 Drew A. Rholl1,2
Drew A. Rholl1,2 Krisztina M. Papp-Wallace3 Andrew P. Tomaras4 Michael L. Vasil4
Krisztina M. Papp-Wallace3 Andrew P. Tomaras4 Michael L. Vasil4 Robert A. Bonomo3,5,6,7
Robert A. Bonomo3,5,6,7 Herbert P. Schweizer1,2*
Herbert P. Schweizer1,2*- 1 Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, CO, USA
- 2 Rocky Mountain Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research, Colorado State University, Fort Collins, CO, USA
- 3 Louis Stokes Cleveland Department of Veterans Affairs Medical Center, Cleveland, OH, USA
- 4 Department of Microbiology, University of Colorado School of Medicine, Aurora, CO, USA
- 5 Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, OH, USA
- 6 Department of Pharmacology, Case Western Reserve University School of Medicine, Cleveland, OH, USA
- 7 Department of Molecular Biology and Microbiology, Case Western Reserve University School of Medicine, Cleveland, OH, USA
Burkholderia pseudomallei is the etiological agent of melioidosis. Because of the bacterium’s intrinsic resistance and propensity to establish latent infections, melioidosis therapy is complicated and prolonged. Newer generation β-lactams, specifically ceftazidime, are used for acute phase therapy, but resistance to this cephalosporin has been observed. The chromosomally encoded penA gene encodes a putative twin arginine translocase (TAT)-secreted β-lactamase, and penA mutations have been implicated in ceftazidime resistance in clinical isolates. However, the role of PenA in resistance has not yet been systematically studied in isogenetic B. pseudomallei mutant backgrounds. We investigated the effects of penA deletion, point mutations, and up-regulation, as well as tat operon deletion and PenA TAT-signal sequence mutations. These experiments were made possible by employing a B. pseudomallei strain that is excluded from Select Agent regulations. Deletion of penA significantly (>4-fold) reduced the susceptibility to six of the nine β-lactams tested and ≥16-fold for ampicillin, amoxicillin, and carbenicillin. Overexpression of penA by single-copy, chromosomal expression of the gene under control of the inducible Ptac promoter, increased resistance levels for all β-lactams tested 2- to 10-fold. Recreation of the C69Y and P167S PenA amino acid substitutions previously observed in resistant clinical isolates increased resistance to ceftazidime by ≥85- and 5- to 8-fold, respectively. Similarly, a S72F substitution resulted in a 4-fold increase in resistance to amoxicillin and clavulanic acid. Susceptibility assays with PenA TAT-signal sequence and ΔtatABC mutants, as well as Western blot analysis, confirmed that PenA is a TAT secreted enzyme and not periplasmic but associated with the spheroplastic cell fraction. Lastly, we determined that two LysR-family regulators encoded by genes adjacent to penA do not play a role in transcriptional regulation of penA expression.
Introduction
Burkholderia pseudomallei, the etiological agent of melioidosis, is a saprophytic Gram negative bacterium endemic to many tropical and subtropical regions of the world although much of the disease and its investigation has historically been confined to Northern Australia and regions of SE Asia, notably NE Thailand, Singapore, and Malaysia (Cheng and Currie, 2005; Wiersinga et al., 2006; Currie et al., 2008). Partially because of its large genome and diverse repertoire of metabolic functions B. pseudomallei can survive hostile conditions and is resilient to many antimicrobial agents, including antibiotics (Holden et al., 2004). This makes choosing effective therapeutic strategies difficult. In the past three decades even the most effective treatment could not prevent a mortality rate of 74% (White et al., 1989). Clinical outcomes improved steadily with implementation of new therapies but the real breakthrough was achieved with the introduction of ceftazidime, an extended-spectrum cephalosporin, which halved the mortality rate compared to the traditional multidrug therapy of chloramphenicol, doxycycline, and trimethoprim–sulfamethoxazole (White et al., 1989). Currently recommended melioidosis treatment involves acute phase therapy followed by a lengthy eradication therapy. Initial parenteral therapy involves ceftazidime or a carbapenem for a minimum of 10–14 days and longer (4–8 weeks) for deep-seated infection. This regimen may be supplemented with trimethoprim–sulfamethoxazole given orally for treatment of patients with neurologic, prostatic, bone, or joint melioidosis. Oral eradication therapy is trimethoprim–sulfamethoxazole with or without doxycycline for at least 3–6 months (Peacock et al., 2008).
Because of the pivotal role that β-lactams play in the acute phase treatment of melioidosis emergence of resistance, though still considered rare, is of concern. It is believed that B. pseudomallei’s resistance to β-lactams is due to chromosomally encoded β-lactamases (Livermore et al., 1987; Godfrey et al., 1991). These include a number of Ambler Class A, B, and D β-lactamases that are encoded by the K96243 and other B. pseudomallei genomes (Holden et al., 2004). The penA gene (K96243 gene BPSS0946 found on chromosome II; Figure 1) encodes a Class A β-lactamase (Cheung et al., 2002; Tribuddharat et al., 2003). This gene is present and expressed in prototype B. pseudomallei strains. PenA confers resistance to numerous β-lactam antibiotics when expressed in Escherichia coli (Cheung et al., 2002; Tribuddharat et al., 2003) and several reports described a role of this enzyme in acquired ceftazidime resistance in patients treated with this antibiotic (Godfrey et al., 1991; Tribuddharat et al., 2003; Sam et al., 2009). Mutations identified in clinical strains included a C69Y substitution leading to high-level ceftazidime resistance (Sam et al., 2009), a P167S substitution leading to medium-level ceftazidime resistance (Tribuddharat et al., 2003) and a S72F mutation that led to resistance to clavulanic acid (Tribuddharat et al., 2003). A Class D Oxa-57 β-lactamase has been studied in vitro but its role in clinically significant β-lactam resistance remains unclear (Keith et al., 2005).
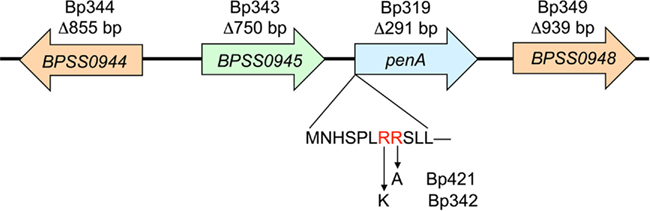
Figure 1. Genomic organization of the B. pseudomallei penA region. The genes and gene order are from sequenced strain K96243 (GenBank accession number NC_006351). The penA region encodes two LysR-type regulators (BPSS0944 and BPSS0948) and a putative peptidase (BPSS0945). The names of the mutants harboring gene deletion and extents of deleted sequences are shown above each gene. The putative PenA twin arginine translocase (TAT) signal sequence is shown below the penA gene with the two conserved arginine residues shown in red letters. Arrows indicate amino acid substitutions, R7K and R8A, in the TAT-signal sequence and the names of the mutants are shown next to the respective amino acids replacing the original arginines.
While B. pseudomallei PenA β-lactamase has been studied in some detail, previously published reports suffered until recently from some unavoidable shortcomings. First, many mutations contributing to clinically significant β-lactam resistance were identified in genetically largely intractable clinical isolates. Thus, it remained unclear whether the mutations were solely responsible for causing the observed resistance. Second, because methods for genetic manipulation of B. pseudomallei were rather rudimentary until recently, most studies involved expression of putative β-lactamase enzymes in E. coli. Third, United States Select Agent and recombinant DNA regulations, as well as dual use concerns, do complicate studies of clinically significant antibiotic resistance mechanisms. To address shortcomings of previous studies, we employed state-of-the-art Select Agent-compliant genetic and biochemical methods and a defined genetic background of a Select Agent excluded B. pseudomallei strain, where applicable, to study the contribution of PenA to B. pseudomallei’s resistance to clinically significant β-lactam antibiotics. The studies also revealed that PenA is secreted via the twin arginine translocase system and that its expression in prototype strains does not seem to be regulated by local transcriptional regulators.
Materials and Methods
Bacterial Strains and Growth Conditions
Burkholderia pseudomallei strains used in this study are listed in Table 1. E. coli strains DH5α (Liss, 1987) and MACH1 (Invitrogen, Carlsbad, CA, USA) were used as general cloning strains, and DB3.1 (Invitrogen) for cloning with Gateway Vectors. RHO3 was used as a mobilizer strain for conjugation of plasmids from E. coli to B. pseudomallei (López et al., 2009). Bacterial strains were grown in Lennox LB (MO BIO Laboratories, Carlsbad, CA, USA) or LB without salt (10 g/L tryptone and 5 g/L yeast extract) at 37°C. Antibiotics were used at the following concentrations: 100 μg/mL ampicillin (Amp), 35 μg/mL kanamycin (Km), and 15 μg/mL zeocin (Zeo) for E. coli and 1,000 μg/mL Km and 2,000 μg/mL Zeo for B. pseudomallei. Antibiotics were purchased from Sigma (St. Louis, MO, USA) except Zeo which was from Invitrogen. The ΔpurM strain Bp82 was grown in media supplemented with 0.6 mM adenine to ensure growth rates comparable to strain 1026b. RHO3 was grown in media containing 400 μg/mL diaminopimelic acid (DAP; LL-, DD-, and meso-isomers; Sigma). Induction of gene expression from Ptac was achieved by adding 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG; Gold Biotechnology, St. Louis, MO, USA) to growth media.

Table 1. Burkholderia pseudomallei strains used in this study.
Isolation of Mutants Containing Chromosomal Deletion or Point Mutations
All deletion and allelic-exchange procedures were based on pEXKm5 (López et al., 2009; plasmids used in this study are listed in Table 2) and were performed using previously described protocols. The desired chromosomal deletions were verified by colony PCR (Choi et al., 2008).
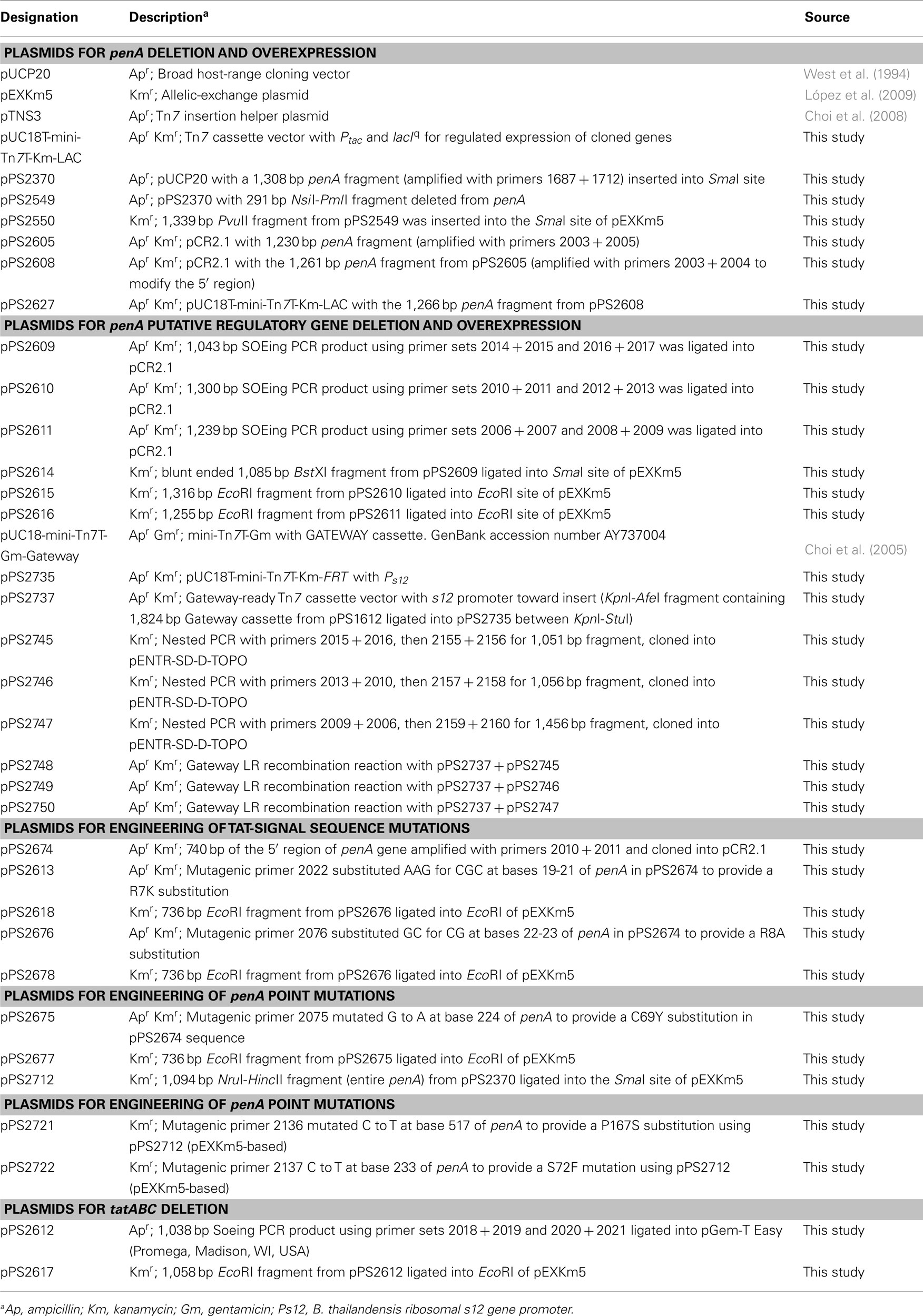
Table 2. Plasmids used in this study.
For construction of penA deletion mutants, PCR with primers 1687 + 1712 (PCR primers and mutagenic oligonucleotides are listed in Table 3) and Taq polymerase (New England BioLabs, Ipswich, MA, USA) was used to amplify a 1,308-bp region containing the BPSS0946 (penA) gene from 1026b chromosomal DNA. The gel-purified PCR fragment was cloned into the SmaI site of pUCP20 to yield pPS2370. This plasmid was then cleaved with NsiI + PmlI, blunt ended with T4 DNA polymerase, followed by re-ligation. This procedure deleted a 291-bp NsiI − PmlI fragment from the penA gene and resulted in pPS2549. A 1,339-bp PvuII ΔpenA fragment was excised from this plasmid and ligated into the SmaI site of pEXKm5 to create pPS2550. This plasmid was used to create Bp82.11 and Bp319 by transferring the plasmid-borne deletion alleles to either Bp82 or 1026b, respectively, via conjugation from RHO3.
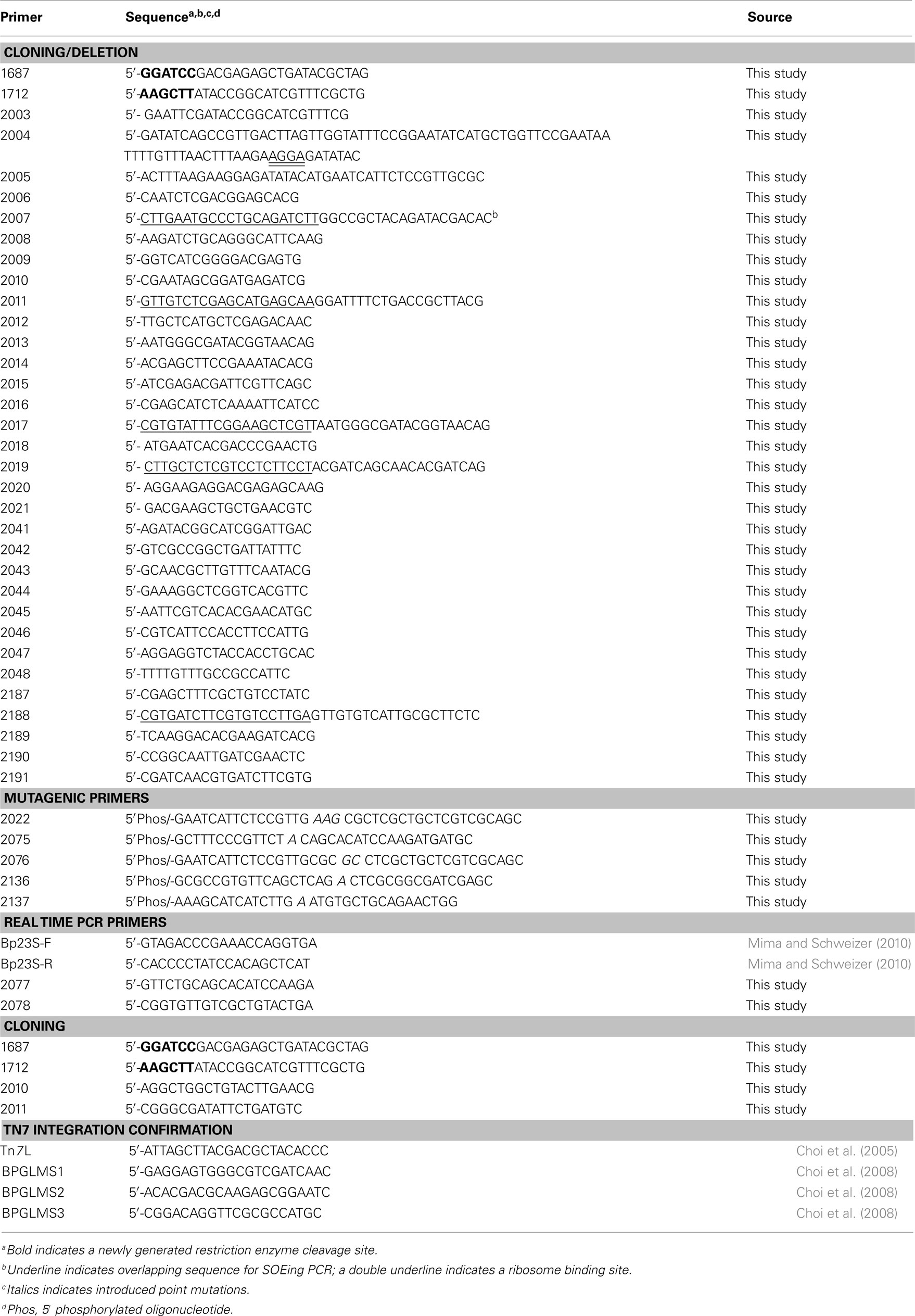
Table 3. Primers used in this study.
For deletion of tatABC, splicing by overlap extension (SOEing) PCR was employed for engineering of deletion constructs. SOEing reactions consisted of separately amplifying two fragments, one using an “internal” primer with overlapping sequence with the internal primer from the other fragment. These bands were gel purified and 50 ng of each product was added to a new PCR reaction where it underwent PCR for five cycles (95°C for 60 s, 54°C 30 s, and 72°C for 60 s). At this point, the two non-overlapping primers were added and the reaction proceeded for another 30 cycles. Using in-house purified Pfu polymerase and primer sets 2018 + 2019 (amplifying a 537-bp tatA 5′ fragment) and 2020 + 2021 (amplifying a 501-bp tatC 3′ fragment), a 1,038-bp SOEing PCR product was generated to delete 1,527 bp from the tatABC gene cluster. This PCR product was ligated into pGem-T Easy (Promega; Madison, WI, USA) to create pPS2612. An EcoRI fragment was rescued from this plasmid and inserted into pEXKm5 to create pPS2617, which was used to create Bp409 and Bp420 by transferring the plasmid-borne deletions to either 1026b or Bp319 (1026b ΔpenA), respectively. PCR using primers 2047 + 2048 was used to confirm the deletion.
Other genes located in the penA region of the chromosome were deleted using a SOEing PCR strategy and pCR2.1 ( Invitrogen) as TA cloning vector. The BPSS0944 deletion construct was created using primer sets 2014 + 2015 and 2016 + 2017 to generate pPS2609, from which a BstXI fragment was excised and inserted into the SmaI site of pEXKm5 to yield pPS2614. The BPSS0945 deletion construct was generated using primer sets 2010 + 2011 and 2012 + 2013 to create pPS2610, from which an EcoRI fragment was excised and inserted into the EcoRI site of pEXKm5 to yield pPS2615. The BPSS0948 deletion construct was created using primer sets 2006 + 2007 and 2008 + 2009 to generate pPS2611, from which an EcoRI fragment was excised and inserted into the EcoRI site of pEXKm5 to yield pPS2616. The plasmid-borne deletion alleles were transferred to the B. pseudomallei 1026b genome which resulted in strains Bp343, Bp344 and Bp349, respectively. Deletions were verified by colony PCR using primer sets 2045 + 2446, 2041 + 2042 and 2043 + 2044, respectively.
Chromosomal penA point mutations were engineered using the QuikChange Multi Kit (Stratagene, La Jolla, CA, USA), 5′-phosphorylated mutagenic oligonucleotides, and plasmid DNA templates. Mutagenic oligonucleotide 2075 was used with pPS2674 to create pPS2675 for the PenA C69Y mutation. A 736-bp EcoRI fragment from pPS2675 was then ligated into the EcoRI site of pEXKm5 to construct pPS2677. Plasmid pPS2712 was created as a platform for other mutations by ligating the NruI–HincII containing penA fragment from pPS2676 into the SmaI site of pEXKm5. Employing pPS2712 template DNA, mutagenic oligonucleotides 2136 and 2137 were used separately to create pPS2721 and pPS2722 carrying PenA P167S and PenA S72F substitutions, respectively. Allelic exchange was carried out by conjugal transfer of pPS2677 (C69Y), pPS2721 (P167S), and pPS2722 (S72F) from RHO3 into Bp82. Mutations were verified by PCR amplifying and sequencing the region containing the expected mutation. TAT-signal sequence mutations were generated using a similar strategy. The R7K mutation was engineered using mutagenic oligonucleotide 2022 and pPS2674 to create pPS2613. The 736-bp EcoRI fragment from this plasmid was ligated into pEXKm5 to yield pPS2618. The R7K allele contained on this fragment was transferred to the 1026b genome which created Bp342. The mutagenic oligonucleotide 2076 was used with pPS2674 to engineer pPS2676 to create an R8A mutation. The 736-bp EcoRI fragment was excised from this plasmid and ligated into pEXKm5 to create pPS2678. The R8A allele contained on this fragment was transferred to the 1026b genome which created Bp421. The presence of the desired point mutations on plasmids and the genome was verified by DNA sequencing.
Gene Complementation and Overexpression Using Single-Copy, Chromosomally Integrated Mini-Tn7 Vectors
The mini-Tn7 system was used for introducing site-specific, stable insertions into the B. pseudomallei genome for purposes of gene complementation or overexpression (Choi et al., 2008). Tn7 transposition was achieved by tri-parental mating involving RHO3 harboring the mini-Tn7 vector, RHO3 containing the helper plasmid pTNS3 and the B. pseudomallei recipient strain, as previously described (Choi et al., 2006). Integration events were verified using primers Tn7L and either BPGLMS1, BPGLMS2, or BPGLMS3 (Choi et al., 2008). All Tn7 mutants retained and used for further experimentation had insertions at the glmS2-associated Tn7 insertion site.
For regulated penA expression and overproduction, the gene was PCR amplified from pPS2370 using primers 2003 + 2005 and Pfu polymerase and the 1,230-bp PCR product cloned into pCR2.1 (Invitrogen) to yield pPS2605. An optimized ribosome binding site (RBS) was introduced upstream of penA to create pPS2608 by PCR amplifying the penA region of pPS2605 with primers 2003 + 2004 and cloning the resulting 1,261 bp fragment into pCR2.1. (The amplicon was expected to be 1,295 bp but the 5′ end was truncated by 34 bp which did not affect the integrity of the penA gene.) An expression construct where penA was transcribed from the inducible Ptac was obtained by cloning the 1,300-bp EcoRI fragment from pPS2608 into pUC18T-mini-Tn7T-Km-LAC to create pPS2627. The mini-Tn7 expression cassette from pPS2627 was integrated into the genome of Bp82 at the glmS2-associated Tn7 integration site to form Bp82.21.
Constitutive expression of genes was achieved from chromosomally integrated mini-Tn7 elements where the respective genes were transcribed from the B. thailandensis s12 promoter (Choi et al., 2008). Nested PCR and Pfu polymerase was used to PCR amplify BPSS0944 (primers 2015 + 2016 and 2155 + 2156), BPSS0945 (primers 2010 + 2013 and 2157 + 2158), and BPSS0948 (primers 2006 + 2009 and 2159 + 2160) from strain 1026b genomic DNA. Each PCR began with three cycles using only the outside set (listed first) then the inner set (listed second) was added for 30 more cycles. Inner primers were designed for use with the pENTR/SD/D-TOPO cloning vector (Invitrogen, Carlsbad, CA, USA), which provides a RBS and directionality, and created pPS2745, pPS2746, and pPS2747, respectively. These plasmids then underwent the Gateway LR recombination reaction (Invitrogen) with pPS2737, a mini-Tn7 vector which enables constitutive expression from the B. thailandensis s12 promoter (this promoter is directed toward the Gateway recombination cassette). To create pPS2737 the 1,824-bp Gateway-cassette-containing the KpnI-AfeI fragment from pUC18-mini-Tn7T-Gm-Gateway was ligated into pPS2735 between the KpnI and StuI sites. This Gateway-compatible mini-Tn7 element was used to create pPS2748, pPS2749, and pPS2750 for constitutive expression of BPSS0944, BPSS0945, and BPSS0948, respectively. The mini-Tn7 elements contained on these plasmids were individually inserted at the glmS2 site of Bp82 with the help of pTNS3 to create Bp82.14, Bp82.15, and Bp82.16.
MIC Determinations
MICs were determined following general procedures recommended by the Clinical and Laboratory Standards Institute (2010). However, since ΔtatABC mutants do not grow well in the presence of salts LB without salt was substituted for Mueller–Hinton Broth. MICs for ampicillin, carbenicillin, and BAL30072 (obtained from Basilea Pharmaceutica, Basel, Switzerland) were determined by the two-fold broth microdilution technique. Etest strips were used to determine MICs for amoxicillin, amoxicillin–clavulanic acid, ceftazidime, imipenem, meropenem, and piperacillin according to manufacturer’s instructions (AB BioMérieux, Marcy l’Etoile, France). When needed, IPTG was added to media at a final concentration of 1 mM. The MICs were recorded after incubation at 37°C for 18–24 h.
Quantification of penA Transcript Levels
Overnight cultures were subcultured into LB medium, grown to an OD600 nm of 0.5 and RNA was extracted with the RNeasy Protect Bacteria Mini Kit (Qiagen, Valencia, CA, USA). cDNA was synthesized using the SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen) and quantified using SYBR GreenER qPCR SuperMix for iCycler Instruments (Invitrogen) and the Bio-Rad iQ5 iCycler. The Bp23S-F + Bp23S-R primer set was used for the 23s rRNA housekeeping gene for data normalization and primers 2077 + 2078 were used to quantify the penA transcript. Data analyses were performed using the iCycler software. For induction studies with both wild-type and the mutants with constitutive regulatory gene expression or deletion several methods were employed. For salt stress testing, strains were grown in media with 150 mM NaCl or no NaCl according to Pumirat et al. (2009). For testing induction by β-lactams at sub-inhibitory levels, strains were subcultured into LB with 4-fold lower than MIC concentrations of either ceftazidime or carbenicillin until an OD600 nm of 0.5 was reached. Induction was also tested with fourfold higher than MIC concentrations of ceftazidime, carbenicillin, imipenem, or penicillin G (2,000 μg/mL for penicillin G) by growing in LB to an OD600 nm of 0.5, then adding β-lactams and shaking for an additional 2 h before RNA extraction, according to Trépanier et al. (1997).
Protein Techniques
Burkholderia pseudomallei cells were fractionated into periplasm and spheroplastic protein fractions (cytosol and membranes) using the PeriPreps™ Periplasting Kit (Epicentre Biotechnologies, Madison, WI, USA) Cells were grown overnight and diluted 1:100 at 37°C in LB medium without NaCl until an optical density of 0.7 (600 nm) was reached. The kit was used according to manufacturer’s protocols, including extended incubation times and higher concentrations of lysozyme (25 μg/reaction), as recommended for hardier bacteria.
Escherichia coli Origami 2 DE3 cells (Novagen, Madison, WI, USA) expressing blapenA minus the first 90 nucleotides (30 amino acids) which encode the N-terminal signal sequence were subcloned in the pET24a(+) vector (Stratagene) and used for protein production and purification. The PenA β-lactamase was extracted from E. coli and purified using preparative isoelectric focusing. After verification of purity by SDS PAGE, 3.0 mg of PenA was sent to New England Peptide (Gardner, MA, USA) where the polyclonal anti-PenA antibodies were raised in rabbits. The antibodies were subsequently isolated from serum using a Protein G column (GE Healthcare Life Sciences, Piscataway, NJ, USA) purification according to the manufacturer’s instructions.
For Western blots, protein samples were separated on NuPAGE® 4–12% Bis–Tris polyacrylamide gels (Invitrogen) alongside Precision Plus Protein Prestained Dual Color Standards (Bio-Rad, Hercules, CA, USA). A goat anti-Rabbit IgG alkaline phosphatase-conjugated antibody (Sigma) was used as a secondary antibody and SIGMAFAST™ BCIP®/NBT tablets (Sigma) as a detection reagent according to the manufacturer’s protocol.
Results
The Role of of penA in β-Lactam Resistance
To assess whether Bp82 and its parent 1026b could interchangeably be used for PenA characterization experiments, the susceptibilities of these strains to various β-lactams were tested. Observable differences in the susceptibilities of these two strains for any of the beta-lactams and clavulanic acid tested (Table 4) were not seen, thereby validating the use of Bp82 in experiments otherwise not feasible under Select Agent regulations. Deletion of the penA gene from 1026b (strain Bp319) and Bp82 (strain Bp82.11) caused a significant (≥4-fold) decrease in the susceptibilities for six of nine β-lactams tested, and ≥16-fold for three of them (ampicillin, amoxicillin, and carbenicillin; Table 4). Likewise, up-regulation of penA by single-copy expression from the IPTG-inducible Ptac (Bp82.21) significantly increased the MIC for seven of the eight β-lactams tested with meropenem showing only slight change. (Amoxicillin could not be tested as the resistance level for the wild-type was already beyond detection.) Quantitative real time PCR experiments showed that in the Ptac-penA strain (Bp82.21) penA transcript levels were 36-fold higher when compared to transcript levels observed in the wild-type strain (data not shown). This increase in transcript levels corresponds to the observed increases in resistance to all β-lactams. These experiments demonstrated that although PenA is a clinically significant β-lactam resistance mechanism, it affects some β-lactams more than others. While mutations in penA can significantly affect the utility of ceftazidime and amoxicillin + clavulanic acid, the enzyme has a lesser effect on the activity of carbapenems and novel experimental drugs such as BAL30072.
Table 4. β-lactam susceptibilities of PenA, TAT-signal sequence, and TAT secretion apparatus mutants.
penA Mutations are Responsible for Clinically Significant Ceftazidime and Amoxicillin + Clavulanic Acid Resistance
Previous studies identified several penA mutations in clinical and laboratory isolates that led to clinically significant ceftazidime or clavulanic acid resistance. Specifically, a C69Y substitution caused high-level ceftazidime resistance (Sam et al., 2009), a P167S substitution medium-level ceftazidime resistance (Tribuddharat et al., 2003), and a S72F mutation resistance to clavulanic acid (Tribuddharat et al., 2003). To assess whether these mutations alone were sufficient to cause the observed resistance phenotypes, they were engineered into the penA gene of strain Bp82 resulting in expression of a mutated PenA from the native penA promoter. Susceptibility studies revealed that the C69Y (Bp82.3) and P167S (Bp82.5) point mutations caused significant increases in ceftazidime resistance of ≥85- and 5- to 8-fold, respectively (Table 4). These changes are clinically significant since the ceftazidime susceptibility, intermediate and resistance breakpoints are defined as MICs of ≤8, 16, and ≥32 μg/mL, respectively. The C69Y mutation sensitized strains to other β-lactams such as amoxicillin, ampicillin, carbenicillin, and imipenem but not amoxicillin + clavulanic acid, piperacillin, meropenem, and BAL30072, whose MICs were already at low levels. The S72F point mutation caused a four-fold increase in resistance to amoxicillin + clavulanic acid in the resulting strain Bp82.4 and did not cause any changes in susceptibility to other β-lactams.
PenA is Secreted via the TAT System
Analysis of the amino-terminal PenA amino acid sequence revealed the presence of a putative TAT-signal sequence indicating that it may be a TAT secreted protein (Figure 1). To test this notion, the two signature arginine residues at positions 7 and 8 were changed to a lysine or alanine, respectively. MIC determinations revealed that disruption of the TAT-signal sequence by an R7K mutation (Bp342) did not affect PenA activity. This is in accordance with previous studies which have shown that mutation of the first arginine to a lysine can either have little effect or be completely inhibitory, depending on the rest of the signal sequence (Stanley et al., 2000). However, an R8A substitution (Bp421) completely abrogated PenA activity, consistent with PenA being a TAT secreted enzyme. This notion was further supported by the finding that a tatABC deletion mutant (Bp409) exhibited a susceptibility profile similar to those of the R8A substitution (Bp421) and ΔpenA deletion strains (Bp319 and Bp82.11). As expected then, a ΔpenA ΔtatABC double mutant (Bp420) was most susceptible to PenA substrates.
Cellular Localization of PenA
We next attempted to localize the PenA protein in the cell envelope using Western blot analysis and polyclonal α-PenA antibodies. Using this method, PenA could not be localized to the periplasmic fraction but rather only to the spheroplastic fraction which contains both cytosolic and membrane proteins. Multiple attempts at isolation of PenA from the periplasmic fraction employing other fractionation methods such as chloroform (Ames et al., 1984) or magnesium chloride (Imperi et al., 2009) extraction yielded the same results. Western blot analysis of the spheroplastic fraction (Figure 2) showed the mature 27 kDa PenA protein is seen only in an extract derived from wild-type 1026b (lane 5). In contrast, the unprocessed 31 kDa PenA protein was observed in extracts from the R8A (lane 1) and ΔtatABC (lane 2) mutants Bp421 and Bp409, respectively. A mixture of mature and unprocessed PenA was seen in the R7K mutant Bp342 extract with the majority being the mature protein (lane 3). As expected, no PenA protein was observed in the extract from the ΔpenA mutant Bp319 (lane 4). This experiment provides biochemical evidence for PenA processing only in 1026b and the R7K mutants, both of which secrete active PenA via the TAT system as judged by β-lactam susceptibility assays. All other strains are susceptible to β-lactams.
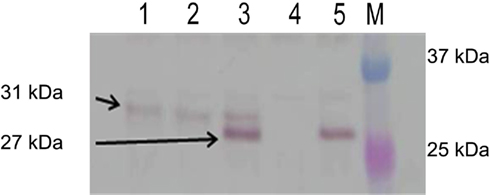
Figure 2. PenA is a TAT secreted protein. Spheroplastic proteins were analyzed by Western blot using anti-PenA polyclonal antibodies. The arrows point to the expected unprocessed (31 kDa) and processed (27 kDa) forms of PenA. Lanes: 1, R8A TAT-signal sequence mutant; 2, ΔtatABC mutant; 3, R7K TAT signal sequence mutant; 4, ΔpenA mutant; 5, wild-type 1026b; M, molecular weight markers (two proteins of the 10- to 250-kDa Precision Plus Protein Dual Color Standards from Bio-Rad, Hercules, CA, USA).
Local Regulators are Not Involved in Regulation of penA Gene Expression
As shown in Figure 1, the B. pseudomallei penA region encodes two LysR-type transcriptional regulators and a putative peptidase. Since chromosomal β-lactamase gene expression was shown to be regulated by products of adjacent regulatory genes in several bacteria, including B. cepacia (Trépanier et al., 1997), the structural genes encoding for these regulators were either deleted or overexpressed and their effects on PenA transcription and activity assessed by either qRT-PCR or MIC determinations. Carbenicillin was used as a “sentry” β-lactam for assessing PenA activity by MIC experiments because it is one of the best PenA substrates (Table 4). MIC determinations showed that neither deletion of putative regulators (Bp343 and Bp349 nor constitutive expression of the regulators (Bp82.14 and Bp82.16) affected PenA activity (data not shown). The same observations were made when ceftazidime was used in susceptibility assays instead of carbenicillin. Likewise, deletion (Bp344 and Bp82.31) or overexpression (Bp82.15) of the putative peptidase gene upstream of penA had no effect on PenA activity (data not shown). Lastly, since bacterial β-lactamase gene expression can either be subject to substrate induction or influenced by environmental factors such as salts (Pumirat et al., 2009), MIC determinations were performed in the presence or absence of substrate and salt. However, presence of ceftazidime, carbenicillin, or high salt and the absence salt had no apparent effect on β-lactam susceptibilities. qRT-PCR assays supported the MIC data, with no change in penA expression levels observed between controls and strains treated with ceftazidime, carbenicillin, imipenem, penicillin G, or high salt (data not shown).
Discussion
The data presented in this study employing isogenetic mutants in a defined genetic background confirm that PenA is a major β-lactam resistance factor in B. pseudomallei. Increased expression of penA conferred increased resistance levels to the majority of β-lactams tested. Conversely, penA deletion resulted in susceptibility to all β-lactams tested. Furthermore, clinically observed penA mutations were responsible for the altered β-lactam substrate spectrum of the enzyme and consequently the new resistance profile. Point mutations near the active site are the most common reason for substrate profile shifting because they accommodate different side chains of various β-lactams by either changing the active site steric properties or locations of the actual active residues (Drawz and Bonomo, 2010). Complete shifts in substrate profiles have been previously documented, such as with a mutant gram negative TEM-1 β-lactamase showing increased ceftazidime hydrolysis but decreased activity against ampicillin (Venkatachalam et al., 1994). The good news is that the B. pseudomallei C69Y PenA mutation sensitizes the cell to other β-lactams thus possibly enabling new therapeutic strategies. Additionally, our studies showed that of all β-lactams tested meropenem is the only β-lactam not affected by penA mutations and overproduction, and thus the superior β-lactam antibiotic for melioidosis treatment with regard to potential emergence of PenA β-lactamase mutants. Unfortunately, current costs limit use of meropenem in many clinical settings, especially resource poor areas of the world (Cheng et al., 2004).
The mutants created and analyzed in this study did not precisely mimic previously documented mutants. In all cases, our strains had an equal or lower resistance level. This may simply be due to variations in MIC methodologies, which can change the measurements significantly (Wuthiekanun et al., 2005, or B. pseudomallei strain variability that may affect resistance patterns (Thibault et al., 2004). Using the S72F clinical mutant as an example, two groups determined the MIC of the parental strain 392a and its mutant, 392f. The first group showed a 16-fold increase in amoxicillin–clavulanic acid resistance (Godfrey et al., 1991) but the other group only showed a two-fold increase (Tribuddharat et al., 2003). These numbers are significantly different from one another. The susceptibilities observed with our isogenetic Bp82 derivatives fell between the two previously reported set of numbers at 4- to 5- fold. Another potential cause for inconsistency was that some of the published experiments cloned the mutant penA gene and expressed it in an unrelated bacterium, E. coli, from a high-copy number plasmid (Ho et al., 2002; Tribuddharat et al., 2003). Our data showed that increased expression of penA from a single-copy, chromosomally inserted expression element with a strong inducible promoter can change the profile from susceptible to resistant. A high-copy number plasmid will obviously lead to higher gene expression and consequently high resistance levels. Additionally, other cellular factors, including outer membrane permeability, can affect the efficacy of resistance mechanisms. For the P167S mutation, Tribuddharat et al. (2003) showed a 16-fold increase in ceftazidime resistance in B. pseudomallei, but only a two-fold change in an E. coli strain expressing recombinant penA genes.
Besides playing a crucial role in the export of virulence factors in many bacteria (De Buck et al., 2008), the TAT system has previously been shown to be required for the export of β-lactamases in Mycobacterium smegmatis (McDonough et al., 2005). Through deletion of the tatABC operon and mutation of a crucial arginine residue of the putative TAT-signal sequence of B. pseudomallei PenA, we showed that PenA is indeed a TAT secreted enzyme. The exact cellular location of PenA β-lactamase could not be pinpointed. Various periplasmic extraction techniques failed to localize the enzyme to the periplasm but rather indicated that it is localized in the spheroplastic compartments of the cell which encompasses the cytosol and the membranes. Since a secreted enzyme is unlikely to be localized in the cytosol this experimental evidence points to the fact that PenA may be a membrane-associated enzyme. This is in agreement with earlier findings by Livermore et al. (1987) who demonstrated that after sonication and centrifugation, the vast majority of β-lactamase activity was present in the membrane fraction. Membrane association of β-lactamases is not common but has been documented. The first account of a membrane-associated β-lactamase in Gram negative bacteria was from work with Moraxella catarrhalis (Bootsma et al., 1999). In this case it was hypothesized that the gene was a lipoprotein of Gram positive origin, as membrane association in Gram positive bacteria had been previously observed. This is unlikely the case for PenA because penA has 70% GC content, comparable to the overall genome (68%). How B. pseudomallei PenA could become membrane associated is unclear since there is no evidence of the enzyme being a lipoprotein.
Further evidence for TAT secretion was obtained by analyzing the PenA processing. Tullman-Ercek et al. (2007) describe the processing of TAT-signal sequences and the cleavage of their amino-termini upon passing through the inner membrane, using MdoD as an example. The authors describe a hydrophobic region before the processing site determined to be an AXA motif. A comparative analysis of the amino-termini of MdoD (MDRRRFKGSMAMAAVCGTSGASLFSQAAFA) and PenA (MNHSPLRRSLLVAAISTPLIGACAOLRGQAKNVAAA) at http://expasy.org/tools/protscale.html using the Kyte and Doolittle hydrophobicity algorithm revealed that the two sequences exhibited a similar hydrophobicity profile and comparable predicted processing sites (underlined AFA in MdoD and AAA in PenA). Using this information, we calculated that the molecular mass of PenA changes from 31 to 27 kDa after processing. Bands corresponding to these sizes were seen observed using Western blot analysis (Figure 2).
Some β-lactamase genes require LysR regulatory factors for expression, with or without a β-lactam inducer. Trépanier et al. (1997) showed that the penA gene from B. cepacia was regulated by a LysR-family regulator encoded by the divergently transcribed penR gene when expressed from plasmids in E. coli. PenA β-lactamase gene expression was not only regulated by PenR but was also inducible in the presence of imipenem in this system. A gene, BPSS0948, homologous to B. cepacia penR is found in B. pseudomallei downstream of penA (Figure 1). An additional LysR-family regulator encoding gene, BPSS0944, is located upstream of penA, but it has less sequence homology to penR and is separated from penA by a putative peptidase gene. Our analyses involving gene deletions, induction experiments, as well as growing cells under conditions such as salt stress (Pumirat et al., 2009 that may trigger β-lactamase induction showed that neither the LysR-type regulators BPSS0944 and BPSS0948 nor the putative peptidase encoded by BPSS0945 are involved in regulation of penA expression in B. pseudomallei strain 1026b, at least not under the experimental conditions employed during these studies. Although many chromosomal β-lactamases are inducible, others are constitutively expressed (Neu and Chin, 1985; Jacoby and Bush, 2005). When multiple β-lactamases are present in a bacterial strain, they can be subject to complex regulation, including co-regulation with penicillin binding protein 2 (Hackbarth and Chambers, 1993; Naas et al., 1995). In this context it is noteworthy that by analysis of clinical strains obtained from patients that failed ceftazidime therapy and studies of recreated 1026b-based mutants we and others recently identified deletion of a B. pseudomallei PBP3 homolog as a mechanism causing high-level ceftazidime resistance (Chantratita et al., unpublished observations).
Definition of the molecular basis of resistance mechanisms for clinically significant β-lactams forms the basis for design of diagnostic tools that allow rapid detection of emergence of resistance and thus redirection (clinical settings) or initiation (biodefense) of proper melioidosis therapy or prophylaxis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grant AI065357 from the National Institutes of Health-National Institute for Allergies and Infectious Diseases to Herbert P. Schweizer and Michael L. Vasil. We thank Lily Trunck for constructing pUC18T-mini-Tn7T-Km-LAC. The Veterans Affairs Career Development Program (Krisztina M. Papp-Wallace), the Veterans Affairs Merit Review Program (Robert A. Bonomo), the National Institutes of Health (RO1 AI063517-01 to Robert A. Bonomo), and the Veterans Integrated Service Network 10 Geriatric Research, Education, and Clinical Center (VISN 10 GRECC) also supported this work. BAL30072 was provided by Basilea Pharmaceutica International, Ltd., Basel, Switzerland.
References
Ames, G. F., Prody, C., and Kustu, S. (1984). Simple, rapid, and quantitative release of periplasmic proteins by chloroform. J. Bacteriol. 160, 1181–1183.
Bootsma, H. J., Aerts, P. C., Posthuma, G., Harmsen, T., Verhoef, J., van Dijk, H., and Mooi, F. R. (1999). Moraxella (Branhamella) catarrhalis BRO β-lactamase: a lipoprotein of Gram-positive origin? J. Bacteriol. 181, 5090–5093.
Cheng, A. C., and Currie, B. J. (2005). Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18, 383–416.
Cheng, A. C., Fisher, D. A., Anstey, N. M., Stephens, D. P., Jacups, S. P., and Currie, B. J. (2004). Outcomes of patients with melioidosis treated with meropenem. Antimicrob. Agents Chemother. 48, 1763–1765.
Cheung, T. K., Ho, P. L., Woo, P. C., Yuen, K. Y., and Chau, P. Y. (2002). Cloning and expression of class A beta-lactamase gene blaA(BPS) in Burkholderia pseudomallei. Antimicrob. Agents Chemother. 46, 1132–1135.
Choi, K.-H., DeShazer, D., and Schweizer, H. P. (2006). mini-Tn7 insertion in bacteria with multiple glmS-linked attTn7 sites: example Burkholderia mallei ATCC 23344. Nat. Protoc. 1, 162–169.
Choi, K.-H., Gaynor, J. B., White, K. G., López, C., Bosio, C. M., Karkhoff-Schweizer, R. R., and Schweizer, H. P. (2005). A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2, 443–448.
Choi, K.-H., Mima, T., Casart, Y., Rholl, D., Kumar, A., Beacham, I. R., and Schweizer, H. P. (2008). Genetic tools for select agent compliant manipulation of Burkholderia pseudomallei. Appl. Environ. Microbiol. 74, 1064–1075.
Clinical and Laboratory Standards Institute. (2010). Performance Standards for Antimicrobial Susceptibility testing: Twentieth Informal Supplement M100-S20. Wayne, PA.
Currie, B. J., Dance, D. A. B., and Cheng, A. C. (2008). The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans. R. Soc. Trop. Med. Hyg. 102, S1–S4.
De Buck, E., Lammertyn, E., and Anné, J. (2008). The importance of the twin-arginine translocation pathway for bacterial virulence. Trends Microbiol. 19, 442–453.
DeShazer, D., Brett, P., Carlyon, R., and Woods, D. (1997). Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J. Bacteriol. 179, 2116–2125.
Drawz, S. M., and Bonomo, R. A. (2010). Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 23, 160–201.
Godfrey, A. J., Wong, S., Dance, D. A., Chaowagul, W., and Bryan, L. E. (1991). Pseudomonas pseudomallei resistance to beta-lactam antibiotics due to alterations in the chromosomally encoded beta-lactamase. Antimicrob. Agents Chemother. 35, 1635–1640.
Hackbarth, C. J., and Chambers, H. F. (1993). blaI and blaR1 regulate beta-lactamase and PBP 2a production in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 37, 1144–1149.
Ho, P. L., Cheung, T. K., Yam, W. C., and Yuen, K. Y. (2002). Characterization of a laboratory-generated variant of BPS beta-lactamase from Burkholderia pseudomallei that hydrolyses ceftazidime. J. Antimicrob. Chemother. 50, 723–726.
Holden, M. T. G., Titball, R. W., Peacock, S. J., Cerdeño-Tárraga, A. M., Atkins, T., Crossman, L. C., Pitt, T., Churcher, C., Mungall, K., Bentley, S. D., Sebaihia, M., Thomson, N. R., Bason, N., Beacham, I. R., Brooks, K., Brown, K. A., Brown, N. F., Challis, G. L., Cherevach, I., Chillingworth, T., Cronin, A., Crossett, B., Davis, P., DeShazer, D., Feltwell, T., Fraser, A., Hance, Z., Hauser, H., Holroyd, S., Jagels, K., Keith, K. E., Maddison, M., Moule, S., Price, C., Quail, M. A., Rabbinowitsch, E., Rutherford, K., Sanders, M., Simmonds, M., Songsivilai, S., Stevens, K., Tumapa, S., Vesaratchavest, M., Whitehead, S., Yeats, C., Barrell, B. G., Oyston, P. C. F., and Parkhill, J. (2004). Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc. Natl. Acad. Sci. U.S.A. 101, 14240–14245.
Imperi, F., Tiburzi, F., and Visca, P. (2009). Molecular basis of pyoverdine siderophore recycling in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 106, 20440–20445.
Jacoby, G. A., and Bush, K. (2005). “Beta-lactam resistance in the 21st century,” in Frontiers in Antimicrobial Resistance: A Tribute to Stuart B. Levy, eds D. G. White, M. N. Alekshun, and P. F. McDermot (Washington, DC: ASM Press), 53–65.
Keith, K. E., Oyston, P. C., Crossett, B., Fairweather, N. F., Titball, R. W., Walsh, T. R., and Brown, K. A. (2005). Functional characterization of OXA-57, a class D beta-lactamase from Burkholderia pseudomallei. Antimicrob. Agents Chemother. 49, 1639–1641.
Livermore, D. M., Chau, P. Y., Wong, A. I., and Leung, Y. K. (1987). Beta-lactamase of Pseudomonas pseudomallei and its contribution to antibiotic resistance. J. Antimicrob. Chemother. 20, 313–321.
López, C. M., Rholl, D. A., Trunck, L. A., and Schweizer, H. P. (2009). Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl. Environ. Microbiol. 75, 6496–6503.
McDonough, J. A., Hacker, K. E., Flores, A. R., Pavelka, M. S. Jr., and Braunstein, M. (2005). The twin-arginine translocation pathway of Mycobacterium smegmatis is functional and required for the export of mycobacterial beta-lactamases. J. Bacteriol. 187, 7667–7679.
Mima, T., and Schweizer, H. P. (2010). The BpeAB-OprB efflux pump of Burkholderia pseudomallei 1026b does not play a role in quorum sensing, virulence factor production, or extrusion of aminoglycosides but is a broad-spectrum drug efflux system. Antimicrob. Agents Chemother. 54, 3113–3120.
Naas, T., Livermore, D. M., and Nordmann, P. (1995). Characterization of an LysR family protein, SmeR from Serratia marcescens S6, its effect on expression of the carbapenem-hydrolyzing beta-lactamase Sme-1, and comparison of this regulator with other beta-lactamase regulators. Antimicrob. Agents Chemother. 39, 629–637.
Neu, H. C., and Chin, N. X. (1985). A perspective on the present contribution of beta-lactamases to bacterial resistance with particular reference to induction of beta-lactamase and its clinical significanceinduction of beta-lactamase and its clinical significance. Chemioterapia 4, 63–70.
Peacock, S. J., Schweizer, H. P., Dance, D. A. B., Smith, T. L., Gee, J. E., Wuthiekanun, V., DeShazer, D., Steinmetz, I., Tan, P., and Currie, B. J. (2008). Consensus guidelines on the management of accidental laboratory exposure to Burkholderia pseudomallei and Burkholderia mallei Em. Infect. Dis. 14, e2.
Propst, K. L., Mima, T., Choi, K.-H., Dow, S. W., and Schweizer, H. P. (2010). A Burkholderia pseudomallei ΔpurM mutant is avirulent in immune competent and immune deficient animals: candidate strain for exclusion from select agent lists. Infect. Immun. 78, 3136–3143.
Pumirat, P., Saetun, P., Sinchaikul, S., Chen, S. T., Korbsrisate, S., and Thongboonkerd, V. (2009). Altered secretome of Burkholderia pseudomallei induced by salt stress. Biochim. Biophys. Acta 1794, 898–904.
Sam, I. C., See, K. H., and Puthucheary, S. D. (2009). Variations in ceftazidime and amoxicillin-clavulanate susceptibilities within a clonal infection of Burkholderia pseudomallei. J. Clin. Microbiol. 47, 1556–1558.
Stanley, N. R., Palmer, T., and Berks, B. C. (2000). The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J. Biol. Chem. 275, 11591–11596.
Thibault, F. M., Hernandez, E., Vidal, D. R., Girardet, M., and Cavallo, J. D. (2004). Antibiotic susceptibility of 65 isolates of Burkholderia pseudomallei and Burkholderia mallei to 35 antimicrobial agents. J. Antimicrob. Chemother. 54, 1134–1138.
Trépanier, S., Prince, A., and Huletzky, A. (1997). Characterization of the penA and penR genes of Burkholderia cepacia 249 which encode the chromosomal class A penicillinase and its LysR-type transcriptional regulator. Antimicrob. Agents Chemother. 41, 2399–2405.
Tribuddharat, C., Moore, R. A., Baker, P., and Woods, D. E. (2003). Burkholderia pseudomallei class a beta-lactamase mutations that confer selective resistance against ceftazidime or clavulanic acid inhibition. Antimicrob. Agents Chemother. 47, 2082–2087.
Tullman-Ercek, D., DeLisa, M. P., Kawarasaki, Y., Iranpour, P., Ribnicky, B., Palmer, T., and Georgiou, G. (2007). Export pathway selectivity of Escherichia coli twin arginine translocation signal peptides. J. Biol. Chem. 282, 8309–8316.
Venkatachalam, K. V., Huang, W., LaRocco, M., and Palzkill, T. (1994). Characterization of TEM-1 beta-lactamase mutants from positions 238 to 241 with increased catalytic efficiency for ceftazidime. J. Biol. Chem. 269, 23444–23450.
West, S. E. H., Schweizer, H. P., Dall, C., Sample, A. K., and Runyen-Janecky, L. J. (1994). Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and the sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 128, 81–86.
White, N. J., Dance, D. A., Chaowagul, W., Wattanagoon, Y., Wuthiekanun, V., and Pitakwatchara, N. (1989). Halving of mortality of severe melioidosis by ceftazidime. Lancet 2, 697–701.
Wiersinga, W. J., van der Poll, T., White, N. J., Day, N. P., and Peacock, S. J. (2006). Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4, 272–282.
Wuthiekanun, V., Cheng, A. C., Chierakul, W., Amornchai, P., Limmathurotsakul, D., Chaowagul, W., Simpson, A. J. H., Short, J. M., Wongsuvan, G., Maharjan, B., White, N. J., and Peacock, S. J. (2005). Trimethoprim/sulfamethoxazole resistance in clinical isolates of Burkholderia pseudomallei. J. Antimicrob. Chemother. 55, 1029–1031.
Keywords: Burkholderia pseudomallei, melioidosis, antibiotic resistance, β-lactams, β-lactamase, TAT secretion
Citation: Rholl DA, Papp-Wallace KM, Tomaras AP, Vasil ML, Bonomo RA and Schweizer HP (2011) Molecular investigations of PenA-mediated β-lactam resistance in Burkholderia pseudomallei. Front. Microbio. 2:139. doi: 10.3389/fmicb.2011.00139
Received: 05 May 2011;
Paper pending published: 01 June 2011;
Accepted: 14 June 2011;
Published online: 01 July 2011.
Edited by:
Alfredo G. Torres, University of Texas Medical Branch, USAReviewed by:
David Aucoin, University of Nevada School of Medicine, USANarisara Chantratita, Mahidol University, Thailand
Copyright: © 2011 Rholl, Papp-Wallace, Tomaras, Vasil, Bonomo and Schweizer. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Herbert P. Schweizer, Department of Microbiology, Immunology and Pathology, Colorado State University, Infectious Disease Research Center at Foothills Campus, 0922 Campus Delivery, Fort Collins, CO 80523, USA.