

- Department of Microbiology and Immunology, University of Michigan Medical School, Ann Arbor, MI, USA
To proliferate within phagocytes, Legionella pneumophila relies on Type IV secretion to modulate host cellular pathways. Autophagy is an evolutionarily conserved degradative pathway that captures and transfers a variety of microbes to lysosomes. Biogenesis of L. pneumophila-containing vacuoles and autophagosomes share several features, including endoplasmic reticulum (ER)-derived membranes, contributions by the host GTPases Rab1, Arf1 and Sar1, and a final destiny in lysosomes. We discuss morphological, molecular genetic, and immunological data that support the model that, although A/J mouse macrophages efficiently engulf L. pneumophila within autophagosomal membranes, the Type IV effector proteins DrrA/SidM, LidA, and RalF prolong association with the ER. By inhibiting immediately delivery to lysosomes, the bacteria persist in immature autophagosomal vacuoles for a period sufficient to differentiate into an acid-resistant, replicative form. Subsequent secretion of the Type IV effector LepB releases the block to autophagosome maturation, and the adapted progeny continue to replicate within autophagolysosomes. Accordingly, L. pneumophila can be exploited as a genetic tool to analyze the recruitment and function of the macrophage autophagy pathway.
Introduction
Legionella pneumophila is an accidental respiratory pathogen that can cause pneumonia in people whose immune defenses are compromised. The natural hosts of L. pneumophila are different species of protozoa that are abundant in aquatic environments (Lau and Ashbolt, 2009). L. pneumophila thrives in natural ecosystems such as ponds, rivers and moist soil, and also in man-made water systems, including cooling towers, whirlpools, and vegetable misters. Although protozoa routinely ingest bacteria as a food source, they can be parasitized by some species of Legionella. Evolutionary pressure to survive and replicate in professional phagocytes of water and soil has led to the emergence of virulence traits that also equip L. pneumophila to proliferate in a similar eukaryotic host, the macrophage. Protozoa and macrophages possess similar anti-microbial defenses, such as production of reactive oxygen and nitrogen species and delivery of invading microbes to the acidic, hydrolytic lysosomes via phagocytosis. Indeed, prior growth in ameba augments subsequent replication in both macrophages and mouse models of infection (Cirillo et al., 1994, 1999; Neumeister et al., 2000).
Upon inhalation within contaminated aerosols, L. pneumophila are phagocytosed by alveolar macrophages. However, the nascent L. pneumophila phagosome avoids the endocytic pathway and instead forms a unique replication vacuole that interacts with particular organelles, including mitochondria and endoplasmic reticulum (ER; Horwitz, 1983a, 1983b; Swanson and Isberg, 1995). After a few rounds of replication in permissive A/J mouse macrophages, the L. pneumophila vacuole acquires lysosomal components, and the progeny continue to replicate in a phagolysosomal compartment (Sturgill-Koszycki and Swanson, 2000). Its mode of entry and replication in host cells require a Type IV secretion system named defect in organelle trafficking/intracellular multiplication (Dot/Icm; Hilbi et al., 2001; Bandyopadhyay et al., 2004, 2007; Hubber and Roy, 2010). To establish a replication niche, intracellular L. pneumophila exploit Type IV secretion to deliver to the host cytosol a large number of effectors predicted to modulate cellular pathways that are highly conserved in ameba and macrophages (Ensminger and Isberg, 2009). Here we focus on interactions between L. pneumophila and the autophagy pathway, an alternate route to the lysosomes of macrophages and amebae.
Autophagy is best known as a catabolic process in which cellular cytoplasm and organelles are degraded as a means to cope with starvation. More than 30 autophagy (Atg) genes in yeast and at least 20 in mammals regulate autophagosome formation and maturation (Mehrpour et al., 2010). Autophagy begins when a double-membraned structure called an isolation membrane, or phagophore, forms around cytoplasm or organelles destined for degradation. The phagophore expands and closes on itself to form a double-membraned vacuole, or autophagosome. In a series of tightly controlled events, the phagophore fuses with vesicles from the endocytic pathway. Maturation is complete when the autophagosome merges with lysosomal vacuoles to form an autophagolysosome, wherein contents of the vacuole are degraded. In addition to its long-established role as a non-selective response to starvation and more recent recognition as a selective mechanism for disposal of damaged organelles or misfolded proteins marked by ubiquitin (Pankiv et al., 2007; Kirkin et al., 2009; Thurston et al., 2009), autophagy is also recruited by the innate and adaptive immune systems (Levine et al., 2011).
Autophagy, an Innate Defense Mechanism Against Intracellular Pathogens
By capturing cytosolic invaders and delivering them to lysosomes, autophagy acts as a barrier against a variety of microbes. When Streptococcus pyogenes, Salmonella enterica, Listeria monocytogenes, or Mycobacterium tuberculosis damage or escape from their phagosomes, some of the microbes are ubiquitinated, recognized by the autophagic surveillance system and trafficked to lysosomes for degradation (Nakagawa et al., 2004; Perrin et al., 2004; Birmingham et al., 2006; Thurston et al., 2009; Yoshikawa et al., 2009; Zheng et al., 2009; Ponpuak et al., 2010). A recent study identified another pathway to capture cargo for autophagy: the cytoskeletal protein Septin traps Shigella flexneri within a meshwork that targets the intracellular bacterium for autophagic degradation (Mostowy et al., 2010).
Bacterial pathogens that reside in endosomal compartments also face death by autophagy, and some of the host regulatory factors have been identified. For example, IFN-γ stimulated cells deliver vacuoles containing M. tuberculosis to lysosomes via autophagy (Gutierrez et al., 2004). Similarly, Chlamydia trachomatis inclusion vacuoles, which typically evade lysosomes, are routed to the autophagic pathway upon IFN-γ activation. This alteration in trafficking is mediated by the host immunity related GTPase (IRG), Irga6 (Al-Younes et al., 2004; Al-Zeer et al., 2009). Infection by the parasite Toxoplasma gondii is also controlled by autophagy in vivo and in vitro. CD40 signaling recruits the autophagy pathway to capture the intracellular parasites, evident from the exacerbated ocular and brain pathology displayed by CD40−/− mice, which are susceptible to chronic toxoplasmosis (Andrade et al., 2006; Ling et al., 2006). S. enterica serovar Typhimurium is captured within autophagosomal membranes derived from the ER by a mechanism that requires Rab1 (Huang et al., 2011), a host GTPase that will be discussed in more detail below.
Selective pressure to circumvent autophagic killing has led to emergence of virulence traits that equip pathogens to survive and replicate in host cells. Coxiella burnetii is an intracellular pathogen that proliferates in spacious vacuoles that eventually fuse with lysosomes. For efficient replication, C. burnetii requires induction of autophagy and inhibition of apoptotic cell death (Beron et al., 2002; Gutierrez et al., 2005; Romano et al., 2007). Francisella tularensis has a remarkable trafficking pattern inside mouse bone marrow-derived macrophages. Soon after infection, F. tularensis breaks out of its phagosome and replicates in the cytoplasm. At later stages of infection, F. tularensis resides in vacuoles with features of autophagosomes. After reentering the endocytic pathway via autophagy, F. tularensis can be exocytosed from the cell (Checroun et al., 2006).
Several lines of evidence have also pointed to autophagy as a strategy for host cells to combat L. pneumophila infection. First, Dictyostelium discoideum that lack the autophagy protein Atg9 are more permissive for infection by L. pneumophila (Tung et al., 2010). Likewise, when expression of Atg5 by A/J mouse peritoneal macrophages is reduced by siRNA, the yield of L. pneumophila increases throughout the 48-h infection period (Matsuda et al., 2009). Conversely, L. pneumophila replication is inhibited when autophagy is induced by treating A/J macrophages with 2-deoxy-d-glucose (Matsuda et al., 2009), a non-hydrolyzable analog of glucose that inhibits glycolysis (Wick et al., 1957). In addition, as discussed in more detail below, the L. pneumophila vacuole traffics along the autophagic pathway more rapidly in restrictive C57Bl/6 mouse macrophages compared with permissive A/J naip5 mutant mouse macrophages (Amer and Swanson, 2005). Next we review morphological, molecular, and immunological data, drawn primarily from studies of L. pneumophila trafficking in A/J mouse macrophages, that are consistent with a model in which L. pneumophila utilizes effectors of the Dot/Icm Type IV secretion system to stall progression of its autophagosomal vacuole to overcome this innate defense against intracellular infection.
ER Contributes to Biogenesis of L. pneumophila Vacuoles and Autophagosomes
The biogenesis of the L. pneumophila replication vacuole is remarkably similar to autophagosome formation. First, the two vacuoles can receive membrane from the same source, the ER. Elegant electron microscopic studies first by Horwitz and later by Swanson and Tilney and their colleagues describe L. pneumophila vacuole biogenesis (Horwitz, 1983b; Swanson and Isberg, 1995; Tilney et al., 2001). Immediately after infection, the L. pneumophila phagosomal membrane resembles the host plasma membrane. Within minutes of uptake by U937 human monocytic cells, osmophilic hair-like projections connect vesicles to the cytoplasmic face of the L. pneumophila phagosome. These vesicles appear to fuse with the phagosomal membrane and with each other to form a double-membraned vacuole, whose thickness is typical of ER. Within the first hours of infection, ribosomes are attached to the cytoplasmic face of the vacuole, which now resembles rough ER. Furthermore, fluorescence microscopy studies of macrophages derived from bone marrow of A/J mice demonstrate that L. pneumophila vacuoles co-localize with a variety of ER markers, including the luminal proteins BiP, glucose-6-phosphatase, and protein disulfide isomerase; the ER membrane proteins calnexin and Sec22b; and yellow fluorescent protein coupled to the ER localization signal KDEL (Swanson and Isberg, 1995; Kagan and Roy, 2002; Derre and Isberg, 2004a; Robinson and Roy, 2006; Arasaki and Roy, 2010).
Several recent reports provide compelling morphological evidence that the ER is also one source of autophagosomal membranes (Axe et al., 2008; Hayashi-Nishino et al., 2009; Yla-Anttila et al., 2009). Autophagosomes originate at phosphatidylinositol-3-phosphate (PI3P)-enriched sections of ER called omegasomes. In addition, autophagosomes are connected to the ER by narrow extensions, and their membranes are a similar thickness (5–7 nm; Arstila and Trump, 1968; Yla-Anttila et al., 2009). Furthermore, immunoelectron microscopy studies of the phagophore membranes identify ER marker proteins and cisternae (Hayashi-Nishino et al., 2009). Thus, in addition to the plasma membrane (Ravikumar et al., 2010) and mitochondria (Hailey et al., 2010), the ER can contribute membrane for autophagosome biogenesis.
Initiation and elongation of the phagophore is coordinated by a cascade of Atg proteins, including two ubiquitin-like conjugation systems (Geng and Klionsky, 2008; Mehrpour et al., 2010). Once Atg7 activates Atg12, then Atg5 is conjugated to Atg12. Atg16L1 is recruited to the growing autophagosome where it forms a multimeric complex with Atg5 and Atg12. Next, cytosolic Atg8, also known as LC3, is cleaved and conjugated to phosphatidylethanolamine in the autophagosomal membrane. Conjugation of LC3 to phosphatidylethanolamine depends on Atg7 and the Atg5–Atg12:Atg16L1 complex. As the phagophore closes to form an autophagosome, Atg16L1 dissociates. Like autophagosomes, newly formed L. pneumophila vacuoles sequentially acquire and then lose Atg7 and Atg8/LC3 (Amer and Swanson, 2005).
During starvation-induced autophagy, the sudden demand for membrane may be readily met by membranous folds of the ER. Similar to L. pneumophila, the intracellular eukaryotic parasite T. gondii resides in a protective vacuole that interacts intimately with the ER (Melo and de Souza, 1997) and that can also be a target of autophagy (Andrade et al., 2006; Ling et al., 2006). Accordingly, we propose that, in response to cytosolic infection, the autophagy machinery recruits the ER to sequester invading microbes.
L. pneumophila Vacuoles and Autophagosomes Merge with the Lysosomes
In the initial stages of infection, the L. pneumophila vacuole does not fuse with lysosomes (Horwitz, 1983b). The vacuole is isolated from the endocytic pathway, as the compartment is inaccessible to exogenous soluble and lipid probes (Joshi et al., 2001). Nutrient cues likely trigger differentiation of intracellular L. pneumophila to the replicative phase (Sauer et al., 2005; Wieland et al., 2005). Since only post-exponential phase L. pneumophila express the virulence factors that inhibit phagosome–lysosome fusion (Byrne and Swanson, 1998; Fernandez-Moreira et al., 2006), the differentiation state of the bacteria impacts the fate of the pathogen’s vacuole. Beginning ~8 h after infection of A/J mouse macrophages, lysosomal markers co-localize with the L. pneumophila vacuole (Sturgill-Koszycki and Swanson, 2000). Although L. pneumophila can replicate at neutral pH in broth and human monocytic cells (Wieland et al., 2004), in A/J macrophages fusion with the lysosomes actually promotes bacterial replication, since pharmacological inhibition of lysosome acidification and phagosome maturation by bafilomycin A inhibits L. pneumophila growth. The period when lysosome fusion is stalled likely allows the bacteria to differentiate to an acid-resistant form (Sturgill-Koszycki and Swanson, 2000). The lysosomal bacteria continue to replicate in A/J macrophages for an additional 10–15 h, until the cell lyses to release progeny that are primed for infection (Byrne and Swanson, 1998).
The fate of a newly formed autophagosome is similar to that of the L. pneumophila replication vacuole. Once formed from membranes of the ER, autophagosomes quickly fuse with early endosomes and late endosomes, generating vacuoles referred to as amphisomes (Liou et al., 1997; Berg et al., 1998; Swanson et al., 2009). These vacuoles acidify and traffic along the microtubular network toward the perinuclear region, where they merge with lysosomes (Kimura et al., 2008).
From the origin of their membrane to their final destination, the L. pneumophila vacuole and the autophagosome share many morphological features. However, differences exist between the two vacuoles. First, unlike L. pneumophila replication vacuoles, autophagosomes do not accumulate ribosomes on their surface. Second, in macrophages derived from the bone marrow of A/J naip5 mutant mice, non-selective autophagosomes induced by starvation or pharmacologically mature at a faster rate than L. pneumophila vacuoles (Amer and Swanson, 2005). We speculate that the capacity of particular Type IV effector proteins, discussed below, to prolong interactions with the ER slows maturation of the L. pneumophila autophagosomal vacuole, enabling ribosomes to accumulate.
Genetic analysis of the contribution of Atg proteins to biogenesis of L. pneumophila replication vacuoles is one approach to test the impact of autophagy on the pathogen’s fate. Otto et al. (2004) analyzed the yield of L. pneumophila over an 8-day period in wild-type D. discoideum or a number of autophagy mutants. The host factors analyzed were Atg1 and Atg6, proteins that contribute to the initial stages of autophagosome formation, and Atg5, Atg7, and Atg8, components of ubiquitin-like conjugation systems that mediate autophagosome elongation (Chen and Klionsky, 2011). Since the mutant ameba supported L. pneumophila replication to levels similar to wild-type, L. pneumophila can replicate independently of several factors that promote autophagosome biogenesis. Whether the bacteria resided in ER-derived. lysosomal, or other compartments when L. pneumophila replication was first evident >48 h after infection of the atg mutant D. discoideum was not analyzed. Subsequent molecular genetic studies in the D. discoideum model reported that the autophagy protein Atg9 equips amebae to restrict infection by L. pneumophila (Tung et al., 2010). Kinetic studies using markers for ER and the endosomal pathway in wild-type and mutant D. discoideum phagocytes are needed to determine when the L. pneumophila vacuole intersects the autophagy pathway, whether the pathogen delays autophagosome maturation, and the composition of vacuoles that support bacterial replication in these environmental host cells.
A Subset of GTPases Direct ER Recruitment by Both L. pneumophila Vacuoles and Autophagosomes
Rab proteins are small GTP-binding proteins that regulate vesicle trafficking. Rab proteins cycle between a cytosolic, inactive GDP-bound state and a membrane-associated, active GTP-bound state (Barr and Lambright, 2010). Cycling between the two states is catalyzed by guanine exchange factors (GEFs), proteins that exchange GDP for GTP, and by GTPase activating proteins (GAPs) that stimulate GTP hydrolysis to inactivate the Rab protein. Additionally, Rab proteins bind SNARE and SNARE-associated proteins, soluble effector proteins that mediate membrane fusion. As vesicles progress along either the secretory and endosomal pathways, Rab proteins dedicated to each distinct compartment are sequentially recruited and then displaced: this so-called “Rab conversion” is critical for maturation of the organelles (Rink et al., 2005).
Legionella pneumophila replication vacuoles and autophagosomes each associate with Rab1, a GTPase of the early secretory pathway that regulates fusion between vesicles exiting the ER and the cis-Golgi (Stenmark, 2009). Immediately after uptake by A/J mouse macrophages or U937 human monocytic cells, Rab1 co-localizes with the L. pneumophila phagosome; by 4 h, it cycles off the vacuole (Derre and Isberg, 2004b; Kagan et al., 2004). Rab1 promotes bacterial replication, since transfection of COS1 and CHO cells with an inactive form of Rab1 reduces the yield of L. pneumophila.
Several L. pneumophila effectors that regulate Rab1 activity have been identified, highlighting the significance of this host protein for L. pneumophila trafficking. For example, two L. pneumophila Type IV secreted proteins, DrrA and LidA, increase the pool of Rab1–GTP, whereas a third, LepB, stimulates GTP hydrolysis to generate Rab1–GDP (Murata et al., 2006; Ingmundson et al., 2007; Machner and Isberg, 2007; Brombacher et al., 2009; Muller et al., 2010). The capacity of this trio of L. pneumophila effectors to modulate trafficking in the host secretory pathway was verified when their ectopic expression disrupted the Golgi network of CHO and COS1 cells (Derre and Isberg, 2005; Machner and Isberg, 2007). LidA may play additional roles because it also interacts with Rab6 and Rab8, and the effector is expressed throughout the bacterial replication period (Conover et al., 2003; Machner and Isberg, 2007).
Time course studies of co-localization of the L. pneumophila effectors that modulate Rab1 suggest that the pathogen vacuole retains secretory vesicles for a defined period. Both DrrA and LidA are detected on L. pneumophila vacuoles shortly after uptake by primary mouse macrophages (Conover et al., 2003; Ingmundson et al., 2007). In contrast, LepB decorates the L. pneumophila vacuole at later times, reaching a high plateau 9 h after infection of A/J mouse macrophages (Ingmundson et al., 2007). Single cell analysis underscored that as LepB accumulates, host Rab1 and bacterial DrrA/SidM disassociate from the L. pneumophila vacuole, becoming undetectable by 4 h. The continued presence of DrrA and LidA during the initial stage of L. pneumophila vacuole maturation is predicted to ensure persistent Rab1 activation and prolong recruitment of ER vesicles. By blocking Rab conversion, retention of active Rab1 by DrrA and LidA may stall L. pneumophila vacuole maturation by inhibiting association of downstream Rab GTPases such as Rab5 and Rab7, which facilitate fusion with early and late endosomes, respectively. Subsequent secretion by L. pneumophila of the effector LepB catalyzes Rab1 to cycle to its inactive form. As a consequence, Rab conversion is predicted to proceed, thereby relieving the block to autophagosome maturation. By this stage of the infection of A/J macrophages, the intracellular bacteria have differentiated to a replicative form that is acid-resistant and equipped to exploit lysosomes as a replication niche (Sturgill-Koszycki and Swanson, 2000).
Consistent with the contribution of ER membrane to autophagosome biogenesis, Rab1 co-localizes with LC3 (Atg8) on autophagosomes generated by starvation of CHO cells (Zoppino et al., 2010). Rab1-positive autophagosomes do not acquire cathepsin D or degradative capacity, indicating that these vesicles represent an early stage of autophagosome maturation. Moreover, over-expression of Rab1 stimulates autophagosome biogenesis, as judged by localization and processing of LC3. Conversely, reducing Rab1 expression by siRNA reduces autophagy (Zoppino et al., 2010). Using similar approaches, Huang et al. (2011) documented that Rab1 contributes to autophagosome formation, clearance of ubiquitinated protein aggregates, and sequestration and degradation of Salmonella typhimurium. In summary, Rab1 association with L. pneumophila vacuoles and with autophagosomes is a critical step in biogenesis of both of these ER-derived vacuoles. By secreting effectors that trap active Rab1, L. pneumophila is predicted to stall maturation, providing the time needed for the pathogen to differentiate to a state that can tolerate and exploit an acidic, hydrolytic autophagolysosomal compartment of A/J macrophages.
A second GTPase in the early secretory pathway that has been implicated in the biogenesis of both L. pneumophila replication vacuoles and autophagosomes is Sar1. CHO FcgRII cells that express a dominant negative form of Sar1 fail to tether ER vesicles to the L. pneumophila phagosome, and their bacterial yield 11 h after infection is reduced (Kagan and Roy, 2002). Likewise, CHO cells that express either a dominant negative form of Sar1 or reduced amounts of wild-type Sar1 protein contain fewer autophagosomes (Zoppino et al., 2010). That Sar1 promotes formation of L. pneumophila replication vacuoles and autophagosomes lends further strength to the model that L. pneumophila is captured by autophagy, but the pathogen utilizes Type IV secretion to stall this host defense pathway, securing time for the intracellular bacterium to adapt to its replication niche.
Arf1 is a third host GTPase known to regulate vesicular traffic in the secretory pathway that contributes to formation of not only L. pneumophila replication vacuoles but also autophagosomes. L. pneumophila-infected CHO FcgRII cells that express a dominant negative form of Arf1 contain a reduced number of intracellular bacteria 11 h after infection (Kagan and Roy, 2002). The L. pneumophila Type IV secretion effector protein RalF acts as a GEF that promotes Arf1 association with the bacterial vacuole (Nagai et al., 2002). However, since RalF mutants do not exhibit the intracellular growth defect observed in cells whose Arf1 function is impaired, other bacterial factors likely regulate the GTPase activity. Recent experiments in yeast determined that Arf1 also contributes to autophagosome biogenesis. In particular, genetic analysis identified this GTPase, as well as the Arf1 GEF protein Sec7, as critical for the Atg8/LC3 processing that promotes autophagosome biogenesis (van der Vaart et al., 2010). Thus, Arf1 is one of three GTPases that function in the secretory pathway and contribute to biogenesis of both L. pneumophila replication vacuoles and autophagosomes.
Polyubiquitinated Proteins are Associated with Autophagosomes and L. pneumophila Vacuoles
Since polyubiquitinated proteins are cargo for autophagosomes and also surround L. pneumophila vacuoles, they provide more clues to understanding the pathogen’s fate. Ubiquitination is an evolutionarily conserved mechanism that tags proteins for degradation by host proteasomes or lysosomes (Clague and Urbe, 2010). Ubiquitin also targets proteins and cytosolic bacteria for selective autophagy. The cytoplasmic adaptor proteins p62, NBR1, or NDP52 bind either ubiquitinated protein aggregates or intracellular S. typhimurium, L. monocytogenes, or M. tuberculosis, marking them as cargo for selective autophagy (Pankiv et al., 2007; Kirkin et al., 2009; Thurston et al., 2009; Yoshikawa et al., 2009; Ponpuak et al., 2010). For example, S. typhimurium that enter the cytosol become ubiquitinated and are degraded by autophagy, and p62 and NDP52 are required for efficient killing of the cytosolic bacteria (Zheng et al., 2009).
Shortly after ingestion by A/J mouse macrophages, U937 human monocytic cells or Acanthamoeba polyphaga, the L. pneumophila vacuole is studded with ubiquitinated proteins, which persist during the bacterial replication period (Dorer et al., 2006; Ivanov and Roy, 2009; Price et al., 2009). A number of proteins translocated by the Dot/Icm Type IV secretion system contain F-box and U-box motifs, hallmarks of E3 ubiquitin ligases, which transfer ubiquitin to proteins to be degraded (Cazalet et al., 2004; Al-Khodor et al., 2008; Kubori et al., 2010). At least two of the five known F-box containing proteins of L. pneumophila exhibit E3 ubiquitin ligase activity and interact with host proteins (Ensminger and Isberg, 2010; Lomma et al., 2010). For example, AnkB (LegAU13) induces ubiquitinated proteins to accumulate on the bacterial vacuole; it also enhances intracellular of growth of one strain of L. pneumophila, but not others (Al-Khodor et al., 2008; Ivanov and Roy, 2009; Ensminger and Isberg, 2010). LubX is a U-box containing E3 ubiquitin ligase that negatively regulates the bacterial Dot/Icm effector SidH by ubiquitinylation and also increases survival of infected D. melanogaster (Kubori et al., 2010). However, LubX is not required for L. pneumophila replication inside murine macrophages or protozoan cells (Kubori et al., 2008). Yet, bacterial replication is attenuated in cells that express reduced amounts of the host proteins Clk1, a substrate of LubX, and Cdc48/p97, a chaperone predicted to enhance translocation of ubiquitinated Type IV secretion substrates (Dorer et al., 2006; Kubori et al., 2008).
At present it is unclear whether ubiquitination of the L. pneumophila vacuole is driven by the pathogen, or the host. The protein modification may aid translocation of Type IV secretion substrates, or modulate maturation of the vacuole. Certain host Atg enzymes catalyze ubiquitin-like reactions during autophagosome biogenesis. However, since none of the Atg proteins have the structure or the HECT- or RING-type motifs typical of E3 ligases (Geng and Klionsky, 2008), it seems unlikely that any of the known L. pneumophila E3 enzymes, which do contain these motifs, act directly as mimics of Atg proteins. An alternative hypothesis that remains to be tested is that, by tagging vacuoles modified or damaged by Type IV secretion with ubiquitin, the host cell directs L. pneumophila to the selective autophagic pathway, perhaps via the adaptor proteins NDP52 or p62 (Thurston et al., 2009; Ponpuak et al., 2010). Consistent with the idea that ubiquitination of the pathogen vacuole is advantageous to the host is the unexpected observation that the F-box bacterial effector AnkB reduces, rather than increases, endogenous ubiquitination of one host target protein (Lomma et al., 2010).
Maturation of the L. pneumophila Vacuole is Governed by the Innate Immune Response
The innate intracellular immune receptor Naip5 plays a critical role in resistance to L. pneumophila infection (Diez et al., 2003; Wright et al., 2003; Derre and Isberg, 2004b; Zamboni et al., 2006). Naip5 is a NOD-like receptor (NLR) protein postulated to detect cytosolic contamination of flagellin delivered to the cytoplasm, perhaps via the Dot/Icm system. Recognition of flagellin by Naip5 initiates host immune responses that control L. pneumophila infection in C57Bl6 macrophages (Amer et al., 2006; Molofsky et al., 2006; Ren et al., 2006; Fortier et al., 2007). Interestingly, in A/J macrophages, which have reduced Naip5 function, maturation of the L. pneumophila vacuole is sluggish, and the pathogen replicates to high numbers (Amer and Swanson, 2005). For example, bone marrow-derived macrophages from A/J mice display maximum co-localization of Atg7 with L. pneumophila phagosomes within minutes, and then the association diminishes by 2 h. LC3 becomes visible on the phagosome at 4 h; by 6 h, 50% of L. pneumophila phagosomes co-localize with LC3. In contrast, in restrictive C57Bl6 macrophages Atg7 and LC3 association with the bacterial vacuole is rapid: by 1 h, some phagosomes first acquire, and then shed, both Atg7 and Atg8 (Amer and Swanson, 2005); by 2 h more vacuoles have merged with lysosomes (Fortier et al., 2007); by 48 h, intracellular bacteria are degraded (Molofsky et al., 2006). Whether Naip5 protein equips mouse macrophages to restrict L. pneumophila infection by promoting a rapid autophagic response to cytosolic contamination remains to be tested directly in isogenic mouse strains. We next propose a model that incorporates these interesting mouse genetics observations in the context of the morphological and molecular genetic studies of the biogenesis of L. pneumophila vacuoles and autophagosomes.
L. pneumophila Vacuoles Resist Maturation Along Autophagic Pathway
We postulate that, when confronted by autophagy as a host defense, L. pneumophila retards autophagosome maturation to establish a productive infection in professional phagocytes. The morphological and molecular similarities between biogenesis of L. pneumophila replication vacuoles and autophagosomes, as well as the capacity of Naip5 to restrict infection of mouse and human macrophages by L. pneumophila and increase delivery of the pathogen to lysosomes (Diez et al., 2003; Wright et al., 2003; Derre and Isberg, 2004b; Amer et al., 2006; Molofsky et al., 2006; Fortier et al., 2007; Vinzing et al., 2008) are consistent with the following working model (Figure 1). After uptake by cells, L. pneumophila effectors along with contaminating flagellin are transported by Type IV secretion to the host cytoplasm. Recognition of flagellin by Naip5 triggers the autophagic defense mechanism, which relies on the GTPases Rab1, Arf1, and Sar1 to deliver secretory vesicles from the ER to envelop the pathogen’s vacuole. To inhibit rapid maturation into a toxic, acidic autophagolysosome, L. pneumophila immediately delivers Type IV effectors to retain ER components. In particular, DrrA and LidA secreted by the pathogen activate and retain Rab1. Likewise, the bacterial effector RalF activates Arf1. Retention of active Rab1 and Arf1 prevents their replacement by a distinct set of Rab proteins needed to recruit vesicles from the endosomal pathway. Thus, L. pneumophila stalls in an ER-derived vacuole that resembles an immature autophagosome. After several hours in permissive A/J macrophages, L. pneumophila expresses a new class of effectors, including LepB, that release Rab1 from the vacuole (Ingmundson et al., 2007). As a consequence, the vacuole resumes maturation to form an autophagolysosome. Presumably, a deliberate, measured pause within an ER-derived immature autophagosome provides L. pneumophila time sufficient to induce acid resistance and other traits critical to exploit lysosomes as a replication niche (Sturgill-Koszycki and Swanson, 2000). For example, L. pneumophila can use as a nutrient source short peptides (Sauer et al., 2005; Wieland et al., 2005), which are likely abundant in lysosomes. Thus, we speculate that residence in the lysosomal compartment provides a constant supply of nutrients and vacuolar membrane to support bacterial replication in A/J macrophages.
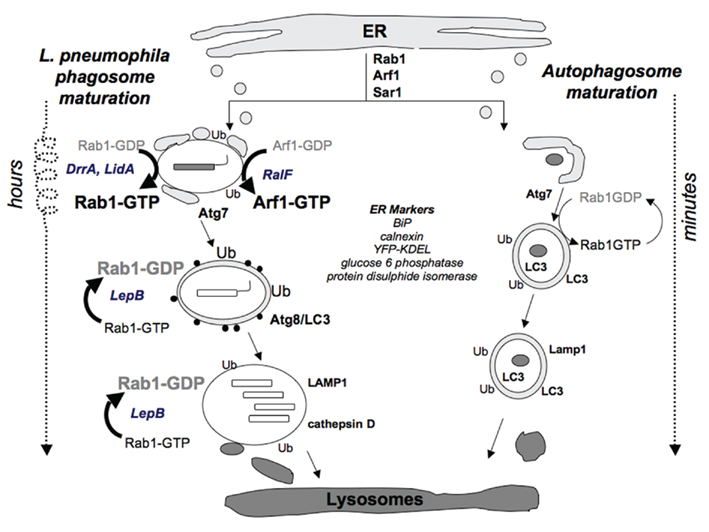
Figure 1. Model for stalled maturation of the L. pneumophila vacuole in the autophagic pathway of permissive phagocytes. ER serves as a membrane source for both L. pneumophila vacuoles and autophagosomes. Early secretory vesicles (gray circles) from the ER are recruited to both organelles in a Rab1-, Arf1-, and Sar1-dependent manner. The double-membraned, LC3 positive, autophagosome sequentially fuses with vesicles from the early endocytic pathway and finally with lysosomes, where its cargo is degraded. Maturation of L. pneumophila is stalled at an early stage when bacterial proteins (italics) DrrA and LidA persistently activate Rab1 and RalF activates Arf1. Thus, the vacuole acquires ER markers BiP, glucose-6-phosphate, calnexin, protein disulfide isomerase, YFP–KDEL, and ribosomes (filled circles). After several hours, L. pneumophila secretes LepB, an effector that inactivates Rab1, releasing it from the membrane. Subsequently, the immature autophagosomal vacuole matures to an autophagolysosome, which accumulates the lysosomal proteins cathepsin D and LAMP1. The deliberate pause coordinated by Type IV secretion effectors enables the pathogen to differentiate into an acid-resistant, replicative form that exploits lysosomes as a replication niche.
Our model predicts that, in response to cytosolic flagellin, the NLR protein Naip5 stimulates macrophage autophagy as a barrier to cytosolic infection. A precedent for NLR-mediated induction of autophagy is the discovery that NOD1 and NOD2 physically interact with Atg16L1 and promote autophagy of intracellular S. flexneri and S. typhimurium (Cooney et al., 2010; Travassos et al., 2010). In particular, we postulate that the wild-type level of Naip5 protein expressed by restrictive C57Bl6 macrophages triggers a robust autophagic response (Amer and Swanson, 2005) that is sufficient to overcome the pathogen’s effector proteins and deliver the vacuole to lysosomes. In contrast, the partial Naip5 function of A/J macrophages elicits a sluggish autophagic response, and the L. pneumophila effectors successfully stall autophagosome maturation. Our model also predicts either that autophagosomes mature more slowly in human macrophages and amebae, which are permissive for infection, compared to mouse phagocytes, which are not. Alternatively, the bacterial effectors DrrA, LidA, and RalF may bind and activate the Rab1 and Arf1 proteins of human and amebae more efficiently than the mouse substrates. Thus, we postulate that the outcome of L. pneumophila infection of environmental or mammalian phagocytes is dictated by a competition: how efficiently host autophagy delivers the microbe to lysosomes versus How effectively the pathogen arsenal unleashed by Type IV secretion stalls autophagosome maturation.
Conclusion
A large number of effectors released by L. pneumophila have eukaryotic-like motifs and functions (Cazalet et al., 2004; Lurie-Weinberger et al., 2010). Remarkably, a number of these proteins equip the pathogen to modulate the activity of host GTPases, first to stall, and then later resume, the exchange of vesicles from the host secretory pathway. By this strategy, L. pneumophila delays its immediate delivery to anti-microbial autophagolysosomes.
Autophagy is modulated by at least two other pathogens to replicate inside cells, C. burnetii and F. tularensis. The C. burnetii phagosome associates with LC3 within minutes, and this association is maintained for up to 48 h in CHO cells (Gutierrez et al., 2005; Romano et al., 2007). Notably, cathepsin D, a lysosomal enzyme, is acquired at a slower rate by phagosomes containing live C. burnetii compared with inactivated bacteria (Romano et al., 2007). The authors propose that association of C. burnetii with autophagosomes delays their delivery with lysosomes, perhaps providing time for infectious C. burnetii to differentiate into their replicative cell type. Later in infection, maturation of the C. burnetii phagosome depends on association with Rab1 and fusion with vesicles from the early secretory pathway, possibly to acquire nutrients and membrane for the expanding vacuole (Campoy et al., 2011).
Similar to L. pneumophila and C. burnetii, delay in progression along the autophagic pathway has been suggested as a successful strategy of F. tularensis. Peripheral blood monocytes infected with F. tularensis down regulate key autophagy genes, as shown by microarray analysis; yet, morphological evidence demonstrates F. tularensis within autophagosomal vacuoles later in infection (Checroun et al., 2006; Butchar et al., 2008). Perhaps by inhibiting expression of autophagy genes, F. tularensis secures the time needed to differentiate to a form that can resist the harsh environment of autophagolysosomes (Cremer et al., 2009). Moreover, F. tularensis may hijack exocytosis of lysosomes to egress out of the cell. In summary, the three pathogens L. pneumophila, C. burnetii, and F. tularensis appear to manipulate the autophagic system to meet their particular nutritional, membrane expansion, and exit requirements. Accordingly, pharmaceutical induction of autophagy may be an effective strategy to combat infection by these and other intracellular pathogens.
Two of the outstanding questions in the autophagy field are how autophagosomes select their cargo and how their maturation is regulated. As a genetically tractable BSL2 microbe, L. pneumophila is an attractive tool to study autophagic processes, which remain challenging to track. Indeed, observations regarding the contribution of secretory pathway membranes and GTPases to autophagosome-like vacuoles were made first by Legionella experimentalists and later by the autophagy field. By exploiting L. pneumophila as a molecular genetic probe, scientists can gain insight to the mechanisms that regulate formation and maturation of autophagosomes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The autophagy research in the Swanson lab was funded by the National Institutes of Health NIAID (R56 AI076300-01).
References
Al-Khodor, S., Price, C. T., Habyarimana, F., Kalia, A., and Abu Kwaik, Y. (2008). A Dot/Icm-translocated ankyrin protein of Legionella pneumophila is required for intracellular proliferation within human macrophages and protozoa. Mol. Microbiol. 70, 908–923.
Al-Younes, H. M., Brinkmann, V., and Meyer, T. F. (2004). Interaction of Chlamydia trachomatis serovar L2 with the host autophagic pathway. Infect. Immun. 72, 4751–4762.
Al-Zeer, M. A., Al-Younes, H. M., Braun, P. R., Zerrahn, J., and Meyer, T. F. (2009). IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS ONE 4, e4588. doi: 10.1371/journal.pone.0004588
Amer, A., Franchi, L., Kanneganti, T. D., Body-Malapel, M., Ozoren, N., Brady, G., Meshinchi, S., Jagirdar, R., Gewirtz, A., Akira, S., and Núñez, G. (2006). Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223.
Amer, A. O., and Swanson, M. S. (2005). Autophagy is an immediate macrophage response to Legionella pneumophila. Cell. Microbiol. 7, 765–778.
Andrade, R. M., Wessendarp, M., Gubbels, M. J., Striepen, B., and Subauste, C. S. (2006). CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Invest. 116, 2366–2377.
Arasaki, K., and Roy, C. R. (2010). Legionella pneumophila promotes functional interactions between plasma membrane syntaxins and Sec22b. Traffic 11, 587–600.
Arstila, A. U., and Trump, B. F. (1968). Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am. J. Pathol. 53, 687–733.
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., Griffiths, G., and Ktistakis, N. T. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701.
Bandyopadhyay, P., Liu, S., Gabbai, C. B., Venitelli, Z., and Steinman, H. M. (2007). Environmental mimics and the Lvh type IVA secretion system contribute to virulence-related phenotypes of Legionella pneumophila. Infect. Immun. 75, 723–735.
Bandyopadhyay, P., Xiao, H., Coleman, H. A., Price-Whelan, A., and Steinman, H. M. (2004). Icm/dot-independent entry of Legionella pneumophila into amoeba and macrophage hosts. Infect. Immun. 72, 4541–4551.
Berg, T. O., Fengsrud, M., Stromhaug, P. E., Berg, T., and Seglen, P. O. (1998). Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J. Biol. Chem. 273, 21883–21892.
Beron, W., Gutierrez, M. G., Rabinovitch, M., and Colombo, M. I. (2002). Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect. Immun. 70, 5816–5821.
Birmingham, C. L., Smith, A. C., Bakowski, M. A., Yoshimori, T., and Brumell, J. H. (2006). Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281, 11374–11383.
Brombacher, E., Urwyler, S., Ragaz, C., Weber, S. S., Kami, K., Overduin, M., and Hilbi, H. (2009). Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284, 4846–4856.
Butchar, J. P., Cremer, T. J., Clay, C. D., Gavrilin, M. A., Wewers, M. D., Marsh, C. B., Schlesinger, L. S., and Tridandapani, S. (2008). Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS ONE 3, e2924. doi: 10.1371/journal.pone.0002924
Byrne, B., and Swanson, M. S. (1998). Expression of Legionella pneumophila virulence traits in response to growth conditions. Infect. Immun. 66, 3029–3034.
Campoy, E. M., Zoppino, F. C., and Colombo, M. I. (2011). The early secretory pathway contributes to the growth of the Coxiella-replicative niche. Infect. Immun. 79, 402–413.
Cazalet, C., Rusniok, C., Bruggemann, H., Zidane, N., Magnier, A., Ma, L., Tichit, M., Jarraud, S., Bouchier, C., Vandenesch, F., Kunst, F., Etienne, J., Glaser, P., and Buchrieser, C. (2004). Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat. Genet. 36, 1165–1173.
Checroun, C., Wehrly, T. D., Fischer, E. R., Hayes, S. F., and Celli, J. (2006). Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. U.S.A. 103, 14578–14583.
Chen, Y., and Klionsky, D. J. (2011). The regulation of autophagy – unanswered questions. J. Cell Sci. 124, 161–170.
Cirillo, J. D., Cirillo, S. L., Yan, L., Bermudez, L. E., Falkow, S., and Tompkins, L. S. (1999). Intracellular growth in Acanthamoeba castellanii affects monocyte entry mechanisms and enhances virulence of Legionella pneumophila. Infect. Immun. 67, 4427–4434.
Cirillo, J. D., Falkow, S., and Tompkins, L. S. (1994). Growth of Legionella pneumophila in Acanthamoeba castellanii enhances invasion. Infect. Immun. 62, 3254–3261.
Clague, M. J., and Urbe, S. (2010). Ubiquitin: same molecule, different degradation pathways. Cell 143, 682–685.
Conover, G. M., Derre, I., Vogel, J. P., and Isberg, R. R. (2003). The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol. Microbiol. 48, 305–321.
Cooney, R., Baker, J., Brain, O., Danis, B., Pichulik, T., Allan, P., Ferguson, D. J., Campbell, B. J., Jewell, D., and Simmons, A. (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97.
Cremer, T. J., Amer, A., Tridandapani, S., and Butchar, J. P. (2009). Francisella tularensis regulates autophagy-related host cell signaling pathways. Autophagy 5, 125–128.
Derre, I., and Isberg, R. R. (2004a). Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect. Immun. 72, 3048–3053.
Derre, I., and Isberg, R. R. (2004b). Macrophages from mice with the restrictive Lgn1 allele exhibit multifactorial resistance to Legionella pneumophila. Infect. Immun. 72, 6221–6229.
Derre, I., and Isberg, R. R. (2005). LidA, a translocated substrate of the Legionella pneumophila type IV secretion system, interferes with the early secretory pathway. Infect. Immun. 73, 4370–4380.
Diez, E., Lee, S. H., Gauthier, S., Yaraghi, Z., Tremblay, M., Vidal, S., and Gros, P. (2003). Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat. Genet. 33, 55–60.
Dorer, M. S., Kirton, D., Bader, J. S., and Isberg, R. R. (2006). RNA interference analysis of Legionella in Drosophila cells: exploitation of early secretory apparatus dynamics. PLoS Pathog. 2, e34. doi:10.1371/journal.ppat.0020034
Ensminger, A. W., and Isberg, R. R. (2009). Legionella pneumophila Dot/Icm translocated substrates: a sum of parts. Curr. Opin. Microbiol. 12, 67–73.
Ensminger, A. W., and Isberg, R. R. (2010). E3 ubiquitin ligase activity and targeting of BAT3 by multiple Legionella pneumophila translocated substrates. Infect. Immun. 78, 3905–3919.
Fernandez-Moreira, E., Helbig, J. H., and Swanson, M. S. (2006). Membrane vesicles shed by Legionella pneumophila inhibit fusion of phagosomes with lysosomes. Infect. Immun. 74, 3285–3295.
Fortier, A., de Chastellier, C., Balor, S., and Gros, P. (2007). Birc1e/Naip5 rapidly antagonizes modulation of phagosome maturation by Legionella pneumophila. Cell. Microbiol. 9, 910–923.
Geng, J., and Klionsky, D. J. (2008). The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. “Protein modifications: beyond the usual suspects” review series. EMBO Rep. 9, 859–864.
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766.
Gutierrez, M. G., Vazquez, C. L., Munafo, D. B., Zoppino, F. C., Beron, W., Rabinovitch, M., and Colombo, M. I. (2005). Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell. Microbiol. 7, 981–993.
Hailey, D. W., Rambold, A. S., Satpute-Krishnan, P., Mitra, K., Sougrat, R., Kim, P. K., and Lippincott-Schwartz, J. (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667.
Hayashi-Nishino, M., Fujita, N., Noda, T., Yamaguchi, A., Yoshimori, T., and Yamamoto, A. (2009). A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 11, 1433–1437.
Hilbi, H., Segal, G., and Shuman, H. A. (2001). Icm/dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol. Microbiol. 42, 603–617.
Horwitz, M. A. (1983a). Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158, 1319–1331.
Horwitz, M. A. (1983b). The Legionnaires’ disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J. Exp. Med. 158, 2108–2126.
Huang, J., Birmingham, C. L., Shahnazari, S., Shiu, J., Zheng, Y. T., Smith, A. C., Campellone, K. G., Heo, W. D., Gruenheid, S., Meyer, T., Welch, M. D., Ktistakis, N. T., Kim, P. K., Klionsky, D. J., and Brumell, J. H. (2011). Antibacterial autophagy occurs at PI(3)P-enriched domains of the endoplasmic reticulum and requires Rab1 GTPase. Autophagy 7, 17–26.
Hubber, A., and Roy, C. R. (2010). Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26, 261–283.
Ingmundson, A., Delprato, A., Lambright, D. G., and Roy, C. R. (2007). Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450, 365–369.
Ivanov, S. S., and Roy, C. R. (2009). Modulation of ubiquitin dynamics and suppression of DALIS formation by the Legionella pneumophila Dot/Icm system. Cell. Microbiol. 11, 261–278.
Joshi, A. D., Sturgill-Koszycki, S., and Swanson, M. S. (2001). Evidence that Dot-dependent and -independent factors isolate the Legionella pneumophila phagosome from the endocytic network in mouse macrophages. Cell. Microbiol. 3, 99–114.
Kagan, J. C., and Roy, C. R. (2002). Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4, 945–954.
Kagan, J. C., Stein, M. P., Pypaert, M., and Roy, C. R. (2004). Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J. Exp. Med. 199, 1201–1211.
Kimura, S., Noda, T., and Yoshimori, T. (2008). Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Func. 33, 109–122.
Kirkin, V., Lamark, T., Sou, Y. S., Bjorkoy, G., Nunn, J. L., Bruun, J. A., Shvets, E., McEwan, D. G., Clausen, T. H., Wild, P., Bilusic, I., Theurillat, J. P., Øvervatn, A., Ishii, T., Elazar, Z., Komatsu, M., Dikic, I., and Johansen, T. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516.
Kubori, T., Hyakutake, A., and Nagai, H. (2008). Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 67, 1307–1319.
Kubori, T., Shinzawa, N., Kanuka, H., and Nagai, H. (2010). Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog. 6, e1001216. doi: 10.1371/journal.ppat.1001216
Lau, H. Y., and Ashbolt, N. J. (2009). The role of biofilms and protozoa in Legionella pathogenesis: implications for drinking water. J. Appl. Microbiol. 107, 368–378.
Levine, B., Mizushima, N., and Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335.
Ling, Y. M., Shaw, M. H., Ayala, C., Coppens, I., Taylor, G. A., Ferguson, D. J., and Yap, G. S. (2006). Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 203, 2063–2071.
Liou, W., Geuze, H. J., Geelen, M. J., and Slot, J. W. (1997). The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 136, 61–70.
Lomma, M., Dervins-Ravault, D., Rolando, M., Nora, T., Newton, H. J., Sansom, F. M., Sahr, T., Gomez-Valero, L., Jules, M., Hartland, E. L., and Buchrieser, C. (2010). The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cell. Microbiol. 12, 1272–1291.
Lurie-Weinberger, M. N., Gomez-Valero, L., Merault, N., Glockner, G., Buchrieser, C., and Gophna, U. (2010). The origins of eukaryotic-like proteins in Legionella pneumophila. Int. J. Med. Microbiol. 300, 470–481.
Machner, M. P., and Isberg, R. R. (2007). A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science 318, 974–977.
Matsuda, F., Fujii, J., and Yoshida, S. (2009). Autophagy induced by 2-deoxy-D-glucose suppresses intracellular multiplication of Legionella pneumophila in A/J mouse macrophages. Autophagy 5, 484–493.
Mehrpour, M., Esclatine, A., Beau, I., and Codogno, P. (2010). Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am. J. Physiol. Cell Physiol. 298, C776–785.
Melo, E. J., and de Souza, W. (1997). Relationship between the host cell endoplasmic reticulum and the parasitophorous vacuole containing Toxoplasma gondii. Cell Struct. Funct. 22, 317–323.
Molofsky, A. B., Byrne, B. G., Whitfield, N. N., Madigan, C. A., Fuse, E. T., Tateda, K., and Swanson, M. S. (2006). Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203, 1093–1104.
Mostowy, S., Bonazzi, M., Hamon, M. A., Tham, T. N., Mallet, A., Lelek, M., Gouin, E., Demangel, C., Brosch, R., Zimmer, C., Sartori, A., Kinoshita, M., Lecuit, M., and Cossart, P. (2010). Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe 8, 433–444.
Muller, M. P., Peters, H., Blumer, J., Blankenfeldt, W., Goody, R. S., and Itzen, A. (2010). The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329, 946–949.
Murata, T., Delprato, A., Ingmundson, A., Toomre, D. K., Lambright, D. G., and Roy, C. R. (2006). The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat. Cell Biol. 8, 971–977.
Nagai, H., Kagan, J. C., Zhu, X., Kahn, R. A., and Roy, C. R. (2002). A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295, 679–682.
Nakagawa, I., Amano, A., Mizushima, N., Yamamoto, A., Yamaguchi, H., Kamimoto, T., Nara, A., Funao, J., Nakata, M., Tsuda, K., Hamada, S., and Yoshimori, T. (2004). Autophagy defends cells against invading group A Streptococcus. Science 306, 1037–1040.
Neumeister, B., Reiff, G., Faigle, M., Dietz, K., Northoff, H., and Lang, F. (2000). Influence of Acanthamoeba castellanii on intracellular growth of different Legionella species in human monocytes. Appl. Environ. Microbiol. 66, 914–919.
Otto, G. P., Wu, M. Y., Clarke, M., Lu, H., Anderson, O. R., Hilbi, H., Shuman, H. A., and Kessin, R. H. (2004). Macroautophagy is dispensable for intracellular replication of Legionella pneumophila in Dictyostelium discoideum. Mol. Microbiol. 51, 63–72.
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., Overvatn, A., Bjorkoy, G., and Johansen, T. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145.
Perrin, A. J., Jiang, X., Birmingham, C. L., So, N. S., and Brumell, J. H. (2004). Recognition of bacteria in the cytosol of mammalian cells by the ubiquitin system. Curr. Biol. 14, 806–811.
Ponpuak, M., Davis, A. S., Roberts, E. A., Delgado, M. A., Dinkins, C., Zhao, Z., Virgin, H. W. t., Kyei, G. B., Johansen, T., Vergne, I., and Deretic, V. (2010). Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32, 329–341.
Price, C. T., Al-Khodor, S., Al-Quadan, T., Santic, M., Habyarimana, F., Kalia, A., and Kwaik, Y. A. (2009). Molecular mimicry by an F-box effector of Legionella pneumophila hijacks a conserved polyubiquitination machinery within macrophages and protozoa. PLoS Pathog. 5, e1000704. doi: 10.1371/journal.ppat.1000704
Ravikumar, B., Moreau, K., Jahreiss, L., Puri, C., and Rubinsztein, D. C. (2010). Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 12, 747–757.
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F., and Vance, R. E. (2006). Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18. doi: 10.1371/journal.ppat.0020018
Rink, J., Ghigo, E., Kalaidzidis, Y., and Zerial, M. (2005). Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749.
Robinson, C. G., and Roy, C. R. (2006). Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell. Microbiol. 8, 793–805.
Romano, P. S., Gutierrez, M. G., Beron, W., Rabinovitch, M., and Colombo, M. I. (2007). The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell. Microbiol. 9, 891–909.
Sauer, J. D., Bachman, M. A., and Swanson, M. S. (2005). The phagosomal transporter A couples threonine acquisition to differentiation and replication of Legionella pneumophila in macrophages. Proc. Natl. Acad. Sci. U.S.A. 102, 9924–9929.
Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525.
Sturgill-Koszycki, S., and Swanson, M. S. (2000). Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J. Exp. Med. 192, 1261–1272.
Swanson, M. S., Byrne, B. G., and Dubuisson, J. F. (2009). Kinetic analysis of autophagosome formation and turnover in primary mouse macrophages. Methods Enzymol. 452, 383–402.
Swanson, M. S., and Isberg, R. R. (1995). Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63, 3609–3620.
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221.
Tilney, L. G., Harb, O. S., Connelly, P. S., Robinson, C. G., and Roy, C. R. (2001). How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114, 4637–4650.
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhaes, J. G., Yuan, L., Soares, F., Chea, E., Le Bourhis, L., Boneca, I. G., Allaoui, A., Jones, N. L., Nuñez, G., Girardin, S. E., and Philpott, D. J. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62.
Tung, S. M., Unal, C., Ley, A., Pena, C., Tunggal, B., Noegel, A. A., Krut, O., Steinert, M., and Eichinger, L. (2010). Loss of Dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of Legionella pneumophila. Cell. Microbiol. 12, 765–780.
van der Vaart, A., Griffith, J., and Reggiori, F. (2010). Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol. Biol. Cell 21, 2270–2284.
Vinzing, M., Eitel, J., Lippmann, J., Hocke, A. C., Zahlten, J., Slevogt, H., N’Guessan, P. D., Gunther, S., Schmeck, B., Hippenstiel, S., Flieger, A., Suttorp, N., and Opitz, B. (2008). NAIP and Ipaf control Legionella pneumophila replication in human cells. J. Immunol. 180, 6808–6815.
Wick, A. N., Drury, D. R., Nakada, H. I., and Wolfe, J. B. (1957). Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 224, 963–969.
Wieland, H., Goetz, F., and Neumeister, B. (2004). Phagosomal acidification is not a prerequisite for intracellular multiplication of Legionella pneumophila in human monocytes. J. Infect. Dis. 189, 1610–1614.
Wieland, H., Ullrich, S., Lang, F., and Neumeister, B. (2005). Intracellular multiplication of Legionella pneumophila depends on host cell amino acid transporter SLC1A5. Mol. Microbiol. 55, 1528–1537.
Wright, E. K., Goodart, S. A., Growney, J. D., Hadinoto, V., Endrizzi, M. G., Long, E. M., Sadigh, K., Abney, A. L., Bernstein-Hanley, I., and Dietrich, W. F. (2003). Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr. Biol. 13, 27–36.
Yla-Anttila, P., Vihinen, H., Jokitalo, E., and Eskelinen, E. L. (2009). 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 5, 1180–1185.
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., Mimuro, H., Nakagawa, I., Yanagawa, T., Ishii, T., Kakizuka, A., Sztul, E., Chakraborty, T., and Sasakawa, C. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240.
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., Kuida, K., Mariathasan, S., Dixit, V. M., Flavell, R. A., Dietrich, W. F., and Roy, C. R. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325.
Zheng, Y. T., Shahnazari, S., Brech, A., Lamark, T., Johansen, T., and Brumell, J. H. (2009). The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916.
Keywords: Legionella pneumophila, Type IV secretion system, autophagy, vacuole maturation, Rab conversion
Citation: Joshi AD and Swanson MS (2011) Secrets of a successful pathogen: Legionella resistance to progression along the autophagic pathway. Front. Microbio. 2:138. doi: 10.3389/fmicb.2011.00138
Received: 16 March 2011; Paper pending published: 13 April 2011;
Accepted: 13 June 2011; Published online: 28 June 2011.
Edited by:
Carmen Buchrieser, Pasteur Institute, FranceReviewed by:
Elizabeth L. Hartland, The University of Melbourne, AustraliaPatrice Coodgno, INSERM, France
Serge Mostowy, Institut Pasteur, France
Copyright: © 2011 Joshi and Swanson. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Michele S. Swanson, Department of Microbiology and Immunology, University of Michigan Medical School, 6733 Medical Science Building II, 1150 West Medical Center Drive, Ann Arbor, MI 48109-5620, USA. e-mail:bXN3YW5zb25AdW1pY2guZWR1