
 Desmond K. Y. Ang2
Desmond K. Y. Ang2
- 1 Department of Microbiology and Immunology, University of Melbourne, Parkville, Victoria, Australia
- 2 Department of Biochemistry and Molecular Biology and the Bio21 Institute, University of Melbourne, Parkville, Victoria, Australia
Legionella pneumophila is an intracellular pathogen that replicates within alveolar macrophages. Through its ability to activate multiple host innate immune components, L. pneumophila has emerged as a useful tool to dissect inflammatory signaling pathways in macrophages. However the resolution of L. pneumophila infection in the lung requires multiple cell types and abundant cross talk between immune cells. Few studies have examined the coordination of events that lead to effective immune control of the pathogen. Here we discuss L. pneumophila interactions with macrophages and dendritic cell subsets and highlight the paucity of knowledge around how these interactions recruit and activate other immune effector cells in the lung.
Introduction
Members of the genus Legionella are Gram-negative, facultative intracellular bacteria of amoebae, including free-living, freshwater, or soil amoebae (Rowbotham, 1980; Tyndall and Domingue, 1982; Fields, 1996). Legionella pneumophila was the first species described and is the known causative agent of an acute form of pneumonia termed Legionnaires’ disease (Fraser et al., 1977; McDade et al., 1977). Humans become secondarily infected after inhaling or aspirating aerosols containing bacteria. Upon its transmission to the human lung, L. pneumophila enters and replicates in alveolar macrophages, leading to inflammation and disease (Horwitz and Silverstein, 1980; Horwitz, 1983a). Replication in macrophages is thus a hallmark of L. pneumophila infection. Within macrophages, the bacteria block phagolysosome fusion and intercept vesicles trafficking in the secretory pathway (Horwitz, 1983b; Kagan and Roy, 2002). The resulting Legionella-containing vacuole (LCV), ultimately takes on properties of the rough endoplasmic reticulum (Roy and Tilney, 2002; Isberg et al., 2009). The formation of the LCV is dependent on a functional Dot/Icm Type IVB secretion system used by the pathogen to deliver effectors into the host cell cytosol (Segal and Shuman, 1997; Segal et al., 1998; Vogel et al., 1998). At least 275 effectors have been identified (Zhu et al., 2011), that target multiple and overlapping host cell functions including host cell GTPase activity, phosphoinositide metabolism, protein secretion, apoptosis, eukaryotic protein translation, ubiquitination, NF-κB activation and mitochondrial function, reviewed in (Franco et al., 2009; Isberg et al., 2009; Weber et al., 2009; Hubber and Roy, 2010; Newton et al., 2010).
Replication of L. Pneumophila in Macrophages
Macrophages and dendritic cells (DC) are important sentinels of the immune system detecting infectious agents by highly conserved microbial motifs, so-called pathogen-associated molecular patterns (PAMPs; Janeway Jr., 1992). Pattern recognition is mediated by a set of invariant pattern-recognition receptors (PRRs) of which four families have been identified: toll-like receptors (TLRs), retinoic acid-inducible gene-I (RIG-I) like receptors (RLRs), C-type lectin receptors (CLRs), and nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs; Takeuchi and Akira, 2010). NLRs comprise a large family of cytoplasmic PRRs of which only a few members have been characterized in detail. Some NLRs form multiprotein complexes called inflammasomes (Schroder and Tschopp, 2010) and activation of these complexes leads to the cleavage of the central effector molecule cysteine protease caspase-1, inducing a form of cell death known as pyroptosis which is accompanied by the release of pyrogenic IL-1ß, IL-18, and IL-33 (Davis et al., 2011).
The flagellin sensing Nlrc4 inflammasome plays a central role in the detection of L. pneumophila, which is the reason most inbred strains of mice are resistant to L. pneumophila infection. The discovery of the Nlrc4 inflammasome began with the observation that macrophages derived from most mouse strains restrict bacterial replication with the notable exception of the A strain (often called A/J, although this terminology refers only to mice derived directly from the Jackson or Janvier laboratories; Yamamoto et al., 1988). Crosses between A mice and non-permissive C57BL/6 mice showed that the susceptibility of the A strain is controlled by a single locus on mouse chromosome 13, designated Lgn1 (Beckers et al., 1995; Dietrich et al., 1995). Genetic studies then identified the new NLR gene, Naip5, within this locus as responsible for the increased susceptibility of A mice to infection (Diez et al., 2003; Wright et al., 2003). Subsequent work showed that Naip5-dependent restriction of L. pneumophila relies on a functional copy of Naip5 as well as Nlrc4 and activation of caspase-1 (Zamboni et al., 2006). Restriction results from the presence of bacterial flagellin in the host cytosol, and recognition of the C-terminus of flagellin is sufficient for activation of the Nlrc4 inflammasome (Molofsky et al., 2006; Ren et al., 2006; Lightfield et al., 2008). Interestingly, the cytosolic localization of flagellin and/or restriction of replication depends on a functional Dot/Icm type 4 secretion system (Amer et al., 2006; Molofsky et al., 2006; Ren et al., 2006; Zamboni et al., 2006; Lamkanfi et al., 2007). However, it is not known how the Dot/Icm system contributes to the translocation of flagellin into the host cytosol and whether the detection of flagellin by the inflammasome occurs directly or indirectly with the help of cofactors.
While formation of the inflammasome leads to the activation of caspase-1, as well as maturation and secretion of IL-1ß and IL-18, neither cytokine makes a major contribution to the restriction of L. pneumophila in vitro or in vivo (Amer et al., 2006; Zamboni et al., 2006; Coers et al., 2007; Akhter et al., 2009; Miao et al., 2010). Nevertheless, caspase-1 knockout macrophages are more permissive for L. pneumophila replication and caspase1-deficient mice are more susceptible to L. pneumophila infection (Amer et al., 2006; Zamboni et al., 2006). Caspase-1 activation upon bacterial infection may also result from an alternative Nlrc4-independent pathway which requires the apoptosis associated speck-like protein (Asc), yet Asc is dispensable for restriction (Zamboni et al., 2006; Case et al., 2009). Although depletion or inhibition of caspase-1 activity leads to decreased targeting of bacteria to lysosomes (Amer et al., 2006; Zamboni et al., 2006), the mechanism of caspase-1-dependent restriction of L. pneumophila replication in macrophages and in vivo is yet to be fully resolved. Activation of the Nlrc4 inflammasome can lead to macrophage cell death through caspase-1 dependent pore formation, which may account for reduced bacterial numbers through macrophage cell lysis (Case et al., 2009; Silveira and Zamboni, 2010). Downstream molecules such as caspase-7, interferon regulatory factor (IRF) 1 and IRF8 also play a significant role in caspase-1 signaling and in the case of caspase-7, this activation leads to increased macrophage apoptosis (Akhter et al., 2009; Fortier et al., 2009). caspase7-deficient mice are also more susceptible to L. pneumophila infection (Akhter et al., 2009). However, the ability of L. pneumophila to replicate within macrophages in vitro does not necessarily equate with virulence in whole animals. For example, type I interferon (IFN-I) receptor-deficient macrophages, support enhanced replication of L. pneumophila yet IFN-I receptor-deficient mice are no more susceptible to infection in vivo (Monroe et al., 2009; Ang et al., 2010).
In contrast to macrophages derived from restrictive mouse strains, human macrophages or monocytes allow robust replication of L. pneumophila despite the presence of Naip and Nlrc4 orthologues. Human Nlrc4 and Naip are functional but only delay L. pneumophila replication when overexpressed (Vinzing et al., 2008) suggesting that the level of inflammasome activity may restrict L. pneumophila replication in humans cells, similar to mice. A recent report also showed that human Asc is able to restrict bacterial growth in a caspase-1-dependent and independent manner but is downregulated during L. pneumophila infection of monocytic THP-1 cells (Abdelaziz et al., 2011). More studies in human cells, ideally in primary macrophages, will provide a useful comparison to the results derived from using mouse infection models.
Immune effector molecules produced by infected macrophages are likely to play an important role in generating a protective immune response and warrant further analysis. In mouse and human macrophages, infection with live L. pneumophila induces the production of inflammatory cytokines and chemokines such as tumor necrosis factor α (TNF), interleukin (IL)-10, IL-6, IL-1β, IL-18, CXCL1 and MCP-1 as well as IFN-I (Shin et al., 2008; Case et al., 2009; Monroe et al., 2009; Plumlee et al., 2009; McCoy-Simandle et al., 2011), whereas other cytokines such as IL-12 and IFN-γ appear to be produced at only very low levels, if at all (Matsunaga et al., 2001, 2003). In whole animals, increased susceptibility to pulmonary L. pneumophila results from cytokine and/or cytokine receptor deficiencies in IL-12, IFN-γ, and TNF (Brieland et al., 1998; Shinozawa et al., 2002; Fujita et al., 2008). This suggests that cytokine production by cell types other than macrophages is important for controlling infection. At this stage a thorough understanding of the role of distinct cytokines and immune cells in combating L. pneumophila lung infection is lacking.
Legionella Pneumophila Interactions with DC
Dendritic cells represent a heterogeneous group of cells with specialized functional properties. DC play a critical role in eliciting adaptive immune responses through their role as primary antigen presentation cells (Heath and Carbone, 2009). Several subsets of DC are now recognized in the mouse, which began with the identification of CD8− and CD8+ DC in the spleen (Heath and Carbone, 2009). Further examination of precursor-product relationships led to the identification of distinct end stage subsets of DC, including CD4−CD8− (double-negative) DC, CD103+, and CD11b + migratory DC and Langerhans cells, as well as plasmacytoid DC (pDC) which are set apart from the other conventional DC by their gene expression profile. The role of pDC in generating adaptive immunity is unclear although evidence for a role in antigen presentation is emerging (Heath and Carbone, 2009).
We recently showed that pDC make an important contribution to the restriction of L. pneumophila infection in vivo (Ang et al., 2010). pDC are known for their ability to combat viral infection through the production of IFN-I (Colonna et al., 2004; Fitzgerald-Bocarsly et al., 2008). However, a role for pDC in resistance to bacterial infection had not been described before. During L. pneumophila infection, pDC are rapidly recruited to the lungs of mice and depletion of pDC significantly increases bacterial burden in the lung (Ang et al., 2010). Currently, the mechanism by which pDC restrict L. pneumophila infection is not known. However, it is clear that IFN-I is not necessary as IFN-I-receptor-deficient (IFNAR−/−) mice are not more severely infected by L. pneumophila compared to wild type mice (Monroe et al., 2009; Ang et al., 2010). Moreover, depletion of pDC in IFNAR−/− mice results in increased bacterial load in the lung, suggesting that IFN-I signaling is dispensable for the anti-bacterial activity of pDC (Ang et al., 2010). Although L. pneumophila can infect pDC (Ang et al., 2010), the number of bacteria per host cell is significantly lower compared to macrophages, suggesting that, similar to conventional DC, bacteria do not replicate intracellularly within pDC (Neild and Roy, 2003). The mechanisms that recruit pDC to the lung are not yet known but as the primary site of L. pneumophila replication, macrophages are a likely source of chemoattractant cytokines. pDC presumably then respond to L. pneumophila infection by producing cytokines that activate neutrophils, NK cells, and/or macrophages to kill intracellular bacteria (Figure 1). Further investigation is needed to determine the mechanisms by which pDC restrict L. pneumophila infection and importantly whether these mechanisms are utilized to combat other bacterial pathogens.
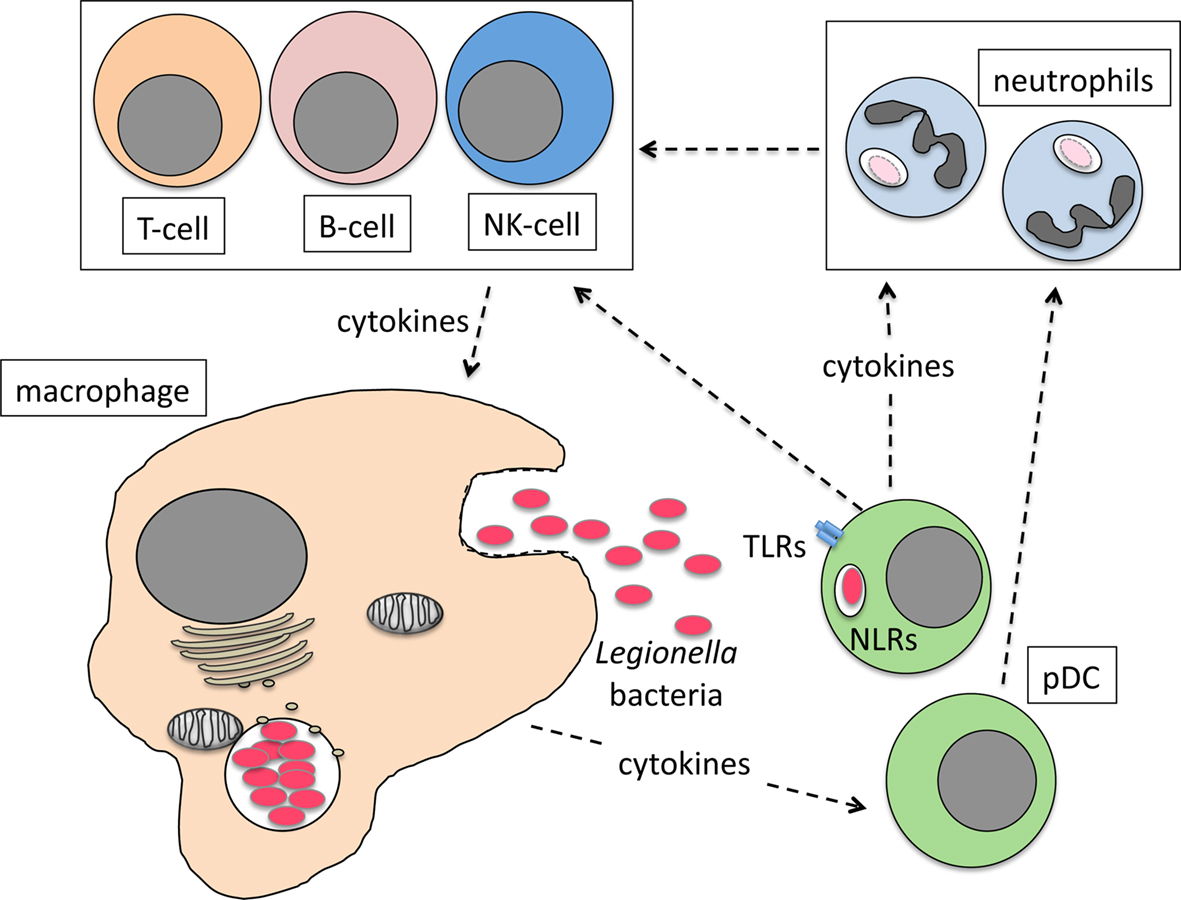
Figure 1. Model for the role of pDC in combating L. pneumophila lung infection. Infected macrophages produce cytokines and chemokines that recruit pDC to the lung. Bacteria activate pDC via TLR/NLR interactions or cytokines from infected macrophages stimulate pDC cytokine production that then activates neutrophils, NK cells and macrophages to kill bacteria directly or indirectly.
In contrast to macrophages, conventional DC do not allow replication of L. pneumophila (Neild and Roy, 2003), even if derived from A strain mice. This is despite the fact that LCV formation in DC appears to be similar to that in macrophages (Neild and Roy, 2003). Restriction of replication by mouse DC is the result of activation of both caspase-1-dependent pyroptosis and classical cell death pathways through Bcl-2-associated X (Bax) and Bcl2 antagonist/killer (Bak) mediated apoptosis (Nogueira et al., 2009). The initiation of the intrinsic (mitochondrial) apoptotic pathway by Bax/Bak leads to early activation of caspase-3 in DC that is delayed in macrophages (Nogueira et al., 2009). L. pneumophila is known to induce the intrinsic pathway in macrophages (Hagele et al., 1998; Gao and Abu Kwaik, 1999; Molmeret et al., 2004; Abu-Zant et al., 2005; Furugen et al., 2008; Nogueira et al., 2009) but counteracts the pro-apoptotic stimuli, in part by triggering NF-κB dependent up-regulation of anti-apoptotic genes (Losick and Isberg, 2006; Abu-Zant et al., 2007; Bartfeld et al., 2009) as well as delivering anti-apoptotic Dot/Icm effectors such as SdhA and SidF (Laguna et al., 2006; Banga et al., 2007). In fact, SidF acts directly on pro-apoptotic Bcl2 family members Bcl-rambo and BNIP3 while the anti-apoptotic mechanism of SdhA seems independent of central components of the apoptosis pathway (Laguna et al., 2006; Banga et al., 2007; Nogueira et al., 2009). It is unclear why these effectors are functional in macrophages but do not have the same impact in conventional DC, despite the fact that SdhA appears to be at least partially functional (Nogueira et al., 2009). Nevertheless, rapid apoptosis is key to the difference between L. pneumophila replication in macrophages and conventional DC because adding the anti-apoptotic Dot/Icm effector, AnkG, from the evolutionarily related pathogen, Coxiella burnetii, inhibits L. pneumophila induced apoptosis of DC and reverses the restriction on bacterial replication (Luhrmann et al., 2010). The importance of conventional DC in controlling L. pneumophila infection in vivo is not known. While DC presumably play a role in antigen presentation and the development of an adaptive response, no direct role for conventional DC in controlling L. pneumophila lung infection has been proven. It has been proposed that DC may act as a dead end for L. pneumophila replication thereby restricting bacterial infection but this hypothesis has not been tested directly in vivo, for example by depletion of conventional DC (Nogueira et al., 2009).
Concluding Remarks
Biopsies from patients with Legionnaire’s disease show bacteria contained within multi-organism vacuoles in alveolar macrophages (Chandler et al., 1977; Glavin et al., 1979; Hernandez et al., 1980). In guinea pig and mouse lung infection models, alveolar macrophages are the first cells infected by L. pneumophila (Winn Jr., 1988; LeibundGut-Landmann et al., 2011). As the initial niche for bacterial replication, macrophages play a pivotal role in initiating the host response to L. pneumophila. Indeed recently, IL-1β production by mouse alveolar macrophages was shown to activate cytokine responses in airway epithelial cells (LeibundGut-Landmann et al., 2011). As such, this initial interaction with macrophages is likely to be crucial for the recruitment of immune effector cells including neutrophils and NK cells. In both intravenous and respiratory infection models, IL-18 is required for IFN-γ production by NK cells (Sporri et al., 2008; Archer et al., 2009), however whereas the intravenous model of L. pneumophila infection suggested increased susceptibility of IL-18 receptor knockout mice (measured by increased splenic bacterial load; Sporri et al., 2008), this result was not validated in the respiratory infection model (Archer et al., 2009). Therefore it appears that the role of cytokines and immune cells during lung infection differs from interactions during systemic responses. T- and B-cells also ultimately contribute to clear the organism (Susa et al., 1998; Kikuchi et al., 2005; Joller et al., 2007) but their recruitment and mechanism of activation has not been closely examined in the context of L. pneumophila infection in vivo. Given that the resolution of L. pneumophila infection requires multiple cell types and abundant cross talk between immune cells, the role of other cell types such as DC as well as the mechanism of action of protective cytokines should be examined. The coordinated functions of these immune components during L. pneumophila infection in vivo is likely to yield important new information about immune defense mechanisms in the lung.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants to Elizabeth L. Hartland and Ian R. van Driel from the Australian National Health and Medical Research Council (NHMRC). Elizabeth L. Hartland is supported by an Australian Research Council Future Fellowship.
References
Abdelaziz, D. H., Gavrilin, M. A., Akhter, A., Caution, K., Kotrange, S., Khweek, A. A., Abdulrahman, B. A., Grandhi, J., Hassan, Z. A., Marsh, C., Wewers, M. D., and Amer, A. O. (2011). Apoptosis-associated speck-like protein (ASC) controls Legionella pneumophila infection in human monocytes. J. Biol. Chem. 286, 3203–3208.
Abu-Zant, A., Jones, S., Asare, R., Suttles, J., Price, C., Graham, J., and Kwaik, Y. A. (2007). Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell. Microbiol. 9, 246–264.
Abu-Zant, A., Santic, M., Molmeret, M., Jones, S., Helbig, J., and Abu Kwaik, Y. (2005). Incomplete activation of macrophage apoptosis during intracellular replication of Legionella pneumophila. Infect. Immun. 73, 5339–5349.
Akhter, A., Gavrilin, M. A., Frantz, L., Washington, S., Ditty, C., Limoli, D., Day, C., Sarkar, A., Newland, C., Butchar, J., Marsh, C. B., Wewers, M. D., Tridandapani, S., Kanneganti, T. D., and Amer, A. O. (2009). Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. 5, e1000361. doi: 10.1371/journal.ppat.1000361
Amer, A., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Ozoren, N., Brady, G., Meshinchi, S., Jagirdar, R., Gewirtz, A., Akira, S., and Nunez, G. (2006). Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223.
Ang, D. K., Oates, C. V., Schuelein, R., Kelly, M., Sansom, F. M., Bourges, D., Boon, L., Hertzog, P. J., Hartland, E. L., and Van Driel, I. R. (2010). Cutting edge: pulmonary Legionella pneumophila is controlled by plasmacytoid dendritic cells but not type I IFN. J. Immunol. 184, 5429–5433.
Archer, K. A., Alexopoulou, L., Flavell, R. A., and Roy, C. R. (2009). Multiple MyD88-dependent responses contribute to pulmonary clearance of Legionella pneumophila. Cell. Microbiol. 11, 21–36.
Banga, S., Gao, P., Shen, X., Fiscus, V., Zong, W. X., Chen, L., and Luo, Z. Q. (2007). Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc. Natl. Acad. Sci. U.S.A. 104, 5121–5126.
Bartfeld, S., Engels, C., Bauer, B., Aurass, P., Flieger, A., Bruggemann, H., and Meyer, T. F. (2009). Temporal resolution of two-tracked NF-kappaB activation by Legionella pneumophila. Cell. Microbiol. 11, 1638–1651.
Beckers, M. C., Yoshida, S., Morgan, K., Skamene, E., and Gros, P. (1995). Natural resistance to infection with Legionella pneumophila: chromosomal localization of the Lgn1 susceptibility gene. Mamm. Genome 6, 540–545.
Brieland, J. K., Remick, D. G., Legendre, M. L., Engleberg, N. C., and Fantone, J. C. (1998). In vivo regulation of replicative Legionella pneumophila lung infection by endogenous interleukin-12. Infect. Immun. 66, 65–69.
Case, C. L., Shin, S., and Roy, C. R. (2009). Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect. Immun. 77, 1981–1991.
Chandler, F. W., Hicklin, M. D., and Blackmon, J. A. (1977). Demonstration of the agent of Legionnaires’ disease in tissue. N. Engl. J. Med. 297, 1218–1220.
Coers, J., Vance, R. E., Fontana, M. F., and Dietrich, W. F. (2007). Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol. 9, 2344–2357.
Colonna, M., Trinchieri, G., and Liu, Y. J. (2004). Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5, 1219–1226.
Davis, B. K., Wen, H., and Ting, J. P. (2011). The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735.
Dietrich, W. F., Damron, D. M., Isberg, R. R., Lander, E. S., and Swanson, M. S. (1995). Lgn1, a gene that determines susceptibility to Legionella pneumophila, maps to mouse chromosome 13. Genomics 26, 443–450.
Diez, E., Lee, S. H., Gauthier, S., Yaraghi, Z., Tremblay, M., Vidal, S., and Gros, P. (2003). Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat. Genet. 33, 55–60.
Fitzgerald-Bocarsly, P., Dai, J., and Singh, S. (2008). Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 19, 3–19.
Fortier, A., Doiron, K., Saleh, M., Grinstein, S., and Gros, P. (2009). Restriction of Legionella pneumophila replication in macrophages requires concerted action of the transcriptional regulators Irf1 and Irf8 and NOD-like receptors Naip5 and Nlrc4. Infect. Immun. 77, 4794–4805.
Franco, I. S., Shuman, H. A., and Charpentier, X. (2009). The perplexing functions and surprising origins of Legionella pneumophila type IV secretion effectors. Cell. Microbiol. 11, 1435–1443.
Fraser, D. W., Tsai, T. R., Orenstein, W., Parkin, W. E., Beecham, H. J., Sharrar, R. G., Harris, J., Mallison, G. F., Martin, S. M., Mcdade, J. E., Shepard, C. C., and Brachman, P. S. (1977). Legionnaires’ disease: description of an epidemic of pneumonia. N. Engl. J. Med. 297, 1189–1197.
Fujita, M., Ikegame, S., Harada, E., Ouchi, H., Inoshima, I., Watanabe, K., Yoshida, S., and Nakanishi, Y. (2008). TNF receptor 1 and 2 contribute in different ways to resistance to Legionella pneumophila-induced mortality in mice. Cytokine 44, 298–303.
Furugen, M., Higa, F., Hibiya, K., Teruya, H., Akamine, M., Haranaga, S., Yara, S., Koide, M., Tateyama, M., Mori, N., and Fujita, J. (2008). Legionella pneumophila infection induces programmed cell death, caspase activation, and release of high-mobility group box 1 protein in A549 alveolar epithelial cells: inhibition by methyl prednisolone. Respir. Res. 9, 39.
Gao, L. Y., and Abu Kwaik, Y. (1999). Activation of caspase 3 during Legionella pneumophila-induced apoptosis. Infect. Immun. 67, 4886–4894.
Glavin, F. L., Winn, W. C. Jr., and Craighead, J. E. (1979). Ultrastructure of lung in Legionnaires’ disease. Observations of three biopsies done during the Vermont epidemic. Ann. Intern. Med. 90, 555–559.
Hagele, S., Hacker, J., and Brand, B. C. (1998). Legionella pneumophila kills human phagocytes but not protozoan host cells by inducing apoptotic cell death. FEMS Microbiol. Lett. 169, 51–58.
Heath, W. R., and Carbone, F. R. (2009). Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 10, 1237–1244.
Hernandez, F. J., Kirby, B. D., Stanley, T. M., and Edelstein, P. H. (1980). Legionnaires’ disease. Postmortem pathologic findings of 20 cases. Am. J. Clin. Pathol. 73, 488–495.
Horwitz, M. A. (1983a). Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158, 1319–1331.
Horwitz, M. A. (1983b). The Legionnaires’ disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J. Exp. Med. 158, 2108–2126.
Horwitz, M. A., and Silverstein, S. C. (1980). Legionnaires’ disease bacterium (Legionella pneumophila) multiples intracellularly in human monocytes. J. Clin. Invest. 66, 441–450.
Hubber, A., and Roy, C. R. (2010). Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26, 261–283.
Isberg, R. R., O’Connor, T. J., and Heidtman, M. (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24.
Janeway, C. A. Jr. (1992). The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Today 13, 11–16.
Joller, N., Sporri, R., Hilbi, H., and Oxenius, A. (2007). Induction and protective role of antibodies in Legionella pneumophila infection. Eur. J. Immunol. 37, 3414–3423.
Kagan, J. C., and Roy, C. R. (2002). Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4, 945–954.
Kikuchi, T., Andarini, S., Xin, H., Gomi, K., Tokue, Y., Saijo, Y., Honjo, T., Watanabe, A., and Nukiwa, T. (2005). Involvement of fractalkine/CX3CL1 expression by dendritic cells in the enhancement of host immunity against Legionella pneumophila. Infect. Immun. 73, 5350–5357.
Laguna, R. K., Creasey, E. A., Li, Z., Valtz, N., and Isberg, R. R. (2006). A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U.S.A. 103, 18745–18750.
Lamkanfi, M., Amer, A., Kanneganti, T. D., Munoz-Planillo, R., Chen, G., Vandenabeele, P., Fortier, A., Gros, P., and Nunez, G. (2007). The NOD-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J. Immunol. 178, 8022–8027.
LeibundGut-Landmann, S., Weidner, K., Hilbi, H., and Oxenius, A. (2011). Nonhematopoietic cells are key players in innate control of bacterial airway infection. J. Immunol. 186, 3130–3137.
Lightfield, K. L., Persson, J., Brubaker, S. W., Witte, C. E., Von Moltke, J., Dunipace, E. A., Henry, T., Sun, Y. H., Cado, D., Dietrich, W. F., Monack, D. M., Tsolis, R. M., and Vance, R. E. (2008). Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178.
Losick, V. P., and Isberg, R. R. (2006). NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203, 2177–2189.
Luhrmann, A., Nogueira, C. V., Carey, K. L., and Roy, C. R. (2010). Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc. Natl. Acad. Sci. U.S.A. 107, 18997–19001.
Matsunaga, K., Klein, T. W., Newton, C., Friedman, H., and Yamamoto, Y. (2001). Legionella pneumophila suppresses interleukin-12 production by macrophages. Infect. Immun. 69, 1929–1933.
Matsunaga, K., Yamaguchi, H., Klein, T. W., Friedman, H., and Yamamoto, Y. (2003). Legionella pneumophila suppresses macrophage interleukin-12 production by activating the p42/44 mitogen-activated protein kinase cascade. Infect. Immun. 71, 6672–6675.
McCoy-Simandle, K., Stewart, C. R., Dao, J., Debroy, S., Rossier, O., Bryce, P. J., and Cianciotto, N. P. (2011). Legionella pneumophila type II secretion dampens the cytokine response of infected macrophages and epithelia. Infect. Immun. 79, 1984–1997.
McDade, J. E., Shepard, C. C., Fraser, D. W., Tsai, T. R., Redus, M. A., and Dowdle, W. R. (1977). Legionnaires’ disease: isolation of a bacterium and demonstration of its role in other respiratory disease. N. Engl. J. Med. 297, 1197–1203.
Miao, E. A., Leaf, I. A., Treuting, P. M., Mao, D. P., Dors, M., Sarkar, A., Warren, S. E., Wewers, M. D., and Aderem, A. (2010). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142.
Molmeret, M., Zink, S. D., Han, L., Abu-Zant, A., Asari, R., Bitar, D. M., and Abu Kwaik, Y. (2004). Activation of caspase-3 by the Dot/Icm virulence system is essential for arrested biogenesis of the Legionella-containing phagosome. Cell. Microbiol. 6, 33–48.
Molofsky, A. B., Byrne, B. G., Whitfield, N. N., Madigan, C. A., Fuse, E. T., Tateda, K., and Swanson, M. S. (2006). Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203, 1093–1104.
Monroe, K. M., Mcwhirter, S. M., and Vance, R. E. (2009). Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5, e1000665. doi: 10.1371/journal.ppat.1000665
Neild, A. L., and Roy, C. R. (2003). Legionella reveal dendritic cell functions that facilitate selection of antigens for MHC class II presentation. Immunity 18, 813–823.
Newton, H. J., Ang, D. K., Van Driel, I. R., and Hartland, E. L. (2010). Molecular pathogenesis of infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 23, 274–298.
Nogueira, C. V., Lindsten, T., Jamieson, A. M., Case, C. L., Shin, S., Thompson, C. B., and Roy, C. R. (2009). Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila. PLoS Pathog. 5, e1000478. doi: 10.1371/journal.ppat.1000478
Plumlee, C. R., Lee, C., Beg, A. A., Decker, T., Shuman, H. A., and Schindler, C. (2009). Interferons direct an effective innate response to Legionella pneumophila infection. J. Biol. Chem. 284, 30058–30066.
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F., and Vance, R. E. (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. doi: 10.1371/journal.ppat.0020018
Rowbotham, T. J. (1980). Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33, 1179–1183.
Roy, C. R., and Tilney, L. G. (2002). The road less traveled: transport of Legionella to the endoplasmic reticulum. J. Cell Biol. 158, 415–419.
Segal, G., Purcell, M., and Shuman, H. A. (1998). Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. U.S.A. 95, 1669–1674.
Segal, G., and Shuman, H. A. (1997). Characterization of a new region required for macrophage killing by Legionella pneumophila. Infect. Immun. 65, 5057–5066.
Shin, S., Case, C. L., Archer, K. A., Nogueira, C. V., Kobayashi, K. S., Flavell, R. A., Roy, C. R., and Zamboni, D. S. (2008). Type IV secretion-dependent activation of host MAP kinases induces an increased proinflammatory cytokine response to Legionella pneumophila. PLoS Pathog. 4, e1000220. doi: 10.1371/journal.ppat.1000220
Shinozawa, Y., Matsumoto, T., Uchida, K., Tsujimoto, S., Iwakura, Y., and Yamaguchi, K. (2002). Role of interferon-gamma in inflammatory responses in murine respiratory infection with Legionella pneumophila. J. Med. Microbiol. 51, 225–230.
Silveira, T. N., and Zamboni, D. S. (2010). Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome-dependent host cell response that precedes pyroptosis. Infect. Immun. 78, 1403–1413.
Sporri, R., Joller, N., Hilbi, H., and Oxenius, A. (2008). A novel role for neutrophils as critical activators of NK cells. J. Immunol. 181, 7121–7130.
Susa, M., Ticac, B., Rukavina, T., Doric, M., and Marre, R. (1998). Legionella pneumophila infection in intratracheally inoculated T cell-depleted or -nondepleted A/J mice. J. Immunol. 160, 316–321.
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820.
Tyndall, R. L., and Domingue, E. L. (1982). Cocultivation of Legionella pneumophila and free-living amoebae. Appl. Environ. Microbiol. 44, 954–959.
Vinzing, M., Eitel, J., Lippmann, J., Hocke, A. C., Zahlten, J., Slevogt, H., N’Guessan P, D., Gunther, S., Schmeck, B., Hippenstiel, S., Flieger, A., Suttorp, N., and Opitz, B. (2008). NAIP and Ipaf control Legionella pneumophila replication in human cells. J. Immunol. 180, 6808–6815.
Vogel, J. P., Andrews, H. L., Wong, S. K., and Isberg, R. R. (1998). Conjugative transfer by the virulence system of Legionella pneumophila. Science 279, 873–876.
Weber, S. S., Ragaz, C., and Hilbi, H. (2009). Pathogen trafficking pathways and host phosphoinositide metabolism. Mol. Microbiol. 71, 1341–1352.
Winn, W. C. Jr. (1988). Legionnaires disease: historical perspective. Clin. Microbiol. Rev. 1, 60–81.
Wright, E. K., Goodart, S. A., Growney, J. D., Hadinoto, V., Endrizzi, M. G., Long, E. M., Sadigh, K., Abney, A. L., Bernstein-Hanley, I., and Dietrich, W. F. (2003). Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr. Biol. 13, 27–36.
Yamamoto, Y., Klein, T. W., Newton, C. A., Widen, R., and Friedman, H. (1988). Growth of Legionella pneumophila in thioglycolate-elicited peritoneal macrophages from A/J mice. Infect. Immun. 56, 370–375.
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., Kuida, K., Mariathasan, S., Dixit, V. M., Flavell, R. A., Dietrich, W. F., and Roy, C. R. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325.
Keywords: Legionnaire’s disease, inflammation, macrophages, plasmacytoid dendritic cells, cytokines
Citation: Schuelein R, Ang DKY, van Driel IR and Hartland EL (2011) Immune control of Legionella infection: an in vivo perspective. Front. Microbio. 2:126. doi: 10.3389/fmicb.2011.00126
Received: 29 April 2011;
Paper pending published: 15 May 2011;
Accepted: 23 May 2011;
Published online: 03 June 2011.
Edited by:
Carmen Buchrieser, Pasteur Institute, FranceReviewed by:
Dario S. Zamboni, Universidade de São Paulo, BrazilHubert Hilbi, Max von Pettenkofer-Institute, Germany
Copyright: © 2011 Schuelein, Ang, Driel and Hartland. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Elizabeth L. Hartland, Department of Microbiology and Immunology, University of Melbourne, Parkville, VIC 3010, Australia. e-mail:aGFydGxhbmRAdW5pbWVsYi5lZHUuYXU=