 Hubert Hilbi*
Hubert Hilbi* Stephen Weber and Ivo Finsel
Stephen Weber and Ivo Finsel
- Max von Pettenkofer Institute, Ludwig-Maximilians University, Munich, Germany
The facultative intracellular pathogen Legionella pneumophila replicates in free-living amoebae and macrophages within a distinct compartment, the “Legionella-containing vacuole” (LCV). LCV formation involves phosphoinositide (PI) glycerolipids, which are key factors controlling vesicle trafficking pathways and membrane dynamics of eukaryotic cells. To govern the interactions with host cells, L. pneumophila employs the Icm/Dot type IV secretion system and more than 250 translocated “effector proteins” that presumably subvert host signaling and vesicle trafficking pathways. Some of the effector proteins anchor through distinct PIs to the cytosolic face of LCVs and promote the interaction with host vesicles and organelles, catalyze guanine nucleotide exchange of small GTPases, or bind to PI-metabolizing enzymes, such as OCRL1. The PI 5-phosphatase OCRL1 and its Dictyostelium homologue Dd5P4 restrict intracellular growth of L. pneumophila. Moreover, OCRL1/Dd5P4, PI 3-kinases (PI3Ks), and PI4KIIIβ regulate LCV formation and localization of the effector protein SidC, which selectively decorates the LCV membrane. SidC and its 20-kDa “P4C” fragment are robust and specific probes for phosphatidylinositol-4-phosphate, and SidC can be targeted to purify intact LCVs by immuno-magnetic separation. Taken together, bacterial PI-binding effectors as well as host PIs and PI-modulating enzymes play a pivotal role for intracellular replication of L. pneumophila, and the PI-binding effectors are valuable tools for the analysis of eukaryotic PI lipids.
Introduction
The causative agent of Legionnaireś pneumonia, Legionella pneumophila, replicates intracellularly in free-living amoebae and macrophages of the innate immune system. Within these phagocytic host cells, the bacteria employ a conserved mechanism to form a unique replication-permissive compartment, the “Legionella-containing vacuole” (LCV). Thus, amoeba and in particular the genetically tractable social soil amoeba Dictyostelium discoideum, are valuable models to study the mechanism of LCV formation on a molecular and cellular level (Solomon and Isberg, 2000; Steinert and Heuner, 2005; Hilbi et al., 2007; Cosson and Soldati, 2008). Within macrophages and amoebae, LCVs avoid fusion with lysosomes, but associate with mitochondria and smooth vesicles and eventually fuse with the endoplasmic reticulum (ER; Horwitz, 1983; Lu and Clarke, 2005; Robinson and Roy, 2006). To accommodate the transfer between host cells and environmental niches, L. pneumophila switches from a replicative to a transmissive growth phase, which involves a complex gene regulation network, including an apparent quorum sensing system (Molofsky and Swanson, 2004; Tiaden et al., 2010; Hilbi et al., 2011).
Intact LCVs from infected D. discoideum amoebae can be isolated and purified using a simple two-step protocol (Urwyler et al., 2010). To this end, D. discoideum producing the LCV and ER marker calnexin-GFP were infected with L. pneumophila fluorescently labeled with DsRed. Subsequently, LCVs in cell-free homogenates were isolated by immuno-magnetic separation using a primary antibody against the L. pneumophila “effector protein” SidC (Substrate of Icm/Dot transporter), which specifically decorates the LCV membrane, and a secondary antibody coupled to magnetic beads. The enriched LCVs were further separated by density gradient centrifugation. The proteome of purified LCVs analyzed by liquid chromatography coupled to tandem mass spectrometry (MS/MS) revealed more than 560 host proteins, including small GTPases, as well as protein or lipid kinases and phosphatases (Urwyler et al., 2009b).
Components of the LCV host cell proteome include several small GTPases of the secretory (Arf1, Rab1, Rab8) or endosomal (Rab7, Rab14) vesicle trafficking pathways (Urwyler et al., 2009b). Using GFP fusion proteins, the recruitment of the Rab GTPases to the LCV membrane was verified. While Rab8 and Rab14 have not been previously identified on LCVs, the proteome data confirmed earlier findings on LCV localization of Arf1 (Kagan and Roy, 2002), Rab1 (Derre and Isberg, 2004; Kagan et al., 2004), and Rab7 (Clemens et al., 2000). The proteome of isolated LCVs was also analyzed in another study that led to the identification of more than 150 host proteins. These include markers of the ER as well as the early and the late endosomal pathways, which are represented by the coatomer or the vacuolar H+-ATPase, respectively (Shevchuk et al., 2009). In agreement with the notion that L. pneumophila modulates phagosome maturation in a sophisticated manner, the effector protein SidK has been shown to inhibit the vacuolar H+-ATPase, thereby preventing acidification of the LCV (Xu et al., 2010). Together, these studies indicate that LCVs communicate extensively not only with the early and late secretory pathway, but also with early and late steps of the endosomal vesicle trafficking pathway (Figure 1).
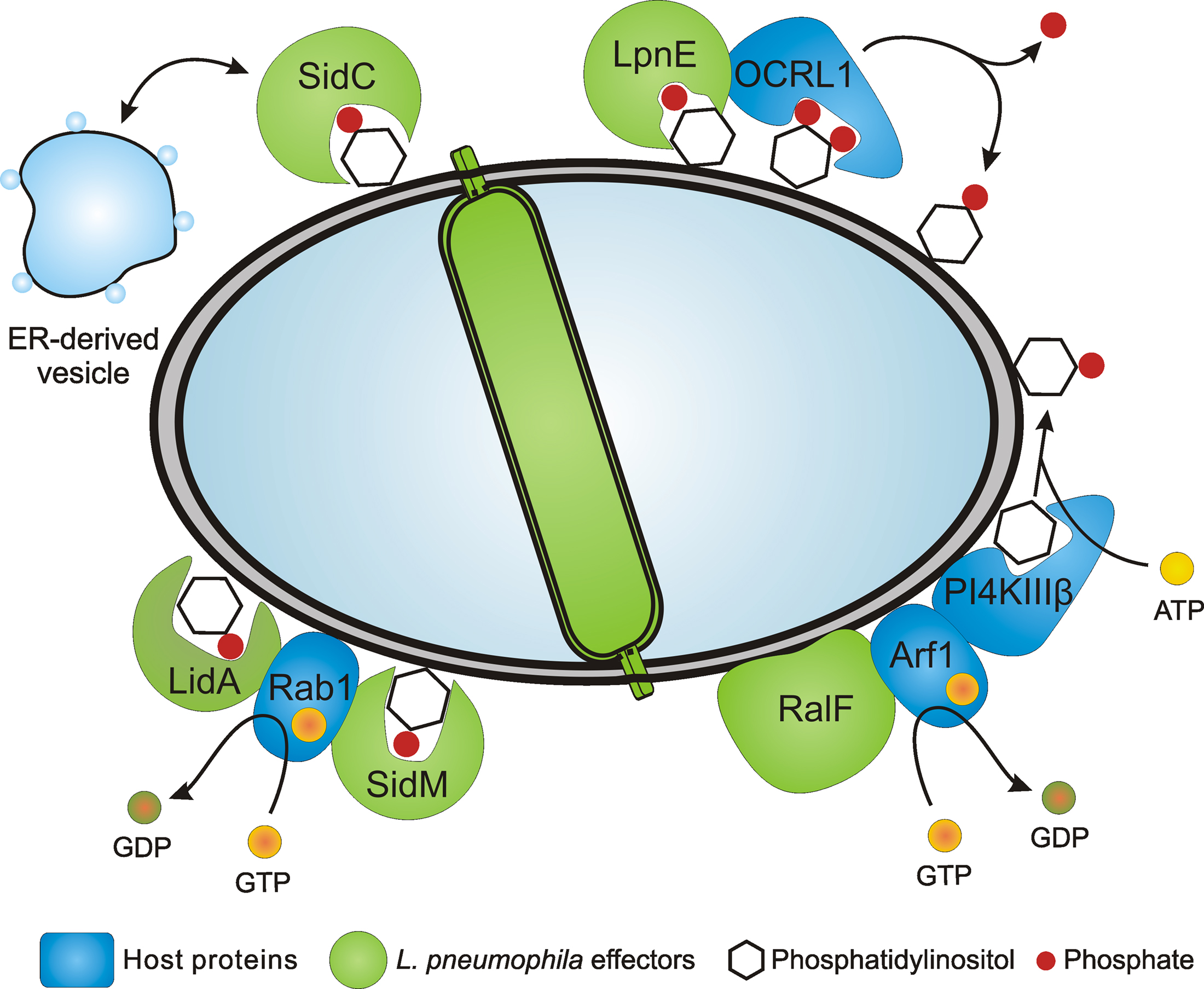
Figure 1. Legionella pneumophila PI-binding effector proteins and LCV formation. L. pneumophila employs the Icm/Dot T4SS to form a replication-permissive LCV that communicates with secretory as well as with endocytic vesicle trafficking pathways and eventually fuses with the ER. Several effector proteins anchor to the LCV membrane through PtdIns(4)P or PtdIns(3)P and promote the interaction with the ER and ER-derived vesicles (SidC), catalyze GEF activity of the small GTPase Rab1 (SidM, LidA), or bind PI-metabolizing enzymes such as the 5-phosphatase OCRL1 (LpnE). The Icm/Dot substrate RalF is an Arf1 GEF that might indirectly recruit PI4KIIIβ.
The formation of LCVs is a robust and complex process that requires the bacterial Icm/Dot (Intracellular multiplication/Defective for organelle trafficking) type IV secretion system (T4SS; Segal et al., 2005). More than 250 different effector proteins are translocated by the Icm/Dot T4SS into the host cell, where they subvert signal transduction and vesicle trafficking pathways by targeting phosphoinositide (PI) metabolism, small GTPases, ubiquitination, microtubuli-dependent trafficking or apoptotic pathways (Brüggemann et al., 2006; Isberg et al., 2009; Urwyler et al., 2009a; Weber et al., 2009b; Hubber and Roy, 2010). While some of the effector proteins target host factors or organelles in a distance from LCVs, many effectors decorate the LCV membrane, thereby directly modulating interactions of this compartment with host vesicles or organelles. In this review, we summarize the current knowledge about how L. pneumophila subverts the host cell’s PI metabolism to form LCVs and replicate intracellularly.
Eukaryotic PI Metabolism and Its Subversion by Intracellular Pathogens
Phosphoinositide glycerolipids play a pivotal role in the regulation of eukaryotic membrane dynamics, cytoskeleton architecture, and signal transduction (De Matteis and Godi, 2004; Di Paolo and De Camilli, 2006; Michell, 2008). The phosphatidylinositol (PtdIns) moiety of these lipids contains glycerol, which is esterified with two fatty acids (usually arachidonic acid and stearic acid) and a myo-inositol 1-phosphate head group. The inositol carbohydrate head group of PI lipids is oriented to the cytoplasmic side of membranes and can be hydrolyzed by PI-specific phospholipase C or reversibly phosphorylated/dephosphorylated at the 3, 4, and/or 5 positions by PI kinases or phosphatases, respectively. The resulting mono- or poly-phosphorylated PIs, jointly with activated small GTPases, recruit distinct effector proteins to specific organelles and thereby co-define the identity and integrity of subcellular compartments as well as cellular membrane dynamics (Shin and Nakayama, 2004; Behnia and Munro, 2005). PtdIns-3-phosphate (PtdIns(3)P) or PtdIns-4-phosphate (PtdIns(4)P), e.g., represent “signposts” of endosomal and secretory trafficking pathways, respectively, and recruit specific effector proteins to membranes involved in these trafficking routes.
In accordance with the importance of PIs for membrane trafficking of eukaryotes, several intracellular bacteria, such as Listeria, Shigella, Salmonella, Brucella, and Mycobacterium spp., exploit PI metabolism to infect host cells, establish a replicative niche and subvert host cell signaling (Pizarro-Cerda and Cossart, 2004; Hilbi, 2006; Weber et al., 2009b). The subversion of PI metabolism by vacuolar pathogens has been studied in some detail in Mycobacterium tuberculosis, the causative agent of the chronic pulmonary disease tuberculosis. The pathogen grows in “Mycobacterium-containing vacuoles” (MCVs), which accumulate the small GTPase Rab5 but not Rab7 and exclude the acidifying vacuolar H+-ATPase as well as lysosomal hydrolases (Russell et al., 2002).
Mycobacterium tuberculosis adopts a dual strategy involving lipid toxins and PI-metabolizing enzymes to keep the levels of PtdIns(3)P on MCVs low, thus arresting the bactericidal endocytic pathway. The glycosylated PI analogue lipoarabinomannan (LAM) and its precursor PtdIns mannoside (PIM) are trafficking toxins (Chua et al., 2004). LAM inhibits a calmodulin kinase II-dependent activation of the class III PtdIns 3-kinase (PI3K) hVps34 (Vergne et al., 2003) and thereby reduces PtdIns(3)P on MCVs and prevents the delivery of the vacuolar H+-ATPase as well as acidic hydrolases (Fratti et al., 2003). Conversely, PIM promotes the homotypic fusion of phagosomes with early endosomes in a PI3K-independent manner, thus allowing continuous communication between MCVs and endosomes, despite the trafficking block imposed by the depletion of PtdIns(3)P (Chua et al., 2004; Vergne et al., 2004). To further reduce PtdIns(3)P on MCVs, M. tuberculosis secretes the PI phosphatases SapM (Vergne et al., 2005) and MptpB (Beresford et al., 2007).
The role of bacterial PI-binding effector proteins, LCV PIs and host PI-modulating enzymes for LCV formation and intracellular replication of L. pneumophila will be discussed in the following sections.
The Icm/Dot Substrate SidC Binds PtdIns(4)P and Promotes LCV-ER Fusion
The L. pneumophila protein SidC was identified as an Icm/Dot substrate by a Cre/loxP-based protein translocation assay, using Icm/Dot-mediated conjugative transport between a donor and a recipient bacterium (Luo and Isberg, 2004). Immuno-fluorescence studies further showed that SidC is translocated to the cytoplasmic side of the vacuole, where the protein decorates the LCV membrane.
The amount of SidC bound to the LCV membrane depends on the presence or absence of PI3Ks in D. discoideum (Weber et al., 2006b), and therefore, we tested in vitro, whether purified GST-SidC fusion protein directly binds PIs. Indeed, purified SidC and its paralogue SdcA (72% identity) were found to selectively bind PtdIns(4)P in protein–lipid overlay assays (Figure 2), as well as in phospholipid vesicle pull down experiments (Weber et al., 2006b). In contrast, the Icm/Dot substrate SidD did not bind to any PIs or other lipids. Further analysis of the PtdIns(4)P-binding domain of SidC revealed that a 20-kDa fragment near the C-terminus was sufficient to selectively bind the PI (Ragaz et al., 2008; Figure 2). The PtdIns(4)P-binding domain was termed “P4C” (PtdIns(4)P-binding domain of SidC) and shows no homology with eukaryotic PI recognition folds, such as the PH (pleckstrin homology), PX (phagocyte oxidase homology), FYVE (Fab1-YotB-Vac1-EEA1), ENTH/ANTH (epsin/AP180 N-terminal homology), FERM (band 4.1-ezrin-radixin-moesin) or KR (lysine/arginine) domains (Downes et al., 2005; Varnai and Balla, 2006; Lemmon, 2008).
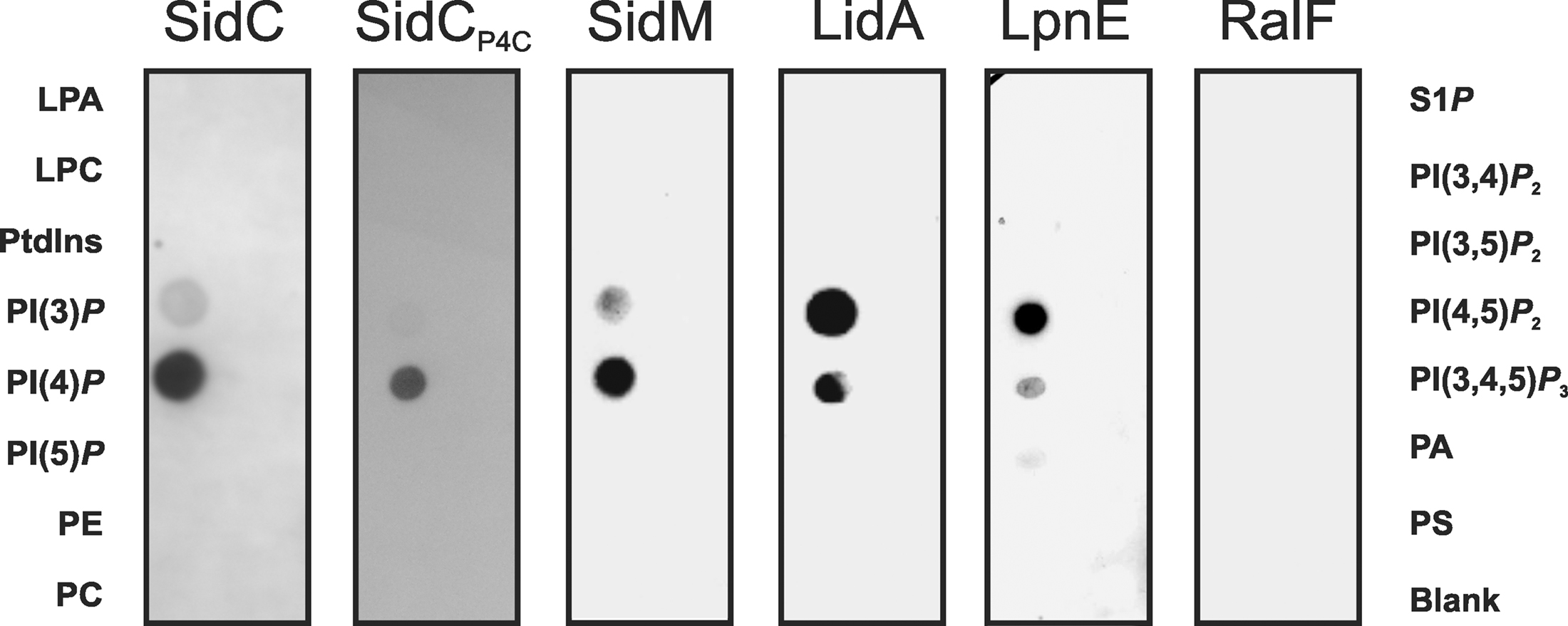
Figure 2. Protein-lipid overlay of L. pneumophila PI-binding effector proteins. GST fusion proteins (200 nM) of SidC, SidCP4C, SidM, LidA, LpnE, and RalF were affinity purified, and binding to different synthetic di-hexadecanoyl-PI lipids (100 pmol) immobilized on nitrocellulose membranes was analyzed by a protein–lipid overlay assay using an anti GST antibody. Left lanes: lysophosphatidic acid (LPA), lysophosphocholine (LPC), phosphatidylinositol (PtdIns), PtdIns phosphate (PI(n)P), phosphatidylethanolamine (PE), phosphatidylcholine (PC). Right lanes: sphingosine-1-phosphate (S1P), PtdIns phosphate (PI(n)P), phosphatidic acid (PA), phosphatidylserine (PS).
Deletion of sidC and the adjacent gene sdcA from the L. pneumophila genome does not impair intracellular replication of the bacteria (Luo and Isberg, 2004; Ragaz et al., 2008). Furthermore, in absence of sidC and sdcA the acquisition of Rab1 or the endosomal marker p80 is not altered; yet, only 20% of LCVs acquire the ER markers calnexin-GFP and GFP-HDEL, indicating that the interaction of LCVs with the ER is severely impaired upon deletion of these genes (Ragaz et al., 2008). The finding that reduced ER acquisition does not impair intracellular replication of the ΔsidC-sdcA strain was unexpected, since defective ER acquisition of LCVs due to a dominant negative form of the small GTPase Sar1 (Kagan and Roy, 2002) or due to the lack of the Icm/Dot substrate SidJ (Liu and Luo, 2007) did inhibit intracellular growth of L. pneumophila.
The ER acquisition phenotype of L. pneumophila lacking sidC and sdcA is complemented by either sidC or sdcA, and the amount of calnexin-GFP and SidC on LCVs is directly proportional. Biochemical experiments revealed that SidC and a 70-kDa N-terminal fragment incubated with lysates of macrophages or D. discoideum bind ER and secretory vesicles (containing calnexin, protein disulfide isomerase and Rab1), but neither lysosomes (containing LAMP-1or “common antigen-1”) nor Golgi fragments (containing giantin). Thus, the N-terminal part of SidC promotes the communication of LCVs with ER-derived vesicles, while the C-terminal part harbors the Icm/Dot translocation determinant and the PtdIns(4)P-binding domain P4C (Figure 1).
SidC and in particular the 20-kDa PtdIns(4)P-binding fragment P4C are stable and can be produced with high yields in E. coli as GST fusion proteins (Weber et al., 2006b; Ragaz et al., 2008). Moreover, P4C can be ectopically produced in D. discoideum, and the probe labels the PtdIns(4)P-positive LCV membrane in amoebae infected with L. pneumophila. Similarly, P4C might be a suitable PtdIns(4)P probe in other eukaryotic cells, including yeast, Drosophila melanogaster and mammalian cells. Taken together, the purified or heterologously produced SidC and P4C proteins are useful as prokaryotic PtdIns(4)P-binding probes in biochemical and cell biological experiments.
The Rab1 GEF SidM is a Major PtdIns(4)P-Binding Effector Protein
To address the question, whether L. pneumophila proteins other than SidC also bind to PIs, bacterial lysates were incubated with agarose beads coated either with one of the seven naturally occurring mono- or poly-phosphorylated PIs or with PtdIns. The eluate from washed beads was separated by SDS-PAGE, and a single protein binding predominantly and specifically to PtdIns(4)P was identified by MS as the effector protein SidM (also termed DrrA; Brombacher et al., 2009). Further analysis showed that SidM is indeed a major L. pneumophila PtdIns(4)P-binding protein (Figure 2), which competes with SidC for binding to this PI on LCVs. The PtdIns(4)P-binding domain of SidM comprises a 12-kDa fragment that was termed “P4M” (PtdIns(4)P-binding of SidM) and is not related to other prokaryotic or eukaryotic PI-binding domains. The high-resolution structure of a SidM fragment including the P4M domain revealed that the effector protein employs a novel fold to bind PtdIns(4)P with an unprecedented high affinity in the nanomolar range (Schoebel et al., 2010). Compared to full length SidM, the affinity of the 12-kDa P4M domain for PtdIns(4)P is reduced (Brombacher et al., 2009), and therefore, the 20-kDa P4C domain, which retains its PI-binding affinity, appears to be the superior PtdIns(4)P probe.
SidM is an Icm/Dot substrate and shows activity as a Rab1 guanine nucleotide exchange factor (GEF), thus activating and recruiting this small GTPase to LCVs (Machner and Isberg, 2006; Murata et al., 2006). The finding that the GEF SidM binds to PtdIns(4)P represents a novel link between the modulation of host GTPases and the exploitation of PIs by pathogenic bacteria (Figure 1). SidM has been suggested to also have activity as a Rab GDP dissociation inhibitor (GDI) displacement factor (GDF), which removes GDI from Rab1-GDP, thus allowing access of the GEF domain to the small GTPase (Ingmundson et al., 2007; Machner and Isberg, 2007). However, the GDF activity turned out to be intrinsic to the GEF activity, rather than a distinct activity (Schoebel et al., 2009).
The membrane cycle of Rab1 is closed by the Icm/Dot substrate LepB, which is a Rab1 GTPase activating protein (GAP) that inactivates and removes Rab1 from membranes (Ingmundson et al., 2007). LepB and another L. pneumophila protein with weak homology to SNAREs and tethering proteins termed LepA have originally been proposed to promote the non-lytic egress of the bacteria from amoebae via a novel pathway, leading to bacteria-filled respirable vesicles (Chen et al., 2004, 2007).
Interestingly, SidM also catalyzes the “AMPylation” (adenosine mono-phosphorylation) of Rab1 at an N-terminal tyrosine residue (Müller et al., 2010). AMPylation of Rab1 “constitutively activates” the small GTPase, since the covalent modification barely affects the GEF activity of SidM but impairs the GAP activity of LepB. In addition to Rab1, SidM AMPylates several other Rab GTPases, including Rab8 and Rab14 (Müller et al., 2010), which are also recruited to LCVs (Urwyler et al., 2009b). Thus, SidM might prolong the activation of several Rab GTPases on the LCV membrane. Rab1-AMP does not bind eukaryotic Rab1 effectors anymore, but still interacts with a SidM auxiliary protein: the L. pneumophila Rab1 effector LidA (Müller et al., 2010).
LidA (Lowered viability in the presence of dotA) is an Icm/Dot substrate that decorates LCVs and promotes the recruitment of early secretory vesicles to LCVs (Conover et al., 2003; Derre and Isberg, 2005). In biochemical experiments LidA was found to interact with several small Rab GTPases (Rab1, Rab6, Rab8) and to support the GEF activity of SidM by binding to GDI-free Rab1 (Machner and Isberg, 2006). In turn, wild-type and constitutively active Rab8 also interact with LidA in L. pneumophila lysates (Urwyler et al., 2009a). LidA represents another effector that localizes to the LCV membrane by interacting with distinct PIs, since it binds to PtdIns(4)P and, with an apparently slightly lower affinity, also to PtdIns(3)P (Brombacher et al., 2009; Figure 2).
Lastly, the Icm/Dot-translocated effector protein RalF does not bind to any PIs or other lipids (Brombacher et al., 2009; Figure 2). RalF (Recruitment of Arf1 to the Legionella phagosome) was the first Icm/Dot substrate identified and characterized as an Arf1-specific GEF, which is required to recruit Arf1 to the LCV membrane (Nagai et al., 2002; Figure 1). Upon deletion of ralF from the L. pneumophila chromosome the small GTPase Arf1 does not localize to LCVs anymore, yet intracellular replication of the bacteria is not impaired. Taken together, these results indicate that two classes of Icm/Dot-translocated L. pneumophila GEFs localize to LCV membranes, one of which by binding to PIs.
The Virulence Factor LpnE Binds PtdIns(3)P and OCRL1
LpnE (Legionella pneumophila entry) is a Sel1 repeat protein of the tetratricopeptide-repeat family (Newton et al., 2006). The lpnE gene was found to be specific for L. pneumophila in a subtractive genomic hybridization screen by comparing L. pneumophila to non-virulent L. micdadei. In agreement with a function for LpnE as a virulence factor, an L. pneumophila lpnE deletion mutant strain is impaired for infection of Acanthamoeba castellanii amoebae, entry into human macrophage-like cells, intracellular trafficking, and virulence in the A/J mouse strain (Newton et al., 2007). While LpnE is secreted into L. pneumophila culture supernatants, the mechanism remains unclear, since neither the Icm/Dot T4SS nor the Lsp T2SS seems to be involved.
Purified recombinant LpnE selectively binds PtdIns(3)P (Weber et al., 2009a; Figure 2), indicating that the Sel1 repeat protein represents another PI-binding virulence factor of L. pneumophila. Moreover, LpnE interacts with the N-terminus of the human enzyme OCRL1 (OCRL11–236) heterologously produced in D. discoideum, and conversely, purified GST-OCRL11–236 binds LpnE in L. pneumophila lysates. OCRL1 (Oculocerebrorenal syndrome of Lowe 1) and its Dictyostelium homologue Dd5P4 (D. discoideum 5-phosphatase 4) are PI-metabolizing enzymes implicated in intracellular replication of L. pneumophila (see below).
In summary, the studies discussed above indicate that L. pneumophila exploits the mono-phosphorylated host PIs PtdIns(4)P and PtdIns(3)P to anchor the effector proteins SidC, SidM, LidA, and LpnE to the LCV membrane. The PI-binding effectors then interfere with host vesicle trafficking and signal transduction (Figure 1).
Host PIs and PI-Metabolizing Enzymes Involved in LCV Formation
Since the Icm/Dot substrates SidC, SidM, and LidA are present on the LCV membrane and bind to PtdIns(4)P in vitro, this PI is expected to be a lipid component of LCVs and enriched on this compartment. Indeed, PtdIns(4)P was identified on LCV membranes by using either an anti-PtdIns(4)P antibody, or purified GST fusion proteins of the eukaryotic PHFAPP1 domain (specifically binding PtdIns(4)P) or prokaryotic SidC as a probe (Weber et al., 2006b).
The mammalian enzyme OCRL1 and D. discoideum Dd5P4 are homologous inositol-polyphosphate 5-phosphatases, which hydrolyze PtdIns(4,5)P2 to yield PtdIns(4)P. GFP fusion proteins of OCRL1 or Dd5P4 localize to LCVs via their N-termini (OCRL11–236, Dd5P41–132) in D. discoideum, and OCRL1 accumulates on LCVs in RAW 264.7 macrophages (Weber et al., 2009a; Figure 1). In D. discoideum lacking Dd5P4 the amount of SidC on LCVs is reduced, suggesting that in absence of Dd5P4 less PtdIns(4)P is produced on the LCV membrane, and consequently, less SidC binds to this compartment. Interestingly, two to three orders of magnitude more L. pneumophila are released from D. discoideum lacking Dd5P4, and therefore, the bacteria grow intracellularly much more efficiently in absence of this inositol-polyphosphate 5-phosphatase. It is currently unknown, how Dd5P4 restricts intracellular growth of L. pneumophila. However, since the mammalian homologue OCRL1 promotes retrograde trafficking from endosomes to the trans-Golgi network (TGN; Johannes and Popoff, 2008), a functional retrograde vesicle trafficking pathway might play a role.
A host PI-metabolizing enzyme termed PtdIns 4-kinase IIIβ (PI4KIIIβ) is likely also involved in the production of PtdIns(4)P on LCVs. Depletion of PI4KIIIβ by RNA interference in Drosophila melanogaster Kc167 phagocytes, which are permissive for intracellular growth of L. pneumophila, significantly reduced the amount of SidC on LCV membranes (Brombacher et al., 2009). The effect was specific for PI4KIIIβ, since depletion of the isoenzymes PI4KIIIα or PI4KIIα did not reduce the amount of LCV-bound SidC. PI4KIIIβ forms PtdIns(4)P in the TGN upon recruitment by the activated small GTPase Arf1 (Godi et al., 1999). Yet, it is currently unknown, whether PI4KIIIβ also localizes to LCVs. Together, these findings suggest that OCRL1/Dd5P4 as well as PI4KIIIβ are implicated in LCV formation and likely catalyze the production of PtdIns(4)P on the LCV membrane (Figure 1).
PI3Ks represent another class of PI-metabolizing enzymes that play important roles for vesicle trafficking and signal transduction in eukaryotic cells. D. discoideum deletion mutants and pharmacological inhibitors revealed that the uptake of icm/dot mutant L. pneumophila depends on PI3Ks. In contrast, the efficient uptake of wild-type L. pneumophila by D. discoideum (Hilbi et al., 2001; Weber et al., 2006a,b) or by human macrophage-like HL-60 cells (Khelef et al., 2001) is barely affected by PI3Ks, indicating that Icm/Dot-proficient bacteria might bypass PI3Ks during entry. In another study, the uptake of L. pneumophila was reported to require PI3Ks (Tachado et al., 2008). Yet, this work used murine J774A.1 macrophage-like cells, which do not support intracellular growth of L. pneumophila. Thus, the formation of a replication-permissive vacuole by L. pneumophila might depend on bypassing PI3K signaling.
Upon deletion or inhibition of PI3Ks in D. discoideum, L. pneumophila replicates more efficiently within the amoebae, and icm/dot mutant bacteria are killed less effectively, in agreement with the well-established role for PI3Ks in the bactericidal endocytic pathway (Weber et al., 2006b). Recent studies confirmed that L. pneumophila interferes with the PI-sensitive fusion of LCVs with acidic vacuoles and indicated that the inhibition of intracellular replication by PI3Ks is restricted to early steps in the infection (Peracino et al., 2010). Notably, the stimulation of intracellular replication of L. pneumophila by pharmacological “PI3K inhibitors” might also be due to an effect on PI4KIIIβ (see above). PI4KIIIβ is a type III PI4K that, in contrast to the type II PI4Ks, is also inhibited by wortmannin (Balla and Balla, 2006).
Finally, the 30-kDa inositol monophosphate phosphatase IMPA was identified as a host component of purified intact LCVs, suggesting that this PI phosphatase might also play a role in LCV formation and intracellular replication of L. pneumophila (Urwyler et al., 2009b). In summary, the PI lipid PtdIns(4)P as well as the host PI-metabolizing enzymes OCRL1/Dd5P4, PI4KIIIβ, PI3Ks, and IMPA are likely constituents of LCVs and implicated in its formation.
Modulation of the PI Pattern on LCVs by L. Pneumophila
PtdIns(4)P accumulates on LCVs harboring wild-type but not icm/dot mutant L. pneumophila (Weber et al., 2006b). Since these vacuoles are vastly different from one another, comparing the two compartments does not indicate, whether the PI pattern on LCVs is modulated directly by the Icm/Dot T4SS. However, upon infection of phagocytes approximately 20% of wild-type L. pneumophila do not reside in a calnexin-positive ER-derived compartment, but rather in a lysosomal compartment, which is positive for the vacuolar H+-ATPase component VatM in D. discoideum and for LAMP-1 in macrophages. The quantification of PtdIns(4)P on VatM-positive vacuoles containing either wild-type or icm/dot mutant L. pneumophila revealed that 42% of the former, but only 6% of the latter accumulated PtdIns(4)P (Weber et al., 2006b). This finding indicates that wild-type L. pneumophila survive in a VatM-positive compartment and interfere with the LCV PI metabolism in an Icm/Dot-dependent manner. Furthermore, modulation of the LCV PI pattern (increasing the amount of PtdIns(4)P) is not sufficient to generate a replication-permissive ER-derived compartment.
To modulate the LCV PI pattern L. pneumophila effectors might (i) activate small GTPases and thus recruit host PI-metabolizing enzymes, (ii) titrate (mask) PIs, (iii) produce factors directly activating or inhibiting host PI-metabolizing enzymes, and/or (iv) translocate bacterial PI-metabolizing enzymes, i.e., PI phosphatases or kinases (Figure 3). Currently, no L. pneumophila factors are known, which directly modify the activity of host PI-metabolizing enzymes or phosphorylate/dephosphorylate PIs.
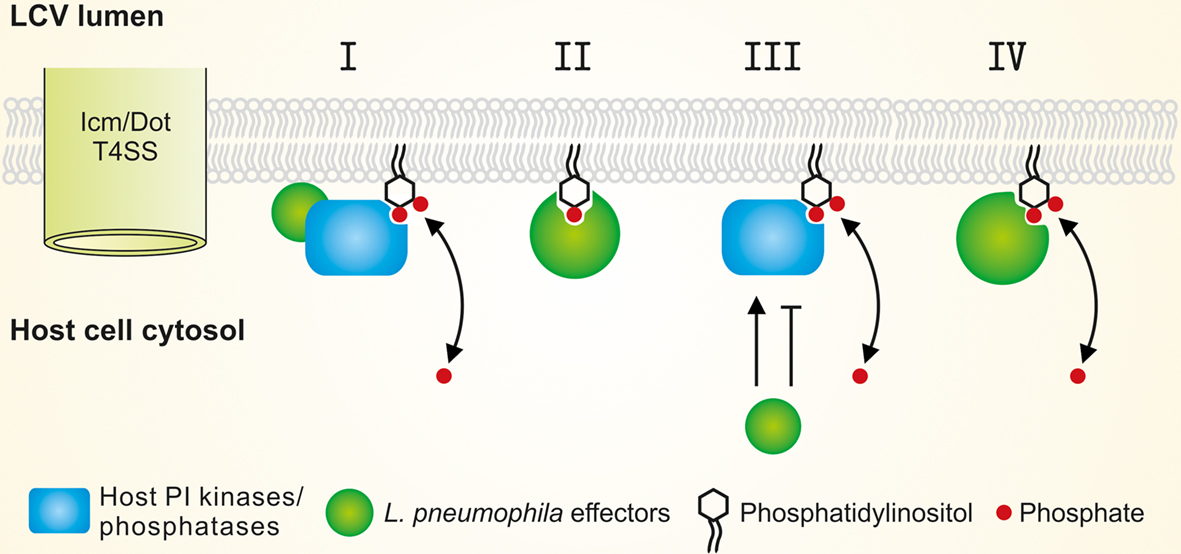
Figure 3. Modulation of the LCV PI pattern by L. pneumophila effectors. L. pneumophila modulates the LCV host PI pattern through the Icm/Dot T4SS. Translocated factors might (I) activate small GTPases and thus recruit host PI-metabolizing enzymes, (II) titrate (mask) PIs, (III) directly activate or inhibit host PI-metabolizing enzymes, and/or (IV) represent bacterial PI phosphatases or kinases.
Legionella pneumophila produces and translocates two GEFs that recruit and activate small GTPases and thus might indirectly modulate the LCV PI pattern: RalF (Nagai et al., 2002) and SidM (Machner and Isberg, 2006; Murata et al., 2006) recruit the small GTPases Arf1 and Rab1to LCVs (Figure 1). Arf1 has been shown to recruit PI4KIIIβ at the TGN (Godi et al., 1999), and a number of small GTPases including Arf1 (Lichter-Konecki et al., 2006) and Rab1 (Hyvola et al., 2006) target OCRL1 to endosomal membranes. Both PI4KIIIβ and OCRL1 produce PtdIns(4)P and are likely enzymatically active on LCVs (Brombacher et al., 2009; Weber et al., 2009a). If L. pneumophila modulates the PI levels on LCVs via the translocation of GEFs, the concentration of PtdIns(4)P on LCVs should decrease in absence of RalF or SidM, and consequently, less SidC should bind to LCVs. However, in absence of RalF the amount of SidC on LCVs remained constant, and in absence of SidM even significantly more SidC bound to LCVs, accounting also for the fact that SidM itself binds to PtdIns(4)P (Brombacher et al., 2009). In light of these results, L. pneumophila likely does not (or at least not exclusively) modulate the LCV PI pattern by the indirect recruitment of host PI-metabolizing enzymes. Thus, while it is clear that L. pneumophila modulates the LCV PI pattern in an Icm/Dot-dependent manner, the mechanistic aspects of this process have not been elucidated yet.
Conclusions and Perspectives
Legionella pneumophila employs the Icm/Dot T4SS to form in a complex and robust process a replication-permissive LCV. Several Icm/Dot-translocated effector proteins anchor to the LCV membrane through PtdIns(4)P or PtdIns(3)P and promote the interaction of LCVs with host vesicles and organelles, small GTPases or PI-metabolizing enzymes. The PI-binding domains of these effectors bind PIs with high affinity and are useful as specific lipid probes. Further research should address the identification and characterization of other L. pneumophila PI-binding effector proteins. Of particular interest is the mechanism, by which L. pneumophila actively modulates the LCV PI pattern in an Icm/Dot-dependent manner. A comprehensive understanding of how L. pneumophila exploits the PI metabolism of host cells will continue to provide mechanistic insights into basic biological processes and shed light on the virulence of this important opportunistic human pathogen.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in our laboratory was funded by the Max von Pettenkofer Institute, Ludwig-Maximilians University Munich, the Deutsche Forschungsgemeinschaft (HI 1511/1-1), the Bundesministerium für Bildung und Forschung (0315834C), and the Swiss National Science Foundation (31003A-125369; CRSI33_130016).
References
Balla, A., and Balla, T. (2006). Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 16, 351–361.
Behnia, R., and Munro, S. (2005). Organelle identity and the signposts for membrane traffic. Nature 438, 597–604.
Beresford, N., Patel, S., Armstrong, J., Szoor, B., Fordham-Skelton, A. P., and Tabernero, L. (2007). MptpB, a virulence factor from Mycobacterium tuberculosis, exhibits triple-specificity phosphatase activity. Biochem. J. 406, 13–18.
Brombacher, E., Urwyler, S., Ragaz, C., Weber, S. S., Kami, K., Overduin, M., and Hilbi, H. (2009). Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284, 4846–4856.
Brüggemann, H., Cazalet, C., and Buchrieser, C. (2006). Adaptation of Legionella pneumophila to the host environment: role of protein secretion, effectors and eukaryotic-like proteins. Curr. Opin. Microbiol. 9, 86–94.
Chen, J., de Felipe, K. S., Clarke, M., Lu, H., Anderson, O. R., Segal, G., and Shuman, H. A. (2004). Legionella effectors that promote nonlytic release from protozoa. Science 303, 1358–1361.
Chen, J., Reyes, M., Clarke, M., and Shuman, H. A. (2007). Host cell-dependent secretion and translocation of the LepA and LepB effectors of Legionella pneumophila. Cell. Microbiol. 9, 1660–1671.
Chua, J., Vergne, I., Master, S., and Deretic, V. (2004). A tale of two lipids: Mycobacterium tuberculosis phagosome maturation arrest. Curr. Opin. Microbiol. 7, 71–77.
Clemens, D. L., Lee, B. Y., and Horwitz, M. A. (2000). Mycobacterium tuberculosis and Legionella pneumophila phagosomes exhibit arrested maturation despite acquisition of Rab7. Infect. Immun. 68, 5154–5166.
Conover, G. M., Derre, I., Vogel, J. P., and Isberg, R. R. (2003). The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol. Microbiol. 48, 305–321.
Cosson, P., and Soldati, T. (2008). Eat, kill or die: when amoeba meets bacteria. Curr. Opin. Microbiol. 11, 271–276.
Derre, I., and Isberg, R. R. (2004). Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect. Immun. 72, 3048–3053.
Derre, I., and Isberg, R. R. (2005). LidA, a translocated substrate of the Legionella pneumophila type IV secretion system, interferes with the early secretory pathway. Infect. Immun. 73, 4370–4380.
Di Paolo, G., and De Camilli, P. (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657.
Downes, C. P., Gray, A., and Lucocq, J. M. (2005). Probing phosphoinositide functions in signaling and membrane trafficking. Trends Cell Biol. 15, 259–268.
Fratti, R. A., Chua, J., Vergne, I., and Deretic, V. (2003). Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc. Natl. Acad. Sci. U.S.A. 100, 5437–5442.
Godi, A., Pertile, P., Meyers, R., Marra, P., Di Tullio, G., Iurisci, C., Luini, A., Corda, D., and De Matteis, M. A. (1999). ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1, 280–287.
Hilbi, H. (2006). Modulation of phosphoinositide metabolism by pathogenic bacteria. Cell. Microbiol. 8, 1697–1706.
Hilbi, H., Hoffmann, C., and Harrison, C. (2011). Legionella spp. outdoors: colonization, communication and persistence. Environ. Microbiol. Rep. doi: 10.1111/j.1758-2229.2011.00247.x. [Epub ahead of print].
Hilbi, H., Segal, G., and Shuman, H. A. (2001). Icm/Dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol. Microbiol. 42, 603–617.
Hilbi, H., Weber, S. S., Ragaz, C., Nyfeler, Y., and Urwyler, S. (2007). Environmental predators as models for bacterial pathogenesis. Environ. Microbiol. 9, 563–575.
Horwitz, M. A. (1983). Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158, 1319–1331.
Hubber, A., and Roy, C. R. (2010). Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26, 261–283.
Hyvola, N., Diao, A., McKenzie, E., Skippen, A., Cockcroft, S., and Lowe, M. (2006). Membrane targeting and activation of the Lowe syndrome protein OCRL1 by Rab GTPases. EMBO J. 25, 3750–3761.
Ingmundson, A., Delprato, A., Lambright, D. G., and Roy, C. R. (2007). Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450, 365–369.
Isberg, R. R., O’Connor, T. J., and Heidtman, M. (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24.
Johannes, L., and Popoff, V. (2008). Tracing the retrograde route in protein trafficking. Cell 135, 1175–1187.
Kagan, J. C., and Roy, C. R. (2002). Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4, 945–954.
Kagan, J. C., Stein, M. P., Pypaert, M., and Roy, C. R. (2004). Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J. Exp. Med. 199, 1201–1211.
Khelef, N., Shuman, H. A., and Maxfield, F. R. (2001). Phagocytosis of wild-type Legionella pneumophila occurs through a wortmannin-insensitive pathway. Infect. Immun. 69, 5157–5161.
Lemmon, M. A. (2008). Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111.
Lichter-Konecki, U., Farber, L. W., Cronin, J. S., Suchy, S. F., and Nussbaum, R. L. (2006). The effect of missense mutations in the RhoGAP-homology domain on ocrl1 function. Mol. Genet. Metab. 89, 121–128.
Liu, Y., and Luo, Z. Q. (2007). The Legionella pneumophila effector SidJ is required for efficient recruitment of endoplasmic reticulum proteins to the bacterial phagosome. Infect. Immun. 75, 592–603.
Lu, H., and Clarke, M. (2005). Dynamic properties of Legionella-containing phagosomes in Dictyostelium amoebae. Cell. Microbiol. 7, 995–1007.
Luo, Z. Q., and Isberg, R. R. (2004). Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U.S.A. 101, 841–846.
Machner, M. P., and Isberg, R. R. (2006). Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev. Cell 11, 47–56.
Machner, M. P., and Isberg, R. R. (2007). A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science 318, 974–977.
Michell, R. H. (2008). Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell Biol. 9, 151–161.
Molofsky, A. B., and Swanson, M. S. (2004). Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 53, 29–40.
Müller, M. P., Peters, H., Blumer, J., Blankenfeldt, W., Goody, R. S., and Itzen, A. (2010). The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329, 946–949.
Murata, T., Delprato, A., Ingmundson, A., Toomre, D. K., Lambright, D. G., and Roy, C. R. (2006). The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat. Cell Biol. 8, 971–977.
Nagai, H., Kagan, J. C., Zhu, X., Kahn, R. A., and Roy, C. R. (2002). A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295, 679–682.
Newton, H. J., Sansom, F. M., Bennett-Wood, V., and Hartland, E. L. (2006). Identification of Legionella pneumophila-specific genes by genomic subtractive hybridization with Legionella micdadei and identification of lpnE, a gene required for efficient host cell entry. Infect. Immun. 74, 1683–1691.
Newton, H. J., Sansom, F. M., Dao, J., McAlister, A. D., Sloan, J., Cianciotto, N. P., and Hartland, E. L. (2007). Sel1 repeat protein LpnE is a Legionella pneumophila virulence determinant that influences vacuolar trafficking. Infect. Immun. 75, 5575–5585.
Peracino, B., Balest, A., and Bozzaro, S. (2010). Phosphoinositides differentially regulate bacterial uptake and Nramp1-induced resistance to Legionella infection in Dictyostelium. J. Cell. Sci. 123, 4039–4051.
Pizarro-Cerda, J., and Cossart, P. (2004). Subversion of phosphoinositide metabolism by intracellular bacterial pathogens. Nat. Cell Biol. 6, 1026–1033.
Ragaz, C., Pietsch, H., Urwyler, S., Tiaden, A., Weber, S. S., and Hilbi, H. (2008). The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell. Microbiol. 10, 2416–2433.
Robinson, C. G., and Roy, C. R. (2006). Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell. Microbiol. 8, 793–805.
Russell, D. G., Mwandumba, H. C., and Rhoades, E. E. (2002). Mycobacterium and the coat of many lipids. J. Cell Biol. 158, 421–426.
Schoebel, S., Blankenfeldt, W., Goody, R. S., and Itzen, A. (2010). High-affinity binding of phosphatidylinositol 4-phosphate by Legionella pneumophila DrrA. EMBO Rep. 11, 598–604.
Schoebel, S., Oesterlin, L. K., Blankenfeldt, W., Goody, R. S., and Itzen, A. (2009). RabGDI displacement by DrrA from Legionella is a consequence of its guanine nucleotide exchange activity. Mol. Cell 36, 1060–1072.
Segal, G., Feldman, M., and Zusman, T. (2005). The Icm/Dot type-IV secretion systems of Legionella pneumophila and Coxiella burnetii. FEMS Microbiol. Rev. 29, 65–81.
Shevchuk, O., Batzilla, C., Hagele, S., Kusch, H., Engelmann, S., Hecker, M., Haas, A., Heuner, K., Glockner, G., and Steinert, M. (2009). Proteomic analysis of Legionella-containing phagosomes isolated from Dictyostelium. Int. J. Med. Microbiol. 299, 489–508.
Shin, H. W., and Nakayama, K. (2004). Dual control of membrane targeting by PtdIns(4)P and ARF. Trends Biochem. Sci. 29, 513–515.
Solomon, J. M., and Isberg, R. R. (2000). Growth of Legionella pneumophila in Dictyostelium discoideum: a novel system for genetic analysis of host-pathogen interactions. Trends Microbiol. 8, 478–480.
Steinert, M., and Heuner, K. (2005). Dictyostelium as host model for pathogenesis. Cell. Microbiol. 7, 307–314.
Tachado, S. D., Samrakandi, M. M., and Cirillo, J. D. (2008). Non-opsonic phagocytosis of Legionella pneumophila by macrophages is mediated by phosphatidylinositol 3-kinase. PLoS ONE 3, e3324. doi: 10.1371/journal.pone.0003324
Tiaden, A., Spirig, T., and Hilbi, H. (2010). Bacterial gene regulation by alpha-hydroxyketone signaling. Trends Microbiol. 18, 288–297.
Urwyler, S., Brombacher, E., and Hilbi, H. (2009a). Endosomal and secretory markers of the Legionella-containing vacuole. Commun Integr Biol 2, 107–109.
Urwyler, S., Finsel, I., Ragaz, C., and Hilbi, H. (2010). Isolation of Legionella-containing vacuoles by immuno-magnetic separation. Curr. Protoc. Cell Biol. 3, 34.
Urwyler, S., Nyfeler, Y., Ragaz, C., Lee, H., Mueller, L. N., Aebersold, R., and Hilbi, H. (2009b). Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic 10, 76–87.
Varnai, P., and Balla, T. (2006). Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim. Biophys. Acta 1761, 957–967.
Vergne, I., Chua, J., and Deretic, V. (2003). Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVps34 cascade. J. Exp. Med. 198, 653–659.
Vergne, I., Chua, J., Lee, H. H., Lucas, M., Belisle, J., and Deretic, V. (2005). Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 102, 4033–4038.
Vergne, I., Fratti, R. A., Hill, P. J., Chua, J., Belisle, J., and Deretic, V. (2004). Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol. Biol. Cell 15, 751–760.
Weber, S. S., Ragaz, C., and Hilbi, H. (2009a). The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell. Microbiol. 11, 442–460.
Weber, S. S., Ragaz, C., and Hilbi, H. (2009b). Pathogen trafficking pathways and host phosphoinositide metabolism. Mol. Microbiol. 71, 1341–1352.
Weber, S. S., Ragaz, C., Reus, K., and Hilbi, H. (2006a). “A role for phosphoinositide metabolism in phagocytosis and intracellular replication of Legionella pneumophila,” in Legionella: State of the Art 30 Years After Its Recognition, eds N. P. Cianciotto, Y. Abu Kwaik, P. H. Edelstein, B. S. Fields, D. F. Geary, T. G. Harrison, C. A. Joseph, R. M. Ratcliff, J. E. Stout, and M. S. Swanson (Washington, DC: ASM Press), 292–296.
Weber, S. S., Ragaz, C., Reus, K., Nyfeler, Y., and Hilbi, H. (2006b). Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2, e46. doi: 10.1371/journal.ppat.0020046
Keywords: amoeba, Dictyostelium, Legionella, macrophage, phosphoinositides, pathogen vacuole, type IV secretion
Citation: Hilbi H, Weber S and Finsel I (2011) Anchors for effectors: subversion of phosphoinositide lipids by Legionella. Front. Microbio. 2:91. doi: 10.3389/fmicb.2011.00091
Received: 28 January 2011;
Paper pending published: 16 February 2011;
Accepted: 14 April 2011;
Published online: 27 April 2011.
Edited by:
Carmen Buchrieser, Pasteur Institute, FranceCopyright: © 2011 Hilbi, Weber and Finsel. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Hubert Hilbi, Max von Pettenkofer Institute, Ludwig-Maximilians University, Pettenkoferstraße 9a, 80336 Munich, Germany.e-mail:aGlsYmlAbXZwLnVuaS1tdWVuY2hlbi5kZQ==