
 David Yin-Wei Lin
David Yin-Wei Lin
- Department of Biological Sciences, Purdue University, West Lafayette, IN, USA
Ubiquitination is a post-translational modification in which one or more 76 amino acid polypeptide ubiquitin molecules are covalently linked to the lysine residues of target proteins. Ubiquitination is the main pathway for protein degradation that governs a variety of eukaryotic cellular processes, including the cell-cycle, vesicle trafficking, antigen presentation, and signal transduction. Not surprisingly, aberrations in the system have been implicated in the pathogenesis of many diseases including inflammatory and neurodegenerative disorders. Recent studies have revealed that viruses and bacterial pathogens exploit the host ubiquitination pathways to gain entry and to aid their survival/replication inside host cells. This review will summarize recent developments in understanding the biochemical and structural mechanisms utilized by bacterial pathogens to interact with the host ubiquitination pathways.
Introduction
Discovery of the ubiquitination system follows an elegant classical biochemistry story, culminating with the award of the 2004 Nobel Prize in Chemistry to three contributors: Dr. Avram Hershko, Dr. Aaron Ciechanover, and Dr. Irwin Rose. The story started with a search for a non-lysosomal pathway for protein degradation. The lysosome was initially given credence for the responsibility of protein turnover in a cell. However, several aspects of the lysosomal mode of proteolysis did not fit data that was emerging, and thus a search for an alternative mechanism began. The breakthrough came with the use of crude extracts from reticulocytes: immature red blood cells which lack lysosomes. Protein degradation was shown to require two complementing fractions resolved from the crude extract: one containing protease activity and the other containing a small heat-stable protein, ATP-dependent proteolytic factor 1 (APF-1), which stimulated proteolysis (Ciehanover et al., 1978). Studying the mechanism of action of APF-1 led to the discovery that this protein is in fact the polypeptide ubiquitin (Goldstein et al., 1975; Ciehanover et al., 1978; Wilkinson et al., 1980). Additional study led to the elucidation of the multi-enzyme cascade which allows ubiquitin modification of proteins, and which firmly established the key steps involved in the ubiquitin–proteasome system (UPS; Ciechanover et al., 1981; Hershko et al., 1983; Reiss et al., 1989).
The UPS consists of three classes of enzymes known as ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin–protein ligases (E3; Figure 1). The ubiquitination reaction is initiated when ubiquitin is activated by the ubiquitin-activating enzyme, E1. A thioester bond forms between the active cysteine residue of E1 and the C-terminus of ubiquitin in an ATP-dependent reaction. Following ubiquitin activation, activated ubiquitin is transferred to a ubiquitin-conjugating enzyme, E2 (also known as Ubc), in another ATP-dependent reaction. Finally, with the help of a ubiquitin–protein ligase, E3, a covalent bond is formed between the C-terminus of ubiquitin and the ε-amino group of a lysine residue on the protein substrate. One round of ubiquitination produces a monoubiquitinated protein. Typically, successive rounds occur, producing a protein with a polyubiquitinated chain. Each additional ubiquitin moiety is attached via the internal lysine of the previously conjugated ubiquitin molecule. There are seven lysine residues within the ubiquitin molecule: K6, K11, K27, K29, K33, K48, and K63, and all seven lysines can be used to link ubiquitin subunits into polyubiquitin chains in vitro (Kim et al., 2007). In vivo, the specific lysine modification used in the polyubiquitin chain translates into the fate of the protein (Figure 1). The formation of a K48-linked polyubiquitin chain usually leads to the substrate being targeted for proteasome degradation (Hough et al., 1986; Ganoth et al., 1988). The formation of a K63-linked polyubiquitin chain provides the substrate with the ability to act as a scaffold, allowing it to assemble signaling complexes and regulate protein localization, protein kinase activation, DNA repair, or transcription through proteasome-independent mechanisms (Pickart, 2001; Weissman, 2001).
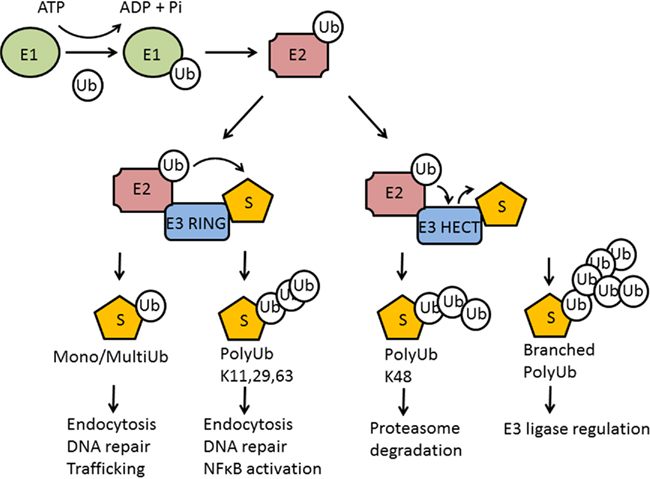
Figure 1. Ubiquitin pathway. Free ubiquitin (Ub) is activated by a ubiquitin-activating enzyme (E1), using ATP to form a complex with ubiquitin. Ubiquitin is transferred from E1 to a ubiquitin-conjugating enzyme (E2). E2 then binds to an E3 molecule, the ubiquitin–protein ligase, which allows the polymerization of one or more ubiquitin molecules on a substrate (S) molecule. One or several ubiquitin molecules can be conjugated to a substrate, determining its cellular fate.
The E1 ubiquitin-activating enzymes are well conserved in eukaryotes (Hershko et al., 1983; Chiu et al., 2007). For many years it was believed that only one E1 existed in eukaryotic cells (Zacksenhaus and Sheinin, 1990; Mcgrath et al., 1991). Recently, however, an E1-like protein, termed E1–L2, was also found to activate ubiquitin (Chiu et al., 2007; Jin et al., 2007; Pelzer et al., 2007). Unlike the E1 family, there are many more members of the E2 and E3 family of proteins, with the E3 family being the largest (Hershko and Ciechanover, 1998). E3s play pivotal roles in defining the specificity of target proteins to be ubiquitinated. There are two major families of E3 ubiquitin ligases; the homologous to E6–AP carboxyl terminus (HECT) domain family and the really interesting new gene (RING) family. The U-box family possess a finger domain that is a close relative of the RING motif and are therefore often categorized as members of the RING family. Each family is differentiated by their distinct sequences, structures, and catalytic properties (Huibregtse et al., 1995; Lorick et al., 1999; Pickart, 2001). The HECT family E3 ubiquitin ligases contain a 350-residue region that maintains a strictly conserved cysteine residue located approximately 35 residues from the C-terminus (Huibregtse et al., 1995; Schwarz et al., 1998). This cysteine forms an essential thioester intermediate during catalysis (Huibregtse et al., 1995). The RING-finger family E3 ubiquitin ligases feature a set of cysteine and histidine residues that have a distinctive spacing to stabilize their globular conformation by interacting with two zinc ions (Lorick et al., 1999). Whereas RING ligases function as molecular scaffolds to bring E2 and the substrates into close proximity, HECT E3s participate directly in the chemistry of ubiquitination by accepting ubiquitin from an E2 enzyme, in the form of a ubiquitin–thioester intermediate, and thus directly catalyze protein ubiquitination (Huibregtse et al., 1995; Figure 1). Both HECT and RING proteins can be modular, single proteins, or multi-protein complexes, such as the Skp, Cullin, F-box (SCF)-containing complexes (Ardley and Robinson, 2005).
Ubiquitin modification of proteins is a process restricted to eukaryotes. Consequently, all of the enzymes that act on ubiquitin were originally believed to be confined to eukaryotes. However, given the importance of ubiquitination to such a large number of cellular processes required for normal cell function, it is perhaps no surprise that pathogens have developed mechanisms to manipulate the host ubiquitin pathway to their advantage. Viruses are known to modulate the host antigen presentation and immune responses by modulating the ubiquitination system (Shackelford and Pagano, 2005; Barry and Fruh, 2006; Gao and Luo, 2006; Lindner, 2007). It is now recognized that bacterial pathogens also manipulate the host ubiquitination system.
Ubiquitin Modulating Effectors Proteins
Many Gram-negative bacteria encode sophisticated protein secretion and translocation systems to deliver bacterial virulence determinants across both the bacterial and host cell membranes into the infected cells. These bacterial virulence factors are often referred to as effectors due to their ability to exploit host cellular functions. Two of the most studied transport apparatuses are the type III and type IV secretion and translocation systems (Cornelis, 2006; Hayes et al., 2010). The biochemical and cellular functions of these effector proteins are subjects of intense study since they are likely to shed light on the molecular mechanisms utilized by bacterial pathogens to mount successful infections. In the last few years, it is becoming increasingly evident that many of these effectors interact with eukaryotic ubiquitination pathways to exploit host functions regulated by ubiquitination. We designate this group of effectors as ubiquitin modulating effectors (UME) Proteins.
E2-Targeting UME
So far, only one example of a bacterial protein targeting a ubiquitin-conjugating/E2 enzyme exists: OspG in Shigella flexneri (Kim et al., 2005). Shigella is the etiological agent of human bacillary dysentery. Virulence is dependent on a type three secretion system (T3SS) encoded on a 200-kb plasmid (Buchrieser et al., 2000). Effectors encoded on the plasmid or on the chromosome are secreted and translocated by this T3SS to enable invasion of the colonic epithelium. Shigella is then able to induce the intense inflammatory response that is characteristic of Shigella infection.
OspG is a 196 residue protein secreted by the Shigella T3SS. A yeast two hybrid screen identified several E2 proteins as binding partners of OspG. This included UbcH5b, a component of the SCFβ-TrCP E3 complex, which promotes ubiquitination of phospho-IκBα and its subsequent degradation by the proteasome (Karin and Ben-Neriah, 2000). IκBα binds the transcription factor NFκB, sequestering it in an inactive state in the cytoplasm. Phosphorylation of IκBα, by IκB kinase (IKK), allows IκBα to be ubiquitinated and targeted for proteasome degradation. This releases NFκB, allowing its translocation to the cell nucleus and the regulation of genes involved in inflammation, immunity, cell survival, and other pathways. OspG was shown to delay the degradation of phospho-IκBα, and consequently reduce the transcription of NFκB-regulated genes (Kim et al., 2005). In vivo, an ospG mutant enhances inflammatory responses. Thus Shigella, by using OspG to bind host E2 proteins, appears to reduce host inflammatory responses at early time points during infection. This may allow Shigella time to establish itself inside the host (Figure 2). An avirulent Salmonella strain has also been shown to block SCFβ-TrCP function and prevent ubiquitination of IκBα (Neish et al., 2000). The authors hypothesized that this may be one of the mechanisms by which gastrointestinal microflora are tolerated by the host. Such a mechanism is therefore likely to be used by other bacteria to promote their colonization of a host. As OspG was also shown to bind other E2 proteins, in addition to UbcH5b, there may be further unknown consequences to the ubiquitination processes during Shigella infection.
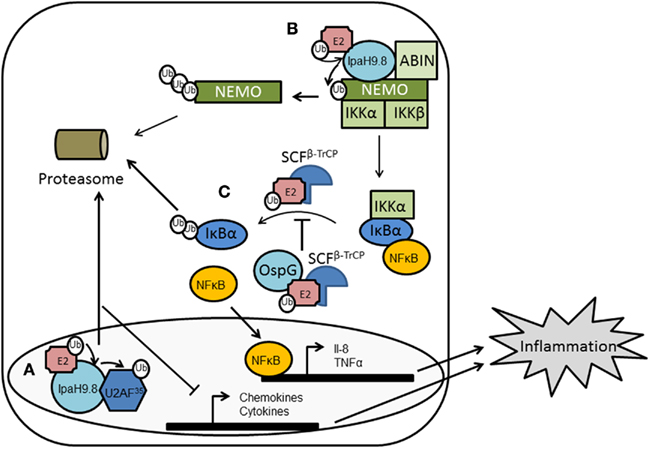
Figure 2. Manipulation of host ubiquitin pathways by Shigella. IpaH9.8 acts as an E3 ligase. (A) Binding and ubiquitination of U2AF35 by IpaH9.8 targets this splicing factor for proteasomal degradation reducing the production of chemokines and cytokines. (B) Binding to NEMO and ABIN allows Ipa9.8 to ubiquitinate NEMO and target it for proteasomal degradation. A consequence of this is the disruption of NF-κB signaling, which is also likely to reduce the inflammatory response during Shigella infection. (C) OspG can bind the E2 UbcH5b, a component of the E3 complex SCFβ-TrCP, thus inhibiting ubiquitination of phospho-IκBα and maintaining the IκBα/NFκB complex. Again, NFκB gene transcription is reduced, decreasing the inflammatory response.
E3-Targeting UME
Salmonella
Salmonella utilizes two T3SS to establish itself inside host cells; the first, encoded by Salmonella Pathogenicity Island-1 (SPI-1), facilitates invasion of non-phagocytic cells, while the second, encoded by SPI-2, enables intracellular survival and replication (Lostroh and Lee, 2001; Knodler and Steele-Mortimer, 2003). Multiple effectors are translocated through each T3SS to exploit host cell functions, and a major focus of Salmonella research is identifying and characterizing the function of each effector.
Salmonella SopA was initially identified as a SPI-1 effector in Salmonella dublin with a key role in the induction of enteritis (Wood et al., 2000; Zhang et al., 2002). However, the biochemical mechanism by which SopA induced this effect was unknown. The first clue suggesting SopA exploits host ubiquitination pathways came from a yeast two hybrid screen searching for host cellular proteins that interact with SopA. The yeast two hybrid assay indicated SopA interacts with human RMA1 (HsRMA1), a membrane-bound RING ubiquitin E3 ligase (Zhang et al., 2005). Bacterial SopA was shown to be ubiquitinated and degraded by the HsRMA1-mediated ubiquitination pathway in vitro and in a cell culture model (Zhang et al., 2005). One consequence of SopA ubiquitination and degradation by the proteasome appeared to be the escape of Salmonella into the cytoplasm of HeLa cells, where it could rapidly replicate. However, it is unclear whether this occurs in vivo during infection since Salmonella remains mostly within a Salmonella-containing vacuole (SCV; Knodler and Steele-Mortimer, 2003).
The E3 ubiquitin ligase activity of SopA itself was discovered accidentally when a mysterious banding pattern was observed in a routine ubiquitination assay in the absence of HsRMA1 (Zhang et al., 2006). This showed SopA possessed auto-ubiquitination activity: a key feature of most E3 ligases. In vitro, the ubiquitination kinetics of SopA is similar to other known E3 ubiquitin ligases (Coscoy et al., 2001; Lostroh and Lee, 2001; Matsuda et al., 2001; You and Pickart, 2001; Yamanaka et al., 2003).
Amino acid comparisons of SopA with HECT domain and RING-finger E3s did not reveal significant primary sequence similarities. However, it was determined that SopA has a cysteine residue (C753) 30 residues away from the C-terminus. Members of the HECT family E3 ubiquitin ligases feature a strictly conserved catalytic cysteine residue positioned approximately 35 residues from the C-terminus (Huibregtse et al., 1995; Scheffner et al., 1995; Dietrich et al., 1997; Hatakeyama et al., 1997; Yamamoto et al., 1997; Bates and Vierstra, 1999; Yanase and Ishi, 1999). This indicated SopA may also belong to this family. Biochemical assays confirmed that Cys753 is the active site of SopA for E3 ubiquitin ligase activity (Zhang et al., 2006). The crystal structure of SopA further confirmed it as a mimic of eukaryotic HECT E3s (Diao et al., 2008). SopA shares structural similarity to HECT E3s, including the sequence of the active site loop, the bilobal architecture, and the conformational flexibility of the C lobe. However, it also possesses many unique characteristics (Figure 3). For example, the folding of the N and C lobes is different and the putative substrate binding domains are in closer proximity than most eukaryotic E3 HECT ligases, which may indicate SopA in fact belongs to a novel class of E3 ligases; only time will tell.
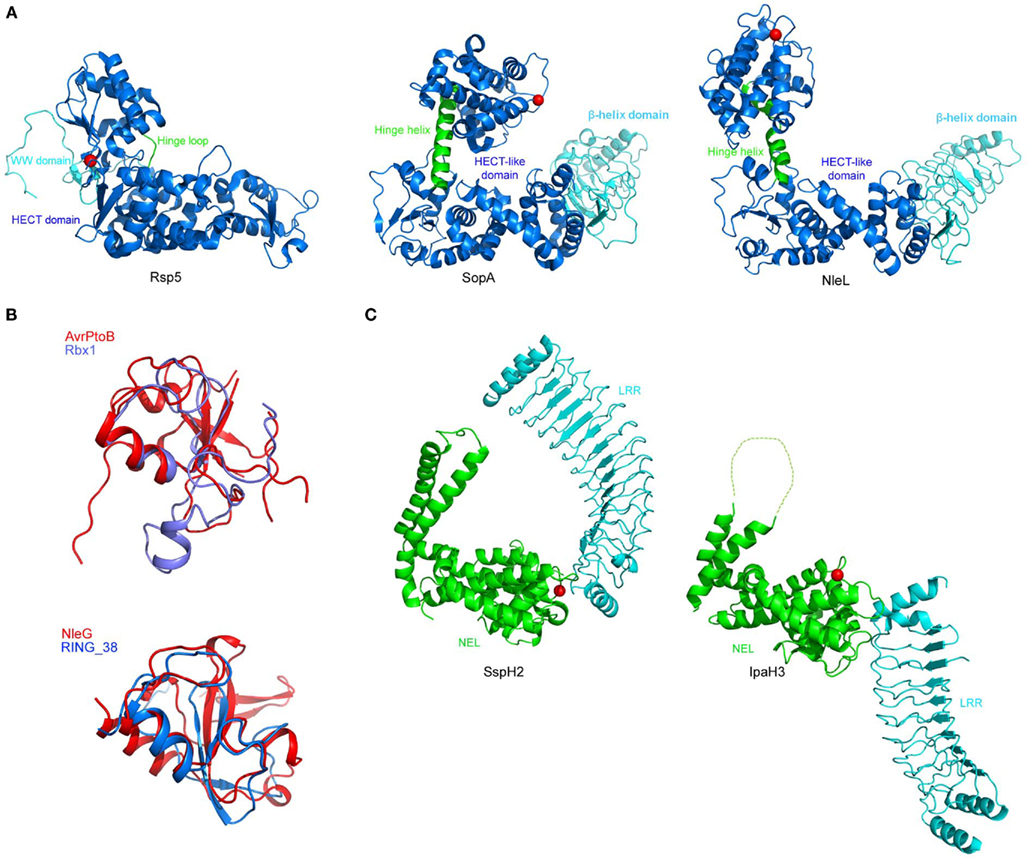
Figure 3. Structures of bacterial E3 ligases. Structural mimicry of effector proteins to E3 ubiquitin ligases. (A) Left, a representative structure of eukaryotic HECT E3 ubiquitin ligase, Rsp5 (PDB 3OLM). Middle, effector protein SopA from Salmonella (PDB 2QYU). Right, effector protein NleL from E. coli O157:H7 (PDB 3NB2). HECT or HECT-like domains are colored in blue with the catalytic cysteines shown in red spheres. The two lobes of the eukaryotic HECT domains are connected by a flexible hinge loop (green); whereas, in SopA and NleL, the two lobes are connected by a flexible hinge helix (green). (B) Superposition of RING-finger and U-box domains of AvrPtoB (PDB 2FD4) and NleG (PDB 2KKX) to eukaryotic RING-finger/U-box domains from Rbx1 (PDB 1LDJ) and RING-finger 38 protein (PDB 1X4J), respectively. (C) Novel E3 ligases. Shown are Salmonella effector SspH2 (PDB 3G06) and Shigella effector IpaH3 (PDB 3CVR). NEL domains are colored in green with the catalytic cysteines shown in red spheres. LRR domains are colored in cyan. Broken line indicated disordered loop regions. Interdomain flexibility between the NEL and LRR domains is apparent between the two structures.
SopA was implicated in the induction of enteritis by Salmonella, in part by stimulating efficient polymorphonuclear leukocyte (PMN) transepithelial migration (Wood et al., 2000). The catalytically inactive SopAC753S mutant strain was found to induce significantly less PMN transepithelial migration as compared to wild-type Salmonella (Zhang et al., 2006). Thus, SopA E3 ligase activity appears to play a role in SopA-mediated enteritis. The SopA substrate(s) remain unknown. It has been postulated that both host proteins and bacterial T3SS translocated effectors are potential targets of SopA. Host proteins may be directly involved in Salmonella-induced inflammatory responses. Alternatively, they may regulate the activity/level of Salmonella effectors that are involved in inducing such inflammation (Zhang et al., 2006).
SopA is not the only ubiquitin E3 ligase Salmonella possesses (Figure 4). It has three others: Salmonella leucine-rich repeat protein (SlrP), Salmonella secreted protein H1 (SspH1) and SspH2 (Rohde et al., 2007). SlrP and SspH2 are found in most Salmonella enterica strains, while SspH1 is limited to serovar Typhimurium strain 14028s (Miao et al., 1999). SlrP, SspH1, and SspH2 were first identified as belonging to a group of T3SS effectors from various human, animal, and plant pathogens that contain LPX repeats; a subtype of the leucine-rich repeat (LRR) superfamily of protein binding domains (Miao et al., 1999; Tsolis et al., 1999). IpaH from Shigella (Venkatesan et al., 1991) and YopM from Yersinia (Boland et al., 1996) also belong to this group. It was assumed LRRs were required for protein–protein interactions, but the precise functions were unclear. SspH1 and SspH2 had together been shown to reduce mortality in calves due to reduced intestinal lesions, although single mutants were not attenuated in this or cell culture models of invasion or replication (Miao et al., 1999). SlrP was required for virulence in mice but not bovine models of infection (Tsolis et al., 1999).
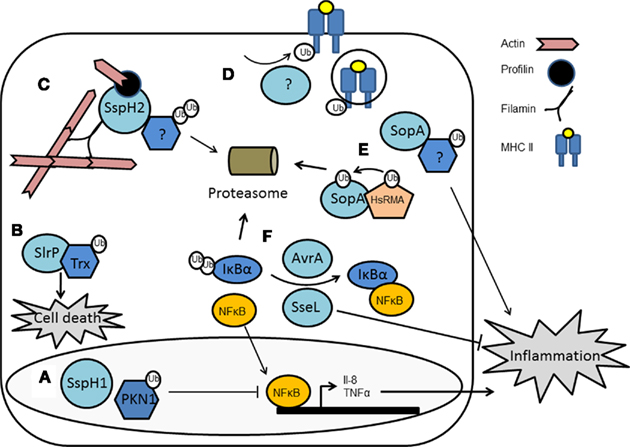
Figure 4. Manipulation of host ubiquitin pathways by Salmonella enterica. Four effector proteins have been identified to be translocated into host cells where they can act as ubiquitin E3 ligases. (A) SspH1 ubiquitinates PKN1 in the cell nucleus which inhibits expression of NFκB-regulated genes. (B) SlrP targets thioredoxin (TRX) to trigger cell death. (C) SspH2 binds profilin and filamin but the substrate of its E3 activity has not been identified. (D) An unknown effector may be responsible for the ubiquitination of MHCII receptors and their internalization, reducing CD4+ T-cell response. (E) SopA ubiquitinates an unknown substrate, the downstream effect of which is induction of inflammation. SopA is also a substrate of the host E3, HsRMA, being targeted for proteasomal degradation. (F) AvrA and SseL both act as DUBs. Both deubiquitinate IκBα and allow it to form an inhibitory complex with NFκB, thus reducing transcription of NFκB genes and therefore decreasing inflammatory responses.
The discovery that Shigella IpaH9.8 was a ubiquitin E3 ligase (Rohde et al., 2007) provided the key to the activity of SlrP, SspH1, and SspH2. Rohde et al. (2007) confirmed SspH1 as an E3 ligase; endowed with the classic activities of E3 ubiquitin ligases, i.e., the ability to remove ubiquitin from ubiquitinated UbcH5B, to autoubiquitinate, and to polyubiquitinate HA-tagged ubiquitin. SspH1 had previously been shown to localize to the nucleus of host cells (Haraga and Miller, 2006). Here, SspH1 binds the human serine/threonine kinase protein kinase PKN1 and leads to NFκB inhibition and reduction in IL-8 secretion (Haraga and Miller, 2006). PKN1 was confirmed as a substrate of SspH1 (Rohde et al., 2007), and thus SspH1 appears to use its E3 activity to down-regulate inflammatory responses.
Salmonella secreted protein H2 and SlrP were also confirmed as ubiquitin E3 ligases. SspH2 is known to localize to the cell periphery, particularly to microvilli (Miao et al., 2003; Quezada et al., 2009). This localization to areas of dynamic actin polymerization may be through the ability of SspH2 to interact with filamin and profilin (Miao et al., 2003). So far, no role has been identified for SspH2 during infection and the substrate(s) for SspH2 are unknown (Quezada et al., 2009). SlrP appears to be involved with inducing cell death. This is achieved through modulating two independent pathways, only one of which is dependent on SlrP E3 activity (Bernal-Bayard and Ramos-Morales, 2009; Bernal-Bayard et al., 2010). SlrP uses cysteine residue 546 to ubiquitinate thioredoxin (Trx), a protein antioxidant. Ubiquitination does not appear to target Trx for proteasome degradation. Instead, it reduces Trx activity, leading to increase in cell death (Bernal-Bayard and Ramos-Morales, 2009). This may be important for the escape of Salmonella should the cell become unviable either through damage or due to maximum numbers of Salmonella being achieved. It is also likely to contribute to the inflammation associated with Salmonella pathogenesis. The non-proteolytic use of ubiquitin by SlrP mimics the interaction between SspH1 and PKN1, indicating these two proteins may use a similar mechanism to manipulate host cell proteins. SspH2 meanwhile, has been shown to synthesize K48-linked poly-Ub chains, implying that its targets are destined for proteasomal destruction (Levin et al., 2010).
The crystal structure of SspH2 has been determined and reveals a two-domain arrangement (Figure 3). The N-terminal domain is composed of 12 LRRs; the C-terminus is globular, with a unique fold unrelated to known HECT or RING-finger E3 ligases, which the authors termed novel E3 ligase (NEL; Quezada et al., 2009). The LRR domain sequesters the catalytic cysteine residue of the NEL domain thereby auto-inhibiting the ligase activity. This may prevent cellular toxicity until SspH2 comes into contact with the appropriate activated-E2 and its substrate, whereupon there is a large conformational shift, reorienting the domains (Quezada et al., 2009). A similar fold and a similar auto-inhibitory mechanism appears to be used by the related Shigella IpaH proteins (Singer et al., 2008; Zhu et al., 2008), and thus likely represents a common feature in this novel class of ubiquitin E3 ligases.
In addition to belonging to a novel group of E3s, SspH2 is further unique in its binding of its cognate E2. Distinct from classical E2–E3 interactions, binding between SspH2 and UbcH5 is via both the ubiquitin and a region in the UbcH5 not previously considered important (Levin et al., 2010). A see-saw mechanism of ubiquitin chain synthesis has been proposed to explain the mechanism of ubiquitin chain formation, whereby the growing ubiquitin chain is reciprocally transferred between the E2 and E3 active sites. To grow the chain, a new E2–Ub is recruited to SspH2–Ubn. The chain is subsequently added to the E2–Ub, creating E2–UBn + 1, before the chain is passed back to SspH2.
In addition to the four E3 ligases already identified in Salmonella, there is the potential that further E3s are encoded on the Salmonella chromosome. In particular, it has been shown that Salmonella enhances the ubiquitination of human leukocyte antigen–DR (HLA–DR). This leads to increased internalization of MHC class II antigens, and subsequent lower expression on the surface of antigen presenting cells, such as DCs (Lapaque et al., 2009). Reducing the ability of cells to present antigen to CD4-restricted T-cells, the most important T-cell subset required for resolution of infection (Hess et al., 1996), may provide a mechanism by which Salmonella can influence the initiation of adaptive immune responses, and enhance its survival. Ubiquitination of HLA–DR and its subsequent internalization were dependent on the T3SS encoded by SPI-2, although no specific effector was identified to be involved (Lapaque et al., 2009). However, this does not rule out the possibility that these or other putative effectors may be involved, especially since, as highlighted above, the currently known Salmonella E3 ubiquitin ligases are mimics of host E3s and share little primary sequence similarity with their host counterparts, and in the case of SlrP, SspH1, and SspH2 share little structural homology. It will be interesting to see as more extensive research is conducted on the Salmonella proteome whether additional E3 ligases exist.
Shigella
The IpaH family of proteins were originally identified as five homologous genes carried on the virulence plasmid: IpaH1.4, IpaH2.5, IpaH4.5, IpaH7.8, and IpaH9.8 (Hartman et al., 1990; Venkatesan et al., 1991). Subsequently, it was shown that there were copies of IpaH on the Shigella chromosome which were also secreted and translocated into host cells and important to pathogenesis (Ashida et al., 2007), bringing the total to nine IpaH proteins. Each IpaH protein has a variable N-terminal domain containing six to eight LRR segments and an approximately 300-residue C-terminal domain (CTD) that is virtually identical in all IpaH proteins (Ashida et al., 2007). It is this CTD which carries the E3 activity.
IpaH E3 ubiquitin ligase activity was identified in a yeast surrogate model. S. flexneri IpaH9.8 was shown to inhibit yeast pheromone response signaling through ubiquitination and targeting of the mitogen-activated protein kinase kinase (MAPKK) Ste7 for degradation by the proteasome (Rohde et al., 2007). Identification of a conserved cysteine residue, C337 in IpaH9.8, within all the IpaH proteins and other homologous LRR-containing bacterial effectors, e.g., Salmonella SspH1 and Yersinia YopM characterized this family of proteins as a novel family of bacterial E3 ubiquitin ligases.
The two-domain architecture of IpaH and the presence of a cysteine residue in the CTD (IpaH–CTD) suggested IpaH may belong to the HECT E3 family. However, IpaH–CTD has no sequence similarity with HECT domain E3s. It also lacks structural similarity with either HECT domain E3s, including Salmonella SopA, or RING-finger proteins (Singer et al., 2008; Zhu et al., 2008). Instead, the IpaH E3 ubiquitin ligase consisted of a new all-helical fold in which the conserved and essential cysteine residue is located in a surface-exposed flexible loop surrounded by conserved acidic residues (CXD motif; Singer et al., 2008; Zhu et al., 2008). This fold shares similarity to the NEL domain identified in Salmonella SspH2 (Quezada et al., 2009). Unsurprisingly, while the cysteine residue acts as a thiol nucleophile to catalyze ubiquitin transfer through a transthiolation reaction, analogous to the catalytic cysteine in HECT E3s, other features of the reaction are different to the HECT family. For example IpaH requires the neighboring Asp365 for thiol-catalyzed polyubiquitin chain formation (Zhu et al., 2008). The N-terminal domain of the protein is also inhibitory for the auto-ubiquitination activity of the full-length IpaH9.8 with substrate binding releasing this inhibition (Singer et al., 2008; Seyedarabi et al., 2010).
How does the IpaH E3 ligase activity contribute to Shigella pathogenesis? Substrates of IpaH9.8 have been identified as U2AF35, a mammalian splicing factor, and NF-κB essential modulator (NEMO)/IKKγ (Ashida et al., 2007, 2010; Seyedarabi et al., 2010; Figure 2). It was previously shown that IpaH9.8 binds U2AF35 and led to down-regulation of pro-inflammatory chemokine and cytokine production during infection (Okuda et al., 2005). Given that Lys48 is the predominant ubiquitin linkage used by IpaH (Zhu et al., 2008), it is likely that U2AF35 is targeted for degradation and falling levels of this splicing factor prevent production of mRNA which can be translated. Interestingly, it has been discovered that IpaH9.8–CTD can exist in vitro as a monomer able to catalyze ubiquitination of U2AF35, and as a dimer that does not possess this activity (Seyedarabi et al., 2010). The dimerization of IpaH9.8–CTD occurs in response to oxidizing conditions and leads to domain swapping which switches off IpaH9.8 E3 ligase activity. Thus it appears the activity of IpaH9.8 can be regulated in response to the level of damage of the host cell, and may allow injured cells to remain in a benign state suitable for bacterial survival and unable to mount an attack (Seyedarabi et al., 2010).
NF-κB essential modulator, also known as inhibitor of NFκB kinase (IKKγ), is the regulatory subunit of the inhibitor of IKK complex, which activates NF-κB. IpaH9.8 interacts with NEMO/IKKγ and A20 binding inhibitor of NF-κB (ABIN-1), a ubiquitin-binding adaptor protein (Ashida et al., 2010). ABIN-1 promotes IpaH9.8-mediated NEMO ubiquitination leading to proteasome degradation of NEMO and disruption of NF-κB signaling. Thus, together with its ubiquitination of U2AF35 it appears the main role of the IpaH9.8 E3 ligase domain is to inhibit the inflammatory response, which presumably allows Shigella to initiate colonization at early stages of infection (Figure 2).
Yersinia
Yersinia pestis is the causative agent of plague, while Yersinia enterocolitica and Yersinia pseudotuberculosis cause gastrointestinal disease. Each utilizes a T3SS to inject virulence proteins, known as Yersinia outer proteins (Yops), into host cells to subvert their function (Cornelis, 1998). Host cells targeted by Yops are typically cells of the immune system such as macrophages, PMNs, and dendritic cells. Impeded in phagocytosis and signaling by the Yop proteins, these cells fail to mount a sufficient immune response to remove the pathogen, subsequently leading to disease.
YopM is one of the six Yop effectors of Yersinia and has an important role for virulence of Yersinia spp. (Trulzsch et al., 2004). The size of YopM differs between different strains and serotypes ranging from 42 to 54 kDa due to a variable number and composition of LRRs (Evdokimov et al., 2001). These LRRs give the protein homology to Salmonella SlrP, SspH1, and SspH2 and the Shigella IpaH proteins. YopM appears to be most similar to SspH1 since it too seems to localize to the nucleus and co-immunoprecipitates with kinases, in this case ribosomal s6 kinase 1 (RSK1) and PKN2 (Mcdonald et al., 2003). However, the role of this interaction is unknown during Yersinia infection, as are other aspects of how YopM aids in eliciting disease.
Soundararajan et al. (2010) using related Salmonella and Shigella effectors as templates, were able to use fold identification and homology-based modeling [protein core atomic interaction network (PCAIN) methodology] to distinguish the structure of full-length YopM. This confirmed the presence of a NEL domain in YopM. The YopM NEL shares the conserved catalytic site, conserved molecular surface electrostatics, and LRR-based auto-inhibition of E3 ligase activity found in Salmonella SspH2 and Shigella IpaH NELs (Soundararajan et al., 2010, 2011). The ability of YopM to act as an E3 ligase has not been empirically tested by biochemical means. Furthermore, it has not been translated into a defined role for YopM during infection. However, given that the YopM homologs from Salmonella and Shigella are involved in the proteolysis of host HLA–DR and NEMO (see above, Figures 2 and 4) it appears plausible that YopM may also modulate human immunity in similar ways (Figure 5).
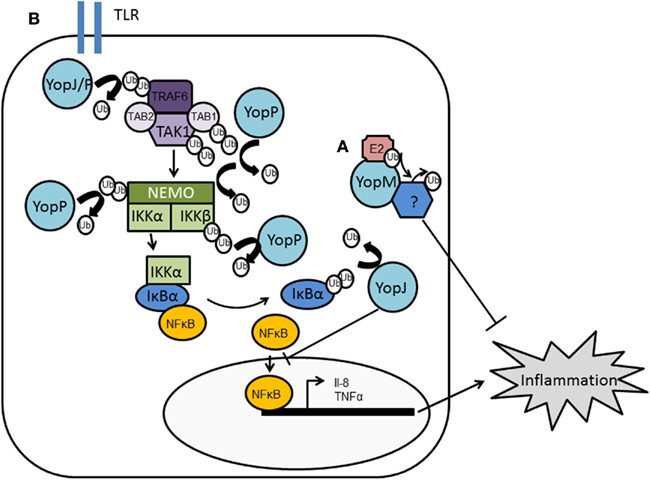
Figure 5. Manipulation of host ubiquitin pathways by Yersinia. Yersinia translocates YopM and YopJ into host cells. (A) YopM is an E3 ligase although its substrate has not been identified but is likely to moderate host cell pathways to reduce immune system function. (B) YopJ is a DUB, with the ability to deubiquitinate TRAF2, TRAF6, and IκBα. In Y. enterocolitica, the homologous protein YopP has been shown to deubiquitinate TAK1, TAB1, IκBβ, and NEMO. Both YopJ and YopP therefore disrupt NFκB signaling and reduce the inflammatory response.
Pseudomonas Syringae
Pseudomonas syringae pathovar tomato causes bacterial speck disease of tomato by using its T3SS to deliver about 30 effectors into the plant cell (Buell et al., 2003). One of these effectors is AvrPtoB, a 59-kDa protein with a modular architecture. The N-terminal region (amino acids 1–387) is recognized by the Pto kinase in tomato varieties that are immune to speck disease. Pto interacts with the resistance (R) protein Prf and upon binding of AvrPtoB activates the hypersensitive response (HR): an event characterized by rapid, localized programmed cell death (PCD), and which ultimately limits pathogen growth. The CTD of AvrPtoB (comprising amino acids 308–553) has anti-PCD activity, and this is correlated with the ability of this domain to act as an E3 ligase (Abramovitch et al., 2006). AvrPtoB–CTD exhibits homology to the eukaryotic RING-finger and U-box families of proteins, and includes conservation of a core fold and a spatially clustered three-amino acid surface patch required for E2-binding (Figure 3). Such a structure suggested AvrPtoB may function as a mimic of host E3 ubiquitin ligases, which was subsequently demonstrated biochemically (Abramovitch et al., 2006).
There appear to be two distinct mechanisms that lead to AvrPtoB-mediated PCD (Figure 6). The first, mentioned above, is dependent on both the Pto and Prf proteins in resistant tomato. AvrPtoB has been shown unable to suppress PCD via this pathway (Pedley and Martin, 2003). The second mechanism, termed “resistance suppressed by AvrPtoB C-terminus” (Rsb), is Pto-independent and is speculated to involve another putative resistance protein and Prf. It was shown by Janjusevic et al. (2006) that in plants lacking Pto and Prf, infection with a mutant of AvrPtoB that lacks E3 activity triggers the HR response, i.e., it can no longer suppress the Rbs response. This indicated that a likely substrate for AvrPtoB was the putative resistance protein that leads to Rsb-mediated PCD. The Pto family member, Fen kinase, was subsequently found to fulfill this role and to physically interact with AvrPtoB1–387 (Rosebrock et al., 2007). The AvrPtoB E3 ligase specifically ubiquitinates Fen, promoting its degradation in a proteasome-dependent manner and leading to disease susceptibility in Fen-expressing tomato lines (Rosebrock et al., 2007).
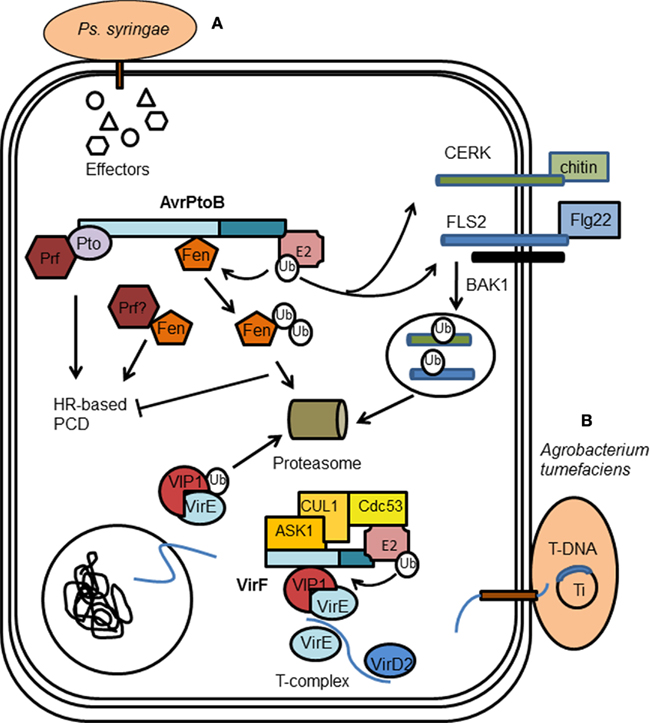
Figure 6. Manipulation of plant cell ubiquitin pathway by bacterial pathogens. (A) Pseudomonas syringae uses the E3 effector AvrPtoB to suppress plant innate immunity responses. AvrPtoB targets the cytoplasmic domains of the receptor-like kinases (RLKs) FLS2 and CERK1 for degradation, preventing downstream signaling which activates plant programmed cell death (PCD) responses. AvrPtoB also prevents PCD by inhibiting the Rbs response; AvrPtoB binds and specifically ubiquitinates Fen kinase, promoting its degradation in a proteasome-dependent manner. (B) Agrobacterium tumefaciens translocates VirF, an F-box protein, into host cells to enable genetic transformation of the host cell. VirF can bind VIP1, a host protein incorporated into the T-complex by bacterial effector VirE2. VirF through binding ASK1 is incorporated into an SCF complex. Consequently, VIP1 is ubiquitinated by the SCF complex and it and any bound VirE2 are targeted for proteasomal degradation. This is proposed to release T-DNA from the DNA–protein complex, allowing it to be integrated into the host chromatin, completing transformation.
Plant innate immunity relies on a subset of receptor-like kinases (RLKs) called pattern recognition receptors (PRRs), which respond to ligands known as pathogen-associated molecular patterns (PAMPs). For example, perception of bacterial flagellin or flg22, a peptide containing the flagellin epitope, is through an induced receptor complex composed of the LRR receptor kinases flagellin-sensing 2 (FLS2) and BRI1-associated kinase 1 (BAK1). Fungal chitin is detected by the Arabidopsis receptor kinase chitin elicitor receptor kinase 1 (CERK1; Figure 6). Upon PAMP-induced activation, PRRs stimulate a plethora of defense responses, one of which is the MAP kinase activation discussed above. AvrPtoB has now also been found to target the cytoplasmic domains of RLKs for degradation (Gohre and Robatzek, 2008; Gimenez-Ibanez et al., 2009). AvrPtoB associates with FLS2 via its N-terminal domain while it uses the E3 ligase activity of its C-terminal to preferentially target activated FLS2 for degradation (Gohre and Robatzek, 2008). AvrPtoB also ubiquitinates the CERK1 kinase domain in vitro and targets CERK1 for degradation in vivo (Gimenez-Ibanez et al., 2009). Since the RLK/Pelle gene family, which includes the known targets of AvrPtoB, makes up 60% of kinases in plants, there are likely to be further potential targets of AvrPtoB within plants. It may also suggest that AvrPtoB targets the plant proteome somewhat non-specifically (Gimenez-Ibanez et al., 2009).
Enterohemorrhagic Escherichia coli
Enterohemorrhagic Escherichia coli (EHEC) causes bloody diarrhea and hemolytic uremic syndrome (HUS). EHEC possesses a T3SS encoded by a region of its chromosome termed the locus of enterocyte effacement (LEE). Effectors encoded in LEE, and also a large number of additional non-LEE-encoded (Nle) effector proteins, are transmitted by the T3SS into host cells, where they interfere with host cellular processes to induce disease. In part this includes subverting actin polymerization to form a “pedestal” on the host cell surface which facilitates adherence and colonization of the pathogen, and leads to attaching and effacing (A/E) lesions.
NleG is one recently identified Nle effector, with 24 homologs identified (mainly in the genomes of pathogenic E. coli and Salmonella). This group of proteins form a distinct family which does not share any significant sequence similarity with other proteins. However, all NleG proteins contain a conserved region of ∼100 residues localized to the C-terminus. Analysis of this region showed the presence of a motif similar to RING-finger/U-box domains, and was shown to act as a functional E3 ligase in the presence of ubiquitin, E1 and E2 (UBE2D2; Wu et al., 2010). E2 interacts with NleG effectors following the general architecture established for E2/eukaryotic RING-finger complexes. Indeed, there is significant structural similarity between NleG and the CTD of AvrPtoB from P. syringae (Figure 3), further indicating NleG may function in part as an E3 ligase (Wu et al., 2010). However, no substrate has been reported for NleG; a major hindrance being that the N-terminal is not conserved between NleG family members and none has an identifiable substrate binding motif. The authors therefore suggest NleG effectors may primarily associate with specific host E2 enzymes rather than transfer ubiquitin onto a substrate protein, and in this way manipulate the host ubiquitination pathway (Wu et al., 2010). This is an intriguing possibility but requires much further work to confirm. Alternatively, the NleG effectors may contain novel binding motifs or use scaffolding proteins to interact with their substrates.
Another Nle effector has been identified in EHEC as an E3 ligase: EspX7, renamed as non-Lee-encoded effector ligase (NleL) to reflect its novel biochemical activity (Piscatelli et al., 2011). This effector was identified as an E3 ligase given its similarity to SopA from Salmonella (Zhang et al., 2006; Lin et al., 2011). Similar to SopA, NleL contains a protease-sensitive N-terminus, a β-helix domain, and a C-terminal catalytic domain that resembles the bilobal architecture of eukaryotic HECT E3s (Figure 3). The ubiquitin ligase activity of NleL is dependent upon a cysteine residue (Cys753) that forms an intermediate with ubiquitin through a thioester bond, thus NleL, like SopA, acts as a bacterial mimic of eukaryotic HECT E3s (Lin et al., 2011; Piscatelli et al., 2011).
In vitro ubiquitination assays show NleL preferentially forms unanchored polyUb chains using Lys6 and Lys48 linkages. This suggests NleL plays a role in moderating signaling pathways rather than targeting proteins for degradation, and is supported by the observation that NleL E3 ubiquitin ligase activity is involved in modulating Tir-mediated pedestal formation (Piscatelli et al., 2011). Interestingly, an EHEC mutant strain deficient in E3 ligase activity induced more pedestals than the wild-type strain. It therefore appears that EHEC may use specific effector proteins to ensure there is control in the manipulation of host pathways so a balance is achieved between bacterial and cell survival. Consistent with the hypothesis that unregulated pedestal formation may alter disease, it was demonstrated that the Citrobacter rodentium NleL homolog, which also acts as an E3 ubiquitin ligase, was required for efficient infection of murine colonic epithelial cells in vivo (Piscatelli et al., 2011). So far the target of NleL is unknown. It appears Tir is not ubiquitinated by NleL (Piscatelli et al., 2011), and thus it is reasonable to suppose that NleL ubiquitinates an unknown bacterial or host protein(s) involved in pedestal formation, with a decrease in protein level or an alteration in localization resulting in a check in pedestal formation.
Legionella
The Legionella chromosome encodes a Dot/Icm type IV secretion system (T4SS) which is essential for a number of virulence traits, including replication within host cells (Segal et al., 1998; Vogel et al., 1998). In Legionella infections, it has been shown that anti-polyubiquitin antibodies decorate Legionella-containing vacuoles (LCVs), suggesting a significant amount of polyubiquitinated proteins exist on LCVs (Dorer et al., 2006). As the proteasome inhibitor MG132 was reported to adversely affect the intracellular growth of Legionella in mouse macrophages, it appears that Legionella may be an additional pathogen that exploits the host UPS (Dorer et al., 2006).
In a search for novel translocated effectors of Legionella, Lpg2830 was identified (Kubori et al., 2008). Lpg2830 contains two-domains with striking similarity to U-boxes (U-box 1 and U-box 2), and therefore was named LubX (Legionella U-box protein). LubX was shown to exhibit E3 ligase activity in vitro, and required U-box 1 for this activity (Kubori et al., 2008). U-box 2 was shown to play a non-canonical role, being required for binding of the LubX substrate: Cdc2-like kinase 1 (Clk1). Clk kinases phosphorylate serine/arginine-rich proteins (SR proteins), which in turn modulate alternative splicing-site selection (Prasad et al., 1999; Schwertz et al., 2006). Clk1 is shown to be required for the growth of Legionella in macrophages, indicating Legionella targets this host protein during infection. While it was demonstrated that LubX does indeed polyubiquitinate Clk1, it was not clear what type of linkage was used and whether Clk1 was targeted for degradation (Kubori et al., 2008).
More recently, it has been shown that another substrate of LubX is the Legionella effector protein SidH (Kubori et al., 2010). SidH levels within host cells were shown to decline over time and were concurrent with increasing levels of LubX. It was then shown that LubX can bind SidH and direct its ubiquitination, targeting it for degradation by the proteasome (Kubori et al., 2010). This is the first example of a bacteria effector targeting and regulating another bacterial effector within host cells, and the authors have designated such an effector as a “metaeffector.” When it is considered that Legionella possesses 275 confirmed Dot/Icm substrates (Zhu et al., 2011), it is perhaps not surprising that some of these may be utilized to coordinate the function of other effector proteins and ensure the infection proceeds as desired by the bacterium. It will not only be interesting to uncover further metaeffectors in the Legionella effector repertoire but to determine what other metaeffectors are used by other bacteria, if at all.
F-Box Proteins
In addition to U-boxes, it is known that proteins containing F-boxes have an important role in protein ubiquitination. The F-box motif is ∼50 amino acids long, although with only a few conserved residues, and mediates protein–protein interactions. F-box proteins form one of the constituents of the SCF complex, a multi-protein E3 ubiquitin ligase (Zimmerman et al., 2010). The SCF complex works in the following way to target proteins for proteasomal degradation. Firstly, F-box proteins bind independently to a target protein via the variable protein–protein interaction domain. S-phase kinase-associated protein 1 (Skp1) binds the F-box protein via the F-box motif and in this way targets the substrate recognition component of the complex to the scaffold protein Cullin-1. Cullin-1 links the Skp domain with the RING-finger domain of Rbx1/Roc1/Roc2. This allows ubiquitin to be passed from the E2 molecule, bound to Rbx1, to the target protein bound by the F-box containing protein.
Agrobacterium
One of the first bacterial F-box proteins identified was VirF, from the plant pathogen Agrobacterium tumefaciens. A. tumefaciens genetically transforms plants during infection leading to the formation of crown galls, which are essentially plant tumors. Genetic transformation is achieved by transporting a single-stranded copy of the bacterial transferred DNA (T-DNA) from the A. tumefaciens tumor-inducing (Ti) plasmid into the plant cell nucleus followed by integration into the host genome (Gelvin, 2000). During genetic transformation, virulence (Vir) proteins are also transferred into the host cell, many of which play a role in mediating T-DNA integration. VirE2, associates with T-DNA to create a nucleoprotein complex (T-complex; Citovsky et al., 2007), and through its association with the host VIP1 protein facilitates nuclear import of the T-complex. Before integration into the host genome, the T-DNA must be uncoated from its cognate proteins. VirF, an F-box protein, had been shown to bind the plant homolog of Skp1, ASK 1 (Schrammeijer et al., 2001). As part of the Skp1–Cdc53/Cullin–F-box (SCF) complex, VirF has been shown to bind VIP1, thereby targeting both VIP1 and its associated VirE2 for proteasomal degradation (Figure 6). This leads to the release of T-DNA from the DNA–protein complex, allowing it to be integrated into the host chromatin (Tzfira et al., 2004).
Interestingly, VirF is not required for genetic transformation of some plant species (Hirooka et al., 1987). Instead, the VirF function is performed by a host plant protein(s). The F-box protein, VIP1-binding F-box (VBF), has recently been identified as fulfilling this role (Zaltsman et al., 2010). VBF recognizes and binds VIP1 and its associated VirE2, forming ternary VBF–VIP1–VirE2 complexes. VBF can then act to destabilize both VIP1 and VirE2 via the SCF–VBF pathway (Zaltsman et al., 2010). Thus, Agrobacterium subverts a host defense pathway induced in response to infection, to facilitate infection.
Ralstonia
Ralstonia solanacearum is another plant pathogen that appears to utilize F-box proteins during infection. A group of seven genes with homology to plant-specific LRRs have been identified and named GALA proteins after a conserved GAxALA sequence in their LRR (Cunnac et al., 2004). These seven GALA family effectors have been shown to contain F-box motifs in their N-terminus and mediate binding to host Skp1 proteins, ASK1 and ASK2 in Arabidopsis (Angot et al., 2006). Thus, GALA family proteins function as SCF E3s, and this was shown to be important for virulence of Ralstonia. A mutant in which all of the seven GALA genes were deleted or mutated did not cause wilt in Arabidopsis, and slowed the progression of wilting in tomato (Angot et al., 2006).
Legionella
Of human pathogens, all strains of Legionella pneumophila sequenced to date encode multiple genes with predicted F-box motifs, including three F-box genes in L. pneumophila strain Paris (Lomma et al., 2010) and five F-box genes in L. pneumophila strain Philadelphia-1 (Ensminger and Isberg, 2010). Since single, double, and triple F-box mutations in the Paris strain F-box genes have been shown to cause impaired intracellular replication and defective colonization of the lungs of A/J mice, it indicates F-box proteins and their potential interactions with host ubiquitin pathways are important to Legionella infection (Lomma et al., 2010).
The L. pneumophila F-box containing proteins have a structure that mimics that of their eukaryotic homologs, with an N-terminal F-box domain and a second C-terminal protein interaction motif. However, they are unique in that the second interaction domain is neither a WD40 repeat, nor a LRR region as reported for eukaryotic F-box proteins (Skaar et al., 2009a,b). One F-box protein in particular has been studied due to its conservation between Legionella strains: AnkB (Lpg2144) in strain AA100/130b, LegAU13 in the Philadelphia-1 strain, and Lpp2082 in the Paris strain. AnkB has been shown to interact with Skp1. The target of the Skp1–Cullin–AnkB complex was found to be ParvB (Lomma et al., 2010). ParvB belongs to the Parvin family of proteins that are involved in linking integrins and associated proteins with intracellular pathways that regulate actin cytoskeletal dynamics and cell survival. Overexpression of AnkB in A549 cells and infection with L. pneumophila wild-type, but not the ankB mutant strain, decreased ubiquitination of ParvB (Lomma et al., 2010). It appears that AnkB inhibits the endogenous ubiquitin ligase of ParvB perhaps by competing for the interaction site (Lomma et al., 2010). The authors also hypothesized that by decreasing ubiquitination of ParvB, AnkB might stabilize the integrin-linked kinase (ILK)–PINCH–ParvB complex leading to caspase-3 activation and apoptosis. Alternatively, ParvB may act as an accessory protein to direct AnkB to the plasma membrane, where the E3 ligase activity of SCF directs the ubiquitination of one or more additional proteins. Since inhibition of ParvB by RNA interference resulted in reduced intracellular growth of L. pneumophila, this reveals a new mechanism by which L. pneumophila may employ translocated effector proteins to promote bacterial survival.
Another Legionella F-box protein, LegU1 (Lpg0171), has also been shown to form a complex with both Skp1 and Cullin, and to therefore possess E3 ligase activity (Ensminger and Isberg, 2010). Using a two-step enrichment strategy in which immunoprecipitates of 3XFLAG-tagged LegU1 were subjected to a second round of immunoprecipitation to isolate ubiquitinated species, HLA–B-associated transcript 3 (BAT3) was identified as a target of the SCF–LegU1 complex (Ensminger and Isberg, 2010). The translocated Legionella effector, Lpg2160, was also found to associate with the SCF–LegU1–BAT3 complex although it does not get ubiquitinated. As two Legionella effectors target BAT3, it appears that modulation of BAT3 activity is crucial during Legionella infection. BAT3 plays a role in the ER stress response and host apoptotic pathways (Desmots et al., 2008; Tsukahara et al., 2009), thus, LegU1 and Lpg2160 may interfere with BAT3 function to mitigate the effects of disrupting normal vesicular trafficking in the host cell.
LicA, is a third translocated L. pneumophila Philadelphia effector with an F-box motif. While it associates with Skp1 it does so in the absence of Cullin1, and lacks auto-ubiquitination activity. This suggests LicA does not form an E3 ligase, but may also suggest the ability of LicA to form a non-canonical SCF complex using alternate eukaryotic components (Ensminger and Isberg, 2010).
Cryptococcus
Another human pathogen utilizing an F-box protein is Cryptococcus neoformans, the leading cause of fungal meningitis in the immunocompromised. Fbp1 in C. neoformans contains a putative F-box domain and 12 LRRs and physically interacts with Skp1, suggesting it may function as part of a SCF E3 ligase (Liu and Shen, 2011). As Fbp1 is essential for C. neoformans virulence, this indicates the SCF E3 ubiquitin ligase-mediated proteolysis pathway is likely to be important during infection, although its substrates remain to be identified.
Non-Pathogenic Bacteria
F-box proteins are not only associated with pathogens. The amebal symbiont Amoebophilus asiaticus encodes 15 proteins with predicted F-box motifs and 9 proteins with E3 ligase-associated U-box domains in its chromosome, indicating it too may interfere with the host ubiquitination system (Schmitz-Esser et al., 2010).
De-Ubiquitinating UME
In addition to being able to moderate the activities of a protein by adding ubiquitin, the removal of ubiquitin can also be a powerful tool to control cellular functions. De-ubiquitinating enzymes (DUBs) cleave the isopeptide bond between ubiquitin and its target protein. This produces free ubiquitin which can then be recycled by the cell. Thus, ubiquitination is a reversible post-translational modification, and as such, is often compared to the post-translational modification of phosphorylation. Both modifications require ATP, are irreversible and alter the recognition of proteins by the cell (Orth, 2002). Since bacteria have evolved to possess effectors with the ability to modify proteins with ubiquitin, it is not surprising that they have also evolved effectors that act as DUBs.
Yersinia
Yersinia effector YopJ (YopP in Y. enterocolitica), is essential for the ability of Yersiniae to kill macrophages and to establish a systemic infection in mice (Monack et al., 1998). YopJ appears to have several activities in infected cells that contribute to its importance to establish infection. YopJ inhibits the pro-inflammatory mitogen-activated protein kinase (MAPK) and NFκB pathways (Palmer et al., 1998; Ruckdeschel et al., 1998; Schesser et al., 1998), both of which play roles in innate and adaptive immunity. Yersinia appears to target both pathways to reduce the inflammatory response during infection; the production of protective cytokines, such as tumor necrosis factor-α (TNF-α) and IL-8, are inhibited, and apoptosis in macrophages is induced. Sequence analysis of YopJ showed no homology to known proteins, but secondary structure analysis indicated similarity to adenovirus protease (AVP; Orth et al., 2000). A conserved catalytic cysteine was identified between these two proteins, mutation of which in YopJ abolishes its ability to inhibit the MAPK and NFκB pathways, and inhibit TNF-α production (Orth et al., 2000). Thus, the protease activity of YopJ is key to its function.
Adenovirus protease had been indicated to resemble the cysteine protease, ubiquitin-like protein protease 1 (Ulp1) in yeast (Li and Hochstrasser, 1999), which is able to remove small ubiquitin-related modifier (SUMO) from modified proteins. YopJ was shown by Orth et al. (2000) to share this activity. Overexpression of YopJ in 293 cells results in a decrease in proteins bound to SUMO. This activity of YopJ has been questioned by Zhou et al. (2005) who found that YopJ did not deSUMOlyate proteins. However, both authors agree that YopJ possesses deubiquintation activity, dependent on cysteine 172, and as such was the first bacterial effector shown with this property (Orth, 2002; Zhou et al., 2005).
YopJ appears to be a potent de-ubiquitinating protease, being able to cleave ubiquitin moieties from many substrates including TNF receptor-associated factor 2 (TRAF2), TRAF6, NF-κB-inducing kinase (NIK), and IκBα (Figure 5). TRAF2 and TRAF6 undergo auto-ubiquitination and acquire K63 polyubiquitin chains which are proposed to promote the assembly of a multi-protein complex that activates the IKK complex and hence, NF-κB signaling. YopJ can remove both K48 and K63 linkages. By reversing the ubiquitination of TRAF2, TRAF6 and/or phosphorylated IκBα during Yersinia infection, YopJ maintains the IκB–NFκB complex. This keeps NFκB sequestered in the cytoplasm thereby attenuating the NFκB pathway which is observed during wild-type Yersinia infections (Figure 5). Y. enterocolitica YopP has also been shown to bind and deubiquitinate IKKb, TRAF6, and NEMO in vitro (Carter et al., 2003; Haase et al., 2005). However, Thiefes et al. (2006) reported TRAF6 was not deubiquitinated by YopP but rather TAK1 and TAB1 are deubiquitinated, inhibiting TAK1 autophosphorylation and activity (Thiefes et al., 2006). There is no evidence that MAPKKs require ubiquitin or SUMO conjugates for activation, and thus it was not clear how YopJ/P could suppress the MAPK pathway simultaneously with the NFκB pathway. By placing the inhibitory mechanism of YopP at TAK1, i.e., downstream from TRAF6 and upstream from MAPKK6/p38 MAPK, it does provide a mechanism whereby YopJ/P can activate both the MAPK and NFκB pathways to down-regulate innate immune signaling. However, it is unclear whether these observations are specific to the IL-1 activation pathway or universal. Recently an alternative mechanism by which YopP/J suppresses the two pathways has been identified: YopJ acts not only as a DUB but as an acetyltransferase (Mittal et al., 2006; Mukherjee et al., 2006). YopJ binds and acetylates critical serine or threonine residues, using acetyl-coenzyme A (CoA), in the activation loop of MAPKKs and IKKb, respectively. This prevents these residues from being phosphorylated and from being activated, blocking signaling through the MAPKK and NFκB pathways (Mukherjee et al., 2006). Inositol hexakisphosphate (IP6) acts as an activation cofactor for YopJ acetyltransferase activity (Mittal et al., 2006). Thus, it appears YopJ has two different enzymatic capabilities and may explain why Yersinia is so efficient at inhibiting the MAPK and NFκB pathways; acting on multiple components in a multitude of ways.
Salmonella
Salmonella possesses two DUBs with homology to YopJ. AvrA is encoded by the SPI-1 region of the Salmonella genome (Hardt and Galán, 1997), and cleaves ubiquitin moieties from IκBα and β-catenin, thus regulating both the NFκB and β-catenin signaling pathways (Collier-Hyams et al., 2002; Sun and Chen, 2004; Ye et al., 2007; Figure 4). Stabilized IκBα leads to inhibition of NF-κB signaling and consequently inhibition of the inflammatory response. Stabilization of β-catenin leads to increased β-catenin transcriptional activity, activating cell proliferation and inhibiting cell apoptosis (Ye et al., 2007). Like YopJ, AvrA also exhibits acetlytransferase activity which is targeted on MAPKKs (Jones et al., 2008). However, interestingly, AvrA is anti-apoptotic not pro-apoptotic like YopJ and its homolog AopP of Aeromonas salmonicida (Jones et al., 2008). These differences may be accounted for by the selectivity of substrates. For example AvrA acts as a c-Jun N-terminal kinase (JNK) inhibitor at the level of MKK4/7, preventing JNK stimulating a pro-apoptotic response (Jones et al., 2008; Du and Galan, 2009), while YopJ has a more promiscuous range of MAPKK inhibitory activity (Mukherjee et al., 2006). Recently, the number of pathways on which AvrA potentially has an impact has been expanded to include: the p53 pathway, mTOR signaling, oxidative phosphorylation, VEGF, and JAK-STAT (Liu et al., 2010; Wu et al., 2010). Their roles in the anti-apoptotic function of AvrA have yet to be characterized.
Examining Salmonella genes expressed under SPI-2 inducing conditions led to the identification and characterization of another YopJ family member in Salmonella: SseL (Coombes et al., 2007; Rytkonen et al., 2007). Biochemical assays show SseL functions as a DUB and, like AvrA, impairs IκBα ubiquitination and degradation, suppressing NFκB activation (Le Negrate et al., 2008). This appears important for the systemic spread of Salmonella in mice, since by competitive index a sseL null mutant is attenuated in virulence, although no defect was observed in vitro (Coombes et al., 2007; Rytkonen et al., 2007). However, while AvrA, through its activity on the β-catenin pathway, inhibits apoptosis, it appears SseL may induce cytotoxicity in macrophages at later stages of infection (Rytkonen et al., 2007); although the importance of SseL in this process has been challenged by Le Negrate et al. (2008) who observed only a slight cytotoxic effect due to SseL. Since both AvrA and SseL act on IκBα, this appears to be an important stage at which the NFκB pathway proteins and Salmonella effectors intersect. The activity of AvrA and SseL is consistent with the in vivo ability of Salmonella to dampen innate immune signaling while preventing the apoptotic elimination of infected, compromised cells. This may allow Salmonella more extensive tissue invasion, extending its long-term survival as it replicates in viable host cells.
In additional to the Salmonella DUBs, several other bacterial proteins have been discovered with similar homology to YopJ (Table 1). This “YopJ family” includes effectors from animal and plant pathogens and plant symbionts. Each shares a conserved catalytic core, consisting of three key amino acid residues (histidine, glutamic acid, and cysteine), and designates them as such to the C55 family of cysteine proteases (Orth et al., 2000; Barrett and Rawlings, 2001). Each protein appears to share at least one catalytic activity prescribed to YopJ, i.e., the ability to deubiquitinate, deSUMOlyate, or acetylate. Interestingly, Xanthomonas possesses not only YopJ-like effectors which exhibit similarity to a ubiquitin-like protein protease (Ulp1), but also the effector XopD which is a bona fide member of the Ulp1 protein family with deSUMOylating activity (Hotson et al., 2003). This may suggest that after acquiring an Ulp1/Ulp-1 like hydrolase, pathogenic bacteria have adapted this protein to increase/modify its range of activity, thus making it a more potent tool during pathogenesis.
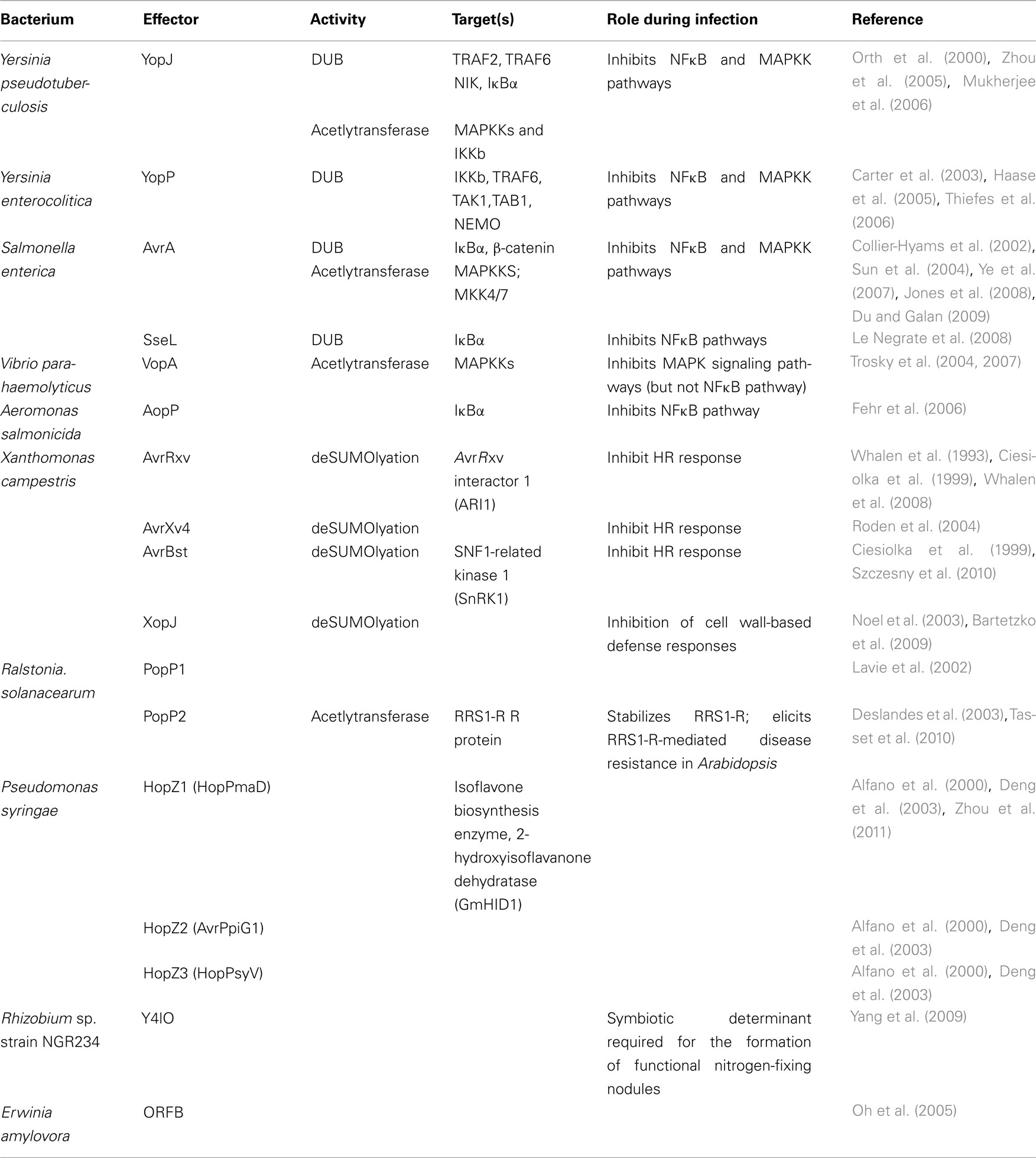
Table 1. YopJ family members.
Chlamydia
Chlamydia trachomatis is a bacterium which possesses DUBs distinct from those of the YopJ family. Instead, the predicted catalytic domains of ChlaDub1 and ChlaDub2 share similarity with the Ulp domain of SUMO-specific proteases (SENPs), a family of proteases that possess either deneddylating or desumoylating activities (Misaghi et al., 2006). ChlaDub1 and ChlaDub2 have been shown in vitro to hydrolyse the thioester bonds that attach ubiquitin and NEDD8 (a ubiquitin-like protein) to target proteins (Misaghi et al., 2006). It appears for ChlaDub1 this translates into impairment of IκBα ubiquitination and its subsequent proteasome-mediated degradation, thereby preventing NFκB activation and potentially providing one mechanism by which Chlamydia is able to evade immune responses and establish long-term carrier states in infected individuals (Le Negrate et al., 2008). Since several viruses encode DUBs (Balakirev et al., 2002; Kattenhorn et al., 2005; Schlieker et al., 2005), it is not surprising that Chlamydia with its similar intracellular replication cycle, has developed tools to modulate the host ubiquitin–proteasome pathway. Further studies into the cellular function of ChlaDub1 and particularly ChlaDub2 will no doubt provide more information as to how C. trachomatis uses DUBs to manipulate its host.
Conclusion
Many bacterial pathogens have developed sophisticated secretion and translocation apparatus to transfer a set of proteins (effectors) directly into the cytoplasm of the host cell. These effectors manipulate host cellular responses, promoting the progression of bacterial infection and pathogenesis. It appears that interfering with ubiquitin signaling in the host is an important and common strategy used by pathogens to promote their survival in the host. In many of the examples reviewed here, exploiting the ubiquitin pathways to evade the host immune system during the course of infection is of particular importance to bacteria; Salmonella, Shigella, and Yersinia all target the NFκB pathway to control the inflammatory response elicited during their infection. However, while each bacterium may target similar pathways, in many instances they appear to have developed different tools, and in doing so may target different stages in a specific host pathway. For example while Shigella uses OspG, an E2 targeting UME, to prevent signaling through the NFκB pathway, Salmonella and Yersinia use DUBs, albeit DUBs which act at different stages of the pathway. Thus, while there are many common themes in how bacteria manipulate host ubiquitin pathways, each bacterium has often developed one or more unique effectors allowing them to exquisitely manipulate the host to suite their particular lifestyle.
An important strategy used by bacteria to manipulate host pathways is the use of effectors that functionally mimic eukaryotic proteins. This is no different for moderation of the ubiquitin pathway. Each of the major subclasses of eukaryotic E3s have bacterial counterparts either in sequence or structure. For example SopA structurally resembles HECT E3s and AvrPtoB resembles a RING E3, while LubX and VirF proteins bear a U-box and an F-box motif, respectively. However, the study of ubiquitin E3 ligases in bacteria has also unearthed a new class of E3s: the NEL family, exemplified by IpaH, SspH2, and YopM (Singer et al., 2008; Zhu et al., 2008; Quezada et al., 2009). This suggests that bacterial and eukaryotic genomes might encode additional novel E3s that have not yet been identified and will no doubt be an area of further study especially as predictive and modeling software becomes more advanced. It is also interesting that most of the bacteria carrying NEL ubiquitin ligases are pathogenic. On the one hand this provides the interesting question as to how these proteins evolved and have been propagated between bacterial species, and on the other provides exciting opportunities for the development of antibacterial therapeutics. Such therapies are likely to be particularly effective given that NEL-containing effectors are often critical virulence determinants and they currently do not have any known mammalian homologs.
In addition to further understanding how bacteria manipulate host ubiquitin pathways, a fruitful area of research is likely to be how bacteria interact with the ubiquitin-like proteins SUMO, NEDD8, ISG-15, and FAT10. These ubiquitin-like proteins contain a C-terminal di-glycine motif that can be covalently attached to protein targets through enzymatic reaction cascades similar to that of ubiquitination. Thus, it will not be surprising if bacteria have also developed mechanisms to manipulate these pathways in cells. Indeed, it has already been shown that the EPEC effector cycle inhibiting factor (Cif) interferes with neddylation-mediated cell-cycle control (Morikawa et al., 2010).
To conclude, the host ubiquitin pathway is carefully moderated by bacteria during infection, indicating its importance to the bacteria–host interaction, and further study will not only educate us in mechanisms used by bacteria during pathogenesis but also in the processes used by cells for survival and maintenance.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramovitch, R. B., Janjusevic, R., Stebbins, C. E., and Martin, G. B. (2006). Type III effector AvrPtoB requires intrinsic E3 ubiquitin ligase activity to suppress plant cell death and immunity. Proc. Natl. Acad. Sci. U.S.A. 103, 2851–2856.
Alfano, J. R., Charkowski, A. O., Deng, W. L., Badel, J. L., Petnicki-Ocwieja, T., Van Dijk, K., and Collmer, A. (2000). The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc. Natl. Acad. Sci. U.S.A. 97, 4856–4861.
Angot, A., Peeters, N., Lechner, E., Vailleau, F., Baud, C., Gentzbittel, L., Sartorel, E., Genschik, P., Boucher, C., and Genin, S. (2006). Ralstonia solanacearum requires F-box-like domain-containing type III effectors to promote disease on several host plants. Proc. Natl. Acad. Sci. U.S.A. 103, 14620–14625.
Ardley, H. C., and Robinson, P. A. (2005). “E3 ubiquitin ligases,” in Essays in Biochemistry, Vol. 41: The Ubiquitin-Proteasome System, R. J. Mayer and R. Layfield (London), 15–30.
Ashida, H., Kim, M., Schmidt-Supprian, M., Ma, A., Ogawa, M., and Sasakawa, C. (2010). A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat. Cell Biol. 12, 66–73.
Ashida, H., Toyotome, T., Nagai, T., and Sasakawa, C. (2007). Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol. Microbiol. 63, 680–693.
Balakirev, M. Y., Jaquinod, M., Haas, A. L., and Chroboczek, J. (2002). Deubiquitinating function of adenovirus proteinase. J. Virol. 76, 6323–6331.
Barrett, A. J., and Rawlings, N. D. (2001). Evolutionary lines of cysteine peptidases. Biol. Chem. 382, 727–733.
Barry, M., and Fruh, K. (2006). Viral modulators of cullin RING ubiquitin ligases: culling the host defense. Sci. STKE 2006, pe21.
Bartetzko, V., Sonnewald, S., Vogel, F., Hartner, K., Stadler, R., Hammes, U. Z., and Bornke, F. (2009). The Xanthomonas campestris pv. vesicatoria type III effector protein XopJ inhibits protein secretion: evidence for interference with cell wall-associated defense responses. Mol. Plant Microbe Interact. 22, 655–664.
Bates, P. W., and Vierstra, R. D. (1999). UPL1 and 2, two 405 kDa ubiquitin-protein ligases from Arabidopsis thaliana related to the HECT-domain protein family. Plant J. 20, 183–195.
Bernal-Bayard, J., Cardenal-Munoz, E., and Ramos-Morales, F. (2010). The Salmonella type III secretion effector, Salmonella leucine-rich repeat protein (SIrP), targets the human chaperone ERdj3. J. Biol. Chem. 285, 16360–16368.
Bernal-Bayard, J., and Ramos-Morales, F. (2009). Salmonella type III secretion effector SlrP is an E3 ubiquitin ligase for mammalian thioredoxin. J. Biol. Chem. 284, 27587–27595.
Boland, A., Sory, M.-P., Iriarte, M., Kerbourch, C., Wattiau, P., and Cornelis, C. R. (1996). Status of YopM and YopN in the Yersinia Yop virulon: YopM of Y. enterocolitica is internalized inside the cytosol of PU5-1.8 macrophages by the YopB, D, N delivery apparatus. EMBO J. 15, 5191–5201.
Buchrieser, C., Glaser, P., Rusniok, C., Nedjari, H., D’hauteville, H., Kunst, F., Sansonetti, P., and Parsot, C. (2000). The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 38, 760–771.
Buell, C. R., Joardar, V., Lindeberg, M., Selengut, J., Paulsen, I. T., Gwinn, M. L., Dodson, R. J., Deboy, R. T., Durkin, A. S., Kolonay, J. F., Madupu, R., Daugherty, S., Brinkac, L., Beanan, M. J., Haft, D. H., Nelson, W. C., Davidsen, T., Zafar, N., Zhou, L. W., Liu, J., Yuan, Q. P., Khouri, H., Fedorova, N., Tran, B., Russell, D., Berry, K., Utterback, T., Van Aken, S. E., Feldblyum, T. V., D’Ascenzo, M., Deng, W. L., Ramos, A. R., Alfano, J. R., Cartinhour, S., Chatterjee, A. K., Delaney, T. P., Lazarowitz, S. G., Martin, G. B., Schneider, D. J., Tang, X., Bender, C. L., White, O., Fraser, C. M., and Collmer, A. (2003). The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. U.S.A. 100, 10181–10186.
Carter, R. S., Pennington, K. N., Ungurait, B. J., Arrate, P., and Ballard, D. W. (2003). Signal-induced ubiquitination of I kappa B kinase-beta. J. Biol. Chem. 278, 48903–48906.
Chiu, Y. H., Sun, Q., and Chen, Z. J. J. (2007). E1-L2 activates both ubiquitin and FAT10. Mol. Cell 27, 1014–1023.
Ciechanover, A., Heller, H., Katz-Etzion, R., and Hershko, A. (1981). Activation of the heat-stable polypeptide of the ATP-dependent proteolytic system. Proc. Natl. Acad. Sci. U.S.A. 78, 761–765.
Ciehanover, A., Hod, Y., and Hershko, A. (1978). A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 81, 1100–1105.
Ciesiolka, L. D., Hwin, T., Gearlds, J. D., Minsavage, G. V., Saenz, R., Bravo, M., Handley, V., Conover, S. M., Zhang, H., Caporgno, J., Phengrasamy, N. B., Toms, A. O., Stall, R. E., and Whalen, M. C. (1999). Regulation of expression of avirulence gene avrRxv and identification of a family of host interaction factors by sequence analysis of avrBsT. Mol. Plant Microbe Interact. 12, 35–44.
Citovsky, V., Kozlovsky, S. V., Lacroix, B., Zaltsman, A., Dafny-Yelin, M., Vyas, S., Tovkach, A., and Tzfira, T. (2007). Biological systems of the host cell involved in Agrobacterium infection. Cell. Microbiol. 9, 9–20.
Collier-Hyams, L. S., Zeng, H., Sun, J., Tomlinson, A. D., Bao, Z. Q., Chen, H., Madara, J. L., Orth, K., and Neish, A. S. (2002). Salmonella AvrA effector inhibits the key proinflammatory, anti-apoptotic NF-kappa B pathway. J. Immunol. 169, 2846–2850.
Coombes, B. K., Lowden, M. J., Bishop, J. L., Wickham, M. E., Brown, N. F., Duong, N., Osborne, S., Gal-Mor, O., and Finlay, B. B. (2007). SseL is a Salmonella-specific translocated effector integrated into the SsrB-controlled Salmonella pathogenicity island 2 type III secretion system. Infect. Immun. 75, 574–580.
Coscoy, L., Sanchez, D. J., and Ganem, D. (2001). A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 155, 1265–1273.
Cunnac, S., Boucher, C., and Genin, S. (2004). Characterization of the cis-acting regulatory element controlling HrpB-mediated activation of the type III secretion system and effector genes in Ralstonia solanacearum. J. Bacteriol. 186, 2309–2318.
Deng, W. L., Rehm, A. H., Charkowski, A. O., Rojas, C. M., and Collmer, A. (2003). Pseudomonas syringae exchangeable effector loci: sequence diversity in representative pathovars and virulence function in P. syringae pv. syringae B728a. J. Bacteriol. 185, 2592–2602.
Deslandes, L., Olivier, J., Peeters, N., Feng, D. X., Khounlotham, M., Boucher, C., Somssich, I., Genin, S., and Marco, Y. (2003). Physical interaction between RRS1-R, a protein conferring resistance to bacterial wilt, and PopP2, a type III effector targeted to the plant nucleus. Proc. Natl. Acad. Sci. U.S.A. 100, 8024–8029.
Desmots, F., Russell, H. R., Michel, D., and Mckinnon, P. J. (2008). Scythe regulates apoptosis-inducing factor stability during endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 283, 3264–3271.
Diao, J., Zhang, Y., Huibregtse, J. M., Zhou, D., and Chen, J. (2008). Crystal structure of SopA, a Salmonella effector protein mimicking a eukaryotic ubiquitin ligase. Nat. Struct. Mol. Biol. 15, 65–70.
Dietrich, F. S., Mulligan, J., Hennessy, K., Yelton, M. A., Allen, E., Araujo, R., Aviles, E., Berno, A., Brennan, T., Carpenter, J., Chen, E., Cherry, J. M., Chung, E., Duncan, M., Guzman, E., Hartzell, G., Hunicke-Smith, S., Hyman, R. W., Kayser, A., Komp, C., Lashkari, D., Lew, H., Lin, D., Mosedale, D., Nakahara, K., Namath, A., Norgren, R., Oefner, P., Oh, C., Petel, F. X., Roberts, D., Sehl, P., Schramm, S., Shogren, T., Smith, V., Taylor, P., Wei, Y., Botstein, D., and Davis, R. W. (1997). The nucleotide sequence of Saccharomyces cerevisiae chromosome V. Nature 387, 78–81.
Dorer, M. S., Kirton, D., Bader, J. S., and Isberg, R. R. (2006). RNA interference analysis of Legionella in Drosophila cells: exploitation of early secretory apparatus dynamics. PLoS Pathog. 2, e34. doi:10.1371/journal.ppat.0020034
Du, F. Y., and Galan, J. E. (2009). Selective inhibition of type III secretion activated signaling by the salmonella effector AvrA. PLoS Pathog. 5, e1000595. doi:10.1371/journal.ppat.1000595
Ensminger, A. W., and Isberg, R. R. (2010). E3 ubiquitin ligase activity and targeting of BAT3 by multiple Legionella pneumophila translocated substrates. Infect. Immun. 78, 3905–3919.
Evdokimov, A. G., Anderson, D. E., Routzahn, K. M., and Waugh, D. S. (2001). Unusual molecular architecture of the Yersinia pestis cytotoxin YopM: a leucine-rich repeat protein with the shortest repeating unit. J. Mol. Biol. 312, 807–821.
Fehr, D., Casanova, C., Liverman, A., Blazkova, H., Orth, K., Dobbelaere, D., Frey, J., and Burr, S. E. (2006). AopP, a type III effector protein of Aeromonas salmonicida, inhibits the NF-kappa B signalling pathway. Microbiology 152, 2809–2818.
Ganoth, D., Leshinsky, E., Eytan, E., and Hershko, A. (1988). A multicomponent system that degrades proteins conjugated to ubiquitin. Resolution of factors and evidence for ATP-dependent complex formation. J. Biol. Chem. 263, 12412–12419.
Gao, G., and Luo, H. L. (2006). The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 84, 5–14.
Gelvin, S. B. (2000). Agrobacterium and plant genes involved in T-DNA transfer and integration. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51, 223–256.
Gimenez-Ibanez, S., Hann, D. R., Ntoukakis, V., Petutschnig, E., Lipka, V., and Rathjen, J. P. (2009). AvrPtoB targets the LysM receptor kinase CERK1 to promote bacterial virulence on plants. Curr. Biol. 19, 423–429.
Gohre, V., and Robatzek, S. (2008). Breaking the barriers: microbial effector molecules subvert plant immunity. Annu. Rev. Phytopathol. 46, 189–215.
Goldstein, G., Scheid, M., Hammerling, U., Schlesinger, D. H., Niall, H. D., and Boyse, E. A. (1975). Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. U.S.A. 72, 11–15.
Haase, R., Richter, K., Pfaffinger, G., Courtois, G., and Ruckdeschel, K. (2005). Yersinia outer protein P suppresses TGF-beta-activated kinase-1 activity to impair innate immune signaling in Yersinia enterocolitica-infected cells. J. Immunol. 175, 8209–8217.
Haraga, A., and Miller, S. I. (2006). A Salmonella type III secretion effector interacts with the mammalian serine/threonine protein kinase PKN1. Cell. Microbiol. 8, 837–846.
Hardt, W. D., and Galán, J. E. (1997). A secreted Salmonella protein with homology to an avirulence determinant of plant pathogenic bacteria. Proc. Natl. Acad. Sci. U.S.A. 94, 9887–9892.
Hartman, A. B., Venkatesan, M., Oaks, E. V., and Buysse, J. M. (1990). Sequence and molecular characterization of a multicopy invasion plasmid antigen gene, ipaH, of Shigella flexneri. J. Bacteriol. 172, 1905–1915.
Hatakeyama, S., Jensen, J. P., and Weissman, A. M. (1997). Subcellular localization and ubiquitin-conjugating enzyme (E2) interactions of mammalian HECT family ubiquitin protein ligases. J. Biol. Chem. 272, 15085–15092.
Hayes, C. S., Aoki, S. K., and Low, D. A. (2010). Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 44, 71–90.
Hershko, A., Heller, H., Elias, S., and Ciechanover, A. (1983). Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258, 8206–8214.
Hess, J., Dreher, A., Gentschev, I., Goebel, W., Ladel, C., Miko, D., and Kaufmann, S. H. (1996). Protein p60 participates in intestinal host invasion by Listeria monocytogenes. Zentralbl. Bakteriol. 284, 263–272.
Hirooka, T., Rogowsky, P. M., and Kado, C. I. (1987). Characterization of the virE locus of Agrobacterium tumefaciens plasmid pTiC58. J. Bacteriol. 169, 1529–1536.
Hotson, A., Chosed, R., Shu, H. J., Orth, K., and Mudgett, M. B. (2003). Xanthomonas type III effector XopD targets SUMO-conjugated proteins in planta. Mol. Microbiol. 50, 377–389.
Hough, R., Pratt, G., and Rechsteiner, M. (1986). Ubiquitin-lysozyme conjugates. Identification and characterization of an ATP-dependent protease from rabbit reticulocyte lysates. J. Biol. Chem. 261, 2400–2408.
Huibregtse, J. M., Scheffner, M., Beaudenon, S., and Howley, P. M. (1995). A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U.S.A. 92, 2563–2567.
Janjusevic, R., Abramovitch, R. B., Martin, G. B., and Stebbins, C. E. (2006). A bacterial inhibitor of host programmed cell death defenses is an E3 ubiquitin ligase. Science 311, 222–226.
Jin, J., Li, X., Gygi, S. P., and Harper, J. W. (2007). Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 447, 1135–1138.
Jones, R. M., Wu, H., Wentworth, C., Luo, L., Collier-Hyams, L., and Neish, A. S. (2008). Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 3, 233–244.
Karin, M., and Ben-Neriah, Y. (2000). Phosphorylation meets ubiquitination: the control of NF-kappa B activity. Annu. Rev. Immunol. 18, 621.
Kattenhorn, L. M., Korbel, G. A., Kessler, B. M., Spooner, E., and Ploegh, H. L. (2005). A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 19, 547–557.
Kim, D. W., Lenzen, G., Page, A. L., Legrain, P., Sansonetti, P. J., and Parsot, C. (2005). The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U.S.A. 102, 14046–14051.
Kim, H. T., Kim, K. P., Lledias, F., Kisselev, A. F., Scaglione, K. M., Skowyra, D., Gygi, S. P., and Goldberg, A. L. (2007). Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 282, 17375–17386.
Knodler, L. A., and Steele-Mortimer, O. (2003). Taking possession: biogenesis of the Salmonella-containing vacuole. Traffic 4, 587–599.
Kubori, T., Hyakutake, A., and Nagai, H. (2008). Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 67, 1307–1319.
Kubori, T., Shinzawa, N., Kanuka, H., and Nagai, H. (2010). Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog. 6, e1001216. doi: 10.1371/journal.ppat.1001216
Lapaque, N., Hutchinson, J. L., Jones, D. C., Meresse, S., Holden, D. W., Trowsdale, J., and Kelly, A. P. (2009). Salmonella regulates polyubiquitination and surface expression of MHC class II antigens. Proc. Natl. Acad. Sci. U.S.A. 106, 14052–14057.
Lavie, M., Shillington, E., Eguiluz, C., Grimsley, N., and Boucher, C. (2002). PopP1, a new member of the YopJ/AvrRxv family of type III effector proteins, acts as a host-specificity factor and modulates aggressiveness of Ralstonia solanacearum. Mol. Plant Microbe Interact. 15, 1058–1068.
Le Negrate, G., Faustin, B., Welsh, K., Loeffler, M., Krajewska, M., Hasegawa, P., Mukherjee, S., Orth, K., Krajewski, S., Godzik, A., Guiney, D. G., and Reed, J. C. (2008). Salmonella secreted factor L deubiquitinase of Salmonella typhimurium inhibits NF-kappa B, suppresses I kappa B alpha ubiquitination and modulates innate immune responses. J. Immunol. 180, 5045–5056.
Levin, I., Eakin, C., Blanc, M. P., Klevit, R. E., Miller, S. I., and Brzovic, P. S. (2010). Identification of an unconventional E3 binding surface on the UbcH5 ~Ub conjugate recognized by a pathogenic bacterial E3 ligase. Proc. Natl. Acad. Sci. U.S.A. 107, 2848–2853.
Li, S. J., and Hochstrasser, M. (1999). A new protease required for cell-cycle progression in yeast. Nature 398, 246–251.
Lin, D. Y., Diao, J., Zhou, D., and Chen, J. (2011). Biochemical and structural studies of a HECT-like ubiquitin ligase from Escherichia coli O157:H7. J. Biol. Chem. 286, 441–449.
Liu, K. H., and Shen, W. C. (2011). Mating differentiation in Cryptococcus neoformans is negatively regulated by the Crk1 protein kinase. Fungal Genet. Biol. 48, 225–240.
Liu, X. Y., Lu, R., Xia, Y. L., Wu, S. P., and Sun, J. (2010). Eukaryotic signaling pathways targeted by Salmonella effector protein AvrA in intestinal infection in vivo. BMC Microbiol. 10, 326. doi:10.1186/1471-2180-10-326
Lomma, M., Dervins-Ravault, D., Rolando, M., Nora, T., Newton, H. J., Sansom, F. M., Sahr, T., Gomez-Valero, L., Jules, M., Hartland, E. L., and Buchrieser, C. (2010). The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cell. Microbiol. 12, 1272–1291.
Lorick, K. L., Jensen, J. P., Fang, S., Ong, A. M., Hatakeyama, S., and Weissman, A. M. (1999). RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 96, 11364–11369.
Lostroh, C. P., and Lee, C. A. (2001). The Salmonella pathogenicity island-1 type III secretion system. Microbes Infect. 3, 1281–1291.
Matsuda, N., Suzuki, T., Tanaka, K., and Nakano, A. (2001). Rma1, a novel type of RING finger protein conserved from Arabidopsis to human, is a membrane-bound ubiquitin ligase. J. Cell. Sci. 114, 1949–1957.
Mcdonald, C., Vacratsis, P. O., Bliska, J. B., and Dixon, J. E. (2003). The Yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J. Biol. Chem. 278, 18514–18523.
Mcgrath, J. P., Jentsch, S., and Varshavsky, A. (1991). UBA 1: an essential yeast gene encoding ubiquitin-activating enzyme. EMBO J. 10, 227–236.
Miao, E. A., Brittnacher, M., Haraga, A., Jeng, R. L., Welch, M. D., and Miller, S. I. (2003). Salmonella effectors translocated across the vacuolar membrane interact with the actin cytoskeleton. Mol. Microbiol. 48, 401–415.
Miao, E. A., Scherer, C. A., Tsolis, R. M., Kingsley, R. A., Adams, L. G., Baumler, A. J., and Miller, S. I. (1999). Salmonella typhimurium leucine-rich repeat proteins are targeted to the SPI1 and SPI2 type III secretion systems. Mol. Microbiol. 34, 850–864.
Misaghi, S., Balsara, Z. R., Catic, A., Spooner, E., Ploegh, H. L., and Starnbach, M. N. (2006). Chlamydia trachomatis-derived deubiquitinating enzymes in mammalian cells during infection. Mol. Microbiol. 61, 142–150.
Mittal, R., Peak-Chew, S. Y., and Mcmahon, H. T. (2006). Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 18574–18579.
Monack, D. M., Mecsas, J., Bouley, D., and Falkow, S. (1998). Yersinia-induced apoptosis in vivo aids in the establishment of a systemic infection of mice. J. Exp. Med. 188, 2127–2137.
Morikawa, H., Kim, M., Mimuro, H., Punginelli, C., Koyama, T., Nagai, S., Miyawaki, A., Iwai, K., and Sasakawa, C. (2010). The bacterial effector Cif interferes with SCF ubiquitin ligase function by inhibiting deneddylation of Cullin1. Biochem. Biophys. Res. Commun. 401, 268–274.
Mukherjee, S., Keitany, G., Li, Y., Wang, Y., Ball, H. L., Goldsmith, E. J., and Orth, K. (2006). Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214.
Neish, A. S., Gewirtz, A. T., Zeng, H., Young, A. N., Hobert, M. E., Karmali, V., Rao, A. S., and Madara, J. L. (2000). Prokaryotic regulation of epithelial responses by inhibition of IκB-α ubiquitination. Science 289, 1560–1563.
Noel, L., Thieme, F., Gabler, J., Buttner, D., and Bonas, U. (2003). XopC and XopJ, two novel type III effector proteins from Xanthomonas campestris pv. vesicatoria. J. Bacteriol. 185, 7092–7102.
Oh, C. S., Kim, J. F., and Beer, S. V. (2005). The Hrp pathogenicity island of Erwinia amylovora and identification of three novel genes required for systemic infection. Mol. Plant Pathol. 6, 125–138.
Okuda, J., Toyotome, T., Kataoka, N., Ohno, M., Abe, H., Shimura, Y., Seyedarabi, A., Pickersgill, R., and Sasakawa, C. (2005). Shigella effector IpaH9.8 binds to a splicing factor U2AF(35) to modulate host immune responses. Biochem. Biophys. Res. Commun. 333, 531–539.
Orth, K., Xu, Z., Mudgett, M. B., Bao, Z. Q., Palmer, L. E., Bliska, J. B., Mangel, W. F., Staskawicz, B., and Dixon, J. E. (2000). Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science 290, 1594–1597.
Palmer, L. E., Hobbie, S., Galán, J. E., and Bliska, J. B. (1998). YopJ of Yersinia pseudotuberculosis is required for inhibition of macrophage TNF-production and downregulation of MAP kinases p38 and JNK. Mol. Microbiol. 27, 953–966.
Pedley, K. F., and Martin, G. B. (2003). Molecular basis of Pto-mediated resistance to bacterial speck disease in tomato. Annu. Rev. Phytopathol. 41, 215–243.
Pelzer, C., Kassner, I., Matentzoglu, K., Singh, R. K., Wollscheid, H. P., Scheffner, M., Schmidtke, G., and Groettrup, M. (2007). UBE1L2, a novel E1 enzyme specific for ubiquitin. J. Biol. Chem. 282, 23010–23014.
Piscatelli, H., Kotkar, S. A., Mcbee, M. E., Muthupalani, S., Schauer, D. B., Mandrell, R. E., Leong, J. M., and Zhou, D. (2011). The EHEC type III effector NleL is an E3 ubiquitin ligase that modulates pedestal formation. PLoS ONE 6, e19331. doi: 10.1371/journal.pone.0019331
Prasad, J., Colwill, K., Pawson, T., and Manley, J. L. (1999). The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol. 19, 6991–7000.
Quezada, C. M., Hicks, S. W., Galan, J. E., and Stebbins, C. E. (2009). A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. U.S.A. 106, 4864–4869.
Reiss, Y., Heller, H., and Hershko, A. (1989). Binding sites of ubiquitin-protein ligase. Binding of ubiquitin-protein conjugates and of ubiquitin-carrier protein. J. Biol. Chem. 264, 10378–10383.
Roden, J., Eardley, L., Hotson, A., Cao, Y., and Mudgett, M. B. (2004). Characterization of the Xanthomonas AvrXv4 effector, a SUMO protease translocated into plant cells. Mol. Plant Microbe Interact. 17, 633–643.
Rohde, J. R., Breitkreutz, A., Chenal, A., Sansonetti, P. J., and Parsot, C. (2007). Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe 1, 77–83.
Rosebrock, T. R., Zeng, L., Brady, J. J., Abramovitch, R. B., Xiao, F., and Martin, G. B. (2007). A bacterial E3 ubiquitin ligase targets a host protein kinase to disrupt plant immunity. Nature 448, 370–374.
Ruckdeschel, K., Harb, S., Roggenkamp, A., Hornef, M., Zumbihl, R., Kohler, S., Heesemann, J., and Rouot, B. (1998). Yersinia enterocolitica impairs activation of transcription factor NF-kappa B: involvement in the induction of programmed cell death and in the suppression of the macrophage tumor necrosis factor alpha production. J. Exp. Med. 187, 1069–1079.
Rytkonen, A., Poh, J., Garmendia, J., Boyle, C., Thompson, A., Liu, M., Freemont, P., Hinton, J. C., and Holden, D. W. (2007). SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 3502–3507.
Scheffner, M., Nuber, U., and Huibregtse, J. M. (1995). Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 373, 81–83.
Schesser, K., Spiik, A. K., Dukuzumuremyi, J. M., Wolf-Watz, H., and Pettersson, S. (1998). Inhibition of NF-kB-mediated signalling by the Yersinia-encoded YopJ protein. FASEB J. 12, 6172.
Schlieker, C., Korbel, G. A., Kattenhorn, L. M., and Ploegh, H. L. (2005). A deubiquitinating activity is conserved in the large tegument protein of the Herpesviridae. J. Virol. 79, 15582–15585.
Schmitz-Esser, S., Tischler, P., Arnold, R., Montanaro, J., Wagner, M., Rattei, T., and Horn, M. (2010). The genome of the amoeba symbiont “ Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J. Bacteriol. 192, 1045–1057.
Schrammeijer, B., Risseeuw, E., Pansegrau, W., Regensburg-Tuink, T. J. G., Crosby, W. L., and Hooykaas, P. J. J. (2001). Interaction of the virulence protein VirF of Agrobacterium tumefaciens with plant homologs of the yeast Skp1 protein. Curr. Biol. 11, 258–262.
Schwarz, S. E., Rosa, J. L., and Scheffner, M. (1998). Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J. Biol. Chem. 273, 12148–12154.
Schwertz, H., Tolley, N. D., Foulks, J. M., Denis, M. M., Risenmay, B. W., Buerke, M., Tilley, R. E., Rondina, M. T., Harris, E. M., Kraiss, L. W., Mackman, N., Zimmerman, G. A., and Weyrich, A. S. (2006). Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenecity of human platelets. J. Exp. Med. 203, 2433–2440.
Segal, G., Purcell, M., and Shuman, H. A. (1998). Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. U.S.A. 95, 1669–1674.
Seyedarabi, A., Sullivan, J. A., Sasakawa, C., and Pickersgill, R. W. (2010). A disulfide driven domain swap switches off the activity of Shigella IpaH9.8E. 3 ligase. FEBS Lett. 584, 4163–4168.
Shackelford, J., and Pagano, J. S. (2005). Targeting of host-cell ubiquitin pathways by viruses. Essays Biochem. 41, 139–156.
Singer, A. U., Rohde, J. R., Lam, R., Skarina, T., Kagan, O., Dileo, R., Chirgadze, N. Y., Cuff, M. E., Joachimiak, A., Tyers, M., Sansonetti, P. J., Parsot, C., and Savchenko, A. (2008). Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat. Struct. Mol. Biol. 15, 1293–1301.
Skaar, J. R., D’angiolella, V., Pagan, J. K., and Pagano, M. (2009a). Snapshot: F box proteins II. Cell 137, 1358–U1100.
Skaar, J. R., Pagan, J. K., and Pagano, M. (2009b). Snapshot: F box proteins I. Cell 137, 1160–1160.e1.
Soundararajan, V., Patel, N., Subramanian, V., Sasisekharan, V., and Sasisekharan, R. (2011). The many faces of the YopM effector from plague causative bacterium Yersinia pestis and its implications for host immune modulation. Innate Immun. . [Epub ahead of print].
Soundararajan, V., Raman, R., Raguram, S., Sasisekharan, V., and Sasisekharan, R. (2010). Atomic interaction networks in the core of protein domains and their native folds. PLoS ONE 5, e9391. doi:10.1371/journal.pone.0009391
Sun, L., and Chen, Z. J. (2004). The novel functions of ubiquitination in signaling. Curr. Opin. Cell Biol. 16, 119–126.
Sun, J., Hobert, M. E., Rao, A. S., Neish, A. S., and Madara, J. L. (2004). Bacterial activation of beta-catenin signaling in human epithelia. Am. J. Physiol.-Gastrointest. Liver Physiol. 287, G220–G227.
Szczesny, R., Buttner, D., Escolar, L., Schulze, S., Seiferth, A., and Bonas, U. (2010). Suppression of the AvrBs1-specific hypersensitive response by the YopJ effector homolog AvrBsT from Xanthomonas depends on a SNF1-related kinase. New Phytol. 187, 1058–1074.
Tasset, C., Bernoux, M., Jauneau, A., Pouzet, C., Briere, C., Kieffer-Jacquinod, S., Rivas, S., Marco, Y., and Deslandes, L. (2010). Autoacetylation of the Ralstonia solanacearum effector PopP2 targets a lysine residue essential for RRS1-R-mediated immunity in Arabidopsis. PLoS Pathog. 6, e1001202. doi: 10.1371/journal.ppat.1001202
Thiefes, A., Wolf, A., Doerrie, A., Grassl, G. A., Matsumoto, K., Autenrieth, I., Bohn, E., Sakurai, H., Niedenthal, R., Resch, K., and Kracht, M. (2006). The Yersinia enterocolitica effector YopP inhibits host cell signalling by inactivating the protein kinase TAK1 in the IL-1 signalling pathway. EMBO Rep. 7, 838–844.
Trosky, J. E., Li, Y., Mukherjee, S., Keitany, G., Ball, H., and Orth, K. (2007). VopA inhibits ATP binding by acetylating the catalytic loop of MAPK kinases. J. Biol. Chem. 282, 34299–34305.
Trosky, J. E., Mukherjee, S., Burdette, D. L., Roberts, M., Mccarter, L., Siegel, R. M., and Orth, K. (2004). Inhibition of MAPK signaling pathways by VopA from Vibrio parahaemolyticus. J. Biol. Chem. 279, 51953–51957.
Trulzsch, K., Sporleder, T., Igwe, E. I., Russmann, H., and Heesemann, J. (2004). Contribution of the major secreted yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect. Immun. 72, 5227–5234.
Tsolis, R. M., Townsend, S. M., Miao, E. A., Miller, S. I., Ficht, T. A., Adams, L. G., and Baumler, A. J. (1999). Identification of a putative Salmonella enterica serotype typhimurium host range factor with homology to IpaH and YopM by signature-tagged mutagenesis. Infect. Immun. 67, 6385–6393.
Tsukahara, T., Kimura, S., Ichimiya, S., Torigoe, T., Kawaguchi, S., Wada, T., Yamashita, T., and Sato, N. (2009). Scythe/BAT3 regulates apoptotic cell death induced by papillomavirus binding factor in human osteosarcoma. Cancer Sci. 100, 47–53.
Tzfira, T., Vaidya, M., and Citovsky, V. (2004). Involvement of targeted proteolysis in plant genetic transformation by Agrobacterium. Nature 431, 87–92.
Venkatesan, M. M., Buysse, J. M., and Hartman, A. B. (1991). Sequence variation in two ipaH genes of Shigella flexneri 5 and homology to the LRG-like family of proteins. Mol. Microbiol. 5, 2435–2445.
Vogel, J. P., Andrews, H. L., Wong, S. K., and Isberg, R. R. (1998). Conjugative transfer by the virulence system of Legionella pneumophila. Science 279, 873–876.
Weissman, A. M. (2001). Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2, 169–178.
Whalen, M. C., Richter, T., Zakhareyvich, K., Yoshikawa, M., Al-Azzeh, D., Adefioye, A., Spicer, G., Mendoza, L. L., Morales, C. Q., Klassen, V., Perez-Baron, G., Toebe, C. S., Tzovolous, A., Gerstman, E., Evans, E., Thompson, C., Lopez, M., and Ronald, P. C. (2008). Identification of a host 14-3-3 protein that interacts with Xanthomonas effector AvrRxv. Physiol. Mol. Plant Pathol. 72, 46–55.
Whalen, M. C., Wang, J. F., Carland, F. M., Heiskell, M. E., Dahlbeck, D., Minsavage, G. V., Jones, J. B., Scott, J. W., Stall, R. E., and Staskawicz, B. J. (1993). Avirulence gene avrRxv from Xanthomonas campestris pv. vesicatoria specifies resistance on tomato line Hawaii 7998. Mol. Plant Microbe Interact. 6, 616–627.
Wilkinson, K. D., Urban, M. K., and Haas, A. L. (1980). Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes. J. Biol. Chem. 255, 7529–7532.
Wood, M. W., Jones, M. A., Watson, P. R., Siber, A. M., Mccormick, B. A., Hedges, S., Rosqvist, R., Wallis, T. S., and Galyov, E. E. (2000). The secreted effector protein of Salmonella dublin, SopA, is translocated into eukaryotic cells and influences the induction of enteritis. Cell. Microbiol. 2, 293–303.
Wu, B., Skarina, T., Yee, A., Jobin, M. C., Dileo, R., Semesi, A., Fares, C., Lemak, A., Coombes, B. K., Arrowsmith, C. H., Singer, A. U., and Savchenko, A. (2010). NleG type 3 effectors from enterohaemorrhagic Escherichia coli are U-Box E3 ubiquitin ligases. PLoS Pathog. 6, e1000960. doi: 10.1371/journal.ppat.1000960
Yamamoto, Y., Huibregtse, J. M., and Howley, P. M. (1997). The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics 41, 263–266.
Yamanaka, K., Ishikawa, H., Megumi, Y., Tokunaga, F., Kanie, M., Rouault, T. A., Morishima, I., Minato, N., Ishimori, K., and Iwai, K. (2003). Identification of the ubiquitin-protein ligase that recognizes oxidized IRP2. Nat. Cell Biol. 5, 336–340.
Yanase, S., and Ishi, N. (1999). Cloning of the oxidative stress-responsive genes in Caenorhabditis elegans. J. Radiat. Res. 40, 39–47.
Yang, F. J., Cheng, L. L., Zhang, L., Dai, W. J., Liu, Z., Yao, N., Xie, Z. P., and Staehelin, C. (2009). Y4lO of Rhizobium sp strain NGR234 is a symbiotic determinant required for symbiosome differentiation. J. Bacteriol. 191, 735–746.
Ye, Z., Petrof, E. O., Boone, D., Claud, E. C., and Sun, J. (2007). Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am. J. Pathol. 171, 882–892.
You, J., and Pickart, C. M. (2001). A HECT domain E3 enzyme assembles novel polyubiquitin chains. J. Biol. Chem. 276, 19871–19878.
Zacksenhaus, E., and Sheinin, R. (1990). Molecular cloning, primary structure and expression of the human X linked A1S9 gene cDNA which complements the ts A1S9 mouse L cell defect in DNA replication. EMBO J. 9, 2923–2929.
Zaltsman, A., Krichevsky, A., Loyter, A., and Citovsky, V. (2010). Agrobacterium induces expression of a host F-box protein required for tumorigenicity. Cell Host Microbe 7, 197–209.
Zhang, S., Santos, R. L., Tsolis, R. M., Stender, S., Hardt, W. D., Baumler, A. J., and Adams, L. G. (2002). The Salmonella enterica serotype typhimurium effector proteins SipA, SopA, SopB, SopD, and SopE2 act in concert to induce diarrhea in calves. Infect. Immun. 70, 3843–3855.
Zhang, Y., Higashide, W., Dai, S., Sherman, D. M., and Zhou, D. (2005). Recognition and ubiquitination of Salmonella type III effector SopA by a ubiquitin E3 ligase, HsRMA1. J. Biol. Chem. 280, 38682–38688.
Zhang, Y., Higashide, W. M., Mccormick, B. A., Chen, J., and Zhou, D. G. (2006). The inflammation-associated Salmonella SopA is a HECT-like E3 ubiquitin ligase. Mol. Microbiol. 62, 786–793.
Zhou, H., Monack, D. M., Kayagaki, N., Wertz, I., Yin, J., Wolf, B., and Dixit, V. M. (2005). Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-kappaB activation. J. Exp. Med. 202, 1327–1332.
Zhou, H. B., Lin, J. A., Johnson, A., Morgan, R. L., Zhong, W. W., and Ma, W. B. (2011). Pseudomonas syringae type III effector HopZ1 targets a host enzyme to suppress isoflavone biosynthesis and promote infection in soybean. Cell Host Microbe 9, 177–186.
Zhu, W. H., Banga, S., Tan, Y. H., Zheng, C., Stephenson, R., Gately, J., and Luo, Z. Q. (2011). Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS ONE 6, e17638. doi: 10.1371/journal.pone.0017638
Zhu, Y., Li, H., Hu, L., Wang, J., Zhou, Y., Pang, Z., Liu, L., and Shao, F. (2008). Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat. Struct. Mol. Biol. 14, 1302–1308.
Keywords: ubiquitin, E3 ligase, F-box, DUB, bacterial pathogen
Citation: Perrett CA, Lin DY-W and Zhou D (2011) Interactions of bacterial proteins with host eukaryotic ubiquitin pathways. Front. Microbio. 2:143. doi: 10.3389/fmicb.2011.00143
Received: 11 May 2011; Accepted: 16 June 2011;
Published online: 04 July 2011.
Edited by:
John S. Gunn, The Ohio State University, USAReviewed by:
Stephanie M. Seveau, The Ohio State University, USAJuan Martinez, University of Chicago, USA
Copyright: © 2011 Perrett, Lin and Zhou. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Daoguo Zhou, Department of Biological Sciences, Purdue University, Lilly Hall, 915 West State Street, West Lafayette, IN, USA. e-mail:emhvdWRAcHVyZHVlLmVkdQ==