

- Department of Microbiology, Molecular Genetics and Immunology, University of Kansas Medical Center, Kansas City, KS, USA
The extraribosomal functions of ribosomal proteins have drawn significant recent attention. Ribosomal protein S3 (RPS3), a component of the eukaryotic 40S ribosomal subunit, is a multifunctional protein that regulates DNA repair, apoptosis, and the innate immune response to bacterial infection. Here we the review the latest findings about RPS3 extraribosomal functions, with special emphasis on their relation to microbial pathogenesis and enteropathogenic Escherichia coli.
Introduction
Ribosomal proteins function not only in protein translation, but also in multiple extraribosomal activities (Blumenthal and Carmichael, 1979). These functions include, but are not limited to, DNA repair, cell death, inflammation, tumorigenesis, and transcriptional regulation (Warner and McIntosh, 2009). Here we focus on a eukaryotic 40S ribosome component, the ribosomal protein S3 (RPS3), and its emerging regulatory roles in DNA repair, apoptosis, and pro-inflammatory signaling during bacterial infection. We propose that RPS3 may play a central role in regulating numerous aspects of host–pathogen interactions.
RPS3 and Microbial Pathogenesis
Ribosomal protein S3 has been directly and indirectly implicated in host–pathogen interactions. A clone of human RPS3 was obtained in a yeast three-hybrid screen designed to identify proteins that bind the 3′ untranslated region (UTR) of hepatitis C virus (Wood et al., 2001). Suppression subtractive hybridization studies of mast cell gene expression modulated by Pseudomonas aeruginosa suggested that RPS3 might be involved in P. aeruginosa pathogenesis (Sun et al., 2005). RPS3 expression levels may also be important to mouse resistance to the H5N1 influenza virus (Boon et al., 2009).
The NF-κB family of transcription factors regulates the expression of genes involved in a variety of cellular functions such as immune responses and cellular proliferation (Lenardo and Baltimore, 1989). NF-κB is normally sequestered in the cytoplasm by inhibitory IκB proteins that mask NF-κB nuclear localization signals (Hacker and Karin, 2006). After a cell recognizes a pathogen-associated molecular pattern (PAMP), the IκB kinase (IKK) complex is activated and subsequently phosphorylates the IκBs, leading to their ubiquitination and degradation by the 26S proteasome, permitting NF-κB subunits to translocate into the nucleus to function in transcription.
It was recently discovered that RPS3 is also inducibly associated with and phosphorylated by IKKβ on serine 209 (S209) in response to NF-κB pathway activation (Wan et al., 2011). This phosphorylation event is essential to the nuclear translocation of RPS3, after it associates with importin-α (Wan et al., 2011). Affinity purification experiments had also revealed that RPS3 interacts with the p65 NF-κB subunit through its K homology (KH) domain (Wan et al., 2007; Figure 1).
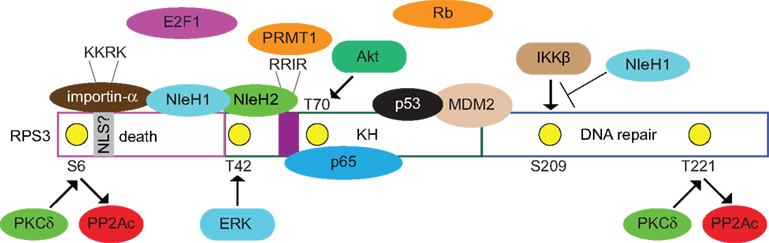
Figure 1. Known and postulated interactions between RPS3 and mammalian or bacterial proteins. Specific RPS3 phosphorylation sites and the protein kinases responsible (PKCδ, ERK, Akt) are indicated. NleH1 and NleH2 are E. coli virulence proteins. NleH1 inhibits the IKKβ-mediated phosphorylation of RSP3 S209. Figure is not drawn to scale and specific protein–protein interactions and/or binding interfaces should not be inferred, except where specifically indicated in the text.
After entering the nucleus, NF-κB binds to κB sites within target gene promoters and regulate transcription by recruiting co-activators/repressors (Wan et al., 2007). This newly discovered NF-κB subunit, RPS3, guides NF-κB to specific κB sites by increasing the affinity of the p65 NF-κB subunit for a subset of target gene promoters (Wan et al., 2007). Likewise, silencing RPS3 expression alters a subset of NF-κB signal transduction pathways. RPS3 thus provides for selective NF-κB recruitment to specific promoters and tailors cellular transcriptional responses to specific stimuli. Interestingly, RPS3 also forms a complex with NF-κB in human islet cells after stimulation with IL-1β (Mokhtari et al., 2009).
The function of type III secretion system (T3SS) effector proteins has been a subject of intense research in recent years (Dean and Kenny, 2009). Some effectors (e.g., NleB, NleC, NleD, NleE, NleH) are key modulators of the innate immune system of intestinal epithelial cells, especially pathways regulated by NF-κB. For example, NleC is a protease that cleaves the NF-κB p65 subunit (Marches et al., 2005; Yen et al., 2010; Baruch et al., 2011; Muhlen et al., 2011; Pearson et al., 2011). NleD cleaves the c-Jun N-terminal kinase (JNK) thus blocking activator protein-1 (AP-1) activation (Baruch et al., 2011). NleE inhibits both p65 nuclear translocation and IκBα degradation (Newton et al., 2010) to block NF-κB activation, in conjunction with NleB (Nadler et al., 2010; Newton et al., 2010).
During attaching/effacing (A/E) pathogen infection, the T3SS effectors NleH1 and NleH2 bind to the N-terminus of RPS3 after their translocation into host cells (Gao et al., 2009). NleH1, but not NleH2, inhibits the nuclear translocation of RPS3, consequently inhibiting the transcription of genes encoding pro-inflammatory cytokines, such as IL-8 and TNF-α, indicating that pathogens target RPS3 to inhibit host immune defenses (Gao et al., 2009).
NleH1 functions by inhibiting the IKKβ-mediated phosphorylation of RPS3 S209 (Wan et al., 2011). NleH1 is an auto-phosphorylated Ser/Thr protein kinase with an active site at lysine 159 (K159; Gao et al., 2009). While the kinase substrate for NleH1 is not yet known, it does not appear to phosphorylate either IKKβ or RPS3. However, NleH1 kinase activity is required to inhibit IKKβ from phosphorylating RPS3, as mutating the NleH1 K159 residue to alanine (K159A) prevented NleH1 from inhibiting RPS3 S209 phosphorylation, both in vitro and in cell culture models (Wan et al., 2011). Studies of gnotobiotic piglets infected with Escherichia coli O157:H7 also demonstrated that RPS3 S209 phosphorylation is inhibited by NleH1 in vivo, possibly to benefit bacterial colonization and transmission (Wan et al., 2011).
It is interesting that IKKβ activation and IκBα degradation appear to be unaffected by NleH1 (Wan et al., 2011) suggesting that it may be beneficial for the pathogen to attenuate the transcription of RPS3-dependent, but not all NF-κB-dependent target genes. It will be important to consider how the apparently selective alteration of NF-κB activity achieved by NleH1 is coordinated with the other enteropathogenic E. coli (EPEC) effectors targeting the NF-κB pathway.
NleH also functions in preventing host cell apoptosis through a mechanism likely to be independent of its interaction with RPS3 (the role of RPS3 in regulating apoptosis will be discussed below). The EPEC effector EspF disrupts host mitochondrial membrane potential and induces the degradation of the anti-apoptotic protein Abcf2 (Nougayrede et al., 2007). Despite the pro-apoptotic function of EspF, EPEC does not induce a large degree of apoptosis, suggesting that other effectors may have anti-apoptotic function. Indeed, an EPEC mutant deleted for both nleH1 and nleH2 reduced host cell survival as compared with wild-type EPEC infection (Hemrajani et al., 2010).
Yeast two-hybrid studies subsequently revealed that NleH1 binds to the Bax inhibitor-1 (BI-1) protein (Hemrajani et al., 2010). This result is interesting because the intrinsic pro-apoptotic pathway involves the activation of Bcl-2-homology 3-only (BH3) proteins, as well as the oligomerization of Bak/Bax proteins. Transfecting NleH1 prevented caspase-3 activation (Hemrajani et al., 2010), as well as Clostridium TcdB-induced apoptosis (Robinson et al., 2010). Interestingly, a pro-apoptotic Bcl-2 protein, the BH3 interacting domain death agonist (BID) was recently shown to interact with the nucleotide-binding oligomerization domain-containing proteins NOD1 and NOD2, as well as the IKK complex, thus integrating apoptosis and NF-κB signaling (Yeretssian et al., 2011).
NleH1 also interacts with the Na+/H+-exchange regulatory factor 2 (NHERF2) at the plasma membrane (Martinez et al., 2010). Because over-expressing NHERF2 reduces the anti-apoptotic function of NleH1, it has been suggested that NHERF2 may serve as a plasma membrane sorting site to bind bacterial effector proteins away from other host targets (Martinez et al., 2010).
DNA Repair
It was determined, after the Drosophila melanogaster RPS3 cDNA was cloned (Wilson et al., 1993), that RPS3 cleaves DNA at apurinic/apyrimidinic (AP) sites of DNA damage (Wilson et al., 1993). The AP site is a DNA lesion which, without removal, can halt mRNA and DNA synthesis and cause cell death (Loeb and Preston, 1986). Drosophila RPS3 possesses an N-glycosylase activity and liberates 8-oxoguanine (8-oxoG) DNA lesions generated during oxidative stress (Yacoub et al., 1996; Deutsch et al., 1997). Transforming RPS3 into E. coli rescues the H2O2 sensitivity of an E. coli mutM strain, as well as the alkylation sensitivity of exo III and endo IV E. coli mutants (Yacoub et al., 1996). Drosophila RPS3 also accelerates the repair of 8-oxoG lesions in both human and mouse cell extracts (Cappelli et al., 2003).
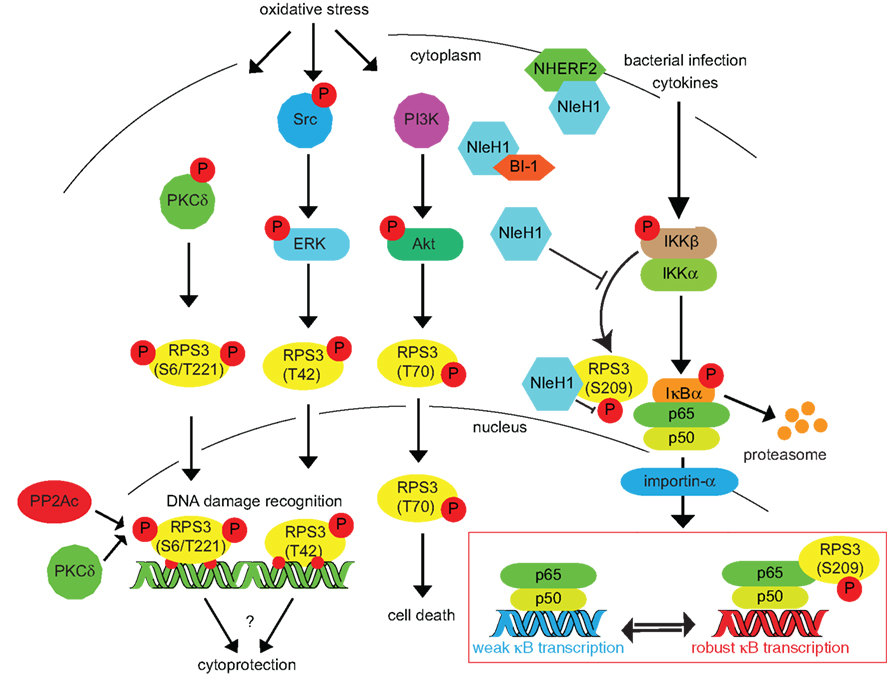
Figure 2. Inducing RPS3 phosphorylation via oxidative stress, DNA damage, and bacterial infection. RPS3 phosphorylation is activated by a variety of cellular stressors. The specific phosphorylation sites and the protein kinases responsible (PKCδ, ERK, Akt) are indicated. Nuclear RPS3 is also bound by the catalytic subunit of protein phosphatase 2A (PP2Ac). IKKβ phosphorylates RPS3 S209 in response to cytokines or bacterial infection. This phosphorylation can be inhibited by the E. coli NleH1 protein, which also binds the BI-1 and NHERF2 proteins.
Both human (Hegde et al., 2004a) and yeast (Jung et al., 2001) RPS3 are also involved in DNA repair. Over-expressing yeast RPS3 overcomes both the osmotic and oxidative stress sensitivity normally observed in a yar1 mutant, a gene encoding an ankyrin-rich repeat protein that serves a stress response function (Loar et al., 2004). The RPS3 gene is found in a single copy in Saccharomyces cerevisiae and its disruption yields non-viable haploid spores (Fingen-Eigen et al., 1996). Human RPS3 binds tightly to both AP and 8-oxoG sites (Hegde et al., 2004b), but appears not to possess its own glycosylase activity (Kim et al., 2005a). Human RPS3 instead functions by binding to and stimulating the activities of a uracil-DNA glycosylase (Ko et al., 2008), as well as the base excision repair (BER) enzymes hOGG1 and APE/Ref-1 (Hegde et al., 2004a), both of which are multifunctional proteins with AP endonuclease activity.
Ribosomal protein S3 nuclear translocation and its subsequent participation in DNA repair are governed by several post-translational modifications. While RPS3 contains a putative nuclear localization sequence (AAs 7–10; KKRK), no data are yet available to indicate definitively whether this sequence motif is essential to RPS3 nuclear translocation. RPS3 nuclear translocation in response to DNA damage (Yadavilli et al., 2007) is regulated by the extracellular signal-regulated kinase 1 (ERK1), which phosphorylates RPS3 on T42 (Kim et al., 2005b; Figure 2). This phosphorylation is critical to regulating RPS3 nuclear translocation, as an RPS3 T42A mutant is significantly reduced in nuclear abundance, even after extensive DNA damage is induced by H2O2 treatment. In contrast, an RPS3 T42D mutant is constitutively localized to the nucleus, even in the absence of DNA damage (Yadavilli et al., 2007).
Protein kinase C delta (PKCδ) is a serine–threonine protein kinase that can function in cellular responses relating to DNA damage (Yoshida, 2007). PKCδ phosphorylates the S6 and T221 residues of RPS3 (Kim et al., 2009a). Chemicals that activate PKCδ [e.g., phorbol myristate acetate (PMA) or H2O2] increase both the extent of phosphorylation and the repair endonuclease activity of RPS3 (Kim et al., 2009a). After nuclear translocation, the N-terminus of RPS3 can be bound by the catalytic subunit of protein phosphatase 2A (PP2Ac; Kim et al., 2009b) and it appears that prior phosphorylation of RPS3 on S6/T221 is necessary for this interaction (Kim et al., 2009b).
A recent study showed that EPEC enters crypts of the human colon (Maddocks et al., 2009). Studies with human colorectal cell cultures demonstrated that EPEC downregulates the expression of the mismatch repair proteins MLH1 and MSH2, in a mechanism independent of apoptosis (Maddocks et al., 2009). It will be interesting to determine the extent to which EPEC effector protein interactions with RPS3 might account for this phenotype.
Apoptosis
Several ribosomal proteins, including RPS3, regulate apoptosis (Naora et al., 1998; Khanna et al., 2003; He and Sun, 2007). Mutational analysis of RPS3 suggests that RPS3 amino acids 15–26, the “death domain,” are critical to the function of RPS3 in inducing apoptosis (Jang et al., 2004). Over-expressing an RPS3–GFP fusion induces DNA condensation and promotes the degradation of both the poly (ADP-ribose) polymerase (PARP) and lamin A/C (Jang et al., 2004; Lee et al., 2010), both of which are hallmarks of apoptotic induction. By contrast, depleting endogenous RPS3 rescues cell survival under oxidative stress conditions (Hegde et al., 2007). Caspase-3, -8, and -9 are activated by over-expressing RPS3 in mouse MPC-11 cells, indicating that RPS3-induced apoptosis is likely to be caspase-dependent (Jang et al., 2004).
In addition, either over-expressing or knocking down RPS3 expression levels can lead to apoptosis, suggesting that the total abundance of RPS3 is important to proper cellular function. Mouse embryonic fibroblasts (MEFs) derived from mice engineered to over-express RPS3 display increased levels of DNA damage after oxidative stress, possibly attributable to RPS3 binding to 8-oxoG and blocking BER activities (Hegde et al., 2009). However, in other systems, in the presence of DNA damaging agents (e.g., H2O2 and methyl methanesulfonate; MMS), knocking down RPS3 actually leads to increased cell survival, by relieving the RPS3-obstacle to liberating 8-oxoG from damaged DNA (Hegde et al., 2007). Knocking down C. elegans RPS3 expression after worms reach adulthood increases lifespan (Curran and Ruvkun, 2007), but the mechanism was not studied in detail.
Ribosomal protein S3 is a substrate for another kinase, Akt (Lee et al., 2010), which can be activated by insulin and pro-survival factors. Activated Akt phosphorylates a variety of cellular proteins involved in the cell cycle, cell survival, and metabolism. Akt phosphorylates the RPS3 T70 residue, also promoting RPS3 nucleus translocation. In this case, T70 phosphorylation is suggested to prevent RPS3-induced apoptosis (Lee et al., 2010). In neuronal cells, RPS3 induces apoptosis by upregulating the expression of pro-apoptotic BH3-only proteins such as Bim and the activator of apoptosis harakiri (Dp5/HRK) by interacting with transcription factor E2F1 (Hershko and Ginsberg, 2004; Lee et al., 2010). Akt-dependent phosphorylation of RPS3 T70 blocks the pro-apoptotic function of RPS3 by inhibiting its interaction with E2F1 while concomitantly enhancing RPS3 endonuclease activity (Lee et al., 2010). These authors also indicate (Lee et al., 2010), in unpublished data, that RPS3 binds to the retinoblastoma (Rb) protein.
A model for RPS3-induced apoptosis has been proposed. Under oxidative stress, RPS3 is phosphorylated by host protein kinases, including ERK, PKCδ, and Akt, consequently translocating into the nucleus to undertake its DNA repair function. Excessive BER may cause irreversible DNA damage and lead to apoptosis (Jang et al., 2004). Thus, increased RPS3 activity at sites of damaged DNA could lead RPS3 to function as an apoptosis signal mediator through a DNA repair enzyme. It is not yet clear what is the mechanism governing cellular apoptosis induced by RPS3 knockdown, though some have speculated that it is related to disrupting protein translation (Lee et al., 2010).
Cancer and p53
Many studies have identified transcripts and proteins that are differentially expressed in cancer. These proteins include many ribosomal proteins and, among them, RPS3. RPS3 expression is increased in adenocarcinomas and in the majority of adenomatous polyps (Pogue-Geile et al., 1991). Suppression subtractive hybridization also identified RPS3 as over-expressed in a leukemia cell line (Zhu et al., 2003). In contrast, RPS3 appears to be under-expressed in squamous cell lung carcinomas (McDoniels-Silvers et al., 2002).
Ribosomal proteins regulate p53 activity, a tumor suppressor involved in arresting the cell cycle and inducing apoptosis (Sulic et al., 2005; Panic et al., 2006; Chakraborty et al., 2009). Zebrafish with heterozygous mutations in genes encoding 17 different ribosomal proteins are impaired in p53 protein production and develop a rare malignant peripheral nerve sheath tumor (Amsterdam et al., 2004). Disrupting the regulation of p53 protein production may lead to tumorigenesis (MacInnes et al., 2008). p53 Levels are normally regulated by MDM2, which possesses an E3 ubiquitin ligase activity that promotes p53 degradation (Honda et al., 1997). However, during nucleolar stress, ribosomal proteins interact with the acidic zinc finger region of MDM2 (Horn and Vousden, 2008), limiting the function of MDM2.
Ribosomal protein S3 interacts, via its KH RNA-binding domain, with both MDM2 and p53 (Yadavilli et al., 2009). p53 and MDM2 levels normally increase and decrease, respectively, after oxidative stress. By contrast, in cells knocked down for RPS3 expression, p53 levels decrease and the E3 ligase activity of MDM2 is lost (Yadavilli et al., 2009), suggesting RPS3 is important to stabilizing p53.
Ribosomal protein S3 can also interact, via its N-terminus, with a nucleoside diphophate kinase, NM23-H1, that is activated during apoptosis and may function as a tumor suppressor (Kim and Kim, 2006). Over-expressing RPS3 reduces invasion by human fibrosarcoma cells and reduced matrix metalloproteinase 9 (MMP-9) secretion and ERK activation in HT1080 cells (Kim and Kim, 2006). NM23-H1 supresses tumor invasiveness by attenuating Ras-Raf-MEK-ERK signaling (Hartsough et al., 2002). As MMP secretion is regulated by ERK and is thought to be critical to tumor cell invasiveness (Lakka et al., 2002), RPS3 interaction with both NM23-H1 and ERK may play an important role in antagonizing cancer development.
Summary
Ribosomal protein S3 is involved in a broad range of physiological activities. It is reasonable to speculate that interrupting ribosomal protein function via environmental stress or infection will lead to changes in host cell survival. Future studies are likely to reveal additional surprises about the extraribosomal functions of ribosomal proteins and the extent to which these functions are targeted for subversion by the T3SS effectors of A/E pathogens.
Conflict of Interest Statement
This research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Acknowledgments
This publication was supported in part by NIH Grants RR016443, AI076227, and AI087686. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We apologize to authors whose work could not be cited due to space limitations.
References
Amsterdam, A., Sadler, K. C., Lai, K., Farrington, S., Bronson, R. T., Lees, J. A., and Hopkins, N. (2004). Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2, E139.
Baruch, K., Gur-Arie, L., Nadler, C., Koby, S., Yerushalmi, G., Ben-Neriah, Y., Yogev, O., Shaulian, E., Guttman, C., Zarivach, R., and Rosenshine, I. (2011). Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J. 30, 221–231.
Blumenthal, T., and Carmichael, G. G. (1979). RNA replication: function and structure of Qbeta-replicase. Annu. Rev. Biochem. 48, 525–548.
Boon, A. C., deBeauchamp, J., Hollmann, A., Luke, J., Kotb, M., Rowe, S., Finkelstein, D., Neale, G., Lu, L., Williams, R. W., and Webby, R. J. (2009). Host genetic variation affects resistance to infection with a highly pathogenic H5N1 influenza A virus in mice. J. Virol. 83, 10417–10426.
Cappelli, E., D’Osualdo, A., Bogliolo, M., Kelley, M. R., and Frosina, G. (2003). Drosophila S3 ribosomal protein accelerates repair of 8-oxoguanine performed by human and mouse cell extracts. Environ. Mol. Mutagen. 42, 50–58.
Chakraborty, A., Uechi, T., Higa, S., Torihara, H., and Kenmochi, N. (2009). Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS ONE 4, e4152.
Curran, S. P., and Ruvkun, G. (2007). Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 3, e56.
Dean, P., and Kenny, B. (2009). The effector repertoire of enteropathogenic E. coli: ganging up on the host cell. Curr. Opin. Microbiol. 12, 101–109.
Deutsch, W. A., Yacoub, A., Jaruga, P., Zastawny, T. H., and Dizdaroglu, M. (1997). Characterization and mechanism of action of Drosophila ribosomal protein S3 DNA glycosylase activity for the removal of oxidatively damaged DNA bases. J. Biol. Chem. 272, 32857–32860.
Fingen-Eigen, M., Domdey, H., and Kohrer, K. (1996). The ribosomal protein gene RPS3 is an essential single copy gene of the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 223, 397–403.
Gao, X., Wan, F., Mateo, K., Callegari, E., Wang, D., Deng, W., Puente, J., Li, F., Chaussee, M. S., Finlay, B. B., Lenardo, M. J., and Hardwidge, P. R. (2009). Bacterial effector binding to ribosomal protein s3 subverts NF-kappaB function. PLoS Pathog. 5, e1000708.
Hacker, H., and Karin, M. (2006). Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13.
Hartsough, M. T., Morrison, D. K., Salerno, M., Palmieri, D., Ouatas, T., Mair, M., Patrick, J., and Steeg, P. S. (2002). Nm23-H1 metastasis suppressor phosphorylation of kinase suppressor of Ras via a histidine protein kinase pathway. J. Biol. Chem. 277, 32389–32399.
He, H., and Sun, Y. (2007). Ribosomal protein S27L is a direct p53 target that regulates apoptosis. Oncogene 26, 2707–2716.
Hegde, V., Wang, M., and Deutsch, W. A. (2004a). Human ribosomal protein S3 interacts with DNA base excision repair proteins hAPE/Ref-1 and hOGG1. Biochemistry 43, 14211–14217.
Hegde, V., Wang, M., and Deutsch, W. A. (2004b). Characterization of human ribosomal protein S3 binding to 7,8-dihydro-8-oxoguanine and abasic sites by surface plasmon resonance. DNA Repair (Amst.) 3, 121–126.
Hegde, V., Yadavilli, S., and Deutsch, W. A. (2007). Knockdown of ribosomal protein S3 protects human cells from genotoxic stress. DNA Repair (Amst.) 6, 94–99.
Hegde, V., Yadavilli, S., McLaughlin, L. D., and Deutsch, W. A. (2009). DNA repair efficiency in transgenic mice over expressing ribosomal protein S3. Mutat Res 666, 16–22.
Hemrajani, C., Berger, C. N., Robinson, K. S., Marches, O., Mousnier, A., and Frankel, G. (2010). NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc. Natl. Acad. Sci. U.S.A. 107, 3129–3134.
Hershko, T., and Ginsberg, D. (2004). Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 279, 8627–8634.
Honda, R., Tanaka, H., and Yasuda, H. (1997). Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 420, 25–27.
Horn, H. F., and Vousden, K. H. (2008). Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene 27, 5774–5784.
Jang, C. Y., Lee, J. Y., and Kim, J. (2004). RpS3, a DNA repair endonuclease and ribosomal protein, is involved in apoptosis. FEBS Lett. 560, 81–85.
Jung, S. O., Lee, J. Y., and Kim, J. (2001). Yeast ribosomal protein S3 has an endonuclease activity on AP DNA. Mol. Cells 12, 84–90.
Khanna, N., Sen, S., Sharma, H., and Singh, N. (2003). S29 ribosomal protein induces apoptosis in H520 cells and sensitizes them to chemotherapy. Biochem. Biophys. Res. Commun. 304, 26–35.
Kim, S. H., and Kim, J. (2006). Reduction of invasion in human fibrosarcoma cells by ribosomal protein S3 in conjunction with Nm23-H1 and ERK. Biochim. Biophys. Acta. 1763, 823–832.
Kim, S. H., Lee, J. Y., and Kim, J. (2005a). Erk phosphorylates threonine 42 residue of ribosomal protein S3. Biochem. Biophys. Res. Commun. 328, 962–967.
Kim, H. D., Lee, J. Y., and Kim, J. (2005b). Characterization of a wide range base-damage-endonuclease activity of mammalian rpS3. Biochem. Biophys. Res. Commun. 328, 927–967.
Kim, T. S., Kim, H. D., and Kim, J. (2009a). PKCdelta-dependent functional switch of rpS3 between translation and DNA repair. Biochim. Biophys. Acta. 1793, 395–405.
Kim, T. S., Kim, H. D., Shin, H. S., and Kim, J. (2009b). Phosphorylation status of nuclear ribosomal protein S3 is reciprocally regulated by protein kinase Cδ and protein phosphatase 2A. J. Biol. Chem. 284, 21201–21208.
Ko, S. I., Park, J. H., Park, M. J., Kim, J., Kang, L. W., and Han, Y. S. (2008). Human ribosomal protein S3 (hRpS3) interacts with uracil-DNA glycosylase (hUNG) and stimulates its glycosylase activity. Mutat. Res. 648, 54–64.
Lakka, S. S., Jasti, S. L., Gondi, C., Boyd, D., Chandrasekar, N., Dinh, D. H., Olivero, W. C., Gujrati, M., and Rao, J. S. (2002). Downregulation of MMP-9 in ERK-mutated stable transfectants inhibits glioma invasion in vitro. Oncogene 21, 5601–5608.
Lee, S. B., Kwon, I. S., Park, J., Lee, K. H., Ahn, Y., Lee, C., Kim, J., Choi, S. Y., Cho, S. W., and Ahn, J. Y. (2010). Ribosomal protein S3, a new substrate of Akt, serves as a signal mediator between neuronal apoptosis and DNA repair. J. Biol. Chem. 285, 29457–29468.
Lenardo, M. J., and Baltimore, D. (1989). NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell 58, 227–229.
Loar, J. W., Seiser, R. M., Sundberg, A. E., Sagerson, H. J., Ilias, N., Zobel-Thropp, P., Craig, E. A., and Lycan, D. E. (2004). Genetic and biochemical interactions among Yar1, Ltv1 and Rps3 define novel links between environmental stress and ribosome biogenesis in Saccharomyces cerevisiae. Genetics 168, 1877–1889.
Loeb, L. A., and Preston, B. D. (1986). Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet. 20, 201–230.
MacInnes, A. W., Amsterdam, A., Whittaker, C. A., Hopkins, N., and Lees, J. A. (2008). Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc. Natl. Acad. Sci. U.S.A. 105, 10408–10413.
Maddocks, O. D., Short, A. J., Donnenberg, M. S., Bader, S., and Harrison, D. J. (2009). Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS ONE 4, e5517.
Marches, O., Wiles, S., Dziva, F., La Ragione, R. M., Schuller, S., Best, A., Phillips, A. D., Hartland, E. L., Woodward, M. J., Stevens, M. P., and Frankel, G. (2005). Characterization of two non-locus of enterocyte effacement-encoded type III-translocated effectors, NleC and NleD, in attaching and effacing pathogens. Infect. Immun. 73, 8411–8417.
Martinez, E., Schroeder, G. N., Berger, C. N., Lee, S. F., Robinson, K. S., Badea, L., Simpson, N., Hall, R. A., Hartland, E. L., Crepin, V. F., and Frankel, G. (2010). Binding to Na(+)/H(+) exchanger regulatory factor 2 (NHERF2) affects trafficking and function of the enteropathogenic Escherichia coli type III secretion system effectors Map, EspI and NleH. Cell. Microbiol. 12, 1718–1731.
McDoniels-Silvers, A. L., Nimri, C. F., Stoner, G. D., Lubet, R. A., and You, M. (2002). Differential gene expression in human lung adenocarcinomas and squamous cell carcinomas. Clin. Cancer Res. 8, 1127–1138.
Mokhtari, D., Barbu, A., Mehmeti, I., Vercamer, C., and Welsh, N. (2009). Overexpression of the Nuclear Factor-κB subunit c-Rel protects against human islet cell death in vitro. Am. J. Physiol. Endocrinol. Metab. E1067–E1077.
Muhlen, S., Ruchaud-Sparagano, M. H., and Kenny, B. (2011). Proteasome-independent degradation of canonical NFkappaB complex components by the NleC protein of pathogenic Escherichia coli. J. Biol. Chem. 286, 5100–5107.
Nadler, C., Baruch, K., Kobi, S., Mills, E., Haviv, G., Farago, M., Alkalay, I., Bartfeld, S., Meyer, T. F., Ben-Neriah, Y., and Rosenshine, I. (2010). The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 6, e1000743.
Naora, H., Takai, I., and Adachi, M. (1998). Altered cellular responses by varying expression of a ribosomal protein gene: sequential coordination of enhancement and suppression of ribosomal protein S3a gene expression induces apoptosis. J. Cell. Biol. 141, 741–753.
Newton, H. J., Pearson, J. S., Badea, L., Kelly, M., Lucas, M., Holloway, G., Wagstaff, K. M., Dunstone, M. A., Sloan, J., Whisstock, J. C., Kaper, J. B., Robins-Browne, R. M., Jans, D. A., Frankel, G., Phillips, A. D., Coulson, B. S., and Hartland, E. L. (2010). The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kappaB p65. PLoS Pathog. 6, e1000898.
Nougayrede, J. P., Foster, G. H., and Donnenberg, M. S. (2007). Enteropathogenic Escherichia coli effector EspF interacts with host protein Abcf2. Cell. Microbiol. 9, 680–693.
Panic, L., Tamarut, S., Sticker-Jantscheff, M., Barkic, M., Solter, D., Uzelac, M., Grabusic, K., and Volarevic, S. (2006). Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol. Cell Biol. 26, 8880–8891.
Pearson, J. S., Riedmaier, P., Marches, O., Frankel, G., and Hartland, E. L. (2011). A type III effector protease NleC from enteropathogenic Escherichia coli targets NF-kappaB for degradation. Mol. Microbiol. 80, 219–230.
Pogue-Geile, K., Geiser, J. R., Shu, M., Miller, C., Wool, I. G., Meisler, A. I., and Pipas, J. M. (1991). Ribosomal protein genes are overexpressed in colorectal cancer: isolation of a cDNA clone encoding the human S3 ribosomal protein. Mol Cell Biol 11, 3842–3849.
Robinson, K. S., Mousnier, A., Hemrajani, C., Fairweather, N., Berger, C. N., and Frankel, G. (2010). The enteropathogenic Escherichia coli effector NleH inhibits apoptosis induced by Clostridium difficile toxin B. Microbiology 156, 1815–1823.
Sulic, S., Panic, L., Barkic, M., Mercep, M., Uzelac, M., and Volarevic, S. (2005). Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev. 19, 3070–3082.
Sun, G., Liu, F., and Lin, T. J. (2005). Identification of Pseudomonas aeruginosa-induced genes in human mast cells using suppression subtractive hybridization: up-regulation of IL-8 and CCL4 production. Clin. Exp. Immunol. 142, 199–205.
Wan, F., Anderson, D. E., Barnitz, R. A., Snow, A., Bidere, N., Zheng, L., Hegde, V., Lam, L. T., Staudt, L. M., Levens, D., Deutsch, W. A., and Lenardo, M. J. (2007). Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell 131, 927–939.
Wan, F., Weaver, A., Gao, X., Bern, M., Hardwidge, P. R., and Lenardo, M. J. (2011). IKKbeta phosphorylation regulates RPS3 nuclear translocation and NF-kappaB function during infection with Escherichia coli strain O157:H7. Nat. Immunol. 12, 335–343.
Warner, J. R., and McIntosh, K. B. (2009). How common are extraribosomal functions of ribosomal proteins? Mol Cell. 34, 3–11.
Wilson, D. M. III, Deutsch, W. A., and Kelley, M. R. (1993). Cloning of the Drosophila ribosomal protein S3: another multifunctional ribosomal protein with AP endonuclease DNA repair activity. Nucleic Acids Res. 21, 2516.
Wood, J., Frederickson, R. M., Fields, S., and Patel, A. H. (2001). Hepatitis C virus 3′X region interacts with human ribosomal proteins. J. Virol. 75, 1348–1358.
Yacoub, A., Augeri, L., Kelley, M. R., Doetsch, P. W., and Deutsch, W. A. (1996). A Drosophila ribosomal protein contains 8-oxoguanine and abasic site DNA repair activities. EMBO J. 15, 2306–2312.
Yadavilli, S., Hegde, V., and Deutsch, W. A. (2007). Translocation of human ribosomal protein S3 to sites of DNA damage is dependant on ERK-mediated phosphorylation following genotoxic stress. DNA Repair (Amst.) 6, 1453–1462.
Yadavilli, S., Mayo, L. D., Higgins, M., Lain, S., Hegde, V., and Deutsch, W. A. (2009). Ribosomal protein S3: A multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair (Amst.) 8, 1215–1224.
Yen, H., Ooka, T., Iguchi, A., Hayashi, T., Sugimoto, N., and Tobe, T. (2010). NleC, a type III secretion protease, compromises NF-kappaB activation by targeting p65/RelA. PLoS Pathog. 6, e1001231.
Yeretssian, G., Correa, R. G., Doiron, K., Fitzgerald, P., Dillon, C. P., Green, D. R., Reed, J. C., and Saleh, M. (2011). Non-apoptotic role of BID in inflammation and innate immunity. Nature 474, 96–99.
Yoshida, K. (2007). PKCdelta signaling: mechanisms of DNA damage response and apoptosis. Cell. Signal. 19, 892–901.
Keywords: apoptosis, DNA repair, EPEC, extraribosomal function, NF-κB, NleH1, ribosomal protein S3
Citation: Gao X and Hardwidge PR (2011) Ribosomal protein S3: a multifunctional target of attaching/effacing bacterial pathogens. Front. Microbio. 2:137. doi: 10.3389/fmicb.2011.00137
Received: 25 March 2011;
Accepted: 13 June 2011;
Published online: 27 June 2011.
Edited by:
Elizabeth L. Hartland, University of Melbourne, AustraliaReviewed by:
Elizabeth L. Hartland, University of Melbourne, AustraliaToru Tobe, Osaka University, Japan
Copyright: © 2011 Gao and Hardwidge. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Philip R. Hardwidge, Department of Microbiology, Molecular Genetics and Immunology, University of Kansas Medical Center, 3901 Rainbow Boulevard, Kansas City, KS 66160, USA. e-mail:cGhhcmR3aWRnZUBrdW1jLmVkdQ==