
- 1 Center for Vascular and Inflammatory Diseases, University of Maryland School of Medicine, Baltimore, MD, USA
- 2 Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD, USA
- 3 University of Maryland at Shady Grove Center, Rockville, MD, USA
- 4 Department of Surgery, University of Maryland School of Medicine, Baltimore, MD USA
Genetically modified B cells are excellent tolerogenic antigen-presenting cells (APCs) in multiple models of autoimmunity. However, the mechanisms of action are still not completely understood. In our models, we generate antigen-specific tolerogenic B cells by transducing naïve or primed B cells with an antigen–immunoglobulin G (peptide–IgG) construct. In order to be transduced, B cells require activation with mitogens such as LPS. We and others have found that LPS stimulation of B cells upregulates the production of IL-10, a key cytokine for maintaining immune tolerance. In the current study, we defined the role of B-cell produced IL-10 in tolerance induction by using IL-10 deficient B cells as donor APCs. We found that peptide–IgG transduced IL-10 KO B cells have the same effects as wt B cells in tolerance induction in an experimental autoimmune encephalomyelitis model. Moreover, we demonstrated that the tolerogenic effect of peptide–IgG B cells was completely abrogated in anti-IL-10 receptor antibody treated recipients. Taken together, our results suggest that tolerance induced by peptide–IgG B-cell gene therapy requires IL-10 from the host but not donor B cells. These data shed important insights into the mechanisms of tolerance induction mediated by B-cell gene therapy.
Introduction
Immune tolerance to self-antigens must be maintained or autoimmunity occurs. Based on the excellent tolerogenicity of IgG (Aldo-Benson and Borel, 1976; Borel et al., 1976) and the capacity of B cells to be tolerogenic antigen-presenting cells (APCs; Eynon and Parker, 1992; Fuchs and Matzinger, 1992; Gilbert and Weigle, 1994), our laboratory has developed a B-cell based gene therapy approach for actively inducing tolerance in animal models of autoimmune diseases and hemophilia (Skupsky et al., 2007). For example, when retrovirally transduced with a fusion construct encoding IgG heavy chain and factor VIII (fVIII) immunodominant domain C2 or A2, B cells are tolerogenic in both naïve and fVIII-primed hemophilic mice (E16, fVIII−/−) as measured by specific cellular response and inhibitory antibody titers (Lei and Scott, 2005). In experimental autoimmune encephalomyelitis (EAE), myelin oligodendrocyte glycoprotein (MOG)–IgG, or myelin basic protein (MBP)–IgG transduced B cells can protect mice from EAE (Xu and Scott, 2004; Zhang et al., 2010). In other models of autoimmune diseases including experimental autoimmune uveitis, type I diabetes, and arthritis, animals that received peptide–IgG transduced B cells exhibited reduced specific humoral and cellular responses, delayed disease onset, and amelioration of ongoing disease (Agarwal et al., 2000; Melo et al., 2002; Soukhareva et al., 2006; Satpute et al., 2007). Achievement of antigen-specific tolerance in these models indicates the promise of this method as therapy for patients with autoimmune diseases or hemophiliacs with fVIII inhibitor formation. Similar results were reported by several other groups where B cells were also utilized for tolerance induction in EAE (Chen et al., 2001, 2004) and in a mouse model of asthma (Ahangarani et al., 2009).
The underlying mechanisms for tolerance induction by B-cell delivered gene therapy remains incompletely understood. IL-10 is a key regulator of immune-suppression and for maintenance of peripheral tolerance (Kuhn et al., 1993). Originally, IL-10 was defined as a Th2 cytokine that inhibits Th1 T-cell responses (Fiorentino et al., 1989). Later studies suggest that IL-10 can be produced by multiple types of T cells including CD4+CD25+ regulatory T cells (Tregs), as well as APCs. In the resting state, B cells produce only low levels of IL-10. However, activated B cells dramatically upregulate the production of IL-10 (Fillatreau et al., 2002; Barr et al., 2007; Lampropoulou et al., 2008). Whether IL-10 is involved in the mechanisms of tolerance induction by B-cell based gene therapy remains controversial. Ahangarani et al. (2009) demonstrated that genetically modified B cells can induce tolerance in an allergy model in an IL-10 dependent manner. On the other hand, Frommer et al. (2008) found that production of IL-10 by MOG presenting B cells plays no role in tolerance induction in an EAE model. To optimize our B-cell based gene therapy protocol for future clinical study, we screened multiple mitogens for B-cell activation and, interestingly, found that the manner of B-cell activation was critical for tolerance induction. Thus, LPS-activated B cells are tolerogenic, but unmethylated DNA CpG stimulated B cells are not (Lei et al., 2005). Further investigation indicated that LPS-treated B cells produce significantly higher amounts of IL-10 than CpG-treated B cells (Skupsky et al., 2007). These findings raise an interesting question regarding the mechanisms of tolerance induction: Do B cells really need IL-10 to induce tolerance?
In the current study, the role of IL-10 was tested by using IL-10 KO mice as B-cell donors or by treating recipient animals with an anti-IL-10 receptor (IL-10R) antibody. We found that IL-10 produced by host but not by donor B cells is critical for tolerance induction. Since we previously showed that CD4+CD25+ Tregs were required for the induction and maintenance of tolerance (Lei and Scott, 2005; Soukhareva et al., 2006), we propose that these cells may be the source of endogenous IL-10 in the host.
Materials and Methods
Mice
C57BL/6, Balb/c, membrane ovalbumin expressing (mOVA) transgenic, and H-2b IL-10 KO mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). H-2d IL-10 KO mice were a kind gift from Dr. Wendy Davidson (University of Maryland School of Medicine, Baltimore, MD, USA). All recipient animals were used at 5–8 weeks of age and housed in pathogen-free micro-isolator cages in the animal facility at the University of Maryland, Baltimore. All animal experiments were approved by IACUC of University of Maryland School of Medicine.
Antibodies
Anti-IL-10R (1B1.3a, rat IgG1) and control mAb (GL113, anti-E.coli β-galactosidase, rat IgG1) hybridomas were originally provided by Dr. Kevin Moore (DNAX, Palo Alto, CA, USA). The anti-IL-10R and control mAbs used in vivo were purified from ascites produced in nude mice by ammonium sulfate precipitation and ion exchange chromatography (Harlan Bioproducts).
Virus-Producing Cell Lines
The cDNA of the peptide–IgG1 heavy chain was subcloned into the murine Moloney leukemia retroviral vector (MBAE), as previously described (Zambidis et al., 1997a,b). Briefly, a DNA fragment of pOVA323–339 or full-length OVA was inserted into the BSSK-IgG vector and then the IgG fusion construct was subsequently subcloned into the MBAE retroviral vector. Virus-producer cell line was prepared by stable transfection of GP + E86 packaging cells with the engineered construct. Viral producing packaging cells were maintained in DMEM medium supplemented with 10% FBS, 2 mM L-glutamine, and 2-mercaptoethanol.
B-Cell Purification and Retroviral Transduction
Splenic B cells from naïve mice were purified to approximately 95% homogeneity with anti-T-cell antibody cocktail (anti-Thy1, anti-CD4, and anti-CD8) followed by complement (Low-Tox-M, Cedarlane Laboratories, Accurate Chemical and Scientific Corporation, Westbury, NY, USA). Purified B cells were pre-stimulated with 1 μg/ml LPS (E. coli 055:B5, Sigma, St. Louis, MO, USA) overnight and then transduced in vitro via co-culture with 1500 rad irradiated virus-producing packaging cells for 24 h in the presence of 6 μg/ml polybrene.
Tolerance Induction to OVA and T-Cell Proliferation Assay
1 × 107 pOVA323–339-IgG or OVA–IgG transduced wt or IL-10 KO B cells were transferred into naïve mice intraperitoneally. One week later, recipient mice were immunized with 25 μg OVA protein emulsified in CFA in one hind footpad and the base of tail. Two weeks after immunization, mice were bled and then sacrificed. Draining inguinal and popliteal LNs were removed for T-cell proliferation assay. Single cell solution was prepared at 5 × 106 cells/ml. One hundred microliter cells were seeded onto 96-well plate in the presence of indicated concentrations of pOVA peptide. 48 h later, cell culture were pulsed with 1 μCi [3H] thymidine (Amersham Life Sciences, Arlington Heights, IL, USA) and incubated for another 16–20 h. Cells were then harvested on glass fiber filters and [3H] thymidine incorporation was counted by using Scintillation Counter. Data were presented as mean Δcpm (count per minute by subtraction of the background) ± SE. Anti-OVA–IgG titers were determined by endpoint ELISA methods.
IL-10R blocking in vivo
Anti-IL-10R (1B1.3a, rat IgG1) mAb was used to block IL-10 function in vivo and anti-E.coli β-galactosidase was used as isotype control. Mice were received B-cell gene therapy (retrovirally transduced wt B cells or mOVA transgenic B cells in different experiments) on day 0 and challenged on day 7. These recipient mice were injected four times on day −3, 0, 7, and 14, respectively. Each time 1 mg/mouse of anti-IL-10R or control mAb was injected intraperitoneally.
B-Cell Gene Therapy for Tolerance Induction in Murine EAE
One week before the active induction of EAE, C57BL/6 (B6) were adoptively transferred with 1 × 107 retrovirally transduced syngeneic tolerogenic B cells expressing MOG35–55–Ig (MOG–Ig), intraperitoneally. For EAE induction, 6-week-old female B6 mice were subcutaneously immunized on the flanks with 200 μg of MOG35–55 peptide emulsified in CFA containing 4 mg/ml of Mycobacterium tuberculosis H37Ra (DIFCO, Detroit, MI). On the day of immunization and 48 h later, the mice also received 200 ng of Pertussis toxin (Sigma-Aldrich) in 0.2 ml PBS intraperitoneally. Clinical signs of EAE were assessed daily with a 0–5 scoring system (Stromnes and Goverman, 2006): 0, normal; 0.5, partially limp tail; 1, paralyzed tail; 2, loss in coordinated movement; 2.5, one hind limb paralyzed; 3, hind limbs paralyzed; 3.5, hind limbs paralyzed and forelimbs weakness; 4, forelimbs paralyzed; 5, moribund.
Statistics
Paired or unequal variances one-tailed Student’s t-test statistics were applied with differences considered significant at *p < 0.05.
Results
IL-10 Produced by Donor B Cells is not Required for Tolerance Induction
Our laboratory has successfully applied the B-cell based peptide–IgG gene therapy protocol in multiple animal models for autoimmunity such as type 1 diabetes, EAE, experimental autoimmune uveitis, and also in a mouse model for hemophilia A. In this gene therapy protocol, B cells have to be activated before retroviral transduction as only dividing cells can be effectively transduced with the peptide–IgG construct. We routinely use LPS to stimulate B cells. Other B-cell mitogens such as anti-IgM and anti-CD40 have similar effects as LPS on B cells in term of tolerance induction. Surprisingly, although it could activate B cells effectively, unmethylated CpG DNA stimulation results in B cells losing their tolerogenic phenotype (Lei et al., 2005). We systematically compared the phenotypic changes of B cells upon LPS and CpG stimulation. Compared to the resting B cells, although both LPS and CpG B-cell blasts have upregulated expression of IL-10 at protein and messenger RNA levels, LPS B cells produce much more IL-10 than CpG B cells (Skupsky et al., 2007). These findings, together with the contradict reports from the literature, led us to ask whether IL-10 is critical for tolerance induction through our B-cell delivered gene therapy protocol. Therefore, in the current study, we first studied the role of B-cell derived IL-10 in our gene therapy system by using H-2d IL-10 KO mice as B-cell donors. We transduced LPS-activated B cells with a retroviral construct encoding an immunodominant epitope 323–339 fragment of ovalbumin (OVA) and IgG1 heavy chain (pOVA323–339–IgG) or a mock control (phospholipid protein, PLP). As expected, mice that received pOVA323–339–IgG transduced wt B cells exhibited reduced anti-OVA antibody. Surprisingly, pOVA323–339–IgG transduced IL-10 KO B cells also induced a significant reduction of antibody to OVA (Figure 1). This suggests that IL-10 KO B cells have normal antigen-presenting function and more importantly, that IL-10 produced by donor B cells is not required for tolerance induction. In addition, both pOVA323–339–IgG transduced wt or IL-10 KO B cells significantly reduced the T-cell response to pOVA323-339 in recipients. We confirmed these results by using H-2b IL-10 KO B cells as donors to induce tolerance to OVA protein in C57BL/6 mice (data not shown).
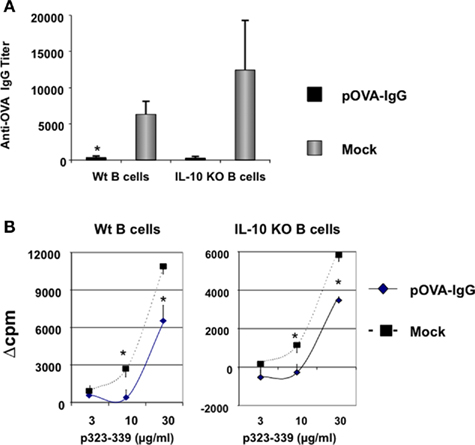
Figure 1. Donor B cells from IL-10 KO mice are tolerogenic. (A) LPS-activated wt or IL-10 KO B cells were transduced with pOVA323–339–IgG or a mock control vector (PLP–IgG) and 1 × 107 B cells per mouse were injected i.p. into naïve Balb/c recipients. Seven days later, mice were immunized with 25 μg OVA protein emulsified in CFA via a hind footpad and the base of tail. Two weeks after immunization, sera were collected and assayed for total anti-OVA–IgG by the endpoint ELISA method (n = 4). (B) Animals were treated as described in Figure 1A. Mice were sacrificed 2 weeks after immunization and draining lymph nodes were removed for CD4+ T-cell proliferation assay. Five × 105 cells from draining LNs were seeded per well in 96-well plates in the presence of indicated concentrations of p323–339. After 48 h, cultures were pulsed with 1 μCi/well of [3H] thymidine (Amersham Life Sciences, Arlington Heights, IL, USA) and incubated for another 16–20 h. Cells were then harvested on glass fiber filters and T-cell proliferation to p323–339 was determined by [3H] thymidine incorporation. Values represent mean Δcpm (count per minute by subtraction of the background) ± SE for four animals. Data are representative of two independent experiments.
To validate that IL-10 produced by tolerogenic B cells is not required for tolerance induction, we used IL-10 KO B cells as tolerogenic APCs to induce tolerance in the EAE model. We have previously showed that MOG–IgG transduced B cells were highly tolerogenic and adoptive transfer of MOG–IgG B cells protected recipients from EAE induction (Zhang et al., 2010). In the current study, we first transduced IL-10 KO C57BL/6 B cells with MOG–IgG or a mock control (OVA–IgG). Transduced B cells (1 × 107) were injected intraperitoneally into naïve C57BL/6 mice 1 week before active induction of EAE with MOG35–55. After EAE induction, we monitored the disease development daily. We found that MOG–IgG transduced wt B cells protected mice from EAE in terms of mean disease score and disease incidence during the early phase of the disease development (Figures 2A,B). However, we observed an overlap of the average clinical scores in the end of this experiment. Based on our previous findings (Zhang et al., 2010), we believe this is due to the relatively short observation period and does not indicate that the disease protection by wt B cells was transient. Interestingly, MOG–IgG transduced IL-10 KO B cells also protected mice from EAE, suggesting that IL-10 deficiency does not affect tolerance inducing property of B cells (Figures 2A,B). Furthermore, in a hemophilia mouse model, we found that fVIII domain C2–IgG transduced IL-10 KO B cells are tolerogenic to C2 peptide in vivo (data not shown). Taken together, we confirmed that tolerance induction by our B-cell gene therapy protocol does not depend on the IL-10 produced by donor B cells in multiple animal models from different genetic background.
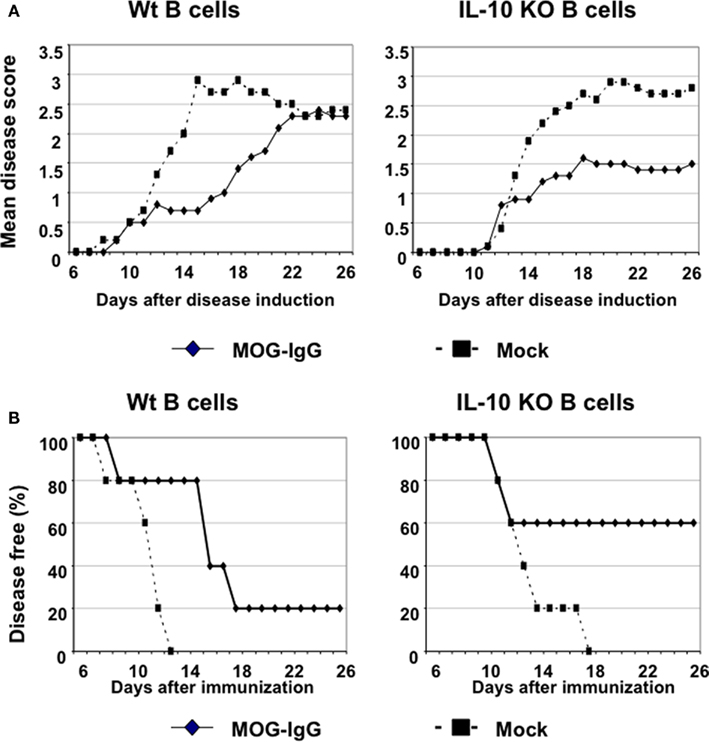
Figure 2. IL-10 deficient B cells are tolerogenic in an EAE model. Female C57BL/6 recipients (n = 5) were adoptively transferred with 1 × 107 retrovirally transduced B cells. One week later, the mice were actively immunized for EAE induction using MOG35–55 (200 μg)/CFA. The mice were also received 200 ng Pertussis toxin in 0.2 ml PBS via i.p. injection at the same day and 2 days after immunization. The clinical score was monitored daily using a 0–5 scoring system. Mean disease score (A) and the disease incidence (B) were shown. As the mice that received MOG–Ig transduced B cells from wt C57BL/6 donors (left panels), the mice that received MOG–Ig transduced IL-10 KO B cells (right panels) also had ameliorated disease symptoms and decreased disease incidence, as compared with the control.
IL-10 Produced by the Host is Required for Tolerance Induced by Peptide–IgG Expressing B Cells
In multiple models of infection and autoimmune diseases, mice that are genetically deficient in IL-10 exhibit dramatically exacerbated diseases (Kuhn et al., 1993; Bettelli et al., 1998; Yin et al., 2002). Since IL-10 plays such a critical role in immune regulation and maintenance of tolerance, we next sought to ask whether IL-10 produced by the host is required for tolerance induction in our model. To this end, we utilized in vivo anti-IL-10R antibody treatment to block IL-10 function in normal mice, rather than using IL-10 KO mice which have an autoimmune syndrome. Following the standard tolerance induction protocol, LPS-activated, OVA–IgG transduced B cells were transferred into naïve mice on day 0. On day −3, 0, 7, and 14, recipients were injected four times with anti-IL-10R or control rat IgG1 mAb. These recipients were immunized with OVA protein in CFA on day 7 and the effect of anti-IL-10R blocking on tolerance induction was evaluated on day 21 by testing in vitro T-cell response and antibody titer. As expected, control mAb treatment has no effect on the hyporesponsiveness induced by OVA–IgG B cells (Figures 3A,B). Mice that received OVA–IgG transduced B cells and control mAb treatment exhibited reduced T-cell and antibody responses against OVA protein. However, in anti-IL-10R treated mice, tolerance induction was completely abrogated, suggesting that IL-10 produced by the host is required for tolerance induction.
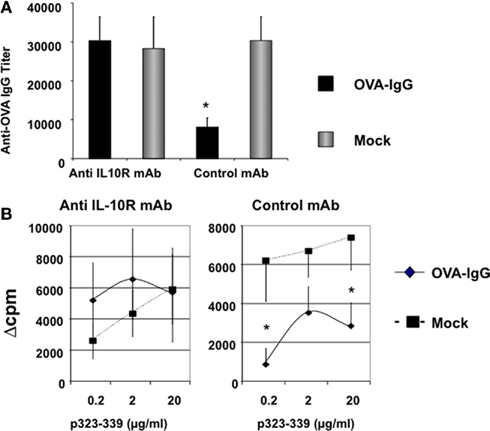
Figure 3. IL-10 produced by the host is required for tolerance induction by peptide–IgG transduced B cells. LPS-activated wt B cells were transduced with OVA–IgG or a mock control vector (PLP) and 1 × 107 B cells per mouse were injected i.p. into naïve C57BL/6 recipients on day 0. Seven days later, mice were immunized with 25 μg OVA protein emulsified in CFA via a hind footpad and the base of tail. Two weeks after immunization, mice were sacrificed and assayed for CD4+ T-cell and antibody responses. On day −3, 0, 7, and 14, mice were injected i.p. four times with 1mg/mouse of anti-IL-10R (1B1.3a, rat IgG1) or control mAb (GL113, anti-E.coli β-galactosidase, rat IgG1). The antibody (A) and T-cell responses (B) were assayed and presented as described in Figure 1 legend. Values represent mean ± SE of antibody titers or mean Δcpm ± SE. (n = 4). Data are representative of two independent experiments.
To confirm these results, we used another model in which mOVA transgenic B cells were used as APCs. We have found that both resting and LPS-activated mOVA B cells are tolerogenic in vivo (Su et al., in preparation). In this study, we first transferred LPS-activated mOVA B cells to wt mice and challenged these recipients 1 week later. The recipients were treated with anti-IL-10R or control mAb four times on day −3, 0, 7, and 14. We assayed T-cell and antibody responses on day 21. Again, the tolerogenicity of mOVA B cells was abrogated by in vivo IL-10R blockade (Figure 4). Together, our data in both systems suggest that host IL-10 production is required for genetic engineered B-cell mediated tolerance induction.
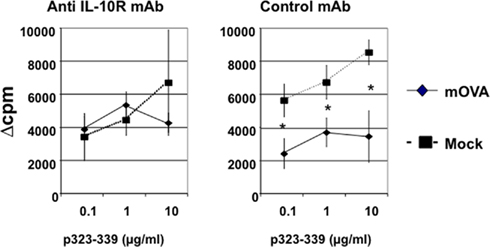
Figure 4. IL-10 produced by the host is required for tolerance induction by transgenic tolerogenic B cells. One × 107 B LPS-activated mOVA or wt B cells were injected into wt C57BL/6 mice. Tolerance induction protocol, anti-IL-10R treatment, and assay method are same as described in Figures 1 and 3 (n = 4). Data are representative of two independent experiments.
Discussion
Adoptive transfer of target-antigen expressing B cells has been demonstrated to be able to induce antigen-specific tolerance in various animal models such as EAE, allergy, and hemophilia. In this study, we defined the role of IL-10 in an established B-cell based gene therapy system for tolerance induction. We demonstrated that antigen-specific tolerance does not need IL-10 generated by peptide–IgG transduced tolerogenic B cells. Rather, IL-10 produced by the host is required for tolerance induction.
B cells play an important role in controlling autoimmunity. For example, B-cell deficient mice lose the ability of recovery from clinical disease symptoms after EAE induction (Wolf et al., 1996). It has been found that IL-10 produced by B cells is critical at the remission stage of EAE as B-cell restricted IL-10 deficiency results in mice that fail to recover from EAE (Fillatreau et al., 2002). Recently, Carter et al. (2011) demonstrated that in an arthritis model, IL-10-producing B-cell deficient mice exhibited an exacerbated disease symptom. In these IL-10-producing B-cell deficient mice, a decrease of FoxP3 + Tregs and an increase of Th1 and Th17 cells were found. These findings indicate that IL-10-producing B cells might have a regulatory cell phenotype and the IL-10 produced by B cells plays an essential role in regulation of autoimmunity.
Several years ago, we demonstrated that bone marrow cells from IL-10 KO mice could be transduced to be tolerogenic for a cognate antigen (El-Amine et al., 2000). In addition, Frommer et al. (2008) showed that IL-10 produced by B cells is not required for tolerance induction in an EAE model. In that study, EAE resistant mice that express MOG on B cells (BMOG) were crossed with IL-10 deficient mice to generate IL-10 KO MOG expressing B cells. They found that IL-10 KO transgenic mice were tolerant and were resistant to MOG-induced EAE. Recently, Calderon-Gomez et al. (2011) validated this result using lentiviral transduction of B cells. In sum, these results suggested that transduced or transgenic B cells tolerize T cells in an IL-10 independent manner.
In contrast, Ahangarani et al. (2009) recently found that IL-10 produced by target-antigen expressing B cells is critical for tolerance in an allergic model. This is of our particular interest because the B-cell gene therapy protocol used in this study is similar as ours. In the allergy model, B cells that express a fusion protein of an endosomal targeting sequence (gp75) and a major T-cell epitope of a common allergen, Der p2, induced tolerance to Der p2 in mice upon peptide immunization. Although detailed mechanisms were not fully elucidated, it was found that genetically modified B cells induced type 1 regulatory T cells (Tr1) in an IL-10 dependent manner (Ahangarani et al., 2009). In a follow-up study, Ahangarani et al. (2011) demonstrated that retroviral particles could bind and activate Toll-like receptor 2 on transduced B cells and in turn, activation of Toll-like receptor 2 results in upregulated production of IL-10 through STAT3 signaling pathway.
In our system, the mechanisms of tolerance induction appear to be different. We previously demonstrated that transduced B cells tolerize recipient animals in a MHC class II dependent manner (Litzinger et al., 2005; Su et al., 2008) and that tolerance induced by peptide–IgG expressing B cells depends on activation of natural CD4+CD25+ Tregs and/or generation of inducible Tregs (Lei and Scott, 2005; Soukhareva et al., 2006; Skupsky et al., 2010). Depletion of CD4+CD25+ Tregs completely abrogates the tolerance induction by peptide–IgG transduced B cells (Lei and Scott, 2005; Soukhareva et al., 2006). We also demonstrated that peptide–IgG transduced B cells could induce an increase of FoxP3+ Tregs in recipient animals and a reduction of the CD4+ effector T-cell population (Skupsky et al., 2010). Moreover, we recently observed by two-photon microscopy that tolerogenic B cells tightly conjugate with target CD4+ T cells and this B:T conjugation is CTLA-4/B7 dependent (Su et al., in preparation). Taken together, these observations suggest that in our system, B cells induce tolerance by induction of antigen-specific CD4+CD25+ Tregs through direct B:T contact. In addition, the IgG heavy chain might also play a role in induction/activation of CD4+CD25+ Tregs. We previously showed that the IgG heavy chain enhances the degree of tolerance and promotes the hyporesponsive state induced by B-cell gene therapy (Kang et al., 1999; Lei et al., 2005).
Recently, several highly promiscuous major histocompatibility complex class II regulatory T-cell epitopes (Tregitopes) in the conserved regions of IgG were identified (De Groot et al., 2008). Indeed, we suggest that these IgG epitopes may enhance the tolerogenic properties of the fusion constructs utilized in our B-cell gene therapy system. We found that in vivo administration of Tregitopes can stimulate natural Tregs in DO11.10 transgenic mice and delivery of these Tregitopes with OVA protein by B cells also protect mice from antigen challenge (Su et al., in preparation). Based on these observations, we propose that the exogenous IL-10 produced by tolerogenic B cells is not required for inducing/activating CD4+CD25+ Tregs in the early stage and maintaining tolerance. Thus, we believe that the distinct roles of IL-10 played in these two studies (our work and Ahangarani et al., 2009) are likely due to the different model systems used, i.e., whether the fusion construct expresses IgG, whether natural Tregs or Tr1 cells are induced, or whether an endosomal targeting sequence is utilized.
Nevertheless, our data suggest that the endogenous IL-10 produced by the host is essential for tolerance induction. We have previously shown that both Th1 and Th2 responses in host mice are reduced by peptide–IgG B-cell gene therapy (Kang et al., 1999). Therefore, it is unlikely that the in vivo IL-10R blockade has skewed the Th1 and Th2 populations directly. Instead, we suggest that the IL-10R blockade affects the activation or function of natural CD4+CD25+ Tregs or inducible Tregs, which we have shown to be integral for the efficacy of B-cell gene therapy. The endogenous IL-10 could be produced by different subsets of lymphocytes including regulatory T cells. One of the suppressive mechanisms of Tregs is through producing IL-10. IL-10 produced by APCs is also important for induction of Tregs. For example, IL-10 produced by macrophages in the lamina propria is critical for conversion of Tregs from convention T cells in mucosal immunity (Denning et al., 2007; Murai et al., 2009; Hadis et al., 2011). In addition, it has been demonstrated recently that transitional two marginal zone B cells, which are IL-10-producing and have regulatory phenotypes, play an important role in inducing FoxP3 Tregs and suppressing Th1 and Th17 cells (Carter et al., 2011). Therefore, although in our system exogenous IL-10 from donor B cells is dispensable for inducing tolerance, blocking endogenous IL-10 from the host interrupt tolerance induced by peptide–IgG B cells possibly due to at least two mechanisms: influence the induction or conversion of Tregs from conventional T cells or interfere the function of Tregs.
In summary, several groups have utilized B cells to deliver tolerance via gene therapy. Here we demonstrate that host IL-10 is critical for the tolerogenicity of B-cell based peptide–IgG gene therapy in vivo and that IL-10 produced by B cells themselves is not required for tolerance induction.
Contribution
Yan Su and Ai-Hong Zhang designed and performed research, analyzed data, and wrote the manuscript; Nancy Noben-Trauth supervised writing of the manuscript; and David W. Scott designed research and supervised data analysis and writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Robert Rossi, Patrick Adair, and Diane Nelson for critical reading of the manuscript. This work was supported by NIH grants RO1 AI035622 and DK068343.
References
Agarwal, R. K., Kang, Y., Zambidis, E., Scott, D. W., Chan, C. C., and Caspi, R. R. (2000). Retroviral gene therapy with an immunoglobulin-antigen fusion construct protects from experimental autoimmune uveitis. J. Clin. Invest. 106, 245–252.
Ahangarani, R. R., Janssens, W., Carlier, V., Vanderelst, L., Vandendriessche, T., Chuah, M., Jacquemin, M., and Saint-Remy, J. M. (2011). Retroviral vectors induce epigenetic chromatin modifications and IL-10 production in transduced B cells via activation of toll-like receptor 2. Mol. Ther. 19, 711–722.
Ahangarani, R. R., Janssens, W., VanderElst, L., Carlier, V., VandenDriessche, T., Chuah, M., Weynand, B., Vanoirbeek, J. A., Jacquemin, M., and Saint-Remy, J. M. (2009). In vivo induction of type 1-like regulatory T cells using genetically modified B cells confers long-term IL-10-dependent antigen-specific unresponsiveness. J. Immunol. 183, 8232–8243.
Aldo-Benson, M., and Borel, Y. (1976). Loss of carrier-determined tolerance in vitro with loss of receptor blockade. J. Immunol. 116, 223–226.
Barr, T. A., Brown, S., Ryan, G., Zhao, J., and Gray, D. (2007). TLR-mediated stimulation of APC: distinct cytokine responses of B cells and dendritic cells. Eur. J. Immunol. 37, 3040–3053.
Bettelli, E., Das, M. P., Howard, E. D., Weiner, H. L., Sobel, R. A., and Kuchroo, V. K. (1998). IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J. Immunol. 161, 3299–3306.
Borel, Y., Kilham, L., Hyslop, N., and Borel, H. (1976). Isologous IgG-induced tolerance to benzyl penicilloyl. Nature 261, 49–50.
Calderon-Gomez, E., Lampropoulou, V., Shen, P., Neves, P., Roch, T., Stervbo, U., Rutz, S., Kuhl, A. A., Heppner, F. L., Loddenkemper, C., Anderton, S. M., Kanellopoulos, J. M., Charneau, P., and Fillatreau, S. (2011). Reprogrammed quiescent B cells provide an effective cellular therapy against chronic experimental autoimmune encephalomyelitis. Eur. J. Immunol. 41, 1696–1708.
Carter, N. A., Vasconcellos, R., Rosser, E. C., Tulone, C., Munoz-Suano, A., Kamanaka, M., Ehrenstein, M. R., Flavell, R. A., and Mauri, C. (2011). Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J. Immunol. 186, 5569–5579.
Chen, C., Rivera, A., Ron, N., Dougherty, J. P., and Ron, Y. (2001). A gene therapy approach for treating T-cell-mediated autoimmune diseases. Blood 97, 886–894.
Chen, C. C., Rivera, A., Dougherty, J. P., and Ron, Y. (2004). Complete protection from relapsing experimental autoimmune encephalomyelitis induced by syngeneic B cells expressing the autoantigen. Blood 103, 4616–4618.
De Groot, A. S., Moise, L., McMurry, J. A., Wambre, E., Van Overtvelt, L., Moingeon, P., Scott, D. W., and Martin, W. (2008). Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes.” Blood 112, 3303–3311.
Denning, T. L., Wang, Y. C., Patel, S. R., Williams, I. R., and Pulendran, B. (2007). Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 8, 1086–1094.
El-Amine, M., Melo, M., Kang, Y., Nguyen, H., Qian, J., and Scott, D. W. (2000). Mechanisms of tolerance induction by a gene-transferred peptide-IgG fusion protein expressed in B lineage cells. J. Immunol. 165, 5631–5636.
Eynon, E. E., and Parker, D. C. (1992). Small B cells as antigen-presenting cells in the induction of tolerance to soluble protein antigens. J. Exp. Med. 175, 131–138.
Fillatreau, S., Sweenie, C. H., McGeachy, M. J., Gray, D., and Anderton, S. M. (2002). B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 3, 944–950.
Fiorentino, D. F., Bond, M. W., and Mosmann, T. R. (1989). Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 170, 2081–2095.
Frommer, F., Heinen, T. J., Wunderlich, F. T., Yogev, N., Buch, T., Roers, A., Bettelli, E., Muller, W., Anderton, S. M., and Waisman, A. (2008). Tolerance without clonal expansion: self-antigen-expressing B cells program self-reactive T cells for future deletion. J. Immunol. 181, 5748–5759.
Fuchs, E. J., and Matzinger, P. (1992). B cells turn off virgin but not memory T cells. Science 258, 1156–1159.
Gilbert, K. M., and Weigle, W. O. (1994). Tolerogenicity of resting and activated B cells. J. Exp. Med. 179, 249–258.
Hadis, U., Wahl, B., Schulz, O., Hardtke-Wolenski, M., Schippers, A., Wagner, N., Muller, W., Sparwasser, T., Forster, R., and Pabst, O. (2011). Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246.
Kang, Y., Melo, M., Deng, E., Tisch, R., El-Amine, M., and Scott, D. W. (1999). Induction of hyporesponsiveness to intact foreign protein via retroviral-mediated gene expression: the IgG scaffold is important for induction and maintenance of immune hyporesponsiveness. Proc. Natl. Acad. Sci. U.S.A. 96, 8609–8614.
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K., and Muller, W. (1993). Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274.
Lampropoulou, V., Hoehlig, K., Roch, T., Neves, P., Calderon Gomez, E., Sweenie, C. H., Hao, Y., Freitas, A. A., Steinhoff, U., Anderton, S. M., and Fillatreau, S. (2008). TLR-activated B cells suppress T cell-mediated autoimmunity. J. Immunol. 180, 4763–4773.
Lei, T. C., and Scott, D. W. (2005). Induction of tolerance to factor VIII inhibitors by gene therapy with immunodominant A2 and C2 domains presented by B cells as Ig fusion proteins. Blood 105, 4865–4870.
Lei, T. C., Su, Y., and Scott, D. W. (2005). Tolerance induction via a B-cell delivered gene therapy-based protocol: optimization and role of the Ig scaffold. Cell. Immunol. 235, 12–20.
Litzinger, M. T., Su, Y., Lei, T. C., Soukhareva, N., and Scott, D. W. (2005). Mechanisms of gene therapy for tolerance: B7 signaling is required for peptide-IgG gene-transferred tolerance induction. J. Immunol. 175, 780–787.
Melo, M. E., Qian, J., El-Amine, M., Agarwal, R. K., Soukhareva, N., Kang, Y., and Scott, D. W. (2002). Gene transfer of Ig-fusion proteins into B cells prevents and treats autoimmune diseases. J. Immunol. 168, 4788–4795.
Murai, M., Turovskaya, O., Kim, G., Madan, R., Karp, C. L., Cheroutre, H., and Kronenberg, M. (2009). Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 10, 1178–1184.
Satpute, S. R., Soukhareva, N., Scott, D. W., and Moudgil, K. D. (2007). Mycobacterial Hsp65-IgG-expressing tolerogenic B cells confer protection against adjuvant-induced arthritis in Lewis rats. Arthritis Rheum. 56, 1490–1496.
Skupsky, J., Su, Y., Lei, T. C., and Scott, D. W. (2007). Tolerance induction by gene transfer to lymphocytes. Curr. Gene Ther. 7, 369–380.
Skupsky, J., Zhang, A. H., Su, Y., and Scott, D. W. (2010). B-cell-delivered gene therapy induces functional T regulatory cells and leads to a loss of antigen-specific effector cells. Mol. Ther. 18, 1527–1535.
Soukhareva, N., Jiang, Y., and Scott, D. W. (2006). Treatment of diabetes in NOD mice by gene transfer of Ig-fusion proteins into B cells: Role of T regulatory cells. Cell. Immunol. 240, 41–46.
Stromnes, I. M., and Goverman, J. M. (2006). Active induction of experimental allergic encephalomyelitis. Nat. Protoc. 1, 1810–1819.
Su, Y., Carey, G., Maric, M., and Scott, D. W. (2008). B cells induce tolerance by presenting endogenous peptide-IgG on MHC class II molecules via an IFN-gamma-inducible lysosomal thiol reductase-dependent pathway. J. Immunol. 181, 1153–1160.
Wolf, S. D., Dittel, B. N., Hardardottir, F., and Janeway, C. A. Jr. (1996). Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J. Exp. Med. 184, 2271–2278.
Xu, B., and Scott, D. W. (2004). A novel retroviral gene therapy approach to inhibit specific antibody production and suppress experimental autoimmune encephalomyelitis induced by MOG and MBP. Clin. Immunol. 111, 47–52.
Yin, Z., Bahtiyar, G., Zhang, N., Liu, L., Zhu, P., Robert, M. E., McNiff, J., Madaio, M. P., and Craft, J. (2002). IL-10 regulates murine lupus. J. Immunol. 169, 2148–2155.
Zambidis, E. T., Barth, R. K., and Scott, D. W. (1997a). Both resting and activated B lymphocytes expressing engineered peptide-Ig molecules serve as highly efficient tolerogenic vehicles in immunocompetent adult recipients. J. Immunol. 158, 2174–2182.
Zambidis, E. T., Kurup, A., and Scott, D. W. (1997b). Genetically transferred central and peripheral immune tolerance via retroviral-mediated expression of immunogenic epitopes in hematopoietic progenitors or peripheral B lymphocytes. Mol. Med. 3, 212–224.
Keywords: gene therapy, B cells, IL-10, IL-10 receptor, tolerance
Citation: Su Y, Zhang A-H, Noben-Trauth N and Scott DW (2011) B-cell gene therapy for tolerance induction: host but not donor B-cell derived IL-10 is necessary for tolerance. Front. Microbio. 2:154. doi: 10.3389/fmicb.2011.00154
Received: 22 June 2011;
Accepted: 01 July 2011;
Published online: 15 July 2011.
Edited by:
Roland W. Herzog, University of Florida, USAReviewed by:
Brad E. Hoffman, Temple University School of Medicine, USAGeorge Aslanidi, University of Florida, USA
Copyright: © 2011 Su, Zhang, Noben-Trauth and Scott. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: David W. Scott, Department of Medicine, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USA. e-mail:ZGF2aWQuc2NvdHRAdXN1aHMuZWR1