
- 1 Geomicrobiology, Federal Institute for Geosciences and Natural Resources, Hannover, Germany
- 2 Faculty of Natural Sciences, Leibniz Universität Hannover, Hannover, Germany
For the first time quantitative data on the abundance of Bacteria, Archaea, and Eukarya in deep terrestrial sediments are provided using multiple methods (total cell counting, quantitative real-time PCR, Q-PCR and catalyzed reporter deposition–fluorescence in situ hybridization, CARD–FISH). The oligotrophic (organic carbon content of ∼0.2%) deep terrestrial sediments in the Chesapeake Bay area at Eyreville, Virginia, USA, were drilled and sampled up to a depth of 140 m in 2006. The possibility of contamination during drilling was checked using fluorescent microspheres. Total cell counts decreased from 109 to 106 cells/g dry weight within the uppermost 20 m, and did not further decrease with depth below. Within the top 7 m, a significant proportion of the total cell counts could be detected with CARD–FISH. The CARD–FISH numbers for Bacteria were about an order of magnitude higher than those for Archaea. The dominance of Bacteria over Archaea was confirmed by Q-PCR. The down core quantitative distribution of prokaryotic and eukaryotic small subunit ribosomal RNA genes as well as functional genes involved in different biogeochemical processes was revealed by Q-PCR for the uppermost 10 m and for 80–140 m depth. Eukarya and the Fe(III)- and Mn(IV)-reducing bacterial group Geobacteriaceae were almost exclusively found in the uppermost meter (arable soil), where reactive iron was detected in higher amounts. The bacterial candidate division JS-1 and the classes Anaerolineae and Caldilineae of the phylum Chloroflexi, highly abundant in marine sediments, were found up to the maximum sampling depth in high copy numbers at this terrestrial site as well. A similar high abundance of the functional gene cbbL encoding for the large subunit of RubisCO suggests that autotrophic microorganisms could be relevant in addition to heterotrophs. The functional gene aprA of sulfate reducing bacteria was found within distinct layers up to ca. 100 m depth in low copy numbers. The gene mcrA of methanogens was not detectable. Cloning and sequencing data of 16S rRNA genes revealed sequences of typical soil Bacteria. The closest relatives of the archaeal sequences were Archaea recovered from terrestrial and marine environments. Phylogenetic analysis of the Crenarchaeota and Euryarchaeota revealed new members of the uncultured South African Gold Mine Group, Deep Sea Hydrothermal Vent Euryarchaeotal Group 6, and Miscellaneous Crenarcheotic Group clusters.
Introduction
The Earth’s deep biosphere includes a variety of subsurface habitats, such as mines and deep aquifer systems in the continental realm, and sediments and igneous rock in the marine realm. It has been estimated that nearly half of total biomass on Earth resides in the deep biosphere (Whitman et al., 1998). However, the existing data used to generate this global census are highly skewed and in reality reflect habitat accessibility. Deeply buried marine sediments are among the best studied deep biosphere habitats. They are populated by a huge number of prokaryotes mainly belonging to uncultivated phylogenetic lineages (Parkes et al., 2000; Teske, 2006; Biddle et al., 2008; Fry et al., 2008; Teske and Sørensen, 2008). The abundance of particular phylogenetic and physiological prokaryotic groups, i.e., Archaea and Bacteria, methanogens or sulfate reducers, in deeply buried marine sediments has been quantified based on 16S rRNA and functional gene analysis by quantitative, real-time PCR (Q-PCR), FISH, and catalyzed reporter deposition–fluorescence in situ hybridization (CARD–FISH; Schippers et al., 2005; Biddle et al., 2006; Inagaki et al., 2006; Schippers and Neretin, 2006; Engelen et al., 2008; Nunoura et al., 2009; Webster et al., 2009).
The terrestrial deep subsurface biosphere has been studied so far only by total cell counting, cultivation techniques as well as by molecular 16S rRNA gene diversity analyses. The hard rock terrestrial deep biosphere in, e.g., granite, basalt, or metabasalt has been mainly explored by groundwater analyses rather than by deep rock drilling (Pedersen, 1993, 1997; Stevens and McKinley, 1995; Fredrickson et al., 1997; Chapelle et al., 2002; Moser et al., 2003; Lin et al., 2006; Hallbeck and Pedersen, 2008; Sahl et al., 2008; Borgonie et al., 2011; Itävaara et al., 2011).
Deep subsurface terrestrial sediments defined as deeper than 30–35 m (Balkwill et al., 1989) have just begun to be studied by molecular techniques. Cell numbers determined by total cell counting or cultivation indicate that a correlation of cell numbers with depth as described for marine sediments (Parkes et al., 1994, 2000) does not exist. Fry et al. (2009) did not find a decrease in cell numbers with depth in a terrestrial drill core of 148 m length including an interbedded coal deposit in New Zealand. Hoos and Schweisfurth (1982) also did not find a decreasing number of colony forming units (CFU) with depth after analyzing cultivable aerobic and anaerobic bacteria up to a sediment depth of 90 m in Lower Saxony, Germany. The lack of decreasing cell numbers with sediment depth is also supported by AODC and CFU numbers in coastal plain and fluvial sediment cores from South Carolina (Savannah River Site) and Washington State (Hanford Site), USA, sampled up to 265 m depth (Balkwill et al., 1989; Sinclair and Ghiorse, 1989; Fredrickson et al., 1991; Kieft et al., 1995) and Cretaceous sedimentary rock in New Mexico, USA at 190 m depth (Takai et al., 2003).
Analysis of the microbial diversity by 16S rRNA gene sequencing revealed the dominance of the following prokaryotic groups in deep terrestrial sediments. Most abundant among the Bacteria were Proteobacteria, Actinobacteria, Firmicutes, Chloroflexi, members of the Geobacteraceae family, sulfate reducers, denitrifiers, fermenters, and acetogens. The most frequently occurring Archaea were the Miscellaneous Crenarchaeotic Group, Methanosarcinales and Methanobacteriales (Boivin-Jahns et al., 1996; Chandler et al., 1997; Detmers et al., 2001, 2004; Takai et al., 2003; Inagaki et al., 2005; Kovacik et al., 2006; Brown and Balkwill, 2009; Fry et al., 2009).
Organic carbon seems to be most important for the long term survival of microorganisms in the terrestrial deep biosphere because a correlation was found between total organic carbon (TOC) and direct counts, basal respiration as well as aerobic glucose mineralization (e.g., Kieft et al., 1995).
The aim of this study was a comprehensive microbial community analysis of deep terrestrial sediments in order to provide missing quantitative data on the abundance of prokaryotes in the terrestrial deep biosphere. As terrestrial study site, deep sediments up to a depth of 140 m in the Chesapeake Bay area at Eyreville, Virginia, USA, were chosen. Total cells stained with SYBR Green were counted with three different methods and the microbial diversity was explored by 16S rRNA gene cloning and sequencing. In addition, Q-PCR and CARD–FISH were applied for the first time to study the deep biosphere in terrestrial sediments. With these quantitative methods, 16S rRNA and functional genes of phylogenetic and physiological prokaryotic groups relevant in deeply buried marine sediments were analyzed.
Materials and Methods
Site and Sediment Description
The Chesapeake Bay impact structure (CBIS), Virginia, USA, was formed during the late Eocene meteoric impact approximately 35.5 million years (Ma) ago. It has been explored by an international team of scientists in a project of the International Continental Scientific Drilling Program (ICDP) and the U.S. Geological Survey (USGS, Gohn et al., 2006, 2008, 2009). A cross-section figure showing main features of the CBIS and the drill site location is shown elsewhere (Gohn et al., 2008). The CBIS project acquired continuously cored sections from three holes drilled to a composite depth of 1766 m at a site within the central zone of the structure at the Eyreville drill site near Cape Charles, Virginia, USA. The drill bit penetrated a 1322-m-thick section of impact-related rocks and sediments and an overlying 444-m-thick section of post-impact sandy and clayish sediments. The latter consist of upper Eocene to Pliocene (∼5.3 to ∼1.8 Ma) continental-shelf sediments and Pleistocene (∼1.8 to ∼0.01 Ma) non-marine sediments. The upper 140 m (Miocene to Pleistocene) studied here were cored in Eyreville hole C during April and May 2006. In this depth interval, the porosity is between 36 and 54%, and the pore water chemistry indicates freshwater conditions, however the NaCl concentration is ∼0.2% at 100 m depth and increases to ∼1% at the bottom of the core (Gohn et al., 2006, 2008, 2009). In this study only post-impact sediments up to 140 m depth not influenced by the meteoric impact were analyzed.
Sediment Sampling
In this study, cores from Eyreville hole C were sampled for terrestrial microbial community analysis. Fifty sediment samples were taken from the surface (arable soil) down to a depth of up to 140 m. To avoid contamination, samples for microbiological analysis were only taken from the center of each sediment core (63.5 mm diameter) using sterilized cut 5 mL syringes or sterilized spatulas. Depth intervals for sampling were selected based on the quality of the cores with a higher depth resolution near the surface and a lower one at greater depth. As a contamination control, fluorescent microspheres were applied during coring for every second core and samples were taken from the periphery as well as the center of the cores for microscopic inspection as previously described (Kallmeyer et al., 2006; Gohn et al., 2009). Four samples could be identified as potentially contaminated and were not further analyzed.
For CARD–FISH and counting total cells with fluorescence microscopy, sediment samples were fixed in 4% formaldehyde–PBS (phosphate buffered saline) as described by Llobet-Brossa et al. (1998) and finally stored at −20°C in PBS–ethanol (1:1). For DNA based molecular as well as geochemical analysis, a parallel set of samples was directly frozen at −20°C. All samples were transported to BGR frozen with dry ice as air-freight, and afterward stored at −20°C until analysis.
Geochemical Analysis
The elemental composition of the solid material was determined by XRF analysis (Philips PW 2400). TOC and the total amount of carbon (TC) and sulfur (TS) were measured with the instrument LECO CS 200 (LECO Corporation). TC and TS were measured after acid removal of carbonates. Reactive iron was extracted with buffered citrate–dithionite as described by Canfield (1989), and measured by ICP-OES (Jobin Yvon Emission 166 Ultrace HR 1000).
Total cell counts and CARD–FISH
Total cell numbers were determined in formaldehyde fixed samples by staining with SYBR Green II following three different protocols. Cells were counted in the sediment matrix as described by Weinbauer et al. (1998) and were detached from sediment particles before counting as described by Kallmeyer et al. (2008) and Lunau et al. (2005). The latter protocol was modified by replacing the ultrasonic bath with an ultrasonic probe. CARD–FISH was carried out as described (Pernthaler et al., 2002; Schippers et al., 2005) and filters were hybridized for Archaea and Bacteria using probes ARCH915 or EUB338 I-III as a mixture. As a negative hybridization control the probe NON-338 was applied. For contamination control fluorescent beads of bacterial size were used and counted.
Quantitative Real-Time PCR Analysis
The quantitative composition of the microbial community was analyzed by Q-PCR after DNA extraction. High-molecular-weight DNA was extracted from 0.5 g of a frozen sediment sample following a modified FastDNA Spin Kit for Soil (Bio101) protocol (Webster et al., 2003). Sterilized quartz sand treated in a muffle furnace for organic carbon removal was used as negative control in the extraction procedure. Extracted DNA was amplified by Q-PCR using the device ABI Prism 7000 (Applied Biosystems) and master mixes from the companies Applied Biosystems, Eurogentec, or Invitrogen. Each DNA extract was measured in triplicate. After each Q-PCR, melting curves were measured for SYBR Green I assays. The copy numbers of the 16S rRNA gene were quantified for Archaea (Takai and Horikoshi, 2000), Bacteria (Nadkarni et al., 2002), the JS-1- and Chloroflexi-related bacteria (Blazejak and Schippers, 2010), and the Fe(III)- and Mn(IV)-reducing family Geobacteraceae (Holmes et al., 2002). The 18S rRNA gene of Eukarya was determined as previously described (Schippers and Neretin, 2006). Functional genes were quantified as described: mcrA for methyl coenzyme M reductase subunit A (Wilms et al., 2007), aprA for adenosine 5′-phosphosulfate reductase subunit A (Blazejak and Schippers, submitted), and cbbL for the large subunit of the enzyme ribulose-1.5-bisphosphate carboxylase/oxygenase (RubisCO, form I “red-like”; Selesi et al., 2007).
Cloning and Sequencing
High-molecular-weight DNA was extracted from 0.5 g of a frozen sediment sample as described above. Four depths (75–108 m) were analyzed for bacterial 16S rRNA gene sequences and two depths (108–125 m) were analyzed for archaeal 16S rRNA gene sequences. PCR reactions were carried out with the 1.1 or 2 Master Mix® (Thermo Scientific). PCR for Bacteria was carried out with the universal primers GM3f (AGA GTT TGA TCM TGG C) and GM4r (TAC CTT GTT ACG ACT T; Muyzer et al., 1995). The following thermocycling conditions were used: one cycle at 95 or 96°C for 5 min; 26–30 cycles at 95 or 96°C for 1 min, 42°C for 1 min, and 72°C for 3 min; and one cycle at 72°C for 7 min. PCR for Archaea was carried out with the primers 109f (ACK GCT CAG TAA CAC GT; Grosskopf et al., 1998) and 912r (CTC CCC CGC CAA TTC CTT TA; Lueders and Friedrich, 2000). These thermocycling conditions were used: one cycle at 95°C for 5 min; 26–30 cycles at 95°C for 1 min, 52°C for 1 min, and 72°C for 3 min; and one cycle at 72°C for 6 min. Cloning in Escherichia coli was carried out with the pGEM-t-Easy Vector Systems (Promega®) Kit following the instruction manual. For screening of 16S rRNA genes, 96 clones per sample were randomly picked. For template DNA, a small amount of cells from each clone colony was picked with a sterile toothpick and suspended in 20 μL of sterile water. One or two microliter of this template DNA, after preheating to 95°C for 2 min, were amplified by PCR as described above by using a 25- to 50-μL (total volume) mixture. PCR products of the correct size (∼1500 bp resp. 850 bp) were purified with the QIAquick PCR Purification Kit (Quiagen®) or directly send for sequencing. Sequencing reactions were carried out by Seqlab Laboratories, Göttingen, Germany. Sequences were edited with BioEdit1. A negative DNA extraction control without sediment was treated in parallel. For Bacteria, PCR products were also obtained for this negative control (presumably contaminants). The negative control of the PCR reaction itself was negative. In conclusion presumably contamination happened during the DNA extraction procedure. Fifty clones resulting from the negative control were analyzed using BLAST. Partial sequences from sediment samples which exhibited more than 98% similarity to the assumed contaminants were not included in the analysis of bacterial 16S rRNA gene sequences. For Archaea, the negative DNA extraction control did not result in a PCR product. All archaeal sequences were aligned by using the SINA Webaligner2 or the integrated Aligner of the ARB software3 (Ludwig et al., 2004) and were manually adjusted. Closest relatives of all bacterial and archaeal 16S rDNA sequences found with BLAST4 were also included in the phylogenetic analysis. For tree construction, sequences were grouped together in a clone family if they exhibited 99% sequence identity using similarity matrix in the ARB software.
Rarefaction curves were calculated with the mothur software5 (Schloss et al., 2009).
Phylogenetic Analysis
Chimera check was done with the Greengenes Bellerophon program6. The closest sequence relatives of the 16S rRNA gene sequences based on BLAST searches were imported into ARB and aligned using the integrated aligner and manually adjusted. The 16S rRNA gene sequence data were analyzed with the ARB software package (see text footnote 3). Phylogenetic trees were calculated by performing distance matrix methods (Neighbor Joining with 1000 bootstrap values, both with the Jukes–Cantor correction, Jukes and Cantor, 1969), maximum parsimony analysis and Maximum-Likelihood analysis. For tree reconstruction only sequences with more than 800 bp were used. Phylogenetic trees were calculated via ARB using the Maximum-Likelihood method. The closest sequence relatives and representatives of the major taxonomic groups were included (Baker et al., 2003). Similarity analysis and clone grouping was done with the ARB similarity matrix with Jukes–Cantor correction (Jukes and Cantor, 1969). The phylogenetic groups were arranged according to Teske and Sørensen (2008) and Spang et al. (2010).
Results
In this study, we analyzed the microbial ecology and bulk geochemistry of 50 samples from the post-impact CBIS terrestrial sediment from land surface (arable soil) up to a depth of 140 m.
Geochemical Results
Data for the geochemical solid phase analysis of 48 sediment samples are summarized in Table 1. TOC as substrate for heterotrophic microorganisms remained low in the complete sediment depth range with a mean value of ∼0.2% and a maximum value of 0.9% (w/w) characterizing the sediments as oligotrophic. Reactive iron, relevant for Fe(III)-reducing microorganisms, was more than twice as high in the uppermost meter than the mean for the total 140 m (data not shown).
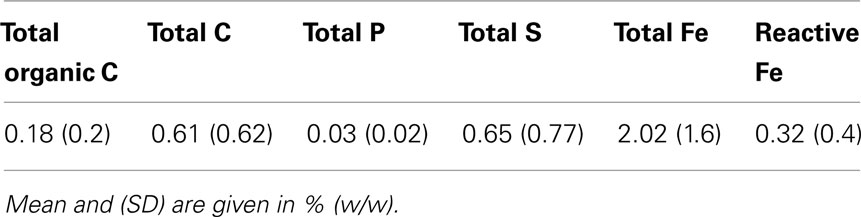
Table 1. Geochemical solid phase analysis of 48 sediment samples.
Microbiological Results Total Cell Counts and CARD–FISH
Total cells stained with SYBR Green were counted following three different protocols. Depth profiles of total cell counts are shown in Figure 1. For all three protocols, the maximal cell counts were detected near the surface. The total cell counts indicate a logarithmic decline with depth within the upper 20 m, and show no significant depth correlation between 20 and 140 m. The method comparison shows that the highest cell counts for all depths were obtained with the protocol without detaching cells from sediment particles after Weinbauer et al. (1998). Maximum cell counts of more than 109 cells/g at the surface declined to about 106 cells/g at 20 m depth. Below 20 m, counts were highly variable and not correlated with depth. In comparison, the overall counts obtained with protocols in which the cells were detached from sediment particles before counting gave about half an order of magnitude (Kallmeyer et al., 2008) and about one order of magnitude (Lunau et al., 2005, modified) lower cell counts.
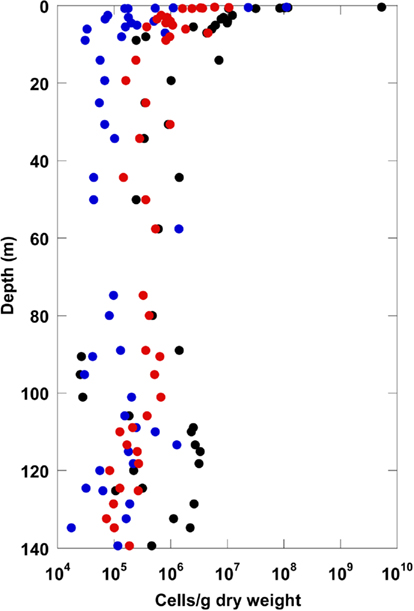
Figure 1. Total cell counts for the terrestrial CBIS post-impact sediment obtained with three different methods (black: method Weinbauer et al., 1998; red: method Kallmeyer et al., 2008; blue: method Lunau et al., 2005).
A comparison of the highest total cell counts after Weinbauer et al. (1998) with numbers of living Bacteria and Archaea obtained by CARD–FISH is given in Figure 2 for the top 7 m sediment depth. A significant proportion of the total cell counts could be detected with CARD–FISH. Interestingly, the CARD–FISH numbers for Bacteria were about an order of magnitude higher than those for the Archaea. At some depth Archaea were not detectable at all. Below 7 m sediment depth the CARD–FISH cell signals were below the detection limit of 105 cells/g.
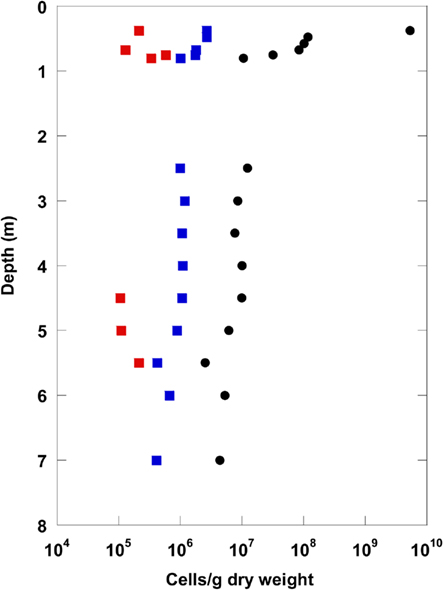
Figure 2. Catalyzed reporter deposition–fluorescence in situ hybridization numbers for Bacteria (blue) and Archaea (red) displayed with total cell counts (black: Weinbauer et al., 1998) for the uppermost 8 m of the terrestrial CBIS post-impact sediment.
Quantitative Microbial Community Analysis by Q-PCR
Results of Q-PCR analysis for the uppermost 10 m depth and for 80–140 m depth are shown in Figure 3. The Q-PCR data on bacterial 16S rRNA gene copy numbers matched well with the total cell counts after Weinbauer et al. (1998). Archaea were found in lower copy numbers than the Bacteria in the top 10 m. At 80–140 m depth, Archaea were detected only at a few depths, and always in lower copy numbers using Q-PCR. Thus, the dominance of Bacteria over Archaea in the CBIS post-impact sediment was confirmed by Q-PCR and CARD–FISH.
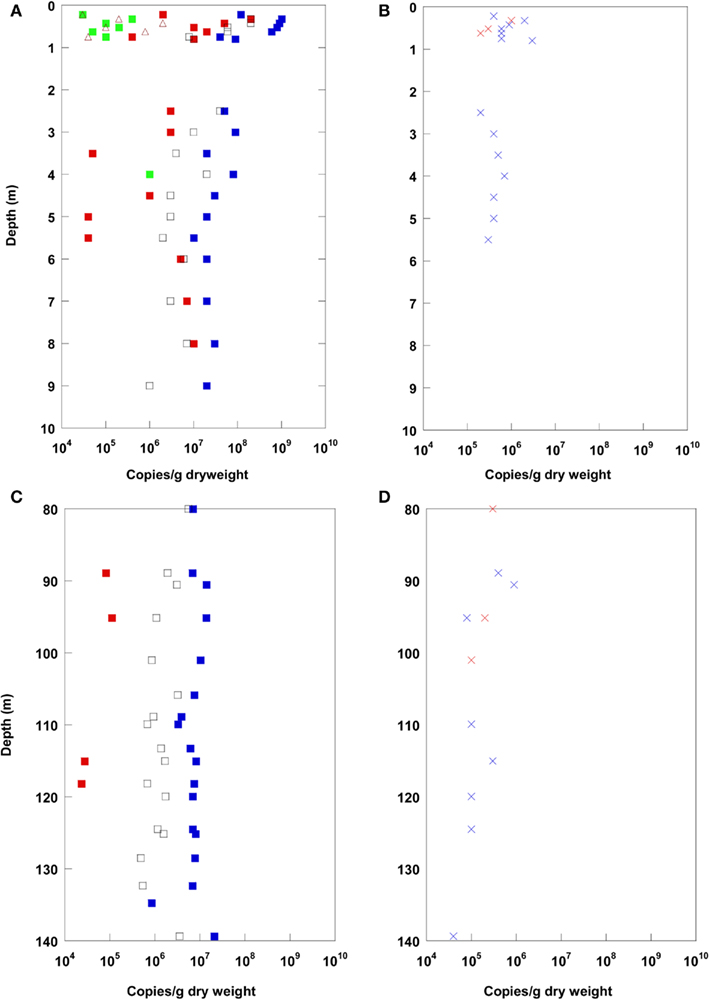
Figure 3. Real-time PCR (Q-PCR) data for the terrestrial CBIS post-impact sediment in the uppermost 10 m depth (A,B), and in 80–140 m depth (C,D). Different phylogenetic groups, blue: Bacteria, red: Archaea, green: Eukarya, brown: Geobacteriaceae, black: JS-1-Chloroflexi(A,C); functional genes, red: aprA, blue: cbbL (B,D).
In addition to the prokaryotic domains Bacteria and Archaea, Eukarya and specific prokaryotic groups were quantified via Q-PCR by 16S rRNA or functional gene quantification. Eukarya and the Fe(III)- and Mn(IV)-reducing bacterial group Geobacteraceae were found in the uppermost meter (arable soil) only (besides at 4 m). In the uppermost meter, reactive iron was detected in higher amounts as a potential electron acceptor for the Geobacteraceae. The bacterial candidate division JS-1 and the classes Anaerolineae and Caldilineae of the phylum Chloroflexi, highly abundant in marine sediments (Blazejak and Schippers, 2010), were found in high copy numbers up to the maximum sampling depth of 140 m. A similar high abundance was found for the functional gene cbbL coding for the large subunit of the form I “red-like” ribulose-1.5-bisphosphate carboxylase/oxygenase (RubisCO) occurring in autotrophic Proteobacteria that fix CO2 via the Calvin–Benson–Bassham (CBB) cycle (Selesi et al., 2007; Badger and Bek, 2008). The functional gene aprA coding for adenosine 5′-phosphosulfate reductase occurring in sulfate reducing bacteria was found within distinct layers up to ca. 100 m depth. The gene mcrA for methyl coenzyme M reductase of methanogenic Archaea could not be detected.
Microbial Diversity
In order to reveal the prokaryotic diversity in the CBIS sediment, a phylogenetic analysis of 16S rRNA gene sequences from four depths for Bacteria and two depths for Archaea was performed. The results for the Bacteria are shown in Table 2, those for the Archaea in Figures 4–7. Overall, the bacterial diversity seems to be very low. This finding may partly be a result of the limited number of reported bacterial clone data. Many bacterial clones had to be excluded since their 16S RNA gene sequences exhibited more than 98% similarity (checked with BLAST) to the 16S rRNA gene sequences obtained from the negative DNA extraction control with no sediment (contaminants). Sequences of the remaining bacterial 16S rRNA genes revealed typical soil bacteria (Table 2).
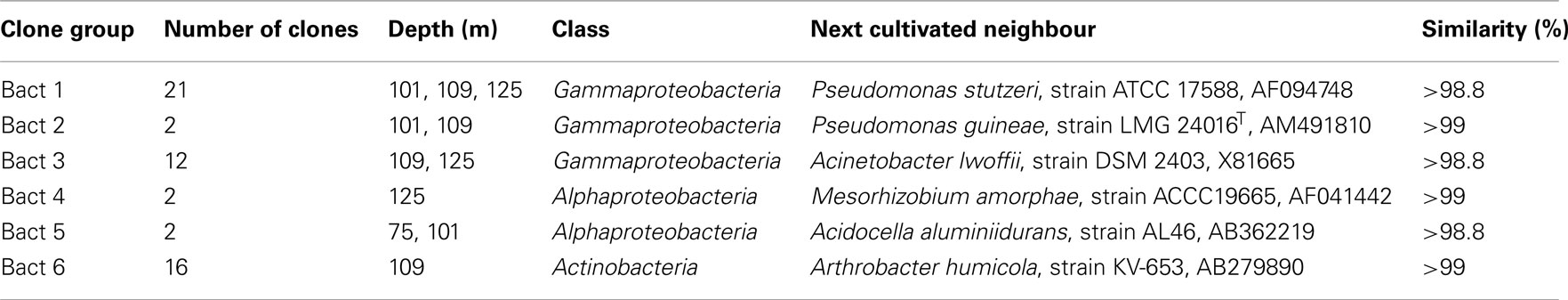
Table 2. Summary of the phylogenetic analysis of bacterial 16S rRNA genes in terrestrial CBIS post-impact sediment.
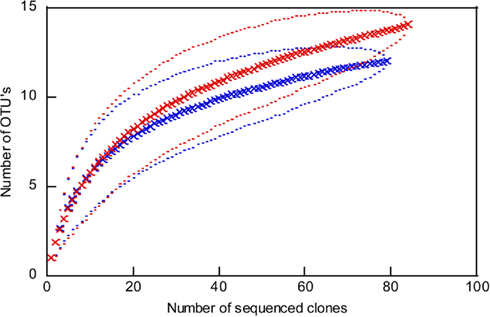
Figure 4. Rarefaction curves for the archaeal 16S rRNA gene sequences from 109 m depth (red) to 125 m depth (blue). Outer and inner lines: high and low confidence interval (95%). OTU, operational taxonomic unit; cut off of OTU’s = 1%.
The analysis of 16S rRNA gene sequences of Archaea from two depths, 109 and 125 m, resulted in 13 and 103 clones which could be allocated to the phyla Euryarchaeota or Crenarchaeota, respectively. The rarefaction curves of the 16S rRNA gene sequences indicate a good coverage of the archaeal diversity as can be seen in the flattening of the two curves (Figure 4). The composition of the archaeal community shows a similar ratio of euryarcheotic and crenarcheotic contingents (109 m: 5 euryarchaeotal sequences, 58 crenarchaeotal sequences, 125 m: 8 euryarchaeotal sequences, 57 crenarchaeotal sequences) for the two analyzed sediment depths (Figure 5).
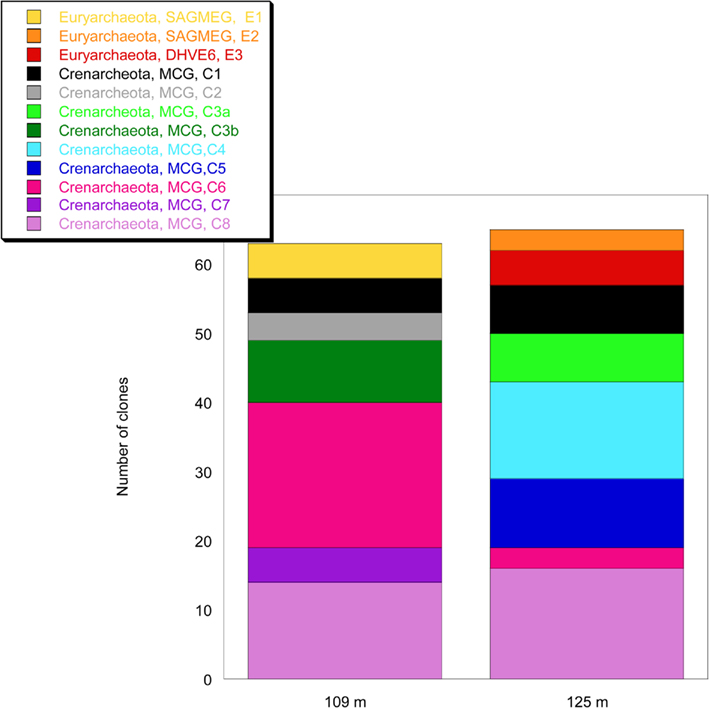
Figure 5. Analysis of the composition of the archaeal community in the terrestrial CBIS post-impact sediment at 109 and 125 m depth.
The phylogenetic analysis with different methods (ARB neighbor joining with 1000 bootstraps, maximum parsimony method, data not shown) gave similar results as the maximum-likelihood analysis. Phylogenetic trees for the two archaeal phyla, Euryarchaeota and Crenarchaeota, are shown in Figures 6 and 7. Two euryarcheotic clone groups, E1 and E2, could be allocated to the South African Gold Mine Group (SAGMEG). Group E1 clustered together with a clone received from deeply buried sediments of the Peru margin (AB177011). Group E2 represents a novel phylogenetic subgroup of archaeal sequences with less than 98% similarity to its closest sequence, AY093454. A third clone group, E3 could be allocated to the Deep Sea Hydrothermal Vent Euryarchaeotal Group 6 (DHVE6). The 16S rRNA gene sequence similarity to its closest sequence EU750878 is less than 89%. Both SAGMEG and DHVE6 contain 16S rRNA gene sequences of terrestrial as well as of marine origin (Teske and Sørensen, 2008). The three clone groups could be allocated to the two different depths. Group E1 includes only sequences from 109 m depth while group E2 and E3 include only sequences from 125 m depth.
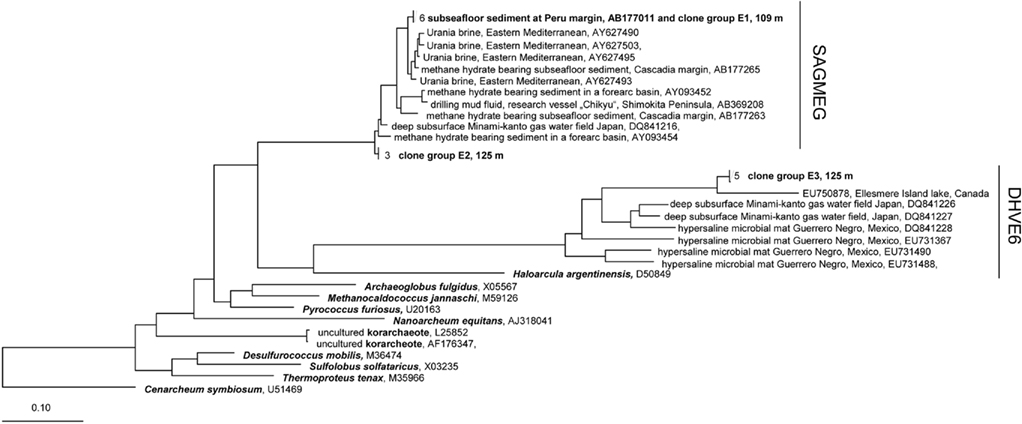
Figure 6. Maximum-Likelihood phylogenetic tree of the Euryarcheota showing clades found in terrestrial CBIS post-impact sediment. The tree was rooted with Cenarchaeum symbiosum as a representative of the deeply branched group “Thaumarcheota.” SAGMEG, South African Gold Mine Group, DHVE6, Deep Sea Hydrothermal Vent Euryarchaeotal Group 6.
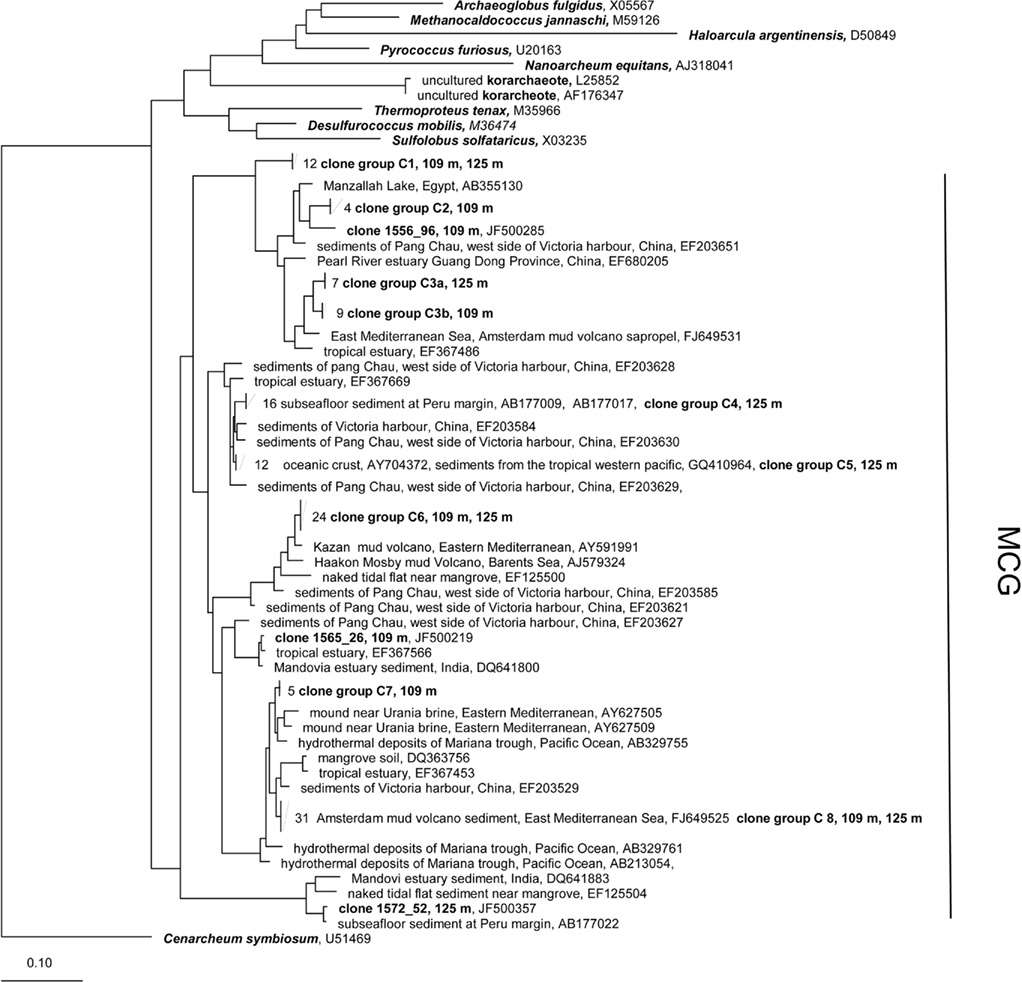
Figure 7. Maximum-Likelihood phylogenetic tree of the Crenarcheota showing clades found in terrestrial CBIS post-impact sediment. The tree was rooted with Cenarchaeum symbiosum as a representative of the deeply branched group “Thaumarcheota.” MCG, Miscellaneous Crenarcheotic Group.
All 16S rRNA gene sequences from the phylum Crenarchaeota belong to the Miscellaneous Crenarcheotic Group (MCG). Sequences received could be grouped into eight different clone groups. Five groups (C1, C2, C3, C6, C7) represent new phylogenetic clusters with less than 99% similarity to their closest related sequences in GenBank. Some clone groups were found in one sample only (e.g., C7, 109 m; C4, 125 m), while others occurred in both samples. Interestingly, the clone groups C3a (125 m) and C3b (109 m) are related to each other and belong together to a new cluster, but the similarity between C3a and C3b is 96.7% and thus below the species level.
Discussion
Abundance of Total and Living Cells
The microbial community in terrestrial sediments up to a depth of 140 m in the Chesapeake Bay area, Virginia, USA, was thoroughly analyzed by SYBR Green total cell counting, Q-PCR, and CARD–FISH, and 16S rRNA gene cloning. The organic carbon content is low (mean ∼0.2%) in these oligotrophic deep terrestrial sediments, thus little substrate is available for sustaining a thriving heterotrophic microbial community. Nevertheless total cell counts after Weinbauer et al. (1998) and Q-PCR data exhibited an average of about 106 cells/g between 20 and 140 m depth without a decrease with depth. In comparison, the overall counts obtained with protocols that detached cells from sediment particles before counting resulted in one (Lunau et al., 2005) to half (Kallmeyer et al., 2008) an order of magnitude lower cell counts. This difference between the protocols can be explained by a loss of cells during the detachment procedure and/or counting of unspecific signals without detaching cells from sediment particles. A comparison of the total cell counts with the 16S rRNA gene copy numbers of Bacteria obtained by Q-PCR gives the best match with the highest cell counts (Weinbauer et al., 1998). Thus, cell loss during the cell detachment procedures seemed to be more relevant than an overestimation by counting unspecific signals. Although the detachment protocols likely result in an underestimate of total cell numbers, the protocols are suitable for sediments in which the numbers of microorganisms are below 105 cells/mL sediment (D’Hondt et al., 2009; Schippers et al., 2010).
The CARD–FISH bacterial cell numbers reflecting living cells were lower than the total cell numbers (Weinbauer et al., 1998) which may indicate a minor proportion of living cells or overlooked CARD–FISH cells due to insufficient cell staining. More evidence for living cells was provided by cultivation experiments (unpublished). In these experiments, fresh samples from various depths were incubated for several months. A strong CO2 release was observed in aerobic and in anaerobic cultures with or without addition of Fe(III) as a terminal electron acceptor, indicating microbial activity under these conditions. Assays with additional sulfate and nitrate did not show evolution of CO2 (Michael Siegert and Martin Krüger, personal communication).
In a previous study of the deeper sediments of the terrestrial CBIS at the same drill site total cell counts were obtained after staining with DAPI and exhibited significantly higher numbers, between 106 and 108 cells/g with high variation over the depth interval 140–444 m of the post-impact CBIS sediment (Gohn et al., 2008). The total cell counts increased with depth below 140 m of the post-impact sediment. One explanation for this finding might be the changing lithology connected to a dramatically changing TOC content with depth. The post-impact CBIS sediments from 140 to 444 m consist of a generally fine-grained upper Eocene to upper Miocene sediment with about a 10 times higher TOC content than the coarser grained upper Miocene to Pleistocene section above 140 m (Gohn et al., 2009). Most likely the higher TOC content at greater depth sustains significantly more cells than in the upper oligotrophic sediment. These data represent the first observation of a significant increase of cell counts with depth in deep terrestrial sediment. The relevance for a lithological control on the deep biosphere has been previously pointed out for deeply buried marine sediments (Coolen et al., 2002; Inagaki et al., 2003; Parkes et al., 2005). Below the post-impact CBIS terrestrial sediments in the geologically different zones of sediment breccias, schist, pegmatite, and granite (444–1766 m depth) the total cell numbers were considerably lower (104 and 106 cells/g or not detectable; Gohn et al., 2008). The novel actinobacterium Tessaracoccus profundi was isolated and described from a depth of 940 m (Finster et al., 2009).
The average total cell numbers of about 106 cells/g between 20 and 140 m depth found in this study are in the same order of magnitude or somewhat higher than those found in other deep terrestrial sediments in a similar depth range by total cell counting or by cultivation (Hoos and Schweisfurth, 1982; Balkwill et al., 1989; Fredrickson et al., 1991; Kieft et al., 1995; Takai et al., 2003; Fry et al., 2009). These studies are in agreement with our study, and did not find a decrease in cell numbers with depth. This is in contrast to marine sediments for which a correlation of cell numbers with depth was described (Parkes et al., 1994, 2000; Schippers et al., 2005). The reason for the difference in cell numbers vs. depth in marine and terrestrial sediments is unknown but has considerable importance for the estimation of the global abundance of prokaryotes on Earth (Whitman et al., 1998) as previously stated (Fry et al., 2009).
Abundance of Bacteria vs. Archaea
This study is the first providing quantitative data on the abundance of Bacteria and Archaea in deep terrestrial sediment. The dominance of Bacteria over Archaea in the CBIS post-impact terrestrial sediment was confirmed by Q-PCR and CARD–FISH. The proportions of Bacteria and Archaea in marine sediments have shown to be highly variable in different sediments and sediment layers and conflicting results have been published for analyses of nucleic-acids (Q-PCR and CARD–FISH) and intact polar lipids (IPL) of cell membranes (Inagaki et al., 2003, 2006; Schippers et al., 2005, 2010; Biddle et al., 2006; Schippers and Neretin, 2006;; Wilms et al., 2007; Engelen et al., 2008; Lipp et al., 2008; Nunoura et al., 2009; Webster et al., 2009). Schouten et al. (2010) and Logemann et al. (2011) reported about a fossilization of archaeal IPL biomarkers in marine sediments indicating that IPL biomarkers detect fossil signals rather than living Archaea, thus putting their proposed dominance in the marine deep biosphere into question.
Another explanation for the conflicting results is given by mismatches of archaeal primers and probes with 16S rRNA gene sequences of the dominant archaeal groups in marine sediments and therefore a potential underestimation of archaeal cell numbers by nucleic-acid based methods (Teske and Sørensen, 2008). A comparison of our archaeal Q-PCR results with our clone library data for the samples at 109 and 125 m revealed a discrepancy of the two methods which used different primers and probes. While in the clone library several different groups were found, Q-PCR did not result in archaeal 16S rRNA gene amplification. For Q-PCR, we used the primers Arch349F and Arch806R, and the TaqMan probe Arch516 (Takai and Horikoshi, 2000). According to Teske and Sørensen (2008) the primer Arch349F has several mismatches within the groups SAGMEG, DHVE6, and MCG. We checked the primer Arch349F against our sequences and found more than five mismatches with some sequences. Similarly, probe Arch516 and primer Arch806R matched only when at least three (probe Arch516F) and two (primer Arch806R) mismatches were allowed. This finding elucidates the necessity for the development of novel archaeal Q-PCR assays.
Abundance of Specific Taxa and of Functional Genes
The detection of the functional gene cbbL coding for the large subunit of the form I “red-like” RubisCO in many samples in relatively high copy numbers in our study indicates that autotrophic Proteobacteria are relevant in the deep terrestrial sediments as well. However, their abundance is at least an order of magnitude lower than the 16S rRNA gene copy number of the dominant Bacteria, thus heterotrophic bacteria play the mayor role in the deep terrestrial sediment despite the low content of organic carbon. However, heterotrophs were also found in oligotrophic deeply buried marine sediments (D’Hondt et al., 2004).
The bacterial candidate division JS-1 and the classes Anaerolineae and Caldilineae of the phylum Chloroflexi comprised a higher proportion of the Bacteria, but these specific groups with almost no cultivated representatives are less abundant than in marine sediments where almost identical 16S rRNA gene copy numbers for the specific groups and the Bacteria were found (Webster et al., 2004, 2011; Blazejak and Schippers, 2010).
Fe(III)-, Mn(IV)-, and sulfate-reducers, methanogens as well as Eukarya quantified via general 18S rRNA genes (Eukarya), specific 16S rRNA genes (Geobacteraceae), or functional genes (aprA, mcrA) play a minor or no role in the deep post-impact CBIS terrestrial sediment while these groups were regularly detected in subsurface marine sediments (Schippers and Neretin, 2006; Wilms et al., 2007; Engelen et al., 2008; Nunoura et al., 2009; Webster et al., 2009; Schippers et al., 2010). Eukarya and Geobacteraceae were found in the uppermost meter of the CBIS drill site where reactive iron and presumably eukaryotic DNA from farming in the arable soil is available. Deeper eukaryotic DNA was detected in one sample only. Due to the low TOC and sulfate content of the terrestrial sediment, sulfate reduction, and methanogenesis are expected to be less relevant than in deeply buried marine continental margin sediments with a higher TOC content (D’Hondt et al., 2004; Parkes et al., 2005; Schippers et al., 2005; Schippers and Neretin, 2006; Teske, 2006). Both processes are also less relevant in oligotrophic deeply buried marine sediments (D’Hondt et al., 2004; Sørensen et al., 2004; Teske, 2006; Nunoura et al., 2009) in agreement with our terrestrial study.
Diversity of Bacteria and Archaea
The bacterial 16S rRNA gene sequences belong to three classes: Alphaproteobacteria, Gammaproteobacteria, and Actinobacteria. All bacterial 16S rRNA gene sequences have more than 98.9% similarity to sequences of cultivated heterotrophic bacteria. Almost all of the identified bacteria were previously found in other deep terrestrial sediments (Balkwill et al., 1989; Boivin-Jahns et al., 1996).
The phylogenetic analysis of the Archaea identified euryarcheotic as well as crenarcheotic 16S rRNA gene sequences including novel phylogenetic clusters related to lineages that do not yet contain cultivated representatives. The euryarcheotic clone groups E1 and E2 belong to the SAGMEG. This group includes 16S rRNA gene sequences found in a South African gold mine and sequences from the deep marine subsurface (Teske and Sørensen, 2008). Fry et al. (2009) also found euryarcheotic sequences belonging to SAGMEG in deep terrestrial sediments including an interbedded coal deposit. Sequences isolated from hot springs (Greece) or dolomite aquifers (South Africa) also belong to the SAGMEG (Figure 6). In conclusion this group seems not to be restricted to the deep subsurface biosphere, and occurs in marine and terrestrial environments. Similarly, the DHVE6 to which clone group E3 belongs includes terrestrial and marine sequences. The DHVE6 group defined by Takai and Horikoshi (1999) includes sequences from deep sea hydrothermal vents in the Eastern Pacific Ocean. Successively, sequences from different habitats could be affiliated to this group; examples are from a hydrothermal field at 13°N, 141°W in the South Pacific Rise (Nercessian et al., 2003) and from ODP Site 1231 at the Peru Basin (Sørensen et al., 2004). The closest relative to the group E3 is a sequence from a highly stratified meromictic lake on Ellesmere Island that is characterized by a high salinity in deeper layers (Poliot et al., 2009). Further related 16S rRNA sequences derive from habitats with high salinity: from a hypersaline microbial mat at Guerrero Negro, Mexico (Robertson et al., 2009) and from a commercial gas–water-producing well water in Japan which contains ancient seawater at depths of 347–1132 m (Mochimaru et al., 2007). In conclusion the novel group E3 seems to be related to clones that derive from environments with higher salinity (Figure 6). The DHVE6 group is affiliated with reduced (metal-) sulfides at vent structures (Takai and Horikoshi, 1999), reduced iron and manganese species (Sørensen et al., 2004), hydrogen sulfide (Robertson et al., 2009), and/or high salinity (Poliot et al., 2009; Robertson et al., 2009).
All crenarcheotic clones found in this study belong to the MCG. This group contains a huge number of diverse phylogenetic lineages from different, partially extreme habitats from terrestrial and marine origin (Teske and Sørensen, 2008). We identified clone groups which have closely related sequences from other environments (C5, C8, Figure 7), and also several novel groups with a relatively high distance to the closest related sequences (C1, C2). As summarized by Teske and Sørensen (2008), the MCG appears to be heterotrophic, which corroborates our Q-PCR data on the dominance of heterotrophic prokaryotes (see above), despite the low TOC content (in particular 0.24% for 109 m and 0.28% for 125 m depth).
Conclusion
For the first time quantitative data on the abundance of Bacteria, Archaea, and Eukarya in deep terrestrial sediments are provided using multiple methods (total cell counting, CARD–FISH, and Q-PCR). This was done together with the description of the bacterial and archaeal lineages and the quantification of specific taxa and of functional genes. The presence of a significant fraction of rRNA containing, viable bacterial and archaeal cells as revealed by CARD–FISH despite low levels of organic carbon is a relevant finding in this study. The dominance of Bacteria over Archaea resulted from CARD–FISH and Q-PCR data. Other major findings are the discovery of new sequence clusters within previously described cren- and euryarchaeotal lineages and the presence of high copy numbers of cbbL encoding for the large subunit of the form I “red-like” RubisCO suggesting that autotrophic Proteobacteria could be relevant in addition to heterotrophs in the terrestrial deep subsurface.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the ICDP, the USGS, and the drilling team for drilling and providing samples, and Mary A. Voytek for giving advice to the application of fluorescent microspheres. We also thank Eastern Shore Laboratory (ESL) in Wachapreague, Virginia, for laboratory space and support during sampling. The 16S rRNA gene sequences of the CBIS post-impact sediment were submitted to NCBI with the accession numbers JF500169-JF500398. This work was funded by the DFG research grant SCHI 535/6 to Axel Schippers.
Footnotes
References
Badger, M. R., and Bek, E. J. (2008). Multiple Rubisco forms in proteobacteria: their functional significance in relation to CO2 acquisition by the CBB cycle. J. Exp. Bot. 59, 1525–1541.
Baker, G. C., Smith, J. J., and Cowan, D. A. (2003). Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 55, 541–555.
Balkwill, D. L., Fredrickson, J. K., and Thomas, J. M. (1989). Vertical and horizontal variations in the physiological diversity of the aerobic chemoheterotrophic bacterial microflora in deep southeast coastal-plain subsurface sediments. Appl. Environ. Microbiol. 55, 1058–1065.
Biddle, J. F., Fitz-Gibbon, S., Schuster, S. C., Brenchley, J. E., and House, C. H. (2008). Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. U.S.A. 105, 10583–10588.
Biddle, J. F., Lipp, J. S., Lever, M., Lloyd, K. G., Sørensen, K. B., Anderson, R., Fredricks, H. F., Elvert, M., Kelly, T. J., Schrag, D. P., Sogin, M. L., Brenchley, J. E., Teske, A, House, C. H., and Hinrichs, K. U. (2006). Heterotrophic Archaea dominate sedimentary subsurface ecosystems off Peru. Proc. Natl. Acad. Sci. U.S.A. 103, 3846–3851.
Blazejak, A., and Schippers, A. (2010). High abundance of JS-1- and Chloroflexi-related bacteria in deeply buried marine sediments revealed by quantitative, real-time PCR. FEMS Microbiol. Ecol. 72, 198–207.
Boivin-Jahns, V., Ruimy, R., Bianchi, A., Daumas, S., and Christen, R. (1996). Bacterial diversity in a deep-subsurface clay environment. Appl. Environ. Microbiol. 62, 3405–3412.
Borgonie, G., García-Moyano, A., Litthauer, D., Bert, W., Bester, A., van Heerden, E., Möller, C., Erasmus, M., and Onstott, T. C. (2011). Nematoda from the terrestrial deep subsurface of South Africa. Nature 474, 79–82.
Brown, M. G., and Balkwill, D. L. (2009). Antibiotic resistance in bacteria from the deep terrestrial subsurface. Microb. Ecol. 57, 484–493.
Chandler, D. P., Li, S.-M., Spadoni, C. M., Drake, G. R., Balkwill, D. L., Fredrickson, J. K., and Brockman, F. J. (1997). A molecular comparison of culturable aerobic heterotrophic bacteria and 16S rDNA clones derived from a deep subsurface sediment. FEMS Microbiol. Ecol. 23, 131–144.
Chapelle, F. H., O’Neill, K., Bradley, P. M., Methé, B. A., Ciufo, S. A., Knobel, L. L., and Lovley, D. R. (2002). A hydrogen-based subsurface microbial community dominated by methanogens. Nature 415, 312–315.
Coolen, M. J. L., Cypionka, H., Sass, A. M., Sass, H., and Overmann, J. (2002). Ongoing modification of Mediterranean Pleistocene sapropels mediated by prokaryotes. Science 296, 2407–2410.
Detmers, J., Schulte, U., Strauss, H., and Kuever, J. (2001). Sulfate reduction at a lignite seam: microbial abundance and activity. Microb. Ecol. 42, 238–247.
Detmers, J., Strauss, H., Schulte, U., Bergmann, A., Knittel, K., and Kuever, J. (2004). FISH shows that Desulfotomaculum spp. are the dominating sulfate-reducing bacteria in a pristine aquifer. Microb. Ecol. 47, 236–242.
D’Hondt, S., Spivack, A. J., Pockalny, R., Ferdelman, T. G., Fischer, J. P., Kallmeyer, J., Abrams, L. J., Smith, D. C., Graham, D., Hasiuk, F., Schrum, H., and Stancin, M. A. (2009). Subseafloor sedimentary life in the South Pacific Gyre. Proc. Natl. Acad. Sci. U.S.A. 106, 11651–11656.
D’Hondt, S. L., Jørgensen, B. B., Miller, D. J., Batzke, A., Blake, R., Cragg, B. A., Cypionka, H., Dickens, G. R., Ferdelman, T. G., Hinrichs, K. U., Holm, N. G., Mitterer, R., Spivack, A., Wang, G., Bekins, B., Engelen, B., Ford, K., Gettemy, G., Rutherford, S. D., Sass, H., Skilbeck, C. G., Aiello, I. W., Guèrin, G., House, C. H., Inagaki, F., Meister, P., Naehr, T., Niitsuma, S., Parkes, R. J., Schippers, A., Smith, D. C., Teske, A., Wiegel, J., Padilla, C. N., and Solis Acosta, J. L. (2004). Distributions of microbial activities in deep subseafloor sediments. Science 306, 2216–2221.
Engelen, B., Ziegelmüller, K., Wolf, L., Köpke, B., Gittel, A., Cypionka, H., Treude, T., Nakagawa, S., Inagaki, F., Lever, M. A., and Steinsbu, B. O. (2008). Fluids from the oceanic crust support microbial activities within the deep biosphere. Geomicrobiol. J. 25, 55–66.
Finster, K. W., Cockell, C. S., Voytek, M. A., Gronstal, A. L., and Kjeldsen, K. U. (2009). Description of Tessaracoccus profundi sp. nov., a deep-subsurface actinobacterium isolated from a Chesapeake impact crater drill core (940 m depth). Antonie Van Leeuwenhoek 96, 515–526.
Fredrickson, J. K., Balkwill, D. L., Zachara, J. M., Li, S. M. W., Brockman, F. J., and Simmons, M. A. (1991). Physiological diversity and distributions of heterotrophic bacteria in deep cretaceous sediments of the Atlantic coastal plain. Appl. Environ. Microbiol. 57, 402–411.
Fredrickson, J. K., McKinley, J. P., Bjornstad, B. N., Long, P. E., Ringelberg, D. B., White, D. C., Krumholz, L. R., Suflita, J. M., Colwell, F. S., Lehman, R. M., Phelps, T. J., and Onstott, T. C. (1997). Pore-size constraints on the activity and survival of subsurface bacteria in a late Cretaceous shale-sandstone sequence, northwestern New Mexico. Geomicrobiol. J. 14, 183–202.
Fry, J. C., Horsfield, B., Sykes, R., Cragg, B. A., Heywood, C., Kim, G. T., Mangelsdorf, K., Mildenhall, D. C., Rinna, J., Vieth, A., Zink, K. G., Sass, H., Weightman, A. J., and Parkes, R. J. (2009). Prokaryotic populations and activities in an interbedded coal deposit, including a previously deeply buried section (1.6-2.3 km) above ~150 Ma basement rock. Geomicrobiol. J. 26, 163–178.
Fry, J. C., Parkes, R. J., Cragg, B. A., Weightman, A. J., and Webster, G. (2008). Prokaryotic diversity and activity in the deep subseafloor biosphere. FEMS Microbiol. Ecol. 66, 181–196.
Gohn, G. S., Koeberl, C., Miller, K. G., and Reimold, W. U. (2009). The ICDP-USGS Deep Drilling Project in the Chesapeake Bay Impact Structure: Results from the Eyreville Core Holes. Boulder, CO: The Geological Society of America Special Paper 458.
Gohn, G. S., Koeberl, C., Miller, K. G., Reimold, W. U., Browning, J. V., Cockell, C. S., Horton, J. W. Jr., Kenkmann, T., Kulpecz, A. A., Powars, D. S. Sanford, W. E., and Voytek, M. A. (2008). Deep drilling into the Chesapeake Bay impact structure. Science 320, 1740–1745.
Gohn, G. S., Koeberl, C., Miller, K. G., Reimold, W. U., Cockell, C. S., Horton, J. W. Jr., Sanford, W. E., and Voytek, M. A. (2006). Chesapeake Bay impact structure drilled. EOS 87, 349, 355.
Grosskopf, R., Janssen, P. H., and Liesack, W. (1998). Diversity and structure of the methanogenic community in anoxic rice paddy soil microcosms as examined by cultivation and direct 16S rRNA gene sequence retrieval. Appl. Environ. Microbiol. 64, 960–969.
Hallbeck, L., and Pedersen, K. (2008). Characterization of microbial processes in deep aquifers of the Fennoscandian Shield. Appl. Geochem. 23, 1796–1819.
Holmes, D. E., Finneran, K. T., O’Neil, R. A., and Lovley, D. R. (2002). Enrichment of members of the family Geobacteraceae associated with stimulation of dissimilatory metal reduction in uranium-contaminated aquifer sediments. Appl. Environ. Microbiol. 68, 2300–2306.
Hoos, E., and Schweisfurth, R. (1982). Untersuchungen über die Verteilung von Bakterien von 10 bis 90 Meter unter Bodenoberkante. Vom Wasser 58, 103–112.
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., Suzuki, M., Takai, K., Delwiche, M., Colwell, F. S., Nealson, K. H., Horikoshi, K., D’Hondt, S., and Jørgensen, B. B. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific ocean margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820.
Inagaki, F., Okada, H., Tsapin, A. I., and Nealson, K. H. (2005). The paleome: a sedimentary genetic record of past microbial communities. Astrobiology 5, 141–153.
Inagaki, F., Suzuki, M., Takai, K., Oida, H., Sakamoto, T., Aoki, K., Nealson, K. H., and Horikoshi, K. (2003). Microbial communities associated with geological horizons in coastal subseafloor sediments from the Sea of Okhotsk. Appl. Environ. Microbiol. 69, 7224–7235.
Itävaara, M., Nyyssönen, M., Kapanen, A., Nousiainen, A., Ahonen, L., and Kukkonen, I. (2011). Characterization of bacterial diversity to a depth of 1500m in the Outokumpu deep borehole, Fennoscandian Shield. FEMS Microbiol. Ecol. 77, 295–309.
Kallmeyer, J., Mangelsdorf, K., Cragg, B. A., and Horsfield, B. (2006). Techniques for contamination assessment during drilling for terrestrial subsurface sediments. Geomicrobiol. J. 23, 227–239.
Kallmeyer, J., Smith, D. C., Spivack, A. J., and D’Hondt, S. L. (2008). New cell extraction procedure applied to deep subsurface sediments. Limnol. Oceanogr. Methods 6, 236–245.
Kieft, T. L, Fredrickson, J. K., McKinley, J. P., Bjornstad, B. N., Rawson, S. A., Phelps, T. J., Brockman, F. J., and Pfiffner, S. M. (1995). Microbiological comparisons within and across contiguous lacustrine, paleosol, and fluvial subsurface sediments. Appl. Environ. Microbiol. 61, 749–757.
Kovacik, W. P. Jr., Takai, K., Mormile, M. R., McKinley, J. P., Brockman, F. J., Fredrickson, J. K., and Holben, W. E. (2006). Molecular analysis of deep subsurface Cretaceous rock indicates abundant Fe(III)- and S°-reducing bacteria in a sulfate-rich environment. Environ. Microbiol. 8, 141–155.
Lin, L. H., Wang, P. L., Rumble, D., Lippmann-Pipke, J., Boice, E., Pratt, L. M., Sherwood Lollar, B., Brodie, E. L., Hazen, T. C., Andersen, G. L., DeSantis, T. Z., Moser, D. P., Kershaw, D., and Onstott, T. C. (2006). Long term sustainability of a high energy, low diversity crustal biome. Science 314, 479–482.
Lipp, J. S., Morono, Y., Inagaki, F., and Hinrichs, K. U. (2008). Significant contribution of Archaea to extent biomass in marine subsurface sediments. Nature 454, 991–994.
Llobet-Brossa, E., Rossello-Mora, R., and Amann, R. (1998). Microbial community composition of Wadden Sea sediments as revealed by fluorescence in situ hybridization. Appl. Environ. Microbiol. 64, 2691–2696.
Logemann, J., Graue, J., Köster, J., Engelen, B., Rullkötter, J., and Cypionka, H. (2011). A laboratory experiment of intact polar lipid degradation in sandy sediments. Biogeosci. Discuss. 8, 3289–3321.
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meiser, H., Yadhukumar, Buchner, A., Lai, T., Steppi, S., Jobb, G., Förster, W., Brettske, I., Gerber, S., Ginhart, A. W., Gross, O., Grumann, S., Hermann, S., Jost, R., König, A., Liss, T., Lüß mann, R., May, M., Nonhoff, B., Reichel, B., Strehlow, R., Stamatakis, A., Stuckmann, N., Vilbig, A., Lenke, M., Ludwig, T., Bode, A., and Schleifer, K. H. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371.
Lueders, T., and Friedrich, M. (2000). Archaeal population dynamics during sequential reduction processes in rice field soil. Appl. Environ. Microbiol. 66, 2732–2742.
Lunau, M., Lemke, A., Walther, K., Martens-Habbena, W., and Simon, M. (2005). An improved method for counting bacteria from sediments and turbid environments by epifluorescence microscopy. Environ. Microbiol. 7, 961–968.
Mochimaru, H., Yoshioka, H., Tamaki, H., Nakamura, K., Kaneko, N., Sakata, S., Imachi, H., Sekiguchi, Y., Uchiyama, H., and Kamagata, Y. (2007). Microbial diversity and methanogenic potential in a high temperature natural gas field in Japan. Extremophiles 11, 453–461.
Moser, D. P., Onstott, T. C., Fredrickson, J. K., Brockman, F. J., Balkwill, D. L., Drake, G. R., Pfiffner, S. M., White, D. C., Takai, K., Pratt, L. M., Fong, J., Sherwood Lollar, B., Slater, G., Phelps, T. J., Spoelstra, N., Deflaun, M., Southam, G., Welty, A. T., Baker, B. J., and Hoek, J. (2003). Temporal shifts in the geochemistry and microbial community structure of an ultradeep mine borehole following isolation. Geomicrobiol. J. 20, 517–548.
Muyzer, G., Teske, A., Wirsen, C. O., and Jannasch, H. W. (1995). Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch. Microbiol. 164, 165–172.
Nadkarni, M., Martin, F. E., Jacques, N. A., and Hunter, N. (2002). Determination of bacterial load by real-time PCR using a broad range (universal) probe and primer set. Microbiology 148, 257–266.
Nercessian, O., Reysenbach, A. L., Prieur, D., and Jeanthon, C. (2003). Archaeal diversity associated with in situ samplers deployed on hydrothermal vents on the East Pacific Rise (13°N). Environ. Microbiol. 5, 492–502.
Nunoura, T., Soffientino, B., Blazejak, A., Kakuta, J., Oida, H., Schippers, A., and Takai, K. (2009). Subseafloor microbial communities associated with rapid turbidite deposition in the Gulf of Mexico continental slope (IODP Expedition 308). FEMS Microbiol. Ecol. 69, 410–424.
Parkes, R. J., Cragg, B. A., Bale, S. J., Getliff, J. M., Goodman, K., Rochelle, P. A., Fry, J. C., Weightman, A. J., and Harvey, S. M. (1994). Deep bacterial biosphere in Pacific Ocean sediments. Nature 371, 410–413.
Parkes, R. J., Cragg, B. A., and Wellsbury, P. (2000). Recent studies on bacterial populations and processes in subseafloor sediments: a review. Hydrogeology J. 8, 11–28.
Parkes, R. J., Webster, G., Cragg, B. A., Weightman, A. J., Newberry, C. J., Ferdelman, T. G., Kallmeyer, J., Jørgensen, B. B., Aiello, I. W., and Fry, J. C. (2005). Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature 436, 390–394.
Pernthaler, A., Pernthaler, J., and Amann, R. (2002). Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl. Environ. Microbiol. 68, 3094–3101.
Poliot, J., Galand, P. E., Loveley, C., and Vincent, W. F. (2009). Vertical structure of archaeal communities and the distribution of ammonia monooxygenase A gene variants in two merocmictic high Arctic lakes. Environ. Microbiol. 11, 687–699.
Robertson, C. E., Spear, J. R., Harris, K., and Pac, N. R. (2009). Diversity and stratification of Archaea in a hypersaline microbial mat. Appl. Environ. Microbiol. 75, 1801–1810.
Sahl, J. W., Schmidt, R., Swanner, E. D., Mandernack, K. W., Templeton, A. S., Kieft, T. L., Smith, R. L., Sanford, W. E., Callaghan, R. L., Mitton, J. B., and Spear, J. R. (2008). Subsurface microbial diversity in deep-granitic-fracture water in Colorado. Appl. Environ. Microbiol. 74, 143–152.
Schippers, A., Köweker, G., Höft, C., and Teichert, B. (2010). Quantification of microbial communities in three forearc sediment basins off Sumatra. Geomicrobiol. J. 27, 170–182.
Schippers, A., and Neretin, L. N. (2006). Quantification of microbial communities in near-surface and deeply buried marine sediments on the Peru continental margin using real-time PCR. Environ. Microbiol. 8, 1251–1260.
Schippers, A., Neretin, L. N., Kallmeyer, J., Ferdelman, T. G., Cragg, B. A., Parkes, R. J., and Jørgensen, B. B. (2005). Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 433, 861–864.
Schloss, P. D., Westcott, S., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. L., Sahl, J. W., Stres, B., Thallinger, G. G., Van Horn, D. L., and Weber, C. F. (2009). Introducing mothur: open-source, platform-independent community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541.
Schouten, S., Middelburg, J. J., Hopmans, E. C., and Sinninghe Damste, J. S. (2010). Fossilization and degradation of intact polar lipids in deep subsurface sediments: a theoretical approach. Geochim. Cosmochim. Acta 74, 3806–3814.
Selesi, D., Pattis, I., Schmid, M., Kandeler, E., and Hartmann, A. (2007). Quantification of bacterial RubisCO genes in soils by cbbL targeted real-time PCR. J. Microbiol. Methods 69, 497–503.
Sinclair, J. L., and Ghiorse, W. C. (1989). Distribution of aerobic bacteria, protozoa, algae and fungi in deep subsurface sediments. Geomicrobiol. J. 7, 15–31.
Sørensen, K. B., Lauer, A., and Teske, A. P. (2004). Archaeal phylotypes in a metal-rich and low-activity deep subsurface sediment of the Peru Basin, ODP Leg 201, Site 1231. Geobiology 2, 151–161.
Spang, A., Hatzenpichler, R., Brochier-Armanet, C., Ratei, T., Tischler, P., Spieck, E., Streit, W., Stahl, D., Wagner, M., and Schleper, C. (2010). Distinct gene set in two different lineges of ammonia oxidizing archaea supports the phylum Thaumarcheota. Trends Microbiol. 18, 331–340.
Stevens, T. O., and McKinley, J. P. (1995). Lithoautotrophic microbial ecosystems in deep basalt aquifers. Science 270, 450–454.
Takai, K., and Horikoshi, K. (1999). Genetic diversity of Archaea in deep-sea hydrothermal vent environments. Genetics 152, 1285–1297.
Takai, K., and Horikoshi, K. (2000). Rapid detection and quantification of members of the archaeal community by quantitative PCR using fluorogenic probes. Appl. Environ. Microbiol. 66, 5066–5072.
Takai, K., Mormile, M. R., McKinley, J. P., Brockman, F. J., Holben, W. E., Kovacik, W. P., and Fredrickson, J. K. (2003). Shifts in archaeal communities associated with lithological and geochemical variations in subsurface Cretaceous rock. Environ. Microbiol. 5, 309–320.
Teske, A. P. (2006). Microbial communities of deep marine subsurface sediments: molecular and cultivation surveys. Geomicrobiol. J. 23, 357–368.
Teske, A. P., and Sørensen, K. B. (2008). Uncultured archaea in deep marine marine subsurface sediments: have we caught them all? ISME J. 2, 3–18.
Webster, G., Blazejak, A., Cragg, B. A., Schippers, A., Sass, H., Rinna, J., Tang, X., Mathes, F., Ferdelman, T. G., Fry, J. C., Weightman, A. J., and Parkes, R. J. (2009). Subsurface microbiology and biogeochemistry of a deep, cold-water carbonate mound from the Porcupine Seabight (IODP Expedition 307). Environ. Microbiol. 11, 239–257.
Webster, G., Newberry, C. J., Fry, J. C., and Weightman, A. J. (2003). Assessment of bacterial community structure in the deep sub-seafloor biosphere by 16S rDNA-based techniques: a cautionary tale. J. Microbiol. Methods 55, 155–164.
Webster, G., Parkes, R. J., Fry, J. C., and Weightman, A. J. (2004). Widespread occurrence of a novel division of bacteria identified by 16S rDNA gene sequences originally found in deep marine sediments. Appl. Environ. Microbiol. 70, 5708–5713.
Webster, G., Sass, H., Cragg, B. A., Gorra, R., Knab, N. J., Green, C. J., Mathes, F., Fry, J. C., Weightman, A. J., and Parkes, R. J. (2011). Enrichment and cultivation of prokaryotes associated with the sulphate-methane transition zone of diffusion-controlled sediments of Aarhus Bay, Denmark, under heterotrophic conditions. FEMS Microbiol. Ecol. 77, 248–263.
Weinbauer, M. G., Beckmann, C., and Höfle, M. G. (1998). Utility of green fluorescent nucleic acid dyes and aluminium oxide membrane filters for rapid epifluorescence enumeration of soil and sediment bacteria. Appl. Environ. Microbiol. 64, 5000–5003.
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583.
Keywords: CARD–FISH, Chesapeake Bay, Crenarchaeota, deep biosphere, Euryarchaeota, real-time PCR, sediments, subsurface
Citation: Breuker A, Köweker G, Blazejak A and Schippers A (2011) The deep biosphere in terrestrial sediments in the chesapeake Bay area, Virginia, USA. Front. Microbio. 2:156. doi: 10.3389/fmicb.2011.00156
Received: 31 March 2011;
Paper pending published: 23 May 2011;
Accepted: 05 July 2011;
Published online: 19 July 2011.
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, USAReviewed by:
Marco J. L. Coolen, Woods Hole Oceanographic Institution, USAKarine Alain, Centre National de la Recherche Scientifique, France
Copyright: © 2011 Breuker, Köweker, Blazejak and Schippers. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Axel Schippers, Bundesanstalt für Geowissenschaften und Rohstoffe, Stilleweg 2, 30655 Hannover, Germany. e-mail:YXhlbC5zY2hpcHBlcnNAYmdyLmRl