-kB pathway. J. Immunol. 169, 2846–2850.
Croswell, A., Amir, E., Teggatz, P., Barman, M., and Salzman, N. H. (2009). Prolonged impact of antibiotics on intestinal microbial ecology and susceptibility to enteric Salmonella infection. Infect. Immun. 77, 2741–2753.
Davis, M. A., Ireton, R. C., and Reynolds, A. B. (2003). A core function for p120-catenin in cadherin turnover. J. Cell Biol. 163, 525–534.
Deplancke, B., and Gaskins, H. R. (2001). Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am. J. Clin. Nutr. 73(Suppl.), 1131S–1141S.
Duan, Y., Liao, A. P., Kuppireddi, S., Ye, Z., Ciancio, M. J., and Sun, J. (2007). β-Catenin activity negatively regulates bacteria-induced inflammation. Lab. Invest. 87, 613–624.
Elphick, D. A., and Mahida, Y. R. (2005). Paneth cells: their role in innate immunity and inflammatory disease. Gut 54, 1802–1809.
Faucher, S. P., Porwollik, S., Dozois, C. M., McClelland, M., and Daigle, F. (2006). Transcriptome of Salmonella enterica serovar Typhi within macrophages revealed through the selective capture of transcribed sequences. Proc. Natl. Acad. Sci. U.S.A. 103, 1906–1911.
Finlay, B., Ruschkowski, S., and Dedhar, S. (1991). Cytoskeletal rearrangements accompanying Salmonella entry into epithelial cells. J. Cell Sci. 99, 283–296.
Forest, C. G., Ferraro, E., Sabbagh, S. C., and Daigle, F. (2010). Intracellular survival of Salmonella enterica serovar Typhi in human macrophages is SPI-2 independent. Microbiology 156(Pt 12), 3689–3698.
Forster, C. (2008). Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 130, 55–70.
Fournier, B., Williams, I. R., Gewirtz, A. T., and Neish, A. S. (2009). Toll-like receptor 5-dependent regulation of inflammation in systemic Salmonella enterica serovar Typhimurium infection. Infect. Immun. 77, 4121–4129.
Frey, A., Giannasca, K. T., Weltzin, R., Giannasca, P. J., Reggio, H., Lencer, W. I., and Neutra, M. R. (1996). Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J. Exp. Med. 184, 1045–1059.
Frost, A. J., Bland, A. P., and Wallis, T. S. (1997). The early dynamic response of the calf ileal epithelium to Salmonella typhimurium. Vet. Pathol. 34, 369–386.
Fu, Y., and Galan, J. E. (1999). A Salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 401, 293–297.
Furuse, M., Itoh, M., Hirase, T., Nagafuchi, A., Yonemura, S., Tsukita, S., and Tsukita, S. (1994). Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J. Cell Biol. 127, 1617–1626.
Furuse, M., Sasaki, H., Fujimoto, K., and Tsukita, S. (1998). A single gene product, Claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 143, 391–401.
Galan, J. E. (1996). Molecular genetic bases of Salmonella entry into host cells. Mol. Microbiol. 20, 263–271.
Geddes, K., Rubino, S., Streutker, C., Cho, J. H., Magalhaes, J. G., Le Bourhis, L., Selvanantham, T., Girardin, S. E., and Philpott, D. J. (2010). Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect. Immun. 78, 5107–5115.
Girardin, S. E., Boneca, I. G., Viala, J., Chamaillard, M., Labigne, A., Thomas, G., Philpott, D. J., and Sansonetti, P. J. (2003). Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278, 8869–8872.
Gonzalez-Escobedo, G., Marshall, J. M., and Gunn, J. S. (2011). Chronic and acute infection of the gall bladder by Salmonella Typhi: understanding the carrier state. Nat. Rev. Microbiol. 9, 9–14.
Gonzalez-Mariscal, L., Betanzos, A., and Avila-Flores, A. (2000). MAGUK proteins: structure and role in the tight junction. Cell Dev. Biol. 11, 315–324.
Graham, S. M., Molyneux, E. M., Walsh, A. L., Cheesbrough, J. S., Molyneux, M. E., and Hart, C. A. (2000). Nontyphoidal Salmonella infections of children in tropical Africa. Pediatr. Infect. Dis. J. 19, 1189–1196.
Green, K. J., and Simpson, C. L. (2007). Desmosomes: new perspectives on a classic. J. Investig. Dermatol. 127, 2499–2515.
Gribar, S. C., Anand, R. J., Sodhi, C. P., and Hackam, D. J. (2008). The role of epithelial toll-like receptor signaling in the pathogenesis of intestinal inflammation. J. Leukoc. Biol. 83, 493–498.
Hapfelmeier, S., Ehrbar, K., Stecher, B., Barthel, M., Kremer, M., and Hardt, W. D. (2004). Role of the Salmonella pathogenicity island 1 effector proteins SipA, SopB, SopE, and SopE2 in Salmonella enterica subspecies 1 Serovar Typhimurium colitis in streptomycin-pretreated mice. Infect. Immun. 72, 795–809.
Hapfelmeier, S., Stecher, B., Barthel, M., Kremer, M., Müller, A. J., Heikenwalder, M., Stallmach, T., Hensel, M., Pfeffer, K., Akira, S., and Hardt, W. D. (2005). The Salmonella pathogenicity island (SPI)-2 and SPI-1 Type III secretion systems allow Salmonella Serovar typhimurium to trigger colitis via MyD88-dependent and MYD88-independent mechanisms. J. Immunol. 174, 1675–1685.
Hardt, W., Chen, L. M., Schuebel, K. E., Bustelo, X. R., and Galán, J. E. (1998). S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93, 815–826.
Hartsock, A., and Nelson, J. W. (2008). Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 1778, 660–669.
Hashizume, T., Togawa, A., Nochi, T., Igarashi, O., Kweon, M. N., Kiyono, H., and Yamamoto, M. (2008). Peyer’s patches are required for intestinal immunoglobulin a responses to Salmonella spp. Infect. Immun. 76, 927–934.
Haskins, J., Gu, L., Wittchen, E. S., Hibbard, J., and Stevenson, B. R. (1998). ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J. Cell Biol. 141, 199–208.
Hayward, R. D., McGhie, E. J., and Koronakis, V. (2000). Membrane fusion activity of purified SipB, a Salmonella surface protein essential for mammalian cell invasion. Mol. Microbiol. 37, 727–739.
Hernandez, L. D., Hueffer, K., Wenk, M. R., and Galan, J. E. (2004). Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304, 1805–1807.
Itoh, M., Morita, K., and Tsukita, S. (1999). Characterization of ZO-2 as a MAGUK family member associated with tight as well as adherens junctions with a binding affinity to occludin and α-catenin. J. Biochem. Chem. 274, 5981–5986.
Jensen, V. B., Harty, J. T., and Jones, B. D. (1998). Interactions of the invasive pathogens Salmonella Typhimurium, Listeria monocytogenes, and Shigella flexneri with M cells and murine Peyer’s patches. Infect. Immun. 66, 3758–3766.
Jiang, X., Rossanese, O. W., Brown, N. F., Kujat-Choy, S., Galán, J. E., Finlay, B. B., and Brumell, J. H. (2004). The related effector proteins SopD and SopD2 from Salmonella enterica serovar Typhimurium contribute to virulence during systemic infection of mice. Mol. Microbiol. 54, 1186–1198.
Jones, B. D., Ghori, N., and Falkow, S. (1994). Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J. Exp. Med. 180, 15–23.
Kaniga, K., Tucker, S., Trollinger, D., and Galan, J. E. (1995). Homologs of the Shigella IpaB and IpaC invasins are required for Salmonella Typhimurium entry into cultured epithelial cells. J. Bacteriol. 177, 3965–3971.
Knodler, L. A., and Steele-Mortimer, O. (2005). The Salmonella effector PipB2 affects late endosome/lysosome distribution to mediate Sif extension. Mol. Biol. Cell 16, 4108–4123.
Knodler, L. A., Vallance, B. A., Celli, J., Winfree, S., Hansen, B., Montero, M., and Steele-Mortimer, O. (2010). Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc. Natl. Acad. Sci. U.S.A. 107, 17733–17738.
Koch, P. J., and Franke, W. W. (1994). Desmosomal cadherins: another growing multigene family of adhesion molecules. Curr. Opin. Cell Biol. 6, 682–687.
Kohler, H., Sakaguchi, T., Hurley, B. P., Kase, B. A., Reinecker, H. C., and McCormick, B. A. (2007). Salmonella enterica serovar Typhimurium regulates intercellular junction proteins and facilitates transepithelial neutrophil and bacterial passage. Am. J. Physiol. Gastroinest. Liver Physiol. 293, G178–G187.
Kraehenbuhl, J., and Neutra, M. R. (2000). Epithelial M cells: differentiation and function. Annu. Rev. Cell Dev. Biol. 16, 301–332.
Kuhle, V., Jackel, D., and Hensel, M. (2004). Effector proteins encoded by Salmonella pathogenicity island 2 interfere with the microtubule cytoskeleton after translocation into host cells. Traffic 5, 356–370.
Lara-Tejero, M., and Galan, J. E. (2009). Salmonella enterica Serovar Typhimurium pathogenicity island 1-encoded type III secretion system translocases mediate intimate attachment to nonphagocytic cells. Infect. Immun. 77, 2635–2642.
Lawley, T. D., Chan, K., Thompson, L. J., Kim, C. C., Govoni, G. R., and Monack, D. M. (2006). Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathogens 2, e11. doi: 10.1371/journal.ppat.0020011
Lawley, T. D., Bouley, D. M., Hoy, Y. E., Gerke, C., Relman, D. A., and Monack, D. M. (2008). Host transmission of Salmonella enterica Serovar typhimurium is controlled by virulence factors and indigenous intestinal microbiota. Infect. Immun. 76, 403–416.
LeLouard, H., Henri, S., De Bovis, B., Mugnier, B., Chollat-Namy, A., Malissen, B., Méresse, S., and Gorvel, J. P. (2010). Pathogenic bacteria and dead cells are internalized by a unique subset of Peyer’s Patch dendritic cells that express lysozyme. Gastroenterology 138, 173–184.
Lesnick, M. L., Reiner, N. E., Fierer, J., and Guiney, D. G. (2001). The Salmonella spvB virulence gene encodes an enzyme that ADP-ribosylates actin and destabilizes the cytoskeleton of eukaryotic cells. Mol. Microbiol. 39, 1464–1470.
Liao, A. P., Petrof, E. O., Kuppireddi, S., Zhao, Y., Xia, Y., Claud, E. C., and Sun, J. (2008). Salmonella Type III effector AvrA stabilizes cell tight junctions to inhibit inflammation in intestinal epithelial cells. PLOS ONE 3, e2369. doi: 10.1371/journal.pone.0002369
Libby, S. J., Brehm, M. A., Greiner, D. L., Shultz, L. D., McClelland, M., Smith, K. D., Cookson, B. T., Karlinsey, J. E., Kinkel, T. L., Porwollik, S., Canals, R., Cummings, L. A., and Fang, F. C. (2010). Humanized nonobese diabetic-scid IL2rynull mice are susceptible to lethal Salmonella Typhi infection. Proc. Natl. Acad. Sci. U.S.A. 107, 15589–15594.
MacDonald, T. T., and Monteleone, G. (2005). Immunity, inflammation, and allergy in the gut. Science 307, 1920–1925.
Madara, J. L. (1990). Maintenance of the macromolecular barrier at cell extrusion sites in intestinal epithelium: physiological rearrangement of tight junctions. J. Membr. Biol. 116, 177–184.
Madara, J. L., Patapoff, T. W., Gillece-Castro, B., Colgan, S. P., Parkos, C. A., Delp, C., Mrsny, R. J. (1993). 5’-Adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J. Clin. Invest. 91, 2320–2325.
Martinez-Argudo, I., and Jepson, M. A. (2008). Salmonella translocates across an in vitro M cell model independently of SPI-1 and SPI-2. Microbiology 154, 3887–3894.
Martinez-Palomo, A., and Erlij, D. (1975). Structure of tight junctions in epithelia with different permeability. Proc. Natl. Acad. Sci. U.S.A. 72, 4487–4491.
Mayhew, T. M., Myklebust, R., Whybrow, A., and Jenkins, R. (1999). Epithelial integrity, cell death and cell loss in mammalian small intestine. Histol. Histolpathol. 14, 257–267.
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O., and Kasper, D. L. (2005). An immunodeficiency molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118.
Mazurkiewicz, P., Thomas, J., Thompson, J. A., Liu, M., Arbibe, L., Sansonetti, P., and Holden, D. W. (2008). SpvC is a Salmonella effector with phosphothreonine lyase activity on host mitogen-activated protein kinases. Mol. Microbiol. 67, 1371–1383.
McCormick, B. A. (2007). Bacterial-induced hepoxilin A3 secretion as a pro-inflammatory mediator. FEBS J. 274, 3513–3518.
McCormick, B. A., Stocker, B. A. D., Laux, D. C., and Cohen, P. S. (1988). Roles of motility, chemotaxis, and penetration through and growth in intestinal mucus in the ability of an avirulent strain of Salmonella typhimurium to colonize the large intestine of streptomycin-treated mice. Infect. Immun. 56, 2209–2217.
McGhie, E. J., Hayward, R. D., and Koronakis, V. (2001). Cooperation between actin-binding proteins of invasive Salmonella: SipA potentiates SipC nucleation and bundling of actin. EMBO J. 20, 2131–2139.
Meyerholz, D. K., and Stabel, T. J. (2003). Comparison of early ileal invasion by Salmonella enterica serovars choleraesuis and typhimurium. Vet. Pathol. 40, 371–375.
Meyerholz, D. K., Stabel, T. J., Ackermann, M. R., Carlson, S. A., Jones, B. D., and Pohlenz, J. (2002). Early epithelial invasion by Salmonella enterica serovar typhimurium DT104 in the swine ileum. Vet. Pathol. 39, 712–720.
Nash, S., Stafford, J., and Madara, J. L. (1987). Effects of Polymorphonuclear leukocyte transmigration on the barrier function of cultured intestinal epithelial monolayers. J. Clin. Invest. 80, 1104–1113.
Nevola, J. J., Laux, D. C., and Cohen, P. S. (1987). In vivo colonization of the mouse large intestine and in vitro penetration of intestinal mucus by an avirulent smooth strain of Salmonella typhimurium and its lipopolysaccharide-deficient mutant. Infect. Immun. 55, 2884–2890.
Nobes, C. D., and Hall, A. (1995). Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62.
Norris, F. A., Wilson, M. P., Wallis, T. S., Galyov, E. E., and Majerus, P. W. (1998). SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc. Natl. Acad. Sci. U.S.A. 95, 14057–14059.
Palazzo, M., Balsari, A., Rossini, A., Selleri, S., Calcaterra, C., Gariboldi, S., Zanobbio, L., Arnaboldi, F., Shirai, Y. F., Serrao, G., and Rumio, C. (2007). Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J. Immunology 178, 4296–4303.
Pazos, M., Siccardi, D., Mumy, K. L., Bien, J. D., Louie, S., Shi, H. N., Gronert, K., Mrsny, R. J., and McCormick, B. A. (2008). Multidrug resistance-associated transporter 2 regulates mucosal inflammation by facilitating the synthesis of hepoxilin A3. J. Immunol. 181, 8044–8052.
Playford, R. J., Marchbank, T., Goodlad, R. A., Chinery, R. A., Poulsom, R., and Hanby, A. M. (1996). Transgenic mice that overexpress the human trefoil factor peptide pS2 have an increased resistance to intestinal damage. Proc. Natl. Acad. Sci. U.S.A. 93, 2137–2142.
Poh, J., Odendall, C., Spanos, A., Boyle, C., Liu, M., Freemont, P., and Holden, D. W. (2008). SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodeling. Cell. Microbiol. 10, 20–30.
Quezada, C. M., Hicks, S. W., Galan, J. E., and Stebbins, C. E. (2009). A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. U.S.A. 106, 4864–4869.
Radtke, A., Wilson, J. W., Sarker, S., and Nickerson, C. A. (2010). Analysis of interactions of Salmonella type three secretion mutants with 3-D intestinal epithelial cells. PLoS ONE 5, e15750. doi: 10.1371/journal.pone.0015750
Raffatellu, M., Wilson, R. P., Chessa, D., Andrews-Polymenis, H., Tran, Q. T., Lawhon, S., Khare, S., Adams, L. G., and Bäumler, A. J. (2005). SipA, SopA, SopB, SopD, and SopE2 contribute to Salmonella enterica serotype typhimurium invasion of epithelial cells. Infect. Immun. 73, 146–154.
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S., and Medzhitov, R. (2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241.
Ramsden, A. E., Mota, L. J., Münter, S., Shorte, S. L., and Holden, D. W. (2007). The SPI-2 type III secretion system restricts motility of Salmonella-containing vacuoles. Cell. Microbiol. 9, 2517–2529.
Ridley, A. J., and Hall, A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399.
Ridley, A. J., Paterson, H. F., Johnston, C. L., Diekmann, D., Hall, A. (1992). The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401–410.
Ruiz-Albert, J., Yu, X. J., Beuzón, C. R., Blakey, A. N., Galyov, E. E., and Holden, D. W. (2002). Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol. Microbiol. 44, 645–661.
Rytkonen, A., and Holden, D. W. (2007). Bacterial interference of ubiquitination and deubiquitination. Cell Host Microbe 1, 13–22.
Rytkonen, A., Poh, J., Garmendia, J., Boyle, C., Thompson, A., Liu, M., Freemont, P., Hinton, J. C. D., and Holden, D. W. (2007). SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 3502–3507.
Sabbagh, S. C., Forest, C. G., Lepage, C., Leclerc, J. M., and Daigle, F. (2010). So similar, yet so different: uncovering distinctive features in the genomes of Salmonella enterica serovars Typhimurium and Typhi. FEMS Microbiol. 305, 1–13.
Salcedo, S. P., and Holden, D. W. (2003). SseG, a virulence protein that targets Salmonella to the golgi network. EMBO J. 22, 5003–5014.
Salzman, N. H., Chou, M. M., de Jong, H., Liu, L., Porter, E. M., and Paterson, Y. (2003). Enteric Salmonella infection inhibits paneth cell antimicrobial peptide expression. Infect. Immun. 71, 1109–1115.
Schroeder, N., Henry, T., de Chastellier, C., Zhao, W., Guilhon, A. A., Gorvel, J. P., and Méresse, S. (2010). The virulence protein SopD2 regulates membrane dynamics of Salmonella-containing vacuoles. PLOS Pathogens 6, e1001002. doi: 10.1371/journal.ppat.1001002
Srikanth, C. V., Wall, D. M., Maldonado-Contreras, A., Shi, H. N., Zhou, D., Demma, Z., Mumy, K. L., and McCormick, B. A. (2010). Salmonella pathogenesis and processing of secreted effectors by caspase-3. Science 330, 390–393.
Stebbins, C. E., and Galan, J. E. (2000). Modulation of host signaling by a bacterial mimic: structure of the Salmonella effector SptP bound to Rac1. Mol. Cell 6, 1449–1460.
Stebbins, C. E., and Galan, J. E. (2003). Priming virulence factors for delivery into the host. Nat. Rev. Mol. Cell Biol. 4, 738–744.
Stecher, B., Robbiani, R., Walker, A. W.,Westendorf, A. M., Barthel, M., Kremer, M., Chaffron, S., Macpherson, A. J., Buer, J., Parkhill, J., Dougan, G., von Mering, C., and Hardt, W. D. (2007). Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLOS Biol. 5, 2177–2189. doi: 10.1371/journal.pbio.0050244
Stein, M., Leung, K. Y., Zwick, M., Garcia-del Portillo, F., and Finlay, B. B. (1996). Identification of a Salmonella virulence gene required for formation of filamentous structures containing lysosomal membrane glycoproteins within epithelial cells. Mol. Microbiol. 20, 151–164.
Stender, S., Friebel, A., Linder, S., Rohde, M., Mirold, S., and Hardt, W. D. (2000). Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol. Microbiol. 36, 1206–1221.
Takeichi, M. (1991). Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251, 1451–1455.
Tam, M. A., Rydstrom, A., Sundquist, M., and Wick, M. J. (2008). Early cellular responses to Salmonella infection: dendritic cells, monocytes, and more. Immunol. Rev. 225, 140–162.
Tsukita, S., Katsuno, T., Yamazaki, Y., Umeda, K., Tamura, A., and Tsukita, S. (2009). Roles of ZO-1 and ZO-2 in establishment of the belt-like adherens and tight junctions with paracellular permselective barrier function. Ann. N.Y. Acad. Sci. 1165, 44–52.
Turner, J. R. (2009). Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809.
Tyrer, P., Foxwell, A. R., Cripps, A. W., Apicella, M. A., and Kyd, J. M. (2006). Microbial pattern recognition receptors mediate M-cell uptake of a gram-negative bacterium. Infect. Immun. 74, 625–631.
Umeda, K., Ikenouchi, J., Katahira-Tayama, S., Furuse, K., Sasaki, H., Nakayama, M., Matsui, T., Tsukita, S., Furuse, M., and Tsukita, S. (2006). ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 126, 741–754.
Vimal, D. B., Khullar, M., Gupta, S., and Ganguly, N. K. (2000). Intestinal mucins: the bindings sites for Salmonella Typhimurium. Mol. Cell. Biochem. 204, 107–117.
Voedisch, S., Koenecke, C., David, S., Herbrand, H., Förster, R., Rhen, M., and Pabst, O. (2009). Mesenteric lymph nodes confine DC-mediated dissemination of Salmonella and limit systemic disease in mice. Infect. Immun. 77, 3170–3180.
Wall, D. M., Nadeau, W. J., Pazos, M. A., Shi, H. N., Galyov, E. E., and McCormick, B. A. (2007). Identification of the Salmonella enterica serotype Typhimurium SipA domain responsible for inducing neutrophil recruitment across the intestinal epithelium. Cell. Microbiol. 9, 2299–2313.
Wood, M. W., Jones, M. A., Watson, P. R., Hedges, S., Wallis, T. S., and Galyov, E. E. (1998). Identification of a pathogenicity island required for Salmonella enteropathogenicity. Mol. Microbiol. 29, 883–891.
Wood, M. W., Jones, M. A., Watson, P. R., Siber, A. M., McCormick, B. A., Hedges, S., Rosqvist, R., Wallis, T. S., and Galyov, E. E. (2000). The secreted effector protein of Salmonella dublin, SopA, is translocated into eukaryotic cells and influences the induction of enteritis. Cell. Microbiol. 2, 293–303.
Ye, Z., Petrof, E. O., Boone, D., Claud, E. C., and Sun, J. (2007). Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am. J. Pathol. 171, 882–892.
Yu, X., Ruiz-Albert, J., Unsworth, K. E., Garvis, S., Liu, M., Holden, D. W. (2002). SpiC is required for secretion of Salmonella pathogenicity island 2 type III secretion system proteins. Cell. Microbiol. 4, 531–540.
Zhang, S., Kingsley, R. A., Santos, R. L., Andrews-Polymenis, H., Raffatellu, M., Figueiredo, J., Nunes, J., Tsolis, R. M., Adams, L. G., and Bäumler, A. J. (2003). Molecular pathogenesis of Salmonella enterica serotype typhimurium-induced diarrhea. Infect. Immun. 71, 1–12.
Zhang, S., Santos, R. L., Tsolis, R. M., Stender, S., Hardt, W. D., Bäumler, A. J., and Adams, L. G. (2002). The Salmonella enterica serotype typhimurium effector proteins SipA, SopA, SopB, SopD, and SopE2 Act in concert to induce diarrhea in calves. Infect. Immun. 70, 3843–3855.
Zhang, Y., Higashide, W. M., McCormick, B. A., Chen, J., and Zhou, D. (2006). The inflammation-associated Salmonella SopA is a HECT-like E3 ubiquitin ligase. Mol. Microbiol. 62, 786–793.
Zhou, D., Chen, L. M., Hernandez, L., Shears, S. B., and Galán, J. E. (2001). A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol. Microbiol. 39, 248–259.
Keywords: Salmonella, intestinal mucosa, tight junctions
Citation: Hallstrom K and McCormick BA (2011) Salmonella interaction with and passage through the intestinal mucosa: through the lens of the organism. Front. Microbio. 2:88. doi: 10.3389/fmicb.2011.00088
Received: 01 February 2011;
Accepted: 13 April 2011;
Published online: 29 April 2011.
Edited by:
John S. Gunn, The Ohio State University, USACopyright: © 2011 Hallstrom and McCormick. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Beth A. McCormick, Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, 55 Lake Avenue North, Worcester, MA 01655, USA.e-mail:YmV0aC5tY2Nvcm1pY2tAdW1hc3NtZWQuZWR1