


- Department of Microbiology, University of Iowa, Iowa City, IA, USA
Pseudomonas aeruginosa is an opportunistic pathogen that is particularly problematic in the healthcare setting where it is a frequent cause of pneumonia, bloodstream, and urinary tract infections. An important determinant of P. aeruginosa virulence is a type III secretion system (T3SS). T3SS-dependent intoxication is a complex process that minimally requires binding of P. aeruginosa to host cells, injection of the cytotoxic effector proteins through the host cell plasma membrane, and induction of T3SS gene expression. The latter process, referred to as contact-dependent expression, involves a well-characterized regulatory cascade that activates T3SS gene expression in response to host cell contact. Although host cell contact is a primary activating signal for T3SS gene expression, the involvement of multiple membrane-bound regulatory systems indicates that additional environmental signals also play a role in controlling expression of the T3SS. These regulatory systems coordinate T3SS gene expression with many other cellular activities including motility, mucoidy, polysaccharide production, and biofilm formation. The signals to which the organism responds are poorly understood but many seem to be coupled to the metabolic state of the cell and integrated within a master circuit that assimilates informational signals from endogenous and exogenous sources. Herein we review progress toward unraveling this complex circuitry, provide analysis of the current knowledge gaps, and highlight potential areas for future studies. Complete understanding of the regulatory networks that control T3SS gene expression will maximize opportunities for the development of strategies to treat P. aeruginosa infections.
Introduction
Pseudomonas aeruginosa is widely appreciated for its ability to survive under adverse conditions. In its native habitat of soil and water, P. aeruginosa is exposed to a myriad of potential growth substrates, competing microorganisms, and outright predators. To cope with these hostile conditions P. aeruginosa has a relatively large genome (6.3 Mb) encoding diverse metabolic pathways and defense mechanisms, and has extensive regulatory networks that reprogram gene expression patterns to suit the prevailing environmental conditions (Stover et al., 2000). Many of the defense mechanisms that provide a selective advantage in soil and water also contribute to virulence in the mammalian host. For instance, the P. aeruginosa type III secretion system (T3SS) contributes to virulence in ameba, insect, zebrafish, and mammalian infection models and protects P. aeruginosa from phagocytosis irrespective of whether the phagocyte is an ameba or a human macrophage (Pukatzki et al., 2002; Miyata et al., 2003; Brannon et al., 2009).
Type III secretion systems contribute to the virulence of many medically relevant Gram-negative organisms and function like a molecular syringe to inject bacterial effector proteins into eukaryotic host cells (Coburn et al., 2007). The injectisome consists of ~25 proteins that form two ring-like structures embedded in the inner and outer membranes, a rod-like structure that spans the periplasmic space, a needle-like structure that extends from the cell surface, and a translocation complex that forms a channel in the host cell membrane (Moraes et al., 2008; Worrall et al., 2011). The effector proteins are transported through the bacterial cell envelope via the hollow rod and needle proteins, and injected through the translocation pore into the host cell. Assembly of the injectisome is dependent upon the T3SS which is located in the inner membrane and is thought to occur in a highly ordered process involving sequential secretion and assembly of the rod, needle, and translocator proteins (Wagner et al., 2010). While the injectisome structure and composition is highly conserved, each pathogen has a unique set of effector proteins suited to its individual lifestyle (Coburn et al., 2007). P. aeruginosa uses the injectisome to translocate at least five effector proteins (ExoS, ExoT, ExoU, ExoY, and FliC) into host cells (Hauser, 2009). The translocated effectors facilitate phagocytic avoidance, tissue invasion and systemic spread, and modulate the inflammatory response (Engel and Balachandran, 2009). Regulation of T3SS transcription is intrinsically controlled by the activity of the injectisome through a well-described and self-contained (encoded) regulatory mechanism (Brutinel and Yahr, 2008). T3SS gene expression is also influenced by extrinsic regulatory genes but in most cases the mechanisms involved are poorly understood. In this review we focus on recent progress toward defining mechanisms that regulate T3SS gene expression.
Intrinsic Regulation of T3SS Gene Expression
ExsA is the Central Regulator of T3SS Gene Expression
The P. aeruginosa T3SS regulon consists of ~40 genes organized within 10 transcriptional units that encode the secretion and translocation machinery, intrinsic regulators of T3SS gene expression (ExsA, ExsD, ExsC, and ExsE), chaperones, and effectors (Frank, 1997). The central regulator of the T3SS regulon is ExsA, a member of the AraC/XylS family of transcriptional activators (Yahr and Frank, 1994; Hovey and Frank, 1995; Yahr et al., 1995). ExsA autoregulates its own expression through the PexsC promoter and is encoded as the last gene of the exsCEBA polycistronic mRNA (Yahr and Frank, 1994). An alignment of the 10 ExsA-dependent promoters has identified a consensus binding site (tAaAAAnwnMyGrCynnnmYTGayAk) with three areas of high sequence conservation: an adenine-rich region, a GxC sequence centered at the -45 position with respect to the transcriptional start site, and a TGxxA sequence located near the -35 position (Figure 1; Hovey and Frank, 1995; Brutinel et al., 2008). Mutagenesis studies have confirmed that each of these elements is required for ExsA binding and activation of the PexoT promoter (Brutinel et al., 2008).
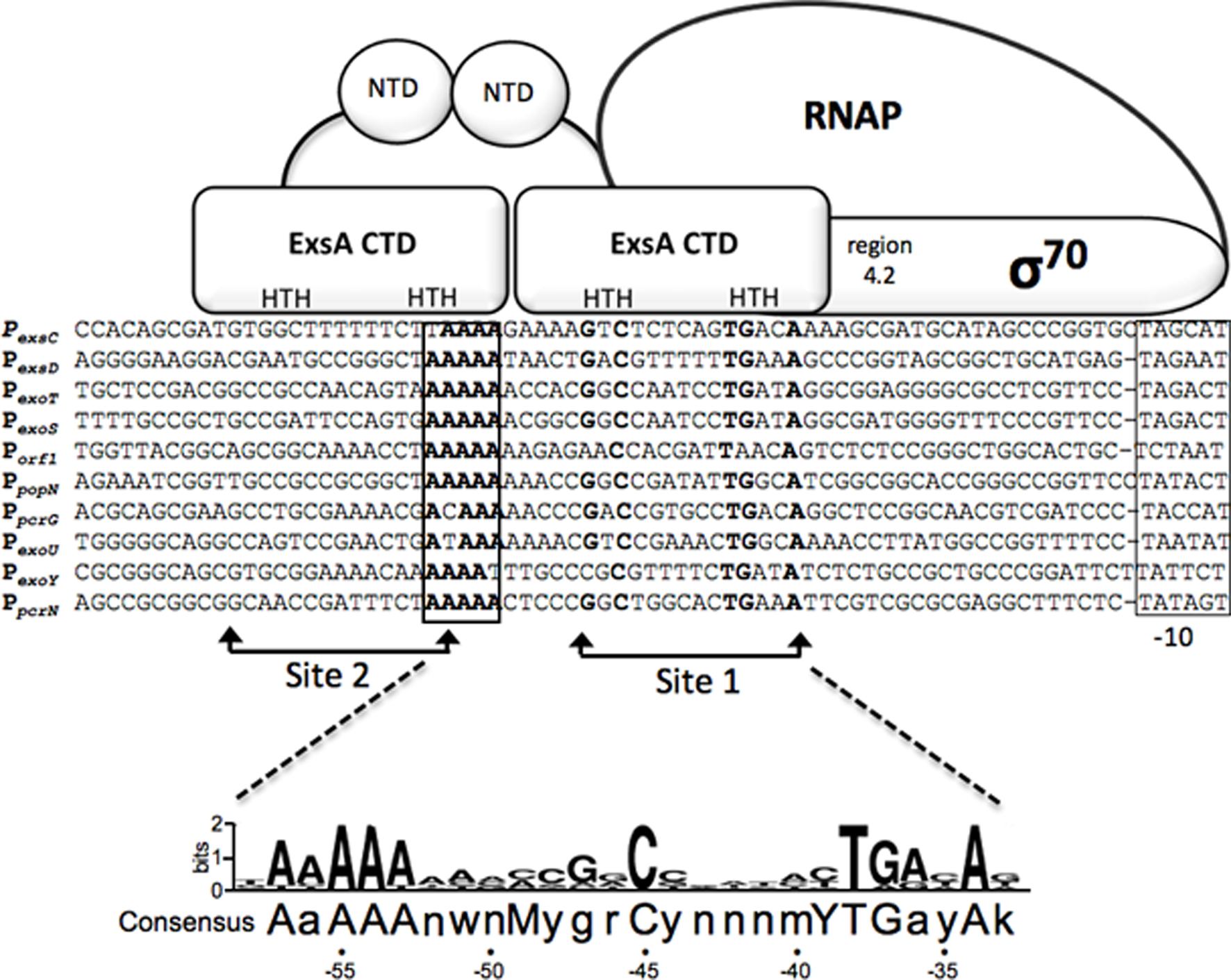
Figure 1. Sequence alignment of ExsA-dependent promoters and working model for promoter recognition and recruitment of σ70-RNAP by ExsA. Nucleotides in bold are highly conserved in all ExsA-dependent promoters and have been shown through mutagenesis and DNA-biding studies to be important for ExsA-dependent activation. The boxed sequences represent the -10 promoter recognition site. Predicted points of interaction for the helix-turn-helix (HTH) DNA-binding motifs of each ExsA monomer are indicated. Studies have shown that ExsA recruits σ70-RNAP through interactions mediated by region 4.2 of σ70. The ExsA consensus binding sequence is indicated at the bottom of the diagram.
Typical of most AraC/XylS family members, ExsA has an amino-terminal domain (NTD) involved in self-association and regulatory functions (discussed further below), and a carboxy-terminal domain (CTD) that contains two helix-turn-helix DNA-binding motifs (Brutinel et al., 2009). In electrophoretic mobility shift assays (EMSA) purified ExsA binds to the PexoT, PexsD, and PexsC promoters with apparent equilibrium constants in the low (1–5) nM range (Brutinel et al., 2008). Two distinct ExsA-promoter complexes are observed: a higher mobility complex representing one ExsA monomer bound to the promoter probe and a lower mobility complex containing two bound ExsA monomers (Brutinel et al., 2008). Mutagenesis and footprinting studies indicate that this binding arrangement represents two adjacent binding sites for monomeric ExsA with binding site 1 overlapping the conserved GxC and TGxxA sequences, and binding site 2 located upstream of site 1 (Figure 1). Whereas binding of ExsA to site 1 of the PexoT promoter is required for occupation of site 2, deletion of binding site 2 has no effect on occupation of site 1 (Brutinel et al., 2008). In addition, occupation of binding site 2 is dependent upon monomer–monomer interactions mediated by the amino-terminal domain of ExsA (Brutinel et al., 2009). These combined data are supportive of a monomer assembly model whereby an ExsA monomer bound to site 1 recruits a second ExsA monomer to binding site 2. The apparent lack of nucleotide sequence conservation between ExsA binding sites 1 and 2, however, raises the possibility that monomer–monomer interactions are the primary determinant for occupation of site 2. While genetic studies are suggestive of cooperative binding to the PexsC, PexoT, and PexsD promoters, the Hill coefficients derived from EMSA experiments are supportive of cooperative binding only at the PexsC promoter (1.9 ± 0.2; Brutinel et al., 2008). This discrepancy may reflect a fundamental difference in the binding of ExsA to the PexsC promoter and is supported by the observation that ExsA elicits a significant bend (78°) upon binding to the PexsC promoter that correlates with increased promoter activity and cooperative binding interactions (Brutinel et al., 2008).
ExsA-dependent promoters possess -35 and -10 sequence elements similar to those of σ70-dependent promoters with one significant difference (Brutinel et al., 2008; Figure 1). Unlike canonical σ70-dependent promoters where the -35 and -10 elements are separated by ~17 bp, the same regions in ExsA-dependent promoters are separated by 21 or 22 bp. In vitro transcription and primer extension assays indicate that the promoters are indeed σ70-dependent and that the -10 regions are functional recognition determinants for σ70 (Vakulskas et al., 2009). The apparent -35 elements, however, are not required for recognition by σ70 but rather serve as determinants for ExsA binding (Vakulskas et al., 2010). ExsA activates transcription by compensating for the lack of functional -35 sites primarily through recruitment of RNA polymerase-σ70 holoenzyme to the promoter but also has a small effect on isomerization to an open complex (Vakulskas et al., 2009). Like many AraC/XylS family members, ExsA interacts with region 4.2 of σ70 to recruit RNAP-σ70 to the promoter (Vakulskas et al., 2010). Somewhat atypical for an AraC/XylS family member, however, is that interactions with the α-subunit of RNAP are not required for ExsA-dependent activation. Although occupation of binding sites 1 and 2 on the promoter is required for ExsA-dependent transcription, it is unclear whether one or both monomers interact with RNAP-σ70 to facilitate recruitment. The proximity of binding site 1 to the expected location for a -35 region, and the fact that ExsA compensates for the lack of a functional -35 region, is consistent with a model in which ExsA bound to site 1 interacts with region 4.2 of σ70 and ExsA bound to site 2 stabilizes ExsA bound to site 1 and/or the ExsA-RNAP-σ70 complex.
Regulation of ExsA Activity by the ExsADCE Regulatory Cascade
Type III secretion systems have been described as contact-dependent secretion systems because translocation of the effector proteins requires intimate contact between the pathogen and the host, and because host cell contact induces T3SS gene expression (Hayes et al., 2010). Growth of P. aeruginosa under Ca2+-limiting conditions, or in the presence of serum, mimics the host-contact signal and is commonly used in the laboratory as a surrogate for the host-contact signal (Frank, 1997; Vallis et al., 1999; Kim et al., 2005; Urbanowski et al., 2007). Low Ca2+, serum, and host cell contact are each thought to convert the injectisome to a secretion-competent state through a poorly defined mechanism (Vallis et al., 1999; McCaw et al., 2002). ExsA activity is intimately coupled to secretion by a partner-switching mechanism involving three additional proteins; ExsC, ExsD, and ExsE (Brutinel and Yahr, 2008). Both ExsC and ExsD have two potential binding partners. ExsD is an anti-activator that inhibits T3SS gene expression by disrupting both the self-association and DNA-binding activities of ExsA (Brutinel et al., 2010). ExsC functions as an anti-anti-activator and forms a 2:2 stoichiometric complex with ExsD (Dasgupta et al., 2004; Lykken et al., 2006; Zheng et al., 2007). ExsC is also a T3SS chaperone and forms a 2:1 complex with ExsE (Zheng et al., 2007). Finally, ExsE is a secreted regulator that prevents ExsC from associating with ExsD (Rietsch et al., 2005; Urbanowski et al., 2005). The current working model is that ExsA-dependent transcription is inactive under non-permissive conditions (i.e., high Ca2+) because the binding equilibria favor formation of the inhibitory ExsD•ExsA and ExsC•ExsE complexes. In contrast, inducing conditions trigger secretion and/or translocation of ExsE (Urbanowski et al., 2007). The resulting decrease in the intracellular concentration of ExsE favors formation of the ExsD•ExsC complex (i.e., partner switching), thereby releasing ExsA to activate transcription. An important feature of the regulatory cascade is that minor changes in the levels of ExsC, ExsE, ExsA, or ExsD (as little as threefold) can have profound effects on T3SS gene expression (Dasgupta et al., 2004).
The partner-switching mechanism predicts that the binding affinities of ExsE, ExsC, ExsD, and ExsA are, in order of strongest affinity, ExsC•ExsE > ExsC•ExsD > ExsD•ExsA. While the binding affinity of the ExsD–ExsA complex has not been determined, the binding constants (Kd) for the ExsC•ExsE and ExsC•ExsD complexes are 1 and 18 nM, respectively (Zheng et al., 2007). Recent analyses of the ExsE•ExsC and ExsD crystal structures provide a basis for the differential binding of ExsC to ExsE and ExsD (Bernhards et al., 2009; Vogelaar et al., 2010). The ExsE monomer contains two ExsC-binding domains located in the amino-and carboxy-terminal portions of the protein and each domain contains an Arg-X-Val-X-Arg motif and a partially overlapping β-motif. In the co-complex the unstructured ExsE monomer wraps around the ExsC homodimer and each Arg-X-Val-X-Arg motif interacts with one of the ExsC monomers, thereby accounting for the 2:1 binding stoichiometry of the ExsC•ExsE complex. Interestingly, the amino-terminal region of ExsD also has an Arg-X-Val-X-Arg motif, a β-motif, and shares 22.5% sequence identity with the amino-terminus of ExsE. Whereas the entire ExsE protein (i.e., both Arg-X-Val-X-Arg motifs) is necessary for formation of the ExsC•ExsE complex, the ExsD amino-terminal region alone is sufficient to bind ExsC. This observation suggests that each ExsD monomer binds to one-half of the ExsC dimer and this likely accounts for the 2:2 binding stoichiometry of the ExsC•ExsD complex. Furthermore, the presence of a common ExsC-binding motif in ExsE and ExsD is consistent with the observation that binding of ExsE and ExsD to ExsC are mutually exclusive (Vogelaar et al., 2010).
ExsD is a dynamic protein capable of forming a homo–trimeric complex in solution, a 1:1 stoichiometric complex with ExsA, and a 2:2 stoichiometric complex with ExsC (Zheng et al., 2007; Bernhards et al., 2009; Thibault et al., 2009). Curiously, the ExsD crystal structure revealed that the amino-terminus of ExsD has structural similarity to the DNA-binding domain of the KorB transcriptional repressor (Bernhards et al., 2009). The predicted DNA-binding interface, however, is buried within the trimeric structure suggesting that dissociation of the trimer might be required for ExsD to exhibit DNA-binding activity. This property might account for the observation that trimeric ExsD lacks DNA-binding activity (Bernhards et al., 2009). Future studies will be required to determine whether monomeric and/or dimeric ExsD has DNA-binding activity and, if so, whether such activity is of biological significance.
Studies of the ExsD•ExsA interaction have relied upon co-expression of the proteins in vivo and subsequent purification as an ExsD•ExsA complex. Consistent with the functional role of ExsD as an anti-activator, the purified ExsA•ExsD complex lacks DNA-binding activity (Thibault et al., 2009; Brutinel et al., 2010). ExsA dissociated from the ExsD•ExsA complex through ExsC addition, however, is able to bind to DNA, indicating that ExsD does not irreversibly inactivate ExsA (Brutinel et al., 2010). The initial and rapid transcriptional response to inducing signals, therefore, likely reflects dissociation of existing ExsD•ExsA complexes by ExsC.
The response to non-inducing signals (i.e., high Ca2+) appears more complicated because neither purified trimeric ExsD, nor ExsD dissociated from the ExsC•ExsD complex, is capable of binding to ExsA in vitro (Thibault et al., 2009; Brutinel et al., 2010). These data, when combined with the observation that ExsD•ExsA complex formation is dependent upon concurrent expression of the proteins in vivo, suggest that complex formation may involve a folding intermediate of either ExsD or ExsA. ExsD dissociated from the ExsC•ExsD complex, therefore, does not contribute to the inhibition of ExsA-dependent transcription. Furthermore, the fact that trimeric ExsD is unable to bind to ExsA suggests two potential pathways for irreversible inactivation of ExsD: homo–trimerization and ExsC•ExsD complex formation. The response to inhibiting signals (i.e., high Ca2+) exhibits a lag period (Diaz and Yahr, unpublished) that may reflect a requisite decay of the existing free ExsA and a requirement for de novo synthesis of ExsD to sequester the newly synthesized ExsA.
ExsD inhibits both the self-association and DNA-binding properties of ExsA (Thibault et al., 2009; Brutinel et al., 2010). Since the amino-terminal domain of ExsA mediates self-association and is sufficient for complex formation with ExsD, ExsD likely inhibits ExsA self-association through steric hindrance. Less clear is how ExsD inhibits the carboxy-terminal DNA-binding domain when bound to the amino-terminus of ExsA. Resolution of this question will require structural studies to determine whether bound ExsD elicits a conformational change to the DNA-binding domain of ExsA or physically occludes one or both of the helix-turn-helix DNA-binding domains.
Physiological Implications of the ExsADCE Regulatory Cascade
The T3SS functions as a primary defense mechanism against phagocytosis and must be rapidly activated following contact with a phagocyte. Prior to host cell contact, P. aeruginosa cells already express low levels of preexisting injectisomes (Rietsch and Mekalanos, 2006) and effectors that initiate the defense response. The ExsADCE regulatory cascade functions to increase the magnitude of the response by rapidly inducing expression of the injectisome components and effectors. While upregulation of T3SS gene expression would be important at the level of individual cells, in the context of an infection this requirement may not extend to the population as a whole. Single cell expression studies have demonstrated that T3SS gene expression exhibits bistability whereby only a fraction of the cells demonstrate increased expression in response to inducing signals (both low Ca2+ and host cell contact; Hornef et al., 2000; Rietsch et al., 2004; Urbanowski et al., 2007). Bistability is a stochastic process generally reflecting noise in the expression of a master regulator (Dubnau and Losick, 2006). Since minor changes in the expression levels of ExsA, ExsD, ExsC, or ExsE have profound effects on T3SS gene expression, bistable expression of the T3SS is likely mediated through the ExsADCE regulatory cascade. The biological significance of bistability is not entirely clear. Bistability might reflect energetic considerations whereby only a subpopulation of T3SS-expressing P. aeruginosa are necessary to neutralize phagocytic clearance or elicit T3SS-dependent cytotoxicity during later stages of colonization. In this context, cells lacking T3SS gene expression could be likened to the social cheaters described in quorum sensing responses (Sandoz et al., 2007). Alternatively, bistability may reflect a state of heightened activation that allows a fraction of the cells to respond rapidly to predators. Finally, there may be a selective advantage for cells that lack T3SS gene expression such as avoidance of antibody responses directed against the injectisome.
Whereas the ExsADCE system allows for rapid induction of T3SS gene expression, there is a notable lag period in down-regulation following exposure to high Ca2+ conditions (as described above) and this lag period likely occurs following the loss of host cell contact as well. This regulatory feature may provide for a slow taper in T3SS gene expression which, during the interim period between full induction and the return to basal expression levels, serves as a safeguard against subsequent encounters with phagocytes.
Extrinsic Regulation of T3SS Gene Expression
The ExsADCE regulatory pathway is central to T3SS gene regulation but does not operate in isolation. As described in a previous review (Yahr and Wolfgang, 2006), many extrinsic regulatory pathways involving no less than 30 gene products influence T3SS gene expression. In nearly every case, the mechanisms by which they exert control over T3SS gene expression is poorly understood. One of the future challenges facing the field is to account for regulation by each of these pathways and identify the signals to which they respond. Despite significant knowledge gaps, molecular details are beginning to emerge for some of these systems.
Global Regulation of T3SS Gene Expression by Vfr
Virulence factor regulator (Vfr) is a cAMP-dependent regulator of transcription that was first identified as an activator of extracellular protease and exotoxin A expression (West et al., 1994). Now appreciated as a global regulator of virulence gene expression, the Vfr regulon consists of ~200 genes including type IV pili, a type II secretion system, quorum sensing, and the T3SS (Wolfgang et al., 2003). In addition to Vfr, the system consists of two adenylate cyclases (CyaA and CyaB) and a cAMP phosphodiesterase (Figure 2; CpdA; Wolfgang et al., 2003; Fuchs et al., 2010). Both high osmolarity (NaCl) and low Ca2+ stimulate cAMP production (Wolfgang et al., 2003), and cAMP homeostasis is tightly controlled by CpdA (Fuchs et al., 2010). Although a Vfr consensus binding sequence has been determined (Kanack et al., 2006), the mechanism by which Vfr controls T3SS gene expression remains unknown and represents a significant milestone for future studies.
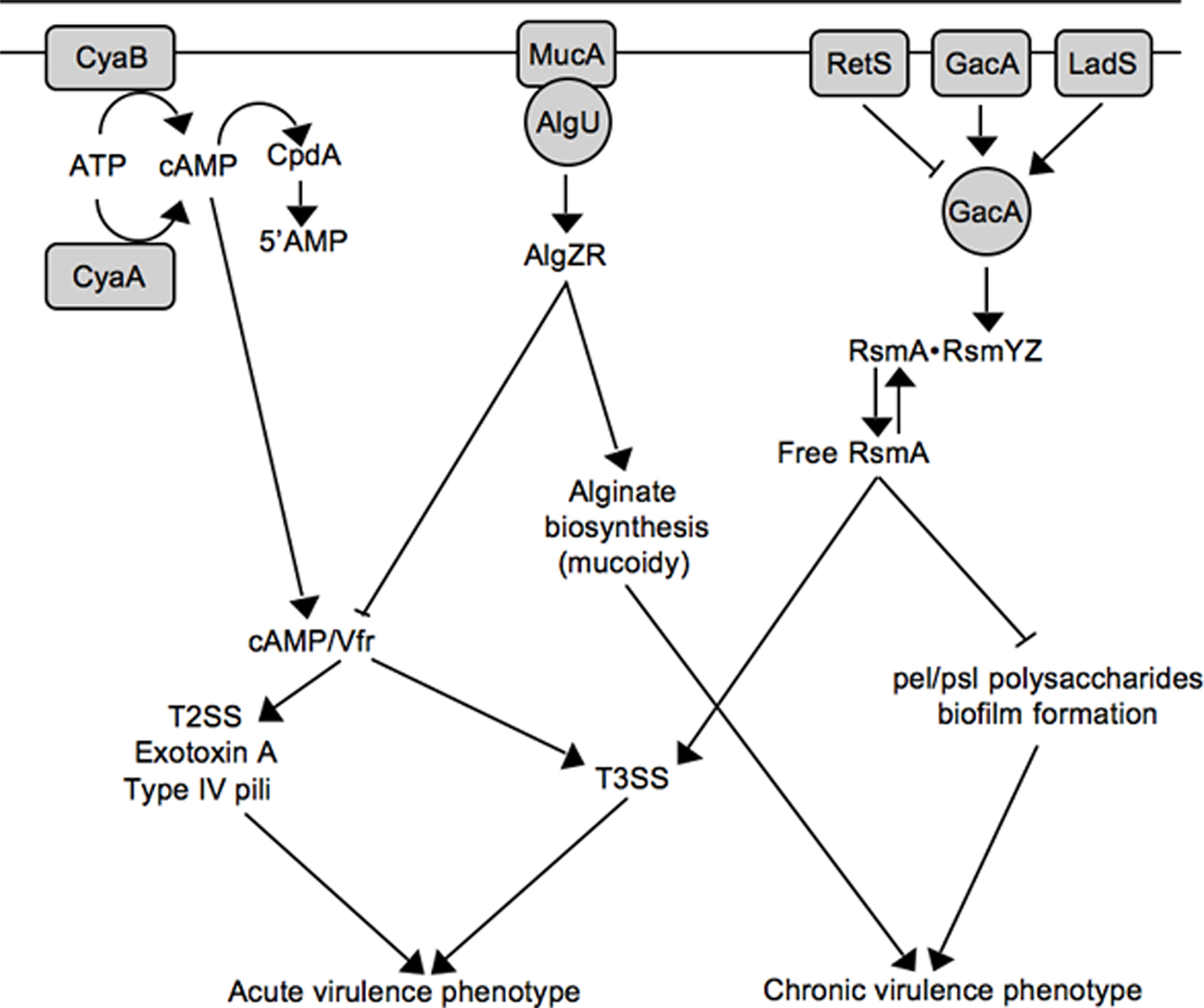
Figure 2. Working model for inverse regulation of T3SS with mucoidy and biofilm formation. See text for details.
Inverse Regulation of Mucoidy and T3SS Gene Expression
The role of the P. aeruginosa T3SS is well established within the context of acute infections where it contributes to tissue damage, sepsis, and mortality (Engel and Balachandran, 2009). P. aeruginosa is also the most common cause of chronic airway infection in individuals with cystic fibrosis (CF). CF infections are characterized by biofilm growth, slow progression, minimal tissue damage, and restriction to the airways (Gomez and Prince, 2007). The mechanistic basis for the striking difference in the progression of acute (non-CF) and chronic CF infections is not well understood. One contributing factor is that many P. aeruginosa virulence determinants are differentially expressed during non-CF and CF infections. Although CF and non-CF patients are initially colonized with nearly identical strains of P. aeruginosa, the majority of chronic CF isolates are mucoid and lack T3SS gene expression (Alonso et al., 1999; Feltman et al., 2001; Allmond et al., 2004). Mucoid conversion results from overproduction of the alginate polysaccharide and is associated with a decline in lung function and clinical prognosis (Pedersen et al., 1992; Li et al., 2005). Mucoid conversion and the loss of T3SS gene expression are now known to be linked events involving mucA mutation (Wu et al., 2004; Jones et al., 2010).
Alginate biosynthesis is regulated by the AlgU alternative sigma factor and the membrane-bound MucA anti-sigma factor (Wozniak and Ohman, 1994; Govan and Deretic, 1996). MucA normally sequesters AlgU to prevent expression of the AlgU regulon (Figure 2; Schurr et al., 1996; Mathee et al., 1997). MucA is (indirectly) responsive to a variety of environmental signals (primarily stress signals) resulting in the release of AlgU (Wood and Ohman, 2009). Liberated AlgU activates many genes including the AlgZR two-component regulatory system and the alginate biosynthetic genes (Wozniak and Ohman, 1994). While environmental signals represent one mechanism for activation of the AlgU regulon, mucoid conversion in CF isolates usually results from nonsense mutations in mucA (Boucher et al., 1997; Mathee et al., 1997). Jin and colleagues were the first to observe that mucA mutants are defective for T3SS gene expression and found the inhibition to be mediated through the AlgZR two-component system (Wu et al., 2004). Wolfgang and colleagues later noted that vfr and mucA mutants share several common phenotypes including reduced expression of the T3SS, T2SS, exotoxin A, and type IV pili, and subsequently confirmed that mucA mutants are defective for vfr transcription (Figure 2; Jones et al., 2010). The lack of T3SS gene expression in mucA mutants, therefore, reflects a defect in cAMP/Vfr signaling. Coincident mucoid conversion and loss of cAMP/Vfr signaling suggests that the selective advantage of mucA mutants in the CF airways may extend beyond mucoidy itself (Rau et al., 2010). Finally, the observation that algZR mutants have increased T3SS gene expression suggests that AlgZR modulates expression of the T3SS even in the presence of functional MucA (Jones et al., 2010). Since the MucA/AlgU system controls a generalized stress response, an interesting question for the future is whether stress signals encountered within the host modulate T3SS gene expression through MucA/AlgU.
T3SS Gene Expression and Biofilm Formation
Like alginate production, P. aeruginosa biofilms are thought to contribute to the chronic nature of CF infections (Singh et al., 2000). Biofilm formation is a complex adaptation subject to multiple layers of regulation (Harmsen et al., 2010). One regulatory mechanism involves the RetS and LadS sensor kinases. Whereas a retS (regulator of exopolysaccharide and type III secretion) mutant is defective for T3SS gene expression and displays enhanced biofilm formation, a ladS mutant has the reciprocal phenotypes (Goodman et al., 2004; Ventre et al., 2006). Mechanistically, RetS and LadS mediate opposite effects on activation of the GacAS system (Figure 2). The GacAS system regulates biofilm formation through control of the Pel and Psl polysaccharides, key components of the biofilm matrix (Ryder et al., 2007). Psl biosynthesis is controlled at the post-transcriptional level by RsmA, an RNA-binding protein that binds directly to the 5’ UTR region of the pslA mRNA to inhibit translational initiation (Irie et al., 2010). In response to unknown signals, GacA activates transcription of two small non-coding regulatory RNAs (RsmY and RsmZ). RsmY and RsmZ each have ~4–6 binding sites for RsmA and function by sequestering free RsmA from target mRNAs (Lapouge et al., 2008). Decreased levels of free RsmA, therefore, are required for Psl expression. In contrast, T3SS gene expression is dependent upon increased levels of free RsmA (Goodman et al., 2004; Mulcahy et al., 2006). The mechanism by which RsmA promotes T3SS gene expression is poorly understood. RsmA is a member of the CsrA family of proteins first described in E. coli (Romeo et al., 1993). Although CsrA family members usually prevent translation by binding to or near the Shine–Dalgarno (SD) sequence of target mRNAs (Lapouge et al., 2008), direct positive regulation by CsrA has also been reported and involves mRNA stabilization (Wei et al., 2001). This leaves several scenarios to account for positive regulation of T3SS gene expression by RsmA. RsmA may function directly at the level of intrinsic regulation by promoting expression of a positive (ExsA or ExsC) acting factor or inhibiting expression of negative (ExsD or ExsE) regulator. Alternatively, RsmA may function indirectly by modulating an extrinsic regulatory pathway that controls the T3SS. With respect to the latter possibility it is noteworthy that the RsmA regulon is extensive and includes rhamnolipid production, lipase production, swarming motility, quorum sensing, lectin, pyocyanin, hydrogen cyanide, and the type VI secretion system (Pessi et al., 2001; Heurlier et al., 2004; Brencic and Lory, 2009). The possibility exists, therefore, that the regulatory activity of RsmA toward T3SS gene expression results from pleiotropic effects that may be difficult to define.
The observation of inverse regulation by RetS and LadS implied that T3SS gene expression and biofilm formation were mutually exclusive events. It is now becoming apparent, however, that such a model is an over-simplification and that the T3SS is possible during biofilm growth (Mikkelsen et al., 2009).
T3SS Gene Expression and Motility
All three forms of P. aeruginosa motility (type IV pili-based twitching, swarming, and flagellar-based swimming,) are coordinated (either positively or negatively) with T3SS gene expression. As described above, type IV pili and T3SS gene expression are positively regulated by Vfr (Jones et al., 2010). The biological significance of this relationship reflects the adherence properties provided by type IV pili, which function as the primary adhesin for host-contact-dependent expression of the T3SS (Sundin et al., 2002). T3SS gene expression is also positively correlated with swarming motility, which might be advantageous during colonization of mucosal surfaces (Overhage et al., 2008). In contrast, flagellar gene expression is inversely regulated with T3SS gene expression. Whereas a fliC mutant is deregulated for T3SS gene expression (three- to ninefold) and displays increased cytotoxicity toward a macrophage cell line, ExsA over-expression reduces motility and expression of a fliA transcriptional reporter (Soscia et al., 2007). The reciprocal nature of T3SS and flagellar gene expression is interesting to contemplate in the context of inflammasome activation. Several studies suggest that flagellin (FliC) translocated into macrophages by the P. aeruginosa T3SS activates the inflammasome leading to a proinflammatory response (Franchi et al., 2007; Galle et al., 2007; Sutterwala et al., 2007; Miao et al., 2008). Despite experimental evidence of inflammasome activation ex vivo, it is unclear whether this occurs in vivo because the ExoS and ExoU effectors both block FliC-dependent inflammasome activation. The additional finding that FliC expression is inversely regulated with T3SS gene expression suggests that inflammasome activation is either relatively unimportant, does not normally occur during infections, or occurs under conditions that remain to be defined.
Environmental Signals that Modulate T3SS Gene Expression
Ca2+-limitation and host cell contact are well-defined signals that activate transcription through the control of T3SS secretory activity. Many additional signals, arising from both endogenous and exogenous sources, also influence T3SS gene expression and are summarized in Table 1. Evident is the fact that these signals are diverse with stress and metabolic signals representing the largest class. Less obvious is that the mechanisms by which these signals influence T3SS gene expression are poorly understood. In fact, besides ExsA only one additional transcriptional factor (PsrA) is known to directly regulate the T3SS. PsrA binds the PexsCpromoter to positively control transcription of the exsCEBA operon (Shen et al., 2006). Interestingly, the DNA-binding activity of PsrA is directly inhibited by long chain fatty acids. Indeed T3SS gene expression is reduced in the presence of long chain fatty acids, although the biological significance of this observation is unclear (Kang et al., 2009). The involvement of PsrA is discussed here to illustrate the point that cross-talk between different regulatory systems coordinates T3SS gene expression with diverse cellular functions. In addition to the T3SS, PsrA regulates a diverse set of genes including type IV pili (a potential link to Vfr), the psl biosynthesis operon (a potential link to RsmA), the stationary phase sigma factor rpoS (a potential link to quorum sensing) and a variety of genes involved in metabolic and energetic metabolism. Furthermore, psrA mutants are defective for swarming motility, biofilm formation, and resistance to some antibiotics. In just this one example, therefore, one begins to appreciate the level of complexity involved in controlling T3SS gene expression. Despite the involvement of numerous signals and gene products, it seems likely that reductionist approaches will ultimately reveal that only a minority (i.e., PsrA, Vfr, RsmA) of the genes implicated in regulation of the T3SS directly participate in the control of T3SS gene expression. Future explorations aimed as examining whether signals and gene products exert their regulatory effects at the level of ExsADCE or through upstream regulatory pathways are required to fully integrate the regulatory pathways leading to T3SS gene expression.
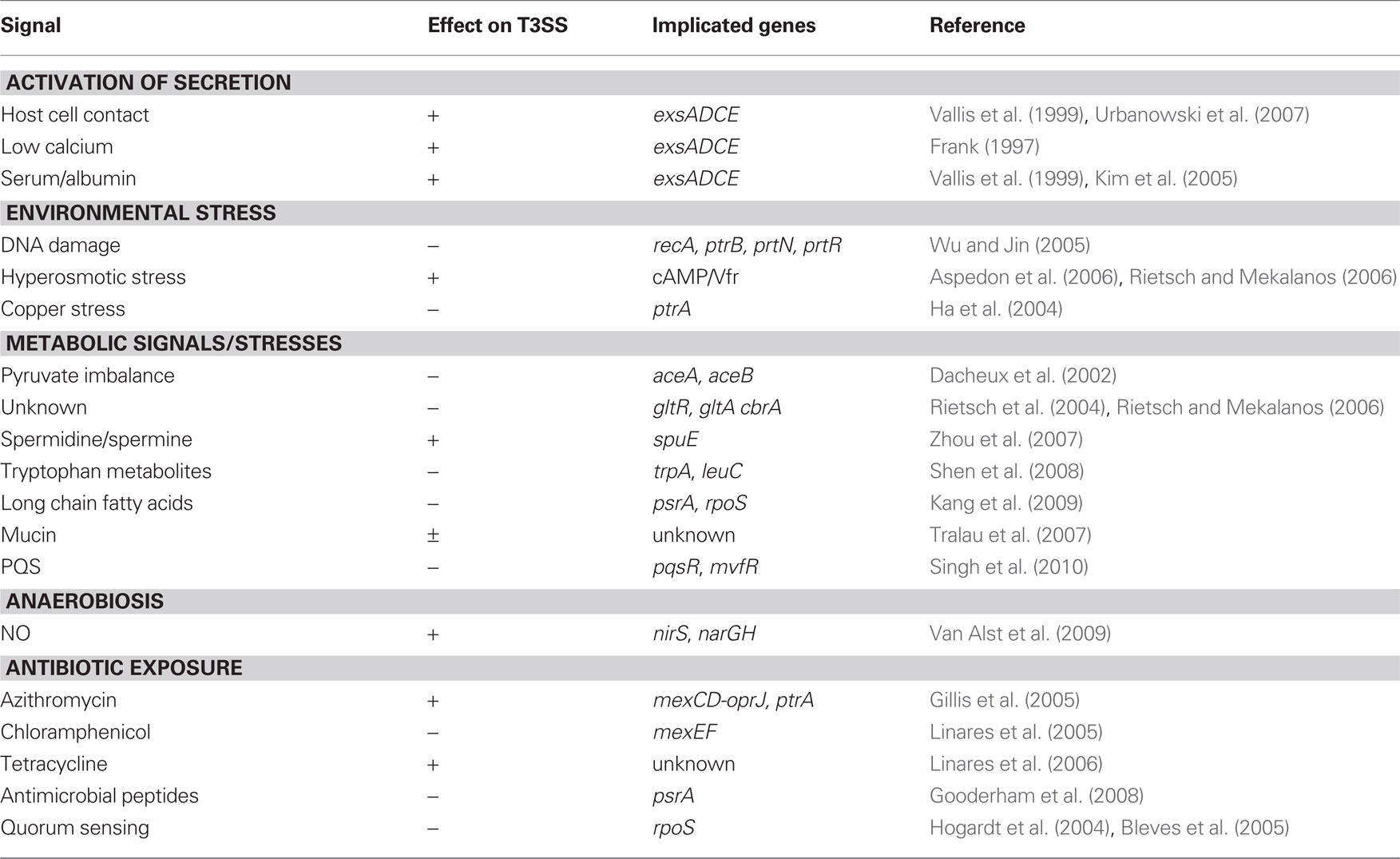
Table 1. Environmental signals that influence T3SS gene expression.
Challenges for the Future
The literature contains a wealth of information regarding genes and conditions that contribute to T3SS gene expression in P. aeruginosa. The challenge ahead lies in assimilating this information into a unified regulatory network supportive of all prior and future observations. In absolute terms this will undoubtedly prove impossible owing to differences in mutant phenotypes, strain backgrounds, growth conditions, and experimental findings. In analyzing the primary literature consideration of the following points is important to keep in mind: (i) Nearly 20 microarray studies have noted alterations in T3SS gene expression. Most of these studies were conducted under non-permissive conditions for T3SS gene expression (i.e., high Ca2+) making it difficult to ascertain whether the observed changes represent minor alterations to the basal level of T3SS transcription or more significant regulatory effects. (ii) To the best of our knowledge, plasmid-expressed ExsA is able to restore transcription to all mutants identified as defective for T3SS transcription. Reports of regulatory effects occurring upstream of ExsA, therefore, must be interpreted with caution since ExsA over-expression can easily obscure the activity of and/or requirement for other regulatory systems. (iii) Although most studies of T3SS gene expression have relied upon Ca2+ chelation as the inducing signal, it is unlikely that Ca2+ chelation and host cell contact are entirely analogous signals. Future studies are required to fully define the physiologically relevant signals encountered in vivo and determine how they modulate or fine-tune T3SS gene expression. (iv) The induction of T3SS gene expression is minimally dependent upon a functional injectisome, the ExsADCE cascade, and the Vfr and RsmA global regulatory systems and is an important consideration in that regulatory phenotypes that alter T3SS gene expression might result from pleiotropic effects without actually contributing to the regulatory process per se.
Therapeutic targeting of the T3SS is an achievable goal with potential intervention points including the inhibition of assembly, secretion, translocation, and T3SS gene expression. Since ExsA-dependent transcription is dependent upon an intact secretion and translocation machinery, each class of inhibitor offers the added benefit of reduced T3SS gene expression. Most efforts to date have logically targeted the highly conserved secretion/translocation machinery with the hope of identifying inhibitors that possess activity against multiple pathogens. A limitation of this approach lies in identification of the inhibitor target since each of the ~25 constituents of the secretion/translocation machinery as well as their interaction interfaces represent potential target sites. Direct targeting of transcription offers the advantage that many promising targets already exist including the ExsADCE regulatory cascade, and the cAMP/Vfr, MucA/AlgU, and GacA/RsmA global regulatory pathways, and in fact inhibitors of ExsA-dependent transcription have already been identified (Grier et al., 2010). Time will tell whether such inhibitors can deliver on the promise of controlling P. aeruginosa infections.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mark Urbanowski for critical discussions and input during the writing of this review. Work in the Yahr laboratory is supported by the National Institutes of Health (RO1-AI055042-08).
References
Allmond, L. R., Ajayi, T., Moriyama, K., Wiener-Kronish, J. P., and Sawa, T. (2004). V-antigen genotype and phenotype analyses of clinical isolates of Pseudomonas aeruginosa. J. Clin. Microbiol. 42, 3857–3860.
Alonso, A., Rojo, F., and Martinez, J. L. (1999). Environmental and clinical isolates of Pseudomonas aeruginosa show pathogenic and biodegradative properties irrespective of their origin. Environ. Microbiol. 1, 421–430.
Aspedon, A., Palmer, K., and Whiteley, M. (2006). Microarray analysis of the osmotic stress response in Pseudomonas aeruginosa. J. Bacteriol. 188, 2721–2725.
Bernhards, R. C., Jing, X., Vogelaar, N. J., Robinson, H., and Schubot, F. D. (2009). Structural evidence suggests that antiactivator ExsD from Pseudomonas aeruginosa is a DNA binding protein. Protein Sci. 18, 503–513.
Bleves, S., Soscia, C., Nogueira-Orlandi, P., Lazdunski, A., and Filloux, A. (2005). Quorum sensing negatively controls type III secretion regulon expression in Pseudomonas aeruginosa PAO1. J. Bacteriol. 187, 3898–3902.
Boucher, J. C., Yu, H., Mudd, M. H., and Deretic, V. (1997). Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect. Immun. 65, 3838–3846.
Brannon, M. K., Davis, J. M., Mathias, J. R., Hall, C. J., Emerson, J. C., Crosier, P. S., Huttenlocher, A., Ramakrishnan, L., and Moskowitz, S. M. (2009). Pseudomonas aeruginosa type III secretion system interacts with phagocytes to modulate systemic infection of zebrafish embryos. Cell. Microbiol. 11, 755–768.
Brencic, A., and Lory, S. (2009). Determination of the regulon and identification of novel mRNA targets of Pseudomonas aeruginosa RsmA. Mol. Microbiol. 72, 612–632.
Brutinel, E. D., Vakulskas, C. A., Brady, K. M., and Yahr, T. L. (2008). Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 68, 657–671.
Brutinel, E. D., Vakulskas, C. A., and Yahr, T. L. (2009). Functional domains of ExsA, the transcriptional activator of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 191, 3811–3821.
Brutinel, E. D., Vakulskas, C. A., and Yahr, T. L. (2010). ExsD inhibits expression of the Pseudomonas aeruginosa type III secretion system by disrupting ExsA self-association and DNA binding activity. J. Bacteriol. 192, 1479–1486.
Brutinel, E. D., and Yahr, T. L. (2008). Control of gene expression by type III secretory activity. Curr. Opin. Microbiol. 11, 128–133.
Coburn, B., Sekirov, I., and Finlay, B. B. (2007). Type III secretion systems and disease. Clin. Microbiol. Rev. 20, 535–549.
Dacheux, D., Epaulard, O., De Groot, A., Guery, B., Leberre, R., Attree, I., Polack, B., and Toussaint, B. (2002). Activation of the Pseudomonas aeruginosa type III secretion system requires an intact pyruvate dehydrogenase aceAB operon. Infect. Immun. 70, 3973–3977.
Dasgupta, N., Lykken, G. L., Wolfgang, M. C., and Yahr, T. L. (2004). A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 53, 297–308.
Engel, J., and Balachandran, P. (2009). Role of Pseudomonas aeruginosa type III effectors in disease. Curr. Opin. Microbiol. 12, 61–66.
Feltman, H., Schulert, G., Khan, S., Jain, M., Peterson, L., and Hauser, A. R. (2001). Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology 147, 2659–2669.
Franchi, L., Stoolman, J., Kanneganti, T. D., Verma, A., Ramphal, R., and Nunez, G. (2007). Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur. J. Immunol. 37, 3030–3039.
Frank, D. W. (1997). The exoenzyme S regulon of Pseudomonas aeruginosa. Mol. Microbiol. 26, 621–629.
Fuchs, E. L., Brutinel, E. D., Klem, E. R., Fehr, A. R., Yahr, T. L., and Wolfgang, M. C. (2010). In vitro and in vivo characterization of the Pseudomonas aeruginosa cyclic AMP (cAMP) phosphodiesterase CpdA, required for cAMP homeostasis and virulence factor regulation. J. Bacteriol. 192, 2779–2790.
Galle, M., Schotte, P., Haegman, M., Wullaert, A., Yang, H. J., Jin, S., and Beyaert, R. (2007). The Pseudomonas aeruginosa type III secretion system plays a dual role in the regulation of caspase-1 mediated IL-1beta maturation. J. Cell. Mol. Med. 12, 1767–1776.
Gillis, R. J., White, K. G., Choi, K. H., Wagner, V. E., Schweizer, H. P., and Iglewski, B. H. (2005). Molecular basis of azithromycin-resistant Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 49, 3858–3867.
Gomez, M. I., and Prince, A. (2007). Opportunistic infections in lung disease: Pseudomonas infections in cystic fibrosis. Curr. Opin. Pharmacol. 7, 244–251.
Gooderham, W. J., Bains, M., Mcphee, J. B., Wiegand, I., and Hancock, R. E. (2008). Induction by cationic antimicrobial peptides and involvement in intrinsic polymyxin and antimicrobial peptide resistance, biofilm formation, and swarming motility of PsrA in Pseudomonas aeruginosa. J. Bacteriol. 190, 5624–5634.
Goodman, A. L., Kulasekara, B., Rietsch, A., Boyd, D., Smith, R. S., and Lory, S. (2004). A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 7, 745–754.
Govan, J. R., and Deretic, V. (1996). Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60, 539–574.
Grier, M. C., Garrity-Ryan, L. K., Bartlett, V. J., Klausner, K. A., Donovan, P. J., Dudley, C., Alekshun, M. N., Tanaka, S. K., Draper, M. P., Levy, S. B., and Kim, O. K. (2010). N-hydroxybenzimidazole inhibitors of ExsA MAR transcription factor in Pseudomonas aeruginosa: in vitro anti-virulence activity and metabolic stability. Bioorg. Med. Chem. Lett. 20, 3380–3383.
Ha, U. H., Kim, J., Badrane, H., Jia, J., Baker, H. V., Wu, D., and Jin, S. (2004). An in vivo inducible gene of Pseudomonas aeruginosa encodes an anti-ExsA to suppress the type III secretion system. Mol. Microbiol. 54, 307–320.
Harmsen, M., Yang, L., Pamp, S. J., and Tolker-Nielsen, T. (2010). An update on Pseudomonas aeruginosa biofilm formation, tolerance, and dispersal. FEMS Immunol. Med. Microbiol. 59, 253–268.
Hauser, A. R. (2009). The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7, 654–665.
Hayes, C. S., Aoki, S. K., and Low, D. A. (2010). Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 44, 71–90.
Heurlier, K., Williams, F., Heeb, S., Dormond, C., Pessi, G., Singer, D., Camara, M., Williams, P., and Haas, D. (2004). Positive control of swarming, rhamnolipid synthesis, and lipase production by the posttranscriptional RsmA/RsmZ system in Pseudomonas aeruginosa PAO1. J. Bacteriol. 186, 2936–2945.
Hogardt, M., Roeder, M., Schreff, A. M., Eberl, L., and Heesemann, J. (2004). Expression of Pseudomonas aeruginosa exoS is controlled by quorum sensing and RpoS. Microbiology 150, 843–851.
Hornef, M. W., Roggenkamp, A., Geiger, A. M., Hogardt, M., Jacobi, C. A., and Heesemann, J. (2000). Triggering the ExoS regulon of Pseudomonas aeruginosa: a GFP-reporter analysis of exoenzyme (Exo) S, ExoT and ExoU synthesis. Microb. Pathog. 29, 329–343.
Hovey, A. K., and Frank, D. W. (1995). Analyses of the DNA-binding and transcriptional activation properties of ExsA, the transcriptional activator of the Pseudomonas aeruginosa exoenzyme S regulon. J. Bacteriol. 177, 4427–4436.
Irie, Y., Starkey, M., Edwards, A. N., Wozniak, D. J., Romeo, T., and Parsek, M. R. (2010). Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol. Microbiol. 78, 158–172.
Jones, A. K., Fulcher, N. B., Balzer, G. J., Urbanowski, M. L., Pritchett, C. L., Schurr, M. J., Yahr, T. L., and Wolfgang, M. C. (2010). Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. J. Bacteriol. 192, 5709–5717.
Kanack, K. J., Runyen-Janecky, L. J., Ferrell, E. P., Suh, S. J., and West, S. E. (2006). Characterization of DNA-binding specificity and analysis of binding sites of the Pseudomonas aeruginosa global regulator, Vfr, a homologue of the Escherichia coli cAMP receptor protein. Microbiology 152, 3485–3496.
Kang, Y., Lunin, V. V., Skarina, T., Savchenko, A., Schurr, M. J., and Hoang, T. T. (2009). The long-chain fatty acid sensor, PsrA, modulates the expression of rpoS and the type III secretion exsCEBA operon in Pseudomonas aeruginosa. Mol. Microbiol. 73, 120–136.
Kim, J., Ahn, K., Min, S., Jia, J., Ha, U., Wu, D., and Jin, S. (2005). Factors triggering type III secretion in Pseudomonas aeruginosa. Microbiology 151, 3575–3587.
Lapouge, K., Schubert, M., Allain, F. H., and Haas, D. (2008). Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol. Microbiol. 67, 241–253.
Li, Z., Kosorok, M. R., Farrell, P. M., Laxova, A., West, S. E., Green, C. G., Collins, J., Rock, M. J., and Splaingard, M. L. (2005). Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 293, 581–588.
Linares, J. F., Gustafsson, I., Baquero, F., and Martinez, J. L. (2006). Antibiotics as intermicrobial signaling agents instead of weapons. Proc. Natl. Acad. Sci. U.S.A. 103, 19484–19489.
Linares, J. F., Lopez, J. A., Camafeita, E., Albar, J. P., Rojo, F., and Martinez, J. L. (2005). Overexpression of the multidrug efflux pumps MexCD-OprJ and MexEF-OprN is associated with a reduction of type III secretion in Pseudomonas aeruginosa. J. Bacteriol. 187, 1384–1391.
Lykken, G. L., Chen, G., Brutinel, E. D., Chen, L., and Yahr, T. L. (2006). Characterization of ExsC and ExsD self-association and heterocomplex formation. J. Bacteriol. 188, 6832–6840.
Mathee, K., Mcpherson, C. J., and Ohman, D. E. (1997). Posttranslational control of the algT (algU)-encoded sigma22 for expression of the alginate regulon in Pseudomonas aeruginosa and localization of its antagonist proteins MucA and MucB (AlgN). J. Bacteriol. 179, 3711–3720.
McCaw, M. L., Lykken, G. L., Singh, P. K., and Yahr, T. L. (2002). ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol. Microbiol. 46, 1123–1133.
Miao, E. A., Ernst, R. K., Dors, M., Mao, D. P., and Aderem, A. (2008). Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. U.S.A. 105, 2562–2567.
Mikkelsen, H., Bind, N. J., Skindersoe, M. E., Givskov, M., Lilley, K. S., and Welch, M. (2009). Biofims and type III secretion are not mutually exclusive in Pseudomonas aeruginosa. Microbiology 155, 687–698.
Miyata, S., Casey, M., Frank, D. W., Ausubel, F. M., and Drenkard, E. (2003). Use of the Galleria mellonella caterpillar as a model host to study the role of the type III secretion system in Pseudomonas aeruginosa pathogenesis. Infect. Immun. 71, 2404–2413.
Moraes, T. F., Spreter, T., and Strynadka, N. C. (2008). Piecing together the type III injectisome of bacterial pathogens. Curr. Opin. Struct. Biol. 18, 258–266.
Mulcahy, H., O’callaghan, J., O’grady, E. P., Adams, C., and O’gara, F. (2006). The posttranscriptional regulator RsmA plays a role in the interaction between Pseudomonas aeruginosa and human airway epithelial cells by positively regulating the type III secretion system. Infect. Immun. 74, 3012–3015.
Overhage, J., Bains, M., Brazas, M. D., and Hancock, R. E. (2008). Swarming of Pseudomonas aeruginosa is a complex adaptation leading to increased production of virulence factors and antibiotic resistance. J. Bacteriol. 190, 2671–2679.
Pedersen, S. S., Hoiby, N., Espersen, F., and Koch, C. (1992). Role of alginate in infection with mucoid Pseudomonas aeruginosa in cystic fibrosis. Thorax 47, 6–13.
Pessi, G., Williams, F., Hindle, Z., Heurlier, K., Holden, M. T., Camara, M., Haas, D., and Williams, P. (2001). The global posttranscriptional regulator RsmA modulates production of virulence determinants and N-acylhomoserine lactones in Pseudomonas aeruginosa. J. Bacteriol. 183, 6676–6683.
Pukatzki, S., Kessin, R. H., and Mekalanos, J. J. (2002). The human pathogen Pseudomonas aeruginosa utilizes conserved virulence pathways to infect the social amoeba Dictyostelium discoideum. Proc. Natl. Acad. Sci. U.S.A. 99, 3159–3164.
Rau, M. H., Hansen, S. K., Johansen, H. K., Thomsen, L. E., Workman, C. T., Nielsen, K. F., Jelsbak, L., Hoiby, N., Yang, L., and Molin, S. (2010). Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ. Microbiol. 12, 1643–1658.
Rietsch, A., and Mekalanos, J. J. (2006). Metabolic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Mol. Microbiol. 59, 807–820.
Rietsch, A., Vallet-Gely, I., Dove, S. L., and Mekalanos, J. J. (2005). ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 102, 8006–8011.
Rietsch, A., Wolfgang, M. C., and Mekalanos, J. J. (2004). Effect of metabolic imbalance on expression of type III secretion genes in Pseudomonas aeruginosa. Infect. Immun. 72, 1383–1390.
Romeo, T., Gong, M., Liu, M. Y., and Brun-Zinkernagel, A. M. (1993). Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J. Bacteriol. 175, 4744–4755.
Ryder, C., Byrd, M., and Wozniak, D. J. (2007). Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Curr. Opin. Microbiol. 10, 644–648.
Sandoz, K. M., Mitzimberg, S. M., and Schuster, M. (2007). Social cheating in Pseudomonas aeruginosa quorum sensing. Proc. Natl. Acad. Sci. U.S.A. 104, 15876–15881.
Schurr, M. J., Yu, H., Martinez-Salazar, J. M., Boucher, J. C., and Deretic, V. (1996). Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J. Bacteriol. 178, 4997–5004.
Shen, D. K., Filopon, D., Chaker, H., Boullanger, S., Derouazi, M., Polack, B., and Toussaint, B. (2008). High-cell-density regulation of the Pseudomonas aeruginosa type III secretion system: implications for tryptophan catabolites. Microbiology 154, 2195–2208.
Shen, D. K., Filopon, D., Kuhn, L., Polack, B., and Toussaint, B. (2006). PsrA is a positive transcriptional regulator of the type III secretion system in Pseudomonas aeruginosa. Infect. Immun. 74, 1121–1129.
Singh, G., Wu, B., Baek, M. S., Camargo, A., Nguyen, A., Slusher, N. A., Srinivasan, R., Wiener-Kronish, J. P., and Lynch, S. V. (2010). Secretion of Pseudomonas aeruginosa type III cytotoxins is dependent on Pseudomonas quinolone signal concentration. Microb. Pathog. 49, 196–203.
Singh, P. K., Schaefer, A. L., Parsek, M. R., Moninger, T. O., Welsh, M. J., and Greenberg, E. P. (2000). Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407, 762–764.
Soscia, C., Hachani, A., Bernadac, A., Filloux, A., and Bleves, S. (2007). Cross talk between type III secretion and flagellar assembly systems in Pseudomonas aeruginosa. J. Bacteriol. 189, 3124–3132.
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., Brinkman, F. S., Hufnagle, W. O., Kowalik, D. J., Lagrou, M., Garber, R. L., Goltry, L., Tolentino, E., Westbrock-Wadman, S., Yuan, Y., Brody, L. L., Coulter, S. N., Folger, K. R., Kas, A., Larbig, K., Lim, R., Smith, K., Spencer, D., Wong, G. K., Wu, Z., Paulsen, I. T., Reizer, J., Saier, M. H., Hancock, R. E., Lory, S., and Olson, M. V. (2000). Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406, 959–964.
Sundin, C., Wolfgang, M. C., Lory, S., Forsberg, A., and Frithz-Lindsten, E. (2002). Type IV pili are not specifically required for contact dependent translocation of exoenzymes by Pseudomonas aeruginosa. Microb. Pathog. 33, 265–277.
Sutterwala, F. S., Mijares, L. A., Li, L., Ogura, Y., Kazmierczak, B. I., and Flavell, R. A. (2007). Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245.
Thibault, J., Faudry, E., Ebel, C., Attree, I., and Elsen, S. (2009). Anti-activator ExsD forms a 1:1 complex with ExsA to inhibit transcription of type III secretion operons. J. Biol. Chem. 284, 15762–15770.
Tralau, T., Vuilleumier, S., Thibault, C., Campbell, B. J., Hart, C. A., and Kertesz, M. A. (2007). Transcriptomic analysis of the sulfate starvation response of Pseudomonas aeruginosa. J. Bacteriol. 189, 6743–6750.
Urbanowski, M. L., Brutinel, E. D., and Yahr, T. L. (2007). Translocation of ExsE into Chinese hamster ovary cells is required for transcriptional induction of the Pseudomonas aeruginosa type III secretion system. Infect. Immun. 75, 4432–4439.
Urbanowski, M. L., Lykken, G. L., and Yahr, T. L. (2005). A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc. Natl. Acad. Sci. U.S.A. 102, 9930–9935.
Vakulskas, C. A., Brady, K. M., and Yahr, T. L. (2009). Mechanism of Transcriptional Activation by Pseudomonas aeruginosa ExsA. J. Bacteriol. 191, 6654–6664.
Vakulskas, C. A., Brutinel, E. D., and Yahr, T. L. (2010). ExsA recruits RNA polymerase to an extended-10 promoter by contacting region 4.2 of Sigma-70. J. Bacteriol. 192, 3597–3607.
Vallis, A. J., Yahr, T. L., Barbieri, J. T., and Frank, D. W. (1999). Regulation of ExoS production and secretion by Pseudomonas aeruginosa in response to tissue culture conditions. Infect. Immun. 67, 914–920.
Van Alst, N. E., Wellington, M., Clark, V. L., Haidaris, C. G., and Iglewski, B. H. (2009). Nitrite reductase NirS is required for type III secretion system expression and virulence in the human monocyte cell line THP-1 by Pseudomonas aeruginosa. Infect. Immun. 77, 4446–4454.
Ventre, I., Goodman, A. L., Vallet-Gely, I., Vasseur, P., Soscia, C., Molin, S., Bleves, S., Lazdunski, A., Lory, S., and Filloux, A. (2006). Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc. Natl. Acad. Sci. U.S.A. 103, 171–176.
Vogelaar, N. J., Jing, X., Robinson, H. H., and Schubot, F. D. (2010). Analysis of the crystal structure of the ExsC. ExsE complex reveals distinctive binding interactions of the Pseudomonas aeruginosa type III secretion chaperone ExsC with ExsE and ExsD. Biochemistry 49, 5870–5879.
Wagner, S., Konigsmaier, L., Lara-Tejero, M., Lefebre, M., Marlovits, T. C., and Galan, J. E. (2010). Organization and coordinated assembly of the type III secretion export apparatus. Proc. Natl. Acad. Sci. U.S.A. 41, 17745–17750.
Wei, B. L., Brun-Zinkernagel, A. M., Simecka, J. W., Pruss, B. M., Babitzke, P., and Romeo, T. (2001). Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol. Microbiol. 40, 245–256.
West, S. E., Sample, A. K., and Runyen-Janecky, L. J. (1994). The vfr gene product, required for Pseudomonas aeruginosa exotoxin A and protease production, belongs to the cyclic AMP receptor protein family. J. Bacteriol. 176, 7532–7542.
Wolfgang, M. C., Lee, V. T., Gilmore, M. E., and Lory, S. (2003). Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev. Cell 4, 253–263.
Wood, L. F., and Ohman, D. E. (2009). Use of cell wall stress to characterize sigma 22 (AlgT/U) activation by regulated proteolysis and its regulon in Pseudomonas aeruginosa. Mol. Microbiol. 72, 183–201.
Worrall, L. J., Lameignere, E., and Strynadka, N. C. (2011). Structural overview of the bacterial injectisome. Curr. Opin. Microbiol. 14, 3–8.
Wozniak, D. J., and Ohman, D. E. (1994). Transcriptional analysis of the Pseudomonas aeruginosa genes algR, algB, and algD reveals a hierarchy of alginate gene expression which is modulated by algT. J. Bacteriol. 176, 6007–6014.
Wu, W., Badrane, H., Arora, S., Baker, H. V., and Jin, S. (2004). MucA-mediated coordination of type III secretion and alginate synthesis in Pseudomonas aeruginosa. J. Bacteriol. 186, 7575–7585.
Wu, W., and Jin, S. (2005). PtrB of Pseudomonas aeruginosa suppresses the type III secretion system under the stress of DNA damage. J. Bacteriol. 187, 6058–6068.
Yahr, T. L., and Frank, D. W. (1994). Transcriptional organization of the trans-regulatory locus which controls exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 176, 3832–3838.
Yahr, T. L., Hovey, A. K., Kulich, S. M., and Frank, D. W. (1995). Transcriptional analysis of the Pseudomonas aeruginosa exoenzyme S structural gene. J. Bacteriol. 177, 1169–1178.
Yahr, T. L., and Wolfgang, M. C. (2006). Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 62, 631–640.
Zheng, Z., Chen, G., Joshi, S., Brutinel, E. D., Yahr, T. L., and Chen, L. (2007). Biochemical characterization of a regulatory cascade controlling transcription of the Pseudomonas aeruginosa type III secretion system. J. Biol. Chem. 282, 6136–6142.
Keywords: Pseudomonas aeruginosa, type III secretion, injectisome, gene regulation, ExsA, cAMP, RsmA, Vfr
Citation: Diaz MR, King JM and Yahr TL (2011) Intrinsic and extrinsic regulation of type III secretion gene expression in pseudomonas aeruginosa. Front. Microbio. 2:89. doi: 10.3389/fmicb.2011.00089
Received: 02 March 2011;
Paper pending published: 30 March 2011;
Accepted: 13 April 2011;
Published online: 25 April 2011.
Edited by:
Dara Frank, Medical College of Wisconsin, USAReviewed by:
William Picking, Oklahoma State University, USAJürgen Heesemann, Max von Pettenkofer-Institute, Germany
Matthew C. Wolfgang, University of North Carolina, USA
Copyright: © 2011 Diaz, King and Yahr. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Timothy L. Yahr, Department of Microbiology, Carver College of Medicine, University of Iowa, 540B EMRB, Iowa City, IA 52242, USA. e-mail:dGltLXlhaHJAdWlvd2EuZWR1