

- Department of Cell Biology, School of Medicine of Ribeirão Preto, University of São Paulo, Ribeirão Preto, SP, Brazil
Innate immune cells, such as macrophages, are highly adapted to rapidly recognize infections by distinct pathogens, including viruses, bacteria, fungi, and protozoa. This recognition is mediated by pattern recognition receptors (PRRs), which are found in host cell surface membranes and the host cell cytoplasm. PRRs include protein families such as the toll-like receptors, nod-like receptors, RIG-I-like receptors, and sensors of cytosolic DNA. The activation of these PRRs by pathogen-associated molecular patterns leads to transcriptional responses and specific forms of cell death. These processes effectively contribute to host resistance to infection either via cell-autonomous processes that lead to the intracellular restriction of microbial replication and/or by activating pathogen-specific adaptive immune responses. Legionella pneumophila, the causative agent of Legionnaires’ disease, is a Gram-negative bacterium that triggers responses by multiple PRRs. Here, we review a set of studies that have contributed to our specific understanding of the molecular mechanisms by which innate immune cells recognize and respond to L. pneumophila and the importance of these processes to the outcome of infection.
Introduction
Activation of innate immune cells is critical for the initiation of adaptive immune responses. This process relies mostly on the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs). Among the canonical PAMPs are molecules such as lipopolysaccharide, peptidoglycan, bacterial lipoproteins, flagellin, and nucleic acids derived from viruses, bacteria, fungi, and protozoa (Janeway and Medzhitov, 2002; Akira et al., 2006; Gazzinelli and Denkers, 2006). Upon direct or indirect ligand recognition, toll-like receptors (TLRs) dimerize and trigger a signaling cascade leading to the activation of proinflammatory responses (Uematsu and Akira, 2006). TLRs are transmembrane proteins containing an extracellular leucine-rich repeat (LRR) domain that facilitates PAMP recognition and an intracellular domain that mediates intracellular signaling via four different adaptor proteins: TRAM, MAL/TIRAP, MyD88, and TRIF (O’Neill, 2008). Depending on the nature of their specific ligands, TLRs are embedded in either the extracellular membrane (TLR-1, -2, -4, -5, -6, -10, -11) or in the membranes of endocytic vacuoles (TLR-3, -7, -8, -9). In addition to TLRs, other PRR families have already been described; these include DNA/RNA-sensing proteins such as the RIG-I-like receptor (RLR) family and sensors of membrane damage and intracellular PAMPs such as the nod-like receptors (NLRs; Creagh and O’Neill, 2006).
The RNA helicase domain-containing proteins retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) comprise a group of cytoplasmic receptors important for the recognition of viral nucleic acids. PAMP recognition by RIG-I and MDA5 triggers the activation of IRF3 via MAVS (IPS-1), culminating in the production of type I interferons (IFN). Recent studies have demonstrated that these receptors also induce type I IFN production upon recognition of nucleic acids from intracellular bacteria (Cao, 2009). In addition, some studies have shown that the recognition of DNA by RIG-I/MDA5 is dependent on cytosolic RNA polymerase III (Pol III; Ablasser et al., 2009; Chiu et al., 2009).
The NLRs comprise a PRR family that can be classified into three sub-groups. The first sub-group is composed of receptors that trigger intracellular signaling pathways leading to the activation of transcriptional factors mediating the expression of inflammatory response genes. Both NOD1 and NOD2 are members of this first group, and they signal via RIP2, a kinase that ubiquitinates NEMO to induce the activation of NF-κB and MAPK (Shaw et al., 2008). The second sub-group comprises NLRs that do not require ASC to trigger caspase-1 activation. Among these proteins are NAIP5 (BIRC1e) and NLRC4 (IPAF), which have been suggested to assembly a unique inflammasome (hereafter referred to as the NLRC4 inflammasome). Activation of this inflammasome triggers a specific form of host cell death called pyroptosis (Lightfield et al., 2008; Case et al., 2009; Broz et al., 2010; Silveira and Zamboni, 2010; Whitfield et al., 2010). The third sub-group of NLRs comprises those that trigger caspase-1 activation via the adaptor protein ASC. These proteins assemble into a multimeric molecular platform known as the “classical” inflammasome. Among the NLRs that trigger the ASC-dependent inflammasome is NALP3, which has been extensively characterized and shown to be important for the recognition of danger-associated molecular patterns (DAMPs) reviewed by Schroder and Tschopp (2010).
In addition to TLRs, NLRs, and RLRs, previous studies have described a class of proteins that recognize cytoplasmic DNA (Ishii and Akira, 2006; Stetson and Medzhitov, 2006). These multiple protein families include DNA-dependent activators of IFN-regulatory factors (DAI; Takaoka et al., 2007), RNA polymerase III, which induces type I IFN production through the RIG-I pathway (Ablasser et al., 2009; Chiu et al., 2009), and the recently described protein absent in melanoma (AIM2), which activates inflammasomes in an ASC-dependentmanner (Burckstummer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Roberts et al., 2009).
Legionella pneumophila, a Gram-negative bacterial pathogen that evolved infecting unicellular protozoa in freshwater reservoirs, may not have encountered strong selective pressure to avoid recognition by mammalian PRRs. Consequently, L. pneumophila triggers multiple PRR and has been a useful model for understanding the biology of PRRs and the induction of appropriate adaptive immune responses against intracellular pathogens. The successful use of L. pneumophila as a tool for studying immunology has been reviewed elsewhere (Vance, 2010). Here, we will review the salient findings that have contributed to our understanding of the molecular mechanisms underlying innate immune cell recognition and response to L. pneumophila infection (Figure 1). Furthermore, we will sumarize studies that have elucidated the importance of these processes to the outcome of L. pneumophila infection.
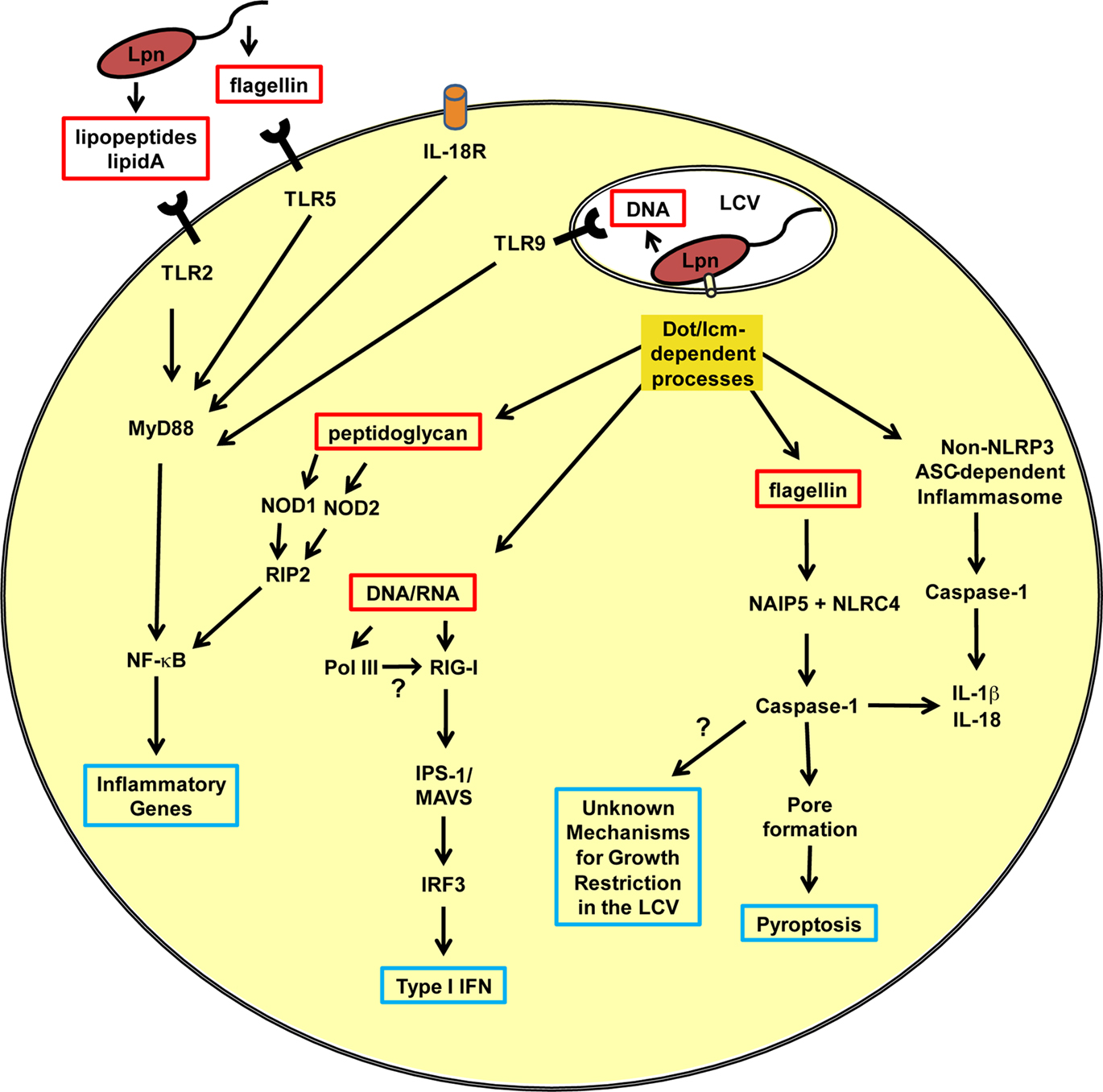
Figure 1. Innate immune responses of a mammalian phagocyte infected with Legionella pneumophila. A schematic representation of the pathways activated in a phagocyte after infection with L. pneumophila. The red boxes indicate L. pneumophila-associated molecular patterns important for the activation of pattern recognition receptors. Blue boxes indicate molecules or processes involved in the cell-autonomous restriction of L. pneumophila replication. LCV, Legionella-containing vacuole; Lpn, L. pneumophila; Dot/Icm, type IVB secretion system; IL, interleukin; IL-18R, IL-18 receptor; TLR, toll-like receptor; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor kappa B; NOD, nucleotide-binding oligomerization domain-containing protein; RIP2, receptor interacting protein 2; Pol III, RNA polymerase III; RIG-I, retinoic-acid-inducible protein I; IPS-I, IFN-β promoter stimulator 1 (also known as MAVS, mitochondrial antiviral signaling); IRF3, interferon regulatory factor 3; NAIP5, neuronal apoptosis inhibitory protein 5; NLRC4, NLR family CARD domain-containing protein 4.
Toll-Like Receptors
It was originally speculated that TLR4, a general LPS sensor, would recognize L. pneumophila. However, work on non-enterobacteriaceae species has indicated that although TLR4/MD2 very efficiently recognizes enterobacterial lipid A, the same is not true for other Gram-negative bacteria. These non-enterobacteriaceae bacterial species often express lipid A containing long fatty acid chains that either fail to trigger TLR4 activation or antagonize the TLR4 receptor (Zamboni et al., 2004). In L. pneumophila, studies performed with TLR4-deficient or TLR4 knockout mice have confirmed that this receptor does not effectively participate in the recognition of L. pneumophila LPS. Even at high MOIs, there is no difference in L. pneumophila infection between wild-type and C3H/HeJ mice, which are defective for TLR4 signaling due to a missense mutation in the Tlr4 gene resulting in the replacement of a proline with a histidine at position 712 (Poltorak et al., 1998; Lettinga et al., 2002). The initial studies on TLR4 function using C3H/HeJ mice were further corroborated in tlr4−/− mouse experiments, which supported the hypothesis that TLR4 deficiency does not influence the outcome of L. pneumophila infection (Akamine et al., 2005; Archer and Roy, 2006; Fuse et al., 2007). Studies by Girard et al. (2003) have shown that lipid A of L. pneumophila signals via TLR2 to induce the expression of CD14. These findings led to the suggestion that L. pneumophila LPS is recognized by TLR2, but the mechanisms underlying the recognition of lipid A by TLR2 have not been completely elucidated; some researchers have speculated that lipid A-mediated TLR2 activation requires either a long chain fatty acid or the presence of a substituent or a branch on the penultimate carbon of a fatty acid chain (Brandenburg et al., 1993). Nonetheless, future studies using a synthetic form of L. pneumophila lipid A may be required to unequivocally confirm that L. pneumophila LPS is a bona fide agonist of TLR2.
Regardless of the proposed role of TLR2 in LPS recognition, other L. pneumophila PAMPs, such as lipopeptides and lipoproteins, are sufficient to activate TLR2. Activation of this receptor is critical to the outcome of L. pneumophila infection in mice. This was unequivocally demonstrated by experiments using tlr2−/− mice, which show impaired cytokine production and are more susceptible to bacterial multiplication in the lungs (Akamine et al., 2005; Archer and Roy, 2006; Hawn et al., 2006).
In addition to TLR2, other TLRs are also important for the host response to L. pneumophila. As a flagellated bacteria, L. pneumophila is recognized by TLR5, and a common polymorphism in the ligand-binding domain of TLR5 causes increased susceptibility to Legionnaires’ disease in humans (Hawn et al., 2003). These data have been corroborated by studies using tlr5−/− mice showing that TLR5 recognition of L. pneumophila in vivo contributes to the recruitment of leukocytes to the pulmonary cavity (Hawn et al., 2007). However, TLR5 deficiency by itself does not render mice more susceptible to infection as measured by CFU counts and cytokine production (Hawn et al., 2007; Archer et al., 2009).
Another TLR important in L. pneumophila infection is TLR9. Mice lacking this receptor exhibit reduced levels of cytokines when challenged with L. pneumophila and are therefore more permissive of L. pneumophila replication in the lungs (Newton et al., 2007; Archer et al., 2009). This observation was corroborated by experiments involving the in vivo administration of CpG oligodeoxynucleotide, a synthetic agonist of TLR9, which protected mice that were pre-infected with L. pneumophila (Bhan et al., 2008).
Importantly, these studies using mice deficient for a single TLR indicate that disruption of a single tlr gene does not result in a striking susceptibility to L. pneumophila; this is possibly due to redundancy in the signaling pathways triggered by these receptors. In contrast, the deletion of the common adaptor protein MyD88, which is important for the signaling of several TLRs, produces mice that are highly susceptible to infection. Mice deficient for MyD88 show impaired cytokine production in response to pulmonary infection with L. pneumophila; they also show high numbers of CFUs in the lungs and succumb to L. pneumophila infection even at low multiplicities of infection (Neild et al., 2005; Archer and Roy, 2006; Hawn et al., 2006; Sporri et al., 2006; Archer et al., 2009, 2010). The increased susceptibility of myd88−/− mice suggests that the deletion of multiple TLR genes will produce mice as susceptible to L. pneumophila infection as those lacking myd88−/−. To test this hypothesis, Archer et al. (2009) constructed mice deficient for multiple TLRs and showed that mice lacking both TLR5 and TLR9 or deficient for TLR2 and either TLR5 or TLR9 are still able to clear L. pneumophila infection. Archer and colleagues elegantly concluded that IL-18 signaling via MyD88 is essential for NK cell production of IFN-γ, a cytokine critical for the restriction of L. pneumophila infection in vivo (Archer et al., 2009). Interestingly, although the authors showed that NK cells signal via IL-18 to produce IFN-γ, they also demonstrated that mice deficient for the IL-18 receptor are no more susceptible to L. pneumophila infection than wild-type animals (Archer et al., 2009). Additional studies will therefore be required to further determine the importance of this pathway in vivo and its redundancy with other pathways.
NOD-Like Receptors: NOD1 and NOD2
The first study addressing NOD1 and NOD2 signaling in response to L. pneumophila infection was performed by Shin et al. (2008). In this study, the authors evaluated the transcriptional responses of macrophages infected with wild-type and dotA mutants of L. pneumophila to identify macrophage genes induced in a Dot/Icm-dependent manner. By comparing macrophages deficient for MyD88 and RIP2 kinase, which impairs both NOD1 and NOD2 signaling, or lacking both MyD88 and TRIF, the authors identified several genes that were regulated by a RIP2-dependent pathway (Shin et al., 2008). Importantly, this study revealed that multiple responses occur after L. pneumophila infection of macrophages; some of these were dependent on MyD88, others on RIP2 and some responses that were induced via unknown sensors were independent of both MyD88 and RIP2 (Shin et al., 2008). This study was further corroborated by in vivo experiments using mice deficient for both RIP2 and MyD88. The rip2−/−/myd88−/− mice were significantly more susceptible to L. pneumophila than the myd88−/− mice, and they succumbed to infection even at low MOIs (Archer et al., 2010). Importantly, this and another study demonstrated that although a RIP2-dependent response was not critical for restricting L. pneumophila infection in vivo, a RIP2-dependent response did contribute to the recruitment of phagocytes to the sites of infection (Archer et al., 2010; Frutuoso et al., 2010). Notably, the RIP2-dependent responses that contributed to the recruitment of neutrophils to the lungs of infected mice were at least partially dependent on the NOD1 and NOD2 receptors (Berrington et al., 2010; Frutuoso et al., 2010). These studies confirmed that NOD1 and NOD2 effectively participate in the pulmonary detection of L. pneumophila infection, but NOD1 and NOD2 deficiency results only in a minor attenuation of bacterial growth restriction in mouse lungs (Berrington et al., 2010; Frutuoso et al., 2010). Importantly, these studies of L. pneumophila infection in mice deficient for TLRs and the NOD/RIP2 pathway demonstrate the substantial redundancy of innate immune receptors in the host response to bacterial infection.
NOD-Like Receptors: NLRC4 and NAIP5
Approximately 30 years ago, it was demonstrated that macrophages from A/J mice fail to restrict the intracellular replication of L. pneumophila (Yamamoto et al., 1988). These phenotypic differences between A/J and other mouse strains provided a useful model for investigating the genes responsible for the phenotypic variations. In later years, the genomic region response for L. pneumophila resistance was mapped to the autosomal recessive locus Lgn1 on chromosome 13 (Beckers et al., 1995; Dietrich et al., 1995). In early 2003, it was finally revealed that the susceptibility gene within the Lgn1 locus was NAIP5 (also known as BIRC1e), a member of the NLR proteins family (Diez et al., 2003; Wright et al., 2003). The mechanisms by which NAIP5 contributes to the host control of infection was unraveled a few years later by the discovery that NAIP5 interfered with caspase-1 activation in response to macrophage infection by L. pneumophila (Zamboni et al., 2006). This response is dependent on the Dot/Icm system and effectively contributes to the restriction of bacterial replication in macrophages in vitro and in vivo (Zamboni et al., 2006). The NAIP5-dependent restriction of L. pneumophila growth required another NLR protein called NLRC4 (IPAF), which contributes for caspase-1 activation upon L. pneumophila infection (Amer et al., 2006; Molofsky et al., 2006; Zamboni et al., 2006). An elegant screening experiment identified the putative agonist of this NAIP5/NLRC4 inflammasome: L. pneumophila deficient for the flagellin gene flaA bypassed the NLRC4 inflammasome and replicated in macrophages harboring the restrictive Lgn1 allele (Molofsky et al., 2006; Ren et al., 2006; Zamboni et al., 2006). Although the role of NAIP5 in caspase-1 activation was questioned (Lamkanfi et al., 2007), assays with naip5−/− mice unequivocally demonstrated the requirement of NAIP5 for caspase-1 activation in response to L. pneumophila flagellin (Lightfield et al., 2008). Furthermore, both NLRC4 and NAIP5 were required for the detection of a carboxy-terminal domain of flagellin, a region not required for TLR5 activation (Lightfield et al., 2008). The same group has recently demonstrated that the N-terminus of L. pneumophila flagellin relieves the requirement for NAIP5 during activation of the NLRC4 inflammasome, which suggests that NAIP5 regulates the specificity of the NLRC4 inflammasome for certain species of bacteria (Lightfield et al., 2011). These data explain why for some species, such as L. pneumophila, the activation of the NLRC4 inflammasome requires NAIP5, whereas for other species, such as Salmonella enterica serovar Typhimurium, NAIP5 is dispensable. Strikingly, activating this NLRC4/NAIP5 inflammasome requires a functional Dot/Icm type IV secretion system. This finding led to the speculation that flagellin may leak from the Legionella cell to the macrophage cytoplasm through the Dot/Icm. However, this hypothesis has not yet been experimentally validated.
The mechanisms by which the NLRC4 inflammasome restricts L. pneumophila replication remains incompletely understood. Activation of these receptors triggers a caspase-1-dependent pore formation in macrophage membranes and leads to a specific form of cell death called pyroptosis (Derre and Isberg, 2004; Case et al., 2009; Silveira and Zamboni, 2010; Whitfield et al., 2010). However, the activation of NAIP5 and NLRC4 also facilitates a process independent of pyroptosis that culminates with the restriction of L. pneumophila multiplication within the replicative vacuole occupied by the bacteria (Swanson and Molofsky, 2005; Amer et al., 2006; Fortier et al., 2007). These processes are possibly dependent on the transcriptional regulation of macrophage genes. This hypothesis has been supported by the demonstration that NAIP5 recognition of L. pneumophila triggers IRF1- and IRF8-mediated upregulation of genes important for macrophage resistance to bacterial infection (Fortier et al., 2009). The inducible nitric oxide synthase (NOS2) gene is a possible candidate, as the NAIP5- and caspase-1-dependent induction of NOS2 expression and nitric oxide production has been observed in macrophages transfected with flagellin (Buzzo et al., 2010).
ASC and the NLRP3-Independent Inflammasome
The adaptor protein called apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC/PYCARD) is a small molecule composed of a PYRIN and a CARD domain. ASC bridges caspase-1 to PYRIN-containing molecules, such as NALP/NLRP family members, via CARD/CARD interactions (Mariathasan et al., 2004). Studies performed with macrophages from ASC-deficient mice have indicated that this molecule is important for the secretion of IL-1β in response to infection but is not required for controlling L. pneumophila replication in C57BL/6 macrophages (Molofsky et al., 2006; Ren et al., 2006; Zamboni et al., 2006). This finding led to the proposition that ASC may participate in the NLRC4-dependent activation of caspase-1 in response to L. pneumophila (Case et al., 2009; Pedra et al., 2009). However, subsequent studies demonstrated that L. pneumophila triggers at least two different inflammasomes: one dependent on NLRC4, NAIP5, and flagellin; and another dependent on ASC and independent of NLRP3 (Sutterwala et al., 2006; Case et al., 2009). Although ASC is dispensable for restricting L. pneumophila replication in mice and murine macrophages, a recent report demonstrated that ASC does contribute to the control of L. pneumophila infection in human monocytes (Abdelaziz et al., 2010). The proteins participating in this ASC-dependent inflammasome and the molecular signals that trigger its activation have not yet been elucidated.
Rig-I-Like Receptor and Induction of Type I Interferon
Several studies have reported the production of type I IFN in response to L. pneumophila infection (Opitz et al., 2006; Stetson and Medzhitov, 2006; Lippmann et al., 2008; Chiu et al., 2009; Monroe et al., 2009), and these experiments have contributed to our understanding of the host cell pathways responsible for type I IFN induction. Although RIG-I and MDA5 were initially reported as sensors of viral infection, recent studies indicate that RLRs are also important for the host cell recognition and response to bacterial infection, and both RIG-I and MDA5 have been implicated in type I IFN production by macrophages in response to L. pneumophila infection (Opitz et al., 2006; Stetson and Medzhitov, 2006; Ablasser et al., 2009; Monroe et al., 2009). The production of type I IFN in response to L. pneumophila infection was shown to be dependent of MAVS and IRF3 (Opitz et al., 2006; Chiu et al., 2009; Monroe et al., 2009). Furthermore, this response accounted to bacterial growth restriction, as the addition of exogenous type I IFN to macrophages lead to the inhibition of L. pneumophila replication in non-permissive macrophages (Schiavoni et al., 2004; Plumlee et al., 2009). Several groups have independently demonstrated that this IRF3-dependent innate immune response does not require the flagellin/NAIP5/NLRC4 axis and occurs in both mice and human cells (Opitz et al., 2006; Coers et al., 2007; Lippmann et al., 2008). Activation of the IRF3 pathway was found to be dependent on a functional bacterial Dot/Icm and to require the presence of bacterial DNA in the host cell cytoplasm (Stetson and Medzhitov, 2006). The current hypothesis is that L. pneumophila DNA leaks into the host cell cytoplasm via the Dot/Icm. However, a recent report has demonstrated that L. pneumophila RNA, but not DNA, is responsible for RIG-I-dependent response to L. pneumophila (Monroe et al., 2009). A subsequent study showed that the host protein RNA polymerase III, which converts poly (dA-dT) DNA into 5′-ppp RNA, is important for IFNβ induction through the RIG-I pathway (Chiu et al., 2009). Future studies are required to determine the ligand and receptor responsible for DNA/RNA recognition. Collectively, the investigations of type I IFN in L. pneumophila infection indicate that type I IFN signaling effectively restricts L. pneumophila replication in phagocytes, thus confirming the importance of this pathway for macrophage resistance to L. pneumophila. Conversely, the importance of type I IFN for murine resistance to L. pneumophila infection is less pronounced. Work from independent groups has demonstrated that mice deficient for IFNAR, which impairs the activation by both IFN-α and IFN-β, show no increased susceptibility to L. pneumophila infection (Monroe et al., 2009; Ang et al., 2010). Nevertheless, further investigation may be required to determine why type I IFN affects macrophage but not mouse resistance to L. pneumophila, and future studies are necessary to identify the ligand and receptors involved in the production of type I IFN in response to L. pneumophila infection.
Concluding Remarks
Different families of PRRs, including TLRs, NLRs, and RLRs, effectively recognize L. pneumophila. This recognition leads to several events important for the outcome of infection: (1) phagocytes activate cell-autonomous mechanisms to restrict bacterial replication; (2) phagocytes trigger the expression of hundreds of inflammatory genes, including those for cytokines and chemokines; (3) phagocytes then express stimulatory and co-stimulatory molecules important for antigen presentation; (4) the secreted cytokines and chemokines recruit additional cells to the sites of the infection; (5) antigen presentation will proceed and the immune system may generate specific acquired responses that are highly protective against reinfection. The continued use of L. pneumophila to dissect these processes will aid us in understanding how the immune system fights to prevent Legionnaire’s disease and will continue to provide important insight into the biological functions of the innate immune responses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all of our laboratory colleagues for their helpful discussions. This work was supported by grants from FAEPA/FMRP, INCTV/CNPq, and FAPESP grants 2006/52867-4 and 2010/50959-4. Liliana M. Massis is supported by a postdoctoral fellowship from FAPESP (2008/56725-5), and Dario S. Zamboni is a research fellow from CNPq.
References
Abdelaziz, D. H., Gavrilin, M. A., Akhter, A., Caution, K., Kotrange, S., Khweek, A. A., Abdulrahman, B. A., Grandhi, J., Hassan, Z. A., Marsh, C., Wewers, M. D., and Amer, A. O. (2010). Apoptosis-associated speck-like protein (ASC) controls Legionella pneumophila infection in human monocytes. J. Biol. Chem. 286, 3203–3208.
Ablasser, A., Bauernfeind, F., Hartmann, G., Latz, E., Fitzgerald, K. A., and Hornung, V. (2009). RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072.
Akamine, M., Higa, F., Arakaki, N., Kawakami, K., Takeda, K., Akira, S., and Saito, A. (2005). Differential roles of toll-like receptors 2 and 4 in in vitro responses of macrophages to Legionella pneumophila. Infect. Immun. 73, 352–361.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Amer, A., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Ozoren, N., Brady, G., Meshinchi, S., Jagirdar, R., Gewirtz, A., Akira, S., and Nunez, G. (2006). Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223.
Ang, D. K., Oates, C. V., Schuelein, R., Kelly, M., Sansom, F. M., Bourges, D., Boon, L., Hertzog, P. J., Hartland, E. L., and Van Driel, I. R. (2010). Cutting edge: pulmonary Legionella pneumophila is controlled by plasmacytoid dendritic cells but not type I IFN. J. Immunol. 184, 5429–5433.
Archer, K. A., Ader, F., Kobayashi, K. S., Flavell, R. A., and Roy, C. R. (2010). Cooperation between multiple microbial pattern recognition systems is important for host protection against the intracellular pathogen Legionella pneumophila. Infect. Immun. 78, 2477–2487.
Archer, K. A., Alexopoulou, L., Flavell, R. A., and Roy, C. R. (2009). Multiple MyD88-dependent responses contribute to pulmonary clearance of Legionella pneumophila. Cell. Microbiol. 11, 21–36.
Archer, K. A., and Roy, C. R. (2006). MyD88-dependent responses involving toll-like receptor 2 are important for protection and clearance of Legionella pneumophila in a mouse model of Legionnaires disease. Infect. Immun. 74, 3325–3333.
Beckers, M. C., Yoshida, S., Morgan, K., Skamene, E., and Gros, P. (1995). Natural resistance to infection with Legionella pneumophila : chromosomal localization of the Lgn1 susceptibility gene. Mamm. Genome 6, 540–545.
Berrington, W. R., Iyer, R., Wells, R. D., Smith, K. D., Skerrett, S. J., and Hawn, T. R. (2010). NOD1 and NOD2 regulation of pulmonary innate immunity to Legionella pneumophila. Eur. J. Immunol. 40, 3519–3527.
Bhan, U., Trujillo, G., Lyn-Kew, K., Newstead, M. W., Zeng, X., Hogaboam, C. M., Krieg, A. M., and Standiford, T. J. (2008). Toll-like receptor 9 regulates the lung macrophage phenotype and host immunity in murine pneumonia caused by Legionella pneumophila. Infect. Immun. 76, 2895–2904.
Brandenburg, K., Mayer, H., Koch, M. H., Weckesser, J., Rietschel, E. T., and Seydel, U. (1993). Influence of the supramolecular structure of free lipid A on its biological activity. Eur. J. Biochem. 218, 555–563.
Broz, P., Von Moltke, J., Jones, J. W., Vance, R. E., and Monack, D. M. (2010). Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483.
Burckstummer, T., Baumann, C., Bluml, S., Dixit, E., Durnberger, G., Jahn, H., Planyavsky, M., Bilban, M., Colinge, J., Bennett, K. L., and Superti-Furga, G. (2009). An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272.
Buzzo, C. L., Campopiano, J. C., Massis, L. M., Lage, S. L., Cassado, A. A., Leme-Souza, R., Cunha, L. D., Russo, M., Zamboni, D. S., Amarante-Mendes, G. P., and Bortoluci, K. R. (2010). A novel pathway for inducible nitric-oxide synthase activation through inflammasomes. J. Biol. Chem. 285, 32087–32095.
Case, C. L., Shin, S., and Roy, C. R. (2009). Asc and Ipaf inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect. Immun. 77, 1981–1991.
Chiu, Y. H., Macmillan, J. B., and Chen, Z. J. (2009). RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591.
Coers, J., Vance, R. E., Fontana, M. F., and Dietrich, W. F. (2007). Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol. 9, 2344–2357.
Creagh, E. M., and O’Neill, L. A. (2006). TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 27, 352–357.
Derre, I., and Isberg, R. R. (2004). Macrophages from mice with the restrictive Lgn1 allele exhibit multifactorial resistance to Legionella pneumophila. Infect. Immun. 72, 6221–6229.
Dietrich, W. F., Damron, D. M., Isberg, R. R., Lander, E. S., and Swanson, M. S. (1995). Lgn1, a gene that determines susceptibility to Legionella pneumophila, maps to mouse chromosome 13. Genomics 26, 443–450.
Diez, E., Lee, S. H., Gauthier, S., Yaraghi, Z., Tremblay, M., Vidal, S., and Gros, P. (2003). Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat. Genet. 33, 55–60.
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513.
Fortier, A., De Chastellier, C., Balor, S., and Gros, P. (2007). Birc1e/Naip5 rapidly antagonizes modulation of phagosome maturation by Legionella pneumophila. Cell. Microbiol. 9, 910–923.
Fortier, A., Doiron, K., Saleh, M., Grinstein, S., and Gros, P. (2009). Restriction of Legionella pneumophila replication in macrophages requires concerted action of the transcriptional regulators Irf1 and Irf8 and nod-like receptors Naip5 and Nlrc4. Infect. Immun. 77, 4794–4805.
Frutuoso, M. S., Hori, J. I., Pereira, M. S., Junior, D. S., Sonego, F., Kobayashi, K. S., Flavell, R. A., Cunha, F. Q., and Zamboni, D. S. (2010). The pattern recognition receptors Nod1 and Nod2 account for neutrophil recruitment to the lungs of mice infected with Legionella pneumophila. Microbes Infect. 12, 819–827.
Fuse, E. T., Tateda, K., Kikuchi, Y., Matsumoto, T., Gondaira, F., Azuma, A., Kudoh, S., Standiford, T. J., and Yamaguchi, K. (2007). Role of toll-like receptor 2 in recognition of Legionella pneumophila in a murine pneumonia model. J. Med. Microbiol. 56, 305–312.
Gazzinelli, R. T., and Denkers, E. Y. (2006). Protozoan encounters with toll-like receptor signaling pathways: implications for host parasitism. Nat. Rev. Immunol. 6, 895–906.
Girard, R., Pedron, T., Uematsu, S., Balloy, V., Chignard, M., Akira, S., and Chaby, R. (2003). Lipopolysaccharides from Legionella and Rhizobium stimulate mouse bone marrow granulocytes via toll-like receptor 2. J. Cell. Sci. 116, 293–302.
Hawn, T. R., Berrington, W. R., Smith, I. A., Uematsu, S., Akira, S., Aderem, A., Smith, K. D., and Skerrett, S. J. (2007). Altered inflammatory responses in TLR5-deficient mice infected with Legionella pneumophila. J. Immunol. 179, 6981–6987.
Hawn, T. R., Smith, K. D., Aderem, A., and Skerrett, S. J. (2006). Myeloid differentiation primary response gene (88)- and toll-like receptor 2-deficient mice are susceptible to infection with aerosolized Legionella pneumophila. J. Infect. Dis. 193, 1693–1702.
Hawn, T. R., Verbon, A., Lettinga, K. D., Zhao, L. P., Li, S. S., Laws, R. J., Skerrett, S. J., Beutler, B., Schroeder, L., Nachman, A., Ozinsky, A., Smith, K. D., and Aderem, A. (2003). A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires’ disease. J. Exp. Med. 198, 1563–1572.
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., Latz, E., and Fitzgerald, K. A. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518.
Ishii, K. J., and Akira, S. (2006). Innate immune recognition of, and regulation by, DNA. Trends Immunol. 27, 525–532.
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216.
Lamkanfi, M., Amer, A., Kanneganti, T. D., Munoz-Planillo, R., Chen, G., Vandenabeele, P., Fortier, A., Gros, P., and Nunez, G. (2007). The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J. Immunol. 178, 8022–8027.
Lettinga, K. D., Florquin, S., Speelman, P., Van Ketel, R., Van Der Poll, T., and Verbon, A. (2002). Toll-like receptor 4 is not involved in host defense against pulmonary Legionella pneumophila infection in a mouse model. J. Infect. Dis. 186, 570–573.
Lightfield, K. L., Persson, J., Brubaker, S. W., Witte, C. E., Von Moltke, J., Dunipace, E. A., Henry, T., Sun, Y. H., Cado, D., Dietrich, W. F., Monack, D. M., Tsolis, R. M., and Vance, R. E. (2008). Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178.
Lightfield, K. L., Persson, J., Trinidad, N. J., Brubaker, S. W., Kofoed, E. M., Sauer, J. D., Dunipace, E. A., Warren, S. E., Miao, E. A., and Vance, R. E. (2011). Differential requirements for NAIP5 in activation of the NLRC4 (IPAF) inflammasome. Infect. Immun. 79, 1606–1614.
Lippmann, J., Rothenburg, S., Deigendesch, N., Eitel, J., Meixenberger, K., Van Laak, V., Slevogt, H., N’guessan P, D., Hippenstiel, S., Chakraborty, T., Flieger, A., Suttorp, N., and Opitz, B. (2008). IFNbeta responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI). Cell. Microbiol. 10, 2579–2588.
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., Roose-Girma, M., Erickson, S., and Dixit, V. M. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218.
Molofsky, A. B., Byrne, B. G., Whitfield, N. N., Madigan, C. A., Fuse, E. T., Tateda, K., and Swanson, M. S. (2006). Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203, 1093–1104.
Monroe, K. M., Mcwhirter, S. M., and Vance, R. E. (2009). Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5, e1000665. doi: 10.1371/journal.ppat.1000665
Neild, A. L., Shin, S., and Roy, C. R. (2005). Activated macrophages infected with Legionella inhibit T cells by means of MyD88-dependent production of prostaglandins. J. Immunol. 175, 8181–8190.
Newton, C. A., Perkins, I., Widen, R. H., Friedman, H., and Klein, T. W. (2007). Role of toll-like receptor 9 in Legionella pneumophila -induced interleukin-12 p40 production in bone marrow-derived dendritic cells and macrophages from permissive and nonpermissive mice. Infect. Immun. 75, 146–151.
O’Neill, L. A. (2008). The interleukin-1 receptor/toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 226, 10–18.
Opitz, B., Vinzing, M., Van Laak, V., Schmeck, B., Heine, G., Gunther, S., Preissner, R., Slevogt, H., N’guessan, P. D., Eitel, J., Goldmann, T., Flieger, A., Suttorp, N., and Hippenstiel, S. (2006). Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J. Biol. Chem. 281, 36173–36179.
Pedra, J. H., Cassel, S. L., and Sutterwala, F. S. (2009). Sensing pathogens and danger signals by the inflammasome. Curr. Opin. Immunol. 21, 10–16.
Plumlee, C. R., Lee, C., Beg, A. A., Decker, T., Shuman, H. A., and Schindler, C. (2009). Interferons direct an effective innate response to Legionella pneumophila infection. J. Biol. Chem. 284, 30058–30066.
Poltorak, A., He, X., Smirnova, I., Liu, M. Y., Van Huffel, C., Du, X., Birdwell, D., Alejos, E., Silva, M., Galanos, C., Freudenberg, M., Ricciardi-Castagnoli, P., Layton, B., and Beutler, B. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088.
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F., and Vance, R. E. (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. doi: 10.1371/journal.ppat.0020018
Roberts, T. L., Idris, A., Dunn, J. A., Kelly, G. M., Burnton, C. M., Hodgson, S., Hardy, L. L., Garceau, V., Sweet, M. J., Ross, I. L., Hume, D. A., and Stacey, K. J. (2009). HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060.
Schiavoni, G., Mauri, C., Carlei, D., Belardelli, F., Pastoris, M. C., and Proietti, E. (2004). Type I IFN protects permissive macrophages from Legionella pneumophila infection through an IFN-gamma-independent pathway. J. Immunol. 173, 1266–1275.
Shaw, M. H., Reimer, T., Kim, Y. G., and Nunez, G. (2008). NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr. Opin. Immunol. 20, 377–382.
Shin, S., Case, C. L., Archer, K. A., Nogueira, C. V., Kobayashi, K. S., Flavell, R. A., Roy, C. R., and Zamboni, D. S. (2008). Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog. 4, e1000220. doi:10.1371/journal.ppat.1000220
Silveira, T. N., and Zamboni, D. S. (2010). Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome-dependent host cell response that precedes pyroptosis. Infect. Immun. 78, 1403–1413.
Sporri, R., Joller, N., Albers, U., Hilbi, H., and Oxenius, A. (2006). MyD88-dependent IFN-gamma production by NK cells is key for control of Legionella pneumophila infection. J. Immunol. 176, 6162–6171.
Stetson, D. B., and Medzhitov, R. (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103.
Sutterwala, F. S., Ogura, Y., Zamboni, D. S., Roy, C. R., and Flavell, R. A. (2006). NALP3: a key player in caspase-1 activation. J. Endotoxin Res. 12, 251–256.
Swanson, M. S., and Molofsky, A. B. (2005). Autophagy and inflammatory cell death, partners of innate immunity. Autophagy 1, 174–176.
Takaoka, A., Wang, Z., Choi, M. K., Yanai, H., Negishi, H., Ban, T., Lu, Y., Miyagishi, M., Kodama, T., Honda, K., Ohba, Y., and Taniguchi, T. (2007). DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505.
Uematsu, S., and Akira, S. (2006). Innate immunity and toll-like receptor. Nippon Naika Gakkai Zasshi 95, 1115–1121.
Whitfield, N. N., Byrne, B. G., and Swanson, M. S. (2010). Mouse macrophages are permissive to motile Legionella species that fail to trigger pyroptosis. Infect. Immun. 78, 423–432.
Wright, E. K., Goodart, S. A., Growney, J. D., Hadinoto, V., Endrizzi, M. G., Long, E. M., Sadigh, K., Abney, A. L., Bernstein-Hanley, I., and Dietrich, W. F. (2003). Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr. Biol. 13, 27–36.
Yamamoto, Y., Klein, T. W., Newton, C. A., Widen, R., and Friedman, H. (1988). Growth of Legionella pneumophila in thioglycolate-elicited peritoneal macrophages from A/J mice. Infect. Immun. 56, 370–375.
Zamboni, D. S., Campos, M. A., Torrecilhas, A. C., Kiss, K., Samuel, J. E., Golenbock, D. T., Lauw, F. N., Roy, C. R., Almeida, I. C., and Gazzinelli, R. T. (2004). Stimulation of toll-like receptor 2 by Coxiella burnetii is required for macrophage production of pro-inflammatory cytokines and resistance to infection. J. Biol. Chem. 279, 54405–54415.
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., Kuida, K., Mariathasan, S., Dixit, V. M., Flavell, R. A., Dietrich, W. F., and Roy, C. R. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325.
Keywords: Legionella pneumophila, innate immunity, pattern recognition receptors, nod-like receptors
Citation: Massis LM and Zamboni DS (2011) Innate immunity to Legionella pneumophila. Front. Microbio. 2:109. doi: 10.3389/fmicb.2011.00109
Received: 11 February 2011;
Paper pending published: 13 March 2011;
Accepted: 03 May 2011;
Published online: 16 May 2011.
Edited by:
Carmen Buchrieser, Pasteur Institute, FranceReviewed by:
Thomas Rudel, University of Wuerzburg, GermanyElizabeth L. Hartland, The University of Melbourne, Australia
Copyright: © 2011 Massis and Zamboni. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Dario S. Zamboni, Department of Cell Biology, School of Medicine, University of São Paulo, Av. Bandeirantes, 3900, Ribeirão Preto, SP 14049-900, Brazil. e-mail:ZHN6YW1ib25pQGZtcnAudXNwLmJy