
 Ashlee V. Moses
Ashlee V. Moses

- Vaccine and Gene Therapy Institute, Oregon Health & Science University, Beaverton, OR, USA
Like the other more well-characterized post-translational modifications (phosphorylation, methylation, acetylation, acylation, etc.), the attachment of the 76 amino acid ubiquitin (Ub) protein to substrates has been shown to govern countless cellular processes. As obligate intracellular parasites, viruses have evolved the capability to commandeer many host processes in order to maximize their own survival, whether it be to increase viral production or to ensure the long-term survival of latently infected host cells. The first evidence that viruses could usurp the Ub system came from the DNA tumor viruses and Adenoviruses, each of which use Ub to dysregulate the host cell cycle (Scheffner et al., 1990; Querido et al., 2001). Today, the list of viruses that utilize Ub includes members from almost every viral class, encompassing both RNA and DNA viruses. Among these, there are examples of Ub usage at every stage of the viral life cycle, involving both ubiquitination and de-ubiquitination. In addition to viruses that merely modify the host Ub system, many of the large DNA viruses encode their own Ub modifying machinery. In this review, we highlight the latest discoveries regarding the myriad ways that viruses utilize Ub to their advantage.
Introduction
In order to discuss the ways in which viruses exploit the host Ub machinery, we must first provide a general outline of that machinery. Prior to its conjugation to targeted proteins, Ub is first activated via an ATP-dependent thioester linkage to an E1 enzyme (reviewed in Schulman and Harper, 2009). Once activated, the energy in that unstable bond is then used to couple the Ub moiety’s C-terminal residue to the catalytic Cys residue of an E2 Ub conjugating enzyme (reviewed in Pickart, 2001). Ub-loaded E2 enzymes are then ready to form conjugates between the Ub C-terminal glycine and either Lys, Ser, Thr, Cys, or N-terminal-MET residues within proteins to be ubiquitinated (Breitschopf et al., 1998; Cadwell and Coscoy, 2005; Wang et al., 2007; Williams et al., 2007). The specificity of that conjugation process is conferred by the over 600 E3 Ub ligases encoded by the human genome (Li et al., 2008). Thus far, four primary types of E3 ligases have been described, and their sub-categorization is based on common structural features. The RING (really interesting new gene, reviewed in Deshaies and Joazeiro, 2009) ligases, which are by far the most commonly occurring E3s, serve as docking complexes that bring E2 and target proteins within close proximity, whereupon the E2 catalyzes the transfer of Ub to the substrate protein. The primary identifying feature of a RING domain is a conserved (Cys)3-His-(Cys)4 sequence that coordinates a pair of zinc atoms to maintain its structure. The plant homeodomain (PHD) ligases are structurally related to the RING ligases and use what appears to be a reversed set of residues [(Cys)4-His-(Cys)3] to coordinate a pair of zinc atoms (reviewed in Bienz, 2006). PHD domains are found in many chromatin-binding proteins, and once their similarity to RING domains was noted, it was expected that at least a subset of these would also function as E3s. However, thus far, only two examples of PHD proteins with E3 activity have surfaced (Dul and Walworth, 2007; Ivanov et al., 2007). The U-box (ubiquitin fusion degradation, UFD2-homology domain) ligases were first identified in yeast (Koegl et al., 1999), and seven human genes encoding such ligases have been identified (reviewed in Marin, 2010). A subset of this family are referred to as E4 ligases, which are distinguished from E3 ligases by virtue of their specific targeting of substrates that have already been modified with 1–3 ubiquitin residues (Hoppe, 2005). Interestingly, U-box ligases are structurally related to RING fingers, but this structure is maintained in the absence of zinc-chelating residues (Aravind and Koonin, 2000; Ohi et al., 2003). Therefore, based on structural features alone, the RING, PHD, and U-box ligases can all be viewed as RING-like variants. Finally, the HECT (homologous to E6-associated protein C-terminus, reviewed in Rotin and Kumar, 2009) ligases harbor their own catalytic cysteine residues to which Ub is first transferred from an E2 enzyme. Once loaded, the HECT ligases themselves catalyze target ubiquitination.
As initially described, lysines were the residues most frequently modified via ubiquitination, and it was found that K48-linked polyubiquitin chains were attached to proteins destined for degradation via the 26S proteasome (reviewed in Hershko and Ciechanover, 1998; Mayer, 2000). Since that time, it is now appreciated that all seven Lys residues within Ub can be used to build chains (Xu et al., 2009), and that this leads to Ub polymers with extremely diverse topologies (reviewed in Komander, 2009), further adding to ubiquitin’s flexibility as a post-translational modification. It is important to note that ubiquitination can be reversed by de-ubiquitinating (DUB) proteases, so, like other post-translational modifications, Ub signals can be precisely controlled. There are also examples of proteins that harbor Ub-binding motifs, which, in combination with Ub ligases and DUBs, enable Ub to serve as a reversible protein-binding surface that allows for regulated generation and subsequent disassembly of protein complexes (reviewed in Hicke et al., 2005; Dikic et al., 2009). Finally, the ubiquitin domain has been found to be encoded within the linear sequence of larger proteins. While several such ubiquitin-like domain (ULD) proteins have been shown to be involved with protein degradation and proteasome function, others participate in functions ranging from metabolism to signal transduction, in both prokaryotes and eukaryotes (reviewed in Burroughs et al., 2007; Grabbe and Dikic, 2009). Thus, the small ubiquitin domain and its cognate ubiquitin-binding domains represent a pair of complimentary structural features that have seen a great deal of use, reuse, and reassortment during evolution.
There are of course many examples in which the host itself uses Ub to limit viral production, particularly via the activation of NF-κB (reviewed in Skaug et al., 2009). Likewise, in addition to Ub, there are other Ub-like (UBL) proteins that are used to modify and control cellular processes (reviewed in Hochstrasser, 2009). However, because it is the most extensively characterized of the Ub-like proteins, we have chosen to focus upon Ub itself and the ways in which viruses exploit this small protein at each stage of the lifecycle. At the end of the review, we have provided a figure that integrates many of the examples discussed herein.
Viral Entry and Nucleocapsid Transport
There have been no reports of a direct link between the Ub-proteasome system (UPS) and viral binding to host cells. However, studies of several viral classes have shown that post-entry steps such as nucleocapsid transport and/or disassembly are impaired in the presence of proteasome inhibitors or in cell lines expressing a temperature sensitive mutant of the Ub-activating enzyme E1 (ts E1; see Table 1). A more direct role for the UPS in influenza virus entry was shown when either depletion of Epsin 1 or expression of a non-ubiquitinable Epsin 1 mutant blocked clathrin-mediated viral transport (Chen and Zhuang, 2008). Interestingly, while the nucleocapsid transport of some parvoviruses (minute virus of mice, MMV and canine parvovirus, CPV) is sensitive to proteasome inhibitors (Ros and Kempf, 2004), the entry of two other family members, bovine parvovirus (BPV) and adeno-associated virus (AAV), were insensitive to proteasome inhibitors (Yan et al., 2002). In fact, previous studies have shown that AAV infections are enhanced by proteasome inhibitors in a process whereby some portion of the intracellular AAV particles are ubiquitinated and degraded (Yan et al., 2002). All of the studies using proteasome inhibitors suggest that the UPS is important for both RNA and DNA virus entry, but the specific viral and cellular players remain to be identified. One exception is a recent report examining adenovirus entry, which has identified ubiquitination of the capsid protein VI as a key step in viral transport to the nucleus. The VI protein contains a PPXY motif (normally associated with viral egress, see below) that recruits a member of the neural-precursor-cell-expressed, developmentally down-regulated (Nedd4) family of E3 Ub ligases, and this interaction is necessary for the microtubule-dependent localization of protein VI. Viruses expressing protein VI with a mutated PPXY domain can exit endosomes, but are unable to transport to the nucleus (Wodrich et al., 2010). Finally, the human immunodeficiency virus (HIV)-1 accessory protein Vpr and its HIV-2/simian immunodeficiency virus (SIV) homolog Vpx appear to improve the ability of their cognate viruses to infect macrophages, and for some time this was thought to be due to an improvement in the translocation of the viral pre-integration complex to the nucleus (references in Casey et al., 2010). However, more recent work has determined that Vpx overcomes a macrophage-specific restriction factor, and that this depends on Vpx’s association with a Cullin (Cul) 4 E3 Ub ligase complex (see below, and Sharova et al., 2008). While it remains to be seen whether Vpr counteracts the same factor in human cells, the observation that Vpx can enhance dendritic cell and macrophage infection by HIV-1 suggests that this will be the case (see references in Ayinde et al., 2010; Table 1).
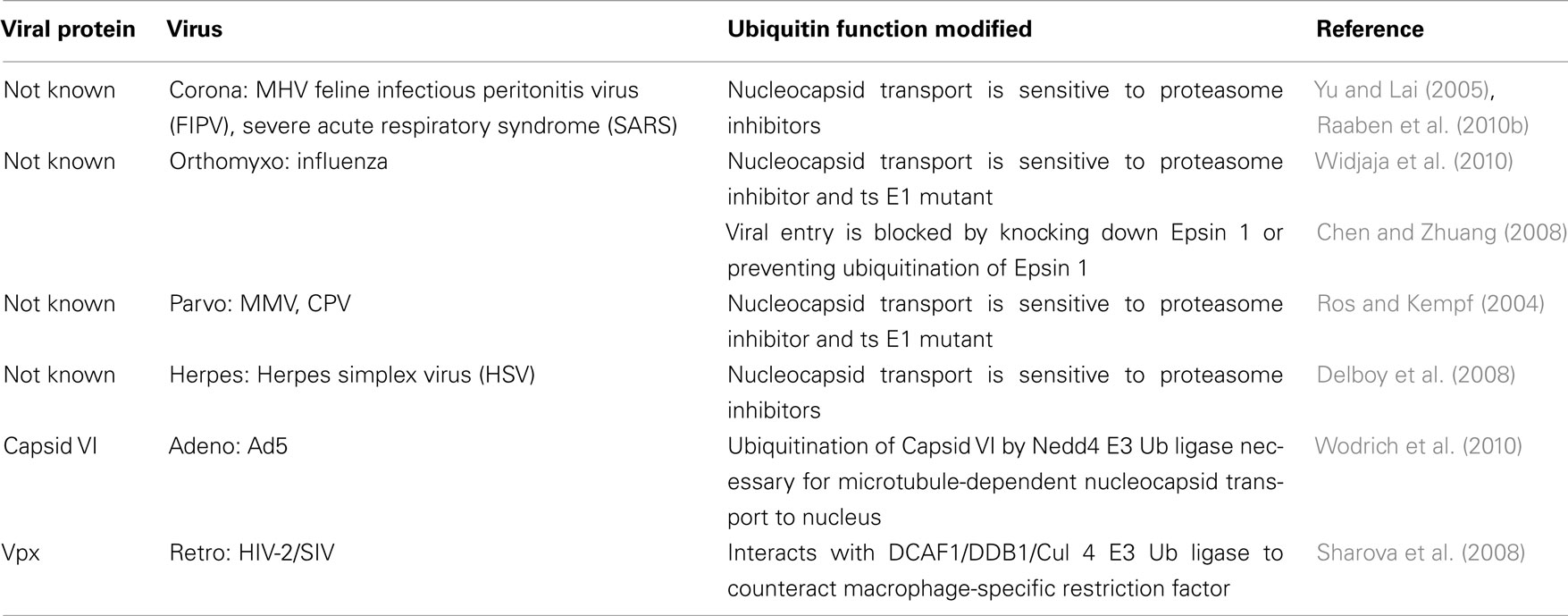
Table 1. Viral entry/nucleocapsid transport.
Proteasome Inhibitors in vivo
Because numerous in vitro studies have shown that multiple points in the viral life cycle can be blocked with proteasome inhibitors, their potential use as anti-viral agents has been an enthusiastic topic of discussion. However, recent in vivo studies suggest no clear consensus for the efficacy of these inhibitors. Several mouse studies evaluating the proteasome inhibitor bortezomib, which is clinically approved for multiple myeloma, found that lymphocytic choriomeningitis virus (LCMV; Basler et al., 2009), mouse hepatitis virus (MHV; Raaben et al., 2010a), and human respiratory syncytial virus (HRSV; Lupfer et al., 2010) each replicated better in the presence of bortezomib, so much so that MHV and HRSV hastened the mortality of infected mice. In contrast, Ma et al. (2010) showed that treatment of MHV-infected mice (pneumonitis model) with three different proteasome inhibitors (including bortezomib) resulted in reduced viral replication and a 40% survival rate. While these studies are not directly comparable due to the variety of viruses and mouse models employed, they suggest that much more research is necessary before these inhibitors can be approved for the treatment of viral infections.
Viral Transcription
There are relatively few examples in which viral transcription is enhanced by manipulation of the UPS. The transactivator proteins encoded by Epstein–Barr virus (EBV), HIV, and human T-lymphotropic virus (HTLV) each appear to interact with the UPS, and this results in the enhancement of transactivator function. The EBV transactivator EBNA1 binds the Ub-specific-processing protease 7 (USP7), a cellular DUB, and this augments binding of EBNA1 to the viral oriP site. This interaction also results in the deubiquitination of histone 2A at the oriP site (Sarkari et al., 2009), although the relevance of this histone modification to viral transactivation has not been evaluated. The HIV-1 transactivator Tat was shown to be ubiquitinated by Hdm2, which did not result in degradation of Tat, but instead enhanced viral transcription from the LTR (Bres et al., 2003). A more recent paper found that basal (Tat-independent) transcription from the HIV LTR requires Ski-interacting protein (SKIP) recruitment by the histone H2B ring finger protein 20 (RNF20) Ub ligase (Bres et al., 2009). Similar to Tat, the HTLV-1 transactivator Tax is also monoubiquitinated (Chiari et al., 2004) and sumoylated (Nasr et al., 2006). These modifications appear to enhance Tax’s ability to activate NF-κB, which in turn is necessary for viral transactivation and is also responsible for the oncogenic properties of the virus (Nasr et al., 2006; Harhaj et al., 2007). Ubiquitination of Tax C-terminal lysine residues is necessary for its role in binding and relocalizing IκB kinase (IKK) from the cytoplasm to perinuclear regions, which in turn modulates NF-κB activation. The sumoylation of Tax on overlapping lysine residues mediates both the development of Tax nuclear bodies (NB) and complete NF-κB activation (Nasr et al., 2006). This same group later found that a single Tax molecule can be both ubiquitinated and sumoylated, and that this differential modification is responsible for shuttling Tax and IKK between the cytoplasm, NB, and the centrosome (Kfoury et al., 2010). The UPS has also been implicated in human cytomegalovirus (HCMV) viral transcription. A delay in both early and late viral gene expression was observed in the presence of proteasome inhibitors, which was likely due to a block in viral RNA transcription. Tran et al. (2010) also observed that the 19s proteasome subunit Rpn2 relocalizes to viral replication centers in a viral DNA replication-dependent manner (Table 2).
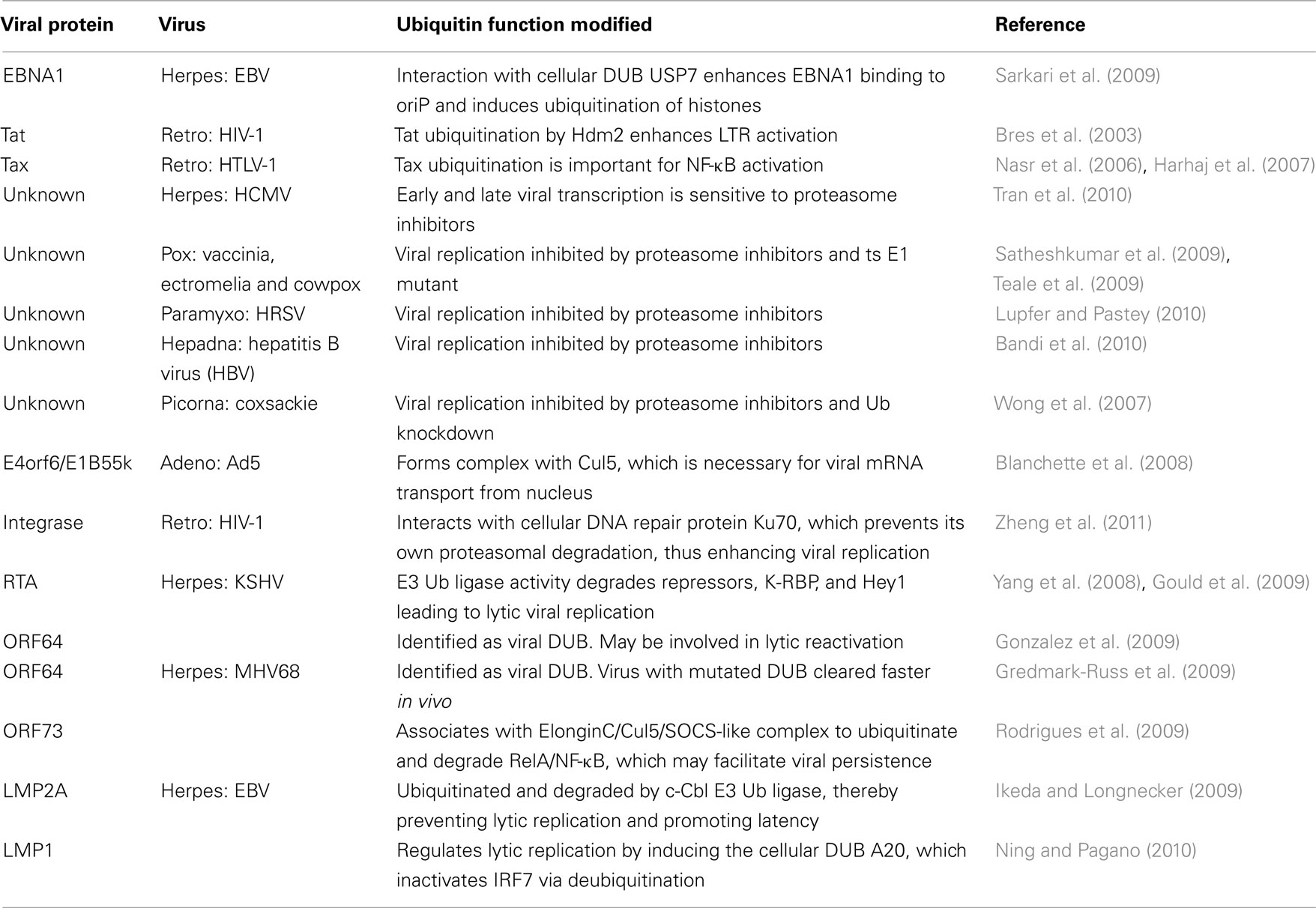
Table 2. Viral transcription, replication, and lytic/latent regulation.
Viral Replication
The use of proteasome inhibitors and ts E1 have been the main tools used to link the UPS to the replication of members of several virus families, including pox, paramyxo, hepadna, and picorna (see Table 2). The importance of the UPS to DNA tumor viruses such as adenovirus during their regulation of the cell cycle and their inhibition of apoptosis are well known and are discussed in detail below. Blanchette et al. (2008) have shown that the same adenoviral E4orf6/E1B55k/Cul5 E3 Ub ligase complex responsible for degrading p53 and Mre11 is also necessary for viral mRNA transport from the nucleus. However, the substrate(s) targeted for ubiquitination in order to achieve this function have yet to be elucidated. A more recent paper has described a novel use of the UPS to enhance HIV replication (Zheng et al., 2011). In a yeast two-hybrid screen using HIV integrase as the “bait,” the cellular protein Ku70, which is involved in DNA repair, the non-homologous end-joining pathway, transcription, apoptosis, and telomere maintenance (reviewed in Downs and Jackson, 2004) was identified (Studamire and Goff, 2008). Zheng et al. (2011) show that Ku70 is incorporated into HIV virions and prevents the proteasomal degradation of HIV integrase. Furthermore, if Ku70 is knocked down with siRNA, integrase levels are diminished and viral replication is decreased (Table 2).
Viral Lytic/Latency Regulation
An important aspect of the herpesviral lifecycle is the regulation of lytic replication and latency. There are now several examples of UPS involvement in various aspects of this regulation. In particular, lytic reactivation of Kaposi’s sarcoma herpesvirus (KSHV) from latency is regulated by at least two UPS-dependent mechanisms. The viral replication and transcription activator (RTA) protein also acts as an E3 Ub ligase that ubiquitinates and degrades its own repressors, including KSHV-RTA binding protein (K-RBP; Yang et al., 2008) and hairy/enhancer-of-split related with YRPW motif 1 (Hey1; Gould et al., 2009), which normally limit lytic replication. Another KSHV protein required for lytic reactivation is the tegument protein ORF64, which has been identified as a viral DUB (Gonzalez et al., 2009). ORF64 cleaves Ub from both K48- and K63-linked chains and requires a specific cysteine for its DUB activity. While knockdown of ORF64 with siRNA lowered lytic reactivation and lytic protein expression, a direct role for DUB activity in this reactivation was not shown. The murine gammaherpesvirus-68 (MHV68) ORF64 homolog has also been identified as a viral DUB. Gredmark-Russ et al. (2009) found that a virus expressing an ORF64 with an alanine substitution for the critical active site cysteine was more rapidly cleared than the wildtype virus in an in vivo mouse model. This suggested an important role for DUB activity in viral replication and possibly persistence, although they observed no differences in viral genome copy numbers (Gredmark-Russ et al., 2009). Another MHV68 protein involved in viral persistence is ORF73. Recent evidence suggests that ORF73’s abilities to both assemble with an ElonginC/Cul5/suppressor of cytokine signaling (SOCS)-like complex and effect the ubiquitination and degradation of RelA/NF-κB may be responsible for maintaining viral infection. In an in vivo mouse model, an MHV68 virus with a mutation of the ORF73 SOCS-box motif was unable to induce B-cell proliferation or set up a persistent infection, suggesting that NF-κB plays a role in these processes (Rodrigues et al., 2009). However, while the degradation of NF-κB also has the potential to impact interferon (IFN) induction (see below), this was not investigated.
An interesting twist on the role of the UPS in the regulation of viral latency is provided by the herpesvirus EBV. The viral latent membrane protein 2A (LMP2A) has been implicated in the regulation of both EBV latency and oncogenesis through its action as a B-cell receptor (BCR) mimic that constitutively activates the Lyn and Syk protein tyrosine kinases (reviewed in Portis et al., 2004). To determine if LMP2A is regulated similarly to BCR, Ikeda and Longnecker (2009) investigated the role that c-Cbl, an E3 Ub ligase that negatively regulates B-cell signaling, might play in LMP2A regulation. They discovered that LMP2A is ubiquitinated and degraded in a c-Cbl-dependent manner and that the Syk kinase is also degraded by c-Cbl in the presence of LMP2A. In addition, when c-Cbl was knocked down with shRNA, LMP2A induced lytic gene expression. Together with previous data showing that LMP2A contains two PPXY motifs and recruits Nedd4 Ub ligase to degrade Lyn kinase (Ikeda et al., 2001), these findings suggest that the cellular UPS is an important factor in the maintenance of EBV latency. In a similar story, the EBV LMP1 protein also appears to modulate latency through its interaction with the UPS. LMP1 stimulates the TNF receptor associated factor 6 (TRAF6) E3 ligase-dependent ubiquitination of IFN response factor (IRF) 7, and this modification is required for the subsequent phosphorylation of IRF7 by RIP1 kinase (Ning et al., 2008). If left unchecked, this would lead to activation of lytic replication and IFN induction. However, LMP1 was also recently shown to induce the cellular DUB A20, which inactivates IRF7 via deubiquitination (Ning and Pagano, 2010). Whether these intricate regulatory mechanisms are more advantageous for the cell or virus remains to be determined.
Cell Cycle Regulation, Inhibition of Apoptosis and Cellular Proliferation
The “high risk” oncogenic human papillomaviruses (HPV) provided the very earliest examples of viruses that utilize the host UPS to destroy host proteins. The HPV-encoded E6 and E7 proteins, which are best known for their ability to degrade p53 and pRb, respectively, can efficiently induce cellular transformation (reviewed in Mammas et al., 2008). The goal of such activities is presumably to extend the lifespan of infected cells, so that the virus can either pursue a latent lifestyle or increase viral proliferation prior to cell death. Since those initial observations, numerous other viruses have been shown to follow a similar program (Table 3).
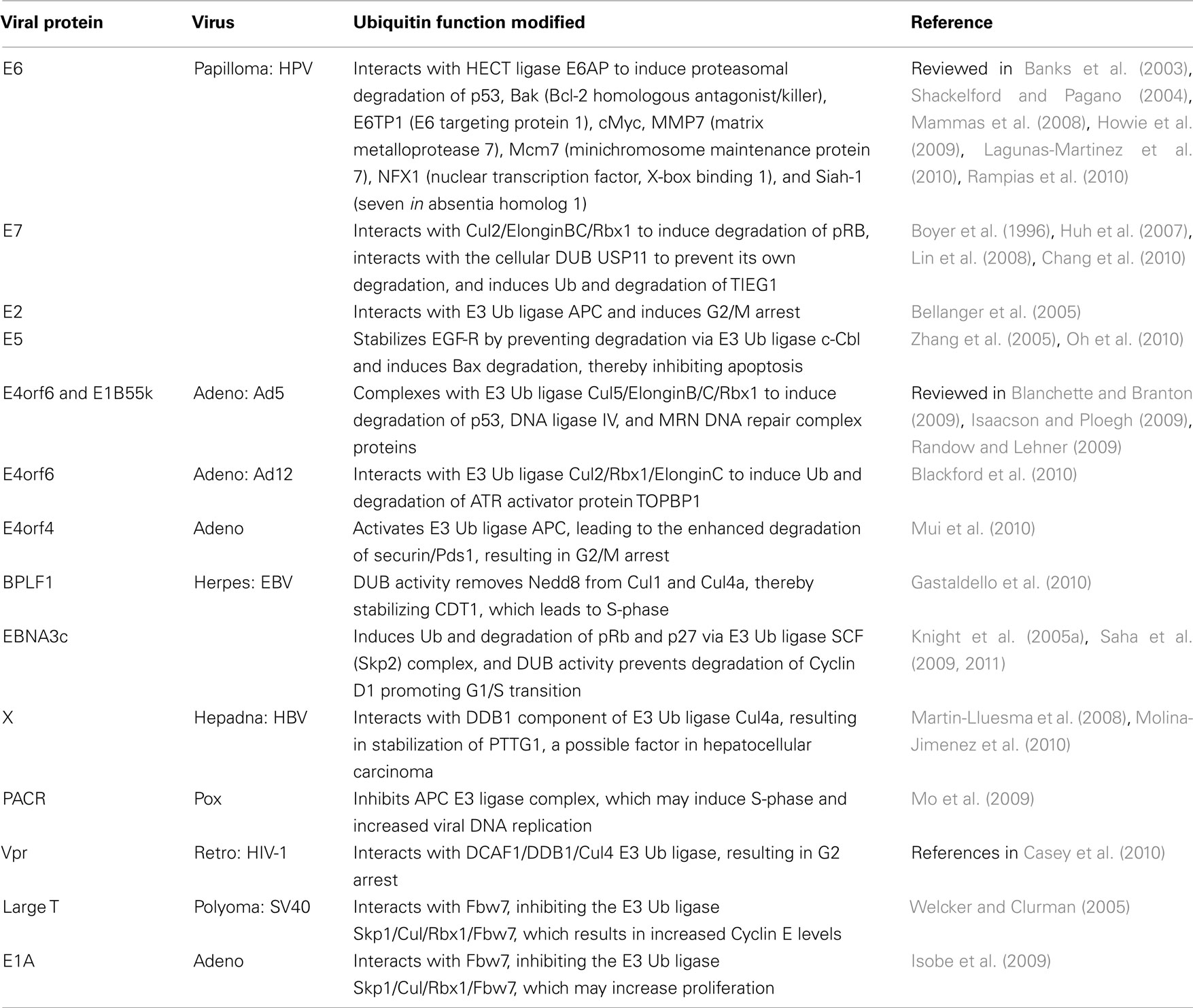
Table 3. Cell cycle regulation/inhibition of apoptosis/cell proliferation.
Human Papillomaviruses E6
Human papillomaviruses E6 can function as an adaptor to redirect the cellular E6-associated protein (E6AP) HECT ligase to target a number of cellular proteins for proteasome-dependent degradation (see Table 3). Chief among these is p53, which appears to be upregulated by the activities of HPV E7 (Demers et al., 1994, and see below). E6 can also target a number of PDZ [post synaptic density protein (PSD95), Drosophila disk large tumor suppressor (DlgA), and zonula occludens-1 protein (zo-1)]-domain proteins for degradation independently of E6AP, including human disks large (hDlg), membrane-associated guanylate kinase with inverted orientation 1, 2, and 3 (MAGI), and hScrib (Mammas et al., 2008). More E6 targets continue to be discovered. For example, E6 has recently been found to augment Wnt signaling in an E6AP-dependent manner, suggesting that Wnts may play a role in HPV carcinogenesis (Lichtig et al., 2010). In a search for the cellular proteins targeted by E6 to stimulate Wnt signals, Rampias et al. (2010) found that the cellular E3 ligase seven in absentia homolog (Siah-1), which is known to promote beta-catenin degradation via the UPS, is inhibited by the combination of HPV E6 and E7. This provides evidence for what may be an interesting example of a virus redirecting a cellular E3 ligase (E6AP) to target another cellular E3 ligase (Siah-1). Yet another player in the p53 pathway, the tumor suppressor Tat-interacting protein 60 kDa (TIP60), is targeted for proteasome-dependent degradation by E6, although this does not require E6AP (Jha et al., 2010). TIP60 was found to bind to the HPV major early promoter, recruiting a cellular repressor of E6 expression in the process. Therefore, degradation of TIP60 serves to derepress both E6 expression as well as cellular genes whose products limit p53-dependent apoptosis. As mentioned above, the cellular E6AP HECT ligase is variably required for E6 phenotypes. A recent study examined the requirement for E6AP in clinical HPV outcomes (Shai et al., 2010). Using a transgenic mouse model of HPV oncogenic phenotypes, they found that while E6AP is dispensable for epithelial hyperplasia and the limitation of DNA damage responses, E6AP is absolutely required for cervical carcinomas. Therefore, while this cellular E3 ligase may not be necessary for all E6 functions, it still has an impact upon the most serious consequence of HPV infection.
Human Papillomaviruses E7
E7 is the other primary contributor to HPV-mediated transformation. Members of the cellular pRb/pocket protein family normally function to control G1/S-phase progression by inhibiting the E2F family of transcriptional activators, which in turn control the transcription of S-phase promoters (Moody and Laimins, 2010). The most well-characterized E7 function is to target pRb family members for UPS-dependent degradation, thereby derepressing E2F-dependent promoters (Boyer et al., 1996). At the same time, E7 is a relatively unstable protein (Selvey et al., 1994) that has been found to be ubiquitinated on its N-terminal residue and then destroyed by the proteasome (Reinstein et al., 2000; Wang et al., 2001). While E7 has been detected in complexes with several E3 ligases (Kamio et al., 2004; Oh et al., 2004), of those, the only E3 complex also shown to contain pRb is Cul2/ElonginBC/Rbx1 (Huh et al., 2007), suggesting that this is the Ub ligase responsible for pRb degradation. Lastly, in what is perhaps an attempt to counter the host’s efforts to destroy it, E7 also interacts with the cellular DUB USP11, which leads to the stabilization of E7 (Lin et al., 2008). While E7 interacts with many cellular proteins to dysregulate the host cell cycle, aside from pRb, few others have been shown to be targeted for degradation. Chang et al. (2010) have recently added to this list the transforming growth factor-beta inducible early gene-1 (TIEG1), a transcription factor that can induce apoptosis in carcinoma cell lines. E7 binds directly to TIEG1, and TIEG1 is subsequently ubiquitinated and degraded via the proteasome, further contributing to E7-induced cellular transformation.
Human Papillomaviruses E2
The HPV protein E2 is a DNA-binding protein that can both activate and repress the transcription of viral promoters (reviewed in Lagunas-Martinez et al., 2010). Interestingly, HPV E2 normally represses the transcription of the genes encoding E6/E7. However, in cells infected with high risk HPV’s, the viral genome frequently integrates into that of the host, and HPV E2 is inactivated in the process. HPV E2 has also been shown to interfere with cellular pathways in a Ub-dependent manner. For example, HPV E2 interacts with the Cdc20 and Cdh1 substrate-specificity subunits of the anaphase promoting complex (APC) E3 ligase. This inhibits normal APC-dependent Cyclin B degradation, thereby leading to G2/M arrest (Bellanger et al., 2005). It has recently been shown that HPV E2 binds to Skp2, an F-box protein that serves as a specificity determinant for the SCF complex. In this way, HPV E2 is targeted for ubiquitination and subsequent proteasomal degradation. However, Skp2 is itself a known APC target, and APC-dependent Skp2 degradation is required to maintain cells in G1. Because HPV E2 interferes with APC (see above), this suggests a criss-crossing autoregulatory loop in which HPV E2 interferes with APC, thus stabilizing Skp2 and promoting G1/S progression. Once Skp2 reaches sufficient levels, Skp2/SCF ubiquitinates and degrades HPV E2, which de-represses HPV E6 and E7 expression. This combination tends to push the host cell toward S-phase, and perhaps into transformation (Bellanger et al., 2010).
Human Papillomaviruses E5
Although it has been shown to induce epithelial cell hyperproliferation when overexpressed in transgenic mice (Maufort et al., 2010), HPV E5 has primarily been characterized as a mere contributor to E6/E7-driven transformation (reviewed in Moody and Laimins, 2010). Due to its ER localization and proposed inhibition of endosome acidification, it is thought that HPV E5 impacts the trafficking of proteins such as EGF-R, which enhances proliferative cell signaling (Leechanachai et al., 1992; Straight et al., 1995). A role for Ub in this process was provided by the observation that E5 binding to EGF-R prevents the receptor from binding to the cellular E3 c-Cbl (Zhang et al., 2005), which is involved in normal EGF-R receptor downregulation. This stabilizes EGF-R and leads to constitutive signaling. In what appears to be yet another Ub-dependent E5 phenotype, Oh et al. (2010) have recently shown that the HPV16 E5 contribution to cervical cancer may lie in its ability to limit Bax-dependent apoptosis of infected cells. Expression of E5 induced the degradation of Bax in a UPS-dependent manner. E5 may therefore function as an adaptor that targets cellular proteins for ubiquitination, and this may be responsible for other established E5 activities as well.
Adenovirus
Like the high risk HPVs, adenoviral infection can also result in host cell transformation. The primary culprits here are the viral proteins E4orf6 and E1B55k, which combine to effect the UPS-dependent degradation of host proteins related to cell cycle regulation and DNA damage repair (see Table 3). E4orf6 functions as an adaptor that links E1B55k to various Cul containing E3 complexes. E1B55k then acts as a substrate recognition subunit that redirects the new E3 complex to various cellular targets. The prevention of apoptosis leads to the accumulation of DNA damage within infected cells. To avoid cellular DNA damage responses, adenovirus blocks the ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3-related) pathways (Carson et al., 2003). Most Adenovirus serotypes, including Ad5 and Ad12, prevent the activation of the ATM pathway by inducing the degradation of the MRN (Mre11, Rad50, and Nbs1) DNA damage complex via the mechanism just described. However, it has recently been shown that Ad5 and Ad12 prevent the activation of the ATR pathway in different ways. Prior to the E4orf6/E1B55K-dependent degradation of the MRN complex, Ad5 utilizes E4orf3 to mislocalize and immobilize the MRN complex, which prevents ATR activation (Carson et al., 2009). In contrast, Ad12 E4orf3 does not appear to have this function. Instead, Ad12 E4orf6 (without E1B55k) interacts with a Cul2/Rbx1/ElonginC ligase to promote the UPS-dependent degradation of the ATR activator protein topoisomerase-ILβ-binding protein-1 (TOPBP1), thereby blocking ATR activation (Blackford et al., 2010). This observation of differential Cul usage by adenoviruses has been extended even further. In a recent survey of all adenoviral subgroups, it was found that while all E4orf6/E1B55k pairs associate with Cul family members, the particular Cul varies. Among the serotypes, Cul5 is most frequently bound, but some serotypes recruit Cul2, and Ad16 E4orf6/E1B55k recruited both Cul2 and Cul5 equally well. While all serotypes degraded DNA ligase IV, complexes from some serotypes failed to degrade Mre11, p53, or integrin α3, suggesting that the choice of Cullin may in part dictate which targets are destroyed (Cheng et al., 2010).
Finally, the adenoviral protein E4orf4 is known to induce cell death via a number of cell-type specific mechanisms (Robert et al., 2002). One commonality amongst these mechanisms is the modification of the APC E3 ligase activity, although it has been reported that E4orf4 can both reduce (Kornitzer et al., 2001) and induce (Mui et al., 2010) APC activity to achieve this end. In the latter case, E4orf4 expression led to degradation of the APC substrate Pds1/securin, a protein that is required for the completion of mitosis. Thus, it appears that yet another adenoviral protein can, albeit indirectly, redirect the host Ub system to target host proteins that disrupt the cell cycle.
Herpesvirus
The herpesviral large tegument proteins harbor a cysteine protease catalytic site that shows little homology to known DUBs, but nevertheless has been shown to display DUB activity (Kattenhorn et al., 2005). As discussed above, the KSHV and MHV68 large tegument homologs (ORF64) have been implicated in the regulation of the lytic/latent balance. Mutation of the active site residues in the Marek’s disease virus homolog lowered viral replication and reduced the number of T-cell lymphomas among infected chickens (Jarosinski et al., 2007). A similar mutation introduced into Pseudorabies virus likewise impaired viral replication and lowered virulence (Bottcher et al., 2008). A bioinformatics approach was used to search the EBV genome for proteins encoding DUB activity, revealing BSLF1, BXLF1, and the EBV large tegument protein BPLF1 (Sompallae et al., 2008). In a follow-up paper, the same group focused on BPLF1 and found that it functions to dysregulate the host cell cycle in an effort to promote viral DNA replication (Gastaldello et al., 2010). This was shown to depend on BPLF1’s ability to proteolytically remove the ubiquitin-like modifier protein NEDD8 from Cul1 and Cul4a. This in turn leads to the stabilization of the Cul substrate CDT1, which then pushes the host cell toward S-phase. The authors found a similar activity encoded by the HSV and MCMV large tegument proteins, suggesting that perhaps all herpesviruses utilize their tegument proteins in this manner.
In addition to BPFL1, EBV encodes numerous other proteins that contribute to cellular transformation. Among these is EBNA3c, which is itself ubiquitinated (Knight et al., 2005b), and targets pRb and p27 for degradation by recruitment of the SCF (Skp2) E3 Ub ligase complex (Knight et al., 2005a; Saha et al., 2009). EBNA3c has also been shown to have a DUB activity that can remove Ub from itself. Interestingly, EBNA3c was found in complexes with both p53 and Mdm2, the cellular E3 responsible for p53 ubiquitination. More recently EBNA3c was shown to prevent the degradation of Cyclin D1, which overrides the activity of the tumor suppressor pRb and thereby promotes G1/S-phase transition (Saha et al., 2011). Thus, EBNA3c appears to both positively and negatively modulate the ubiquitin status of a number of proteins whose dysregulation can promote tumorigenesis.
Hepadnaviruses
Hepatitis B virus (HBV) is one of the primary causes of hepatocellular carcinoma (HCC), and the viral protein HBx has been shown to promote cell cycle progression via a number of mechanisms (reviewed in Kew, 2011). While a role for ubiquitin was not initially appreciated, it was found that HBx binds to damage-specific DNA-binding protein 1 (DDB1; Lee et al., 1995), and that numerous HBx phenotypes depend upon this interaction (see references in Martin-Lluesma et al., 2008). The later finding that DDB1 is a part of Cul4a E3 ligase complex (Shiyanov et al., 1999) suggested that HBx functions to interfere with such complexes, resulting in many of the HBx phenotypes. One recent paper has shown that HBx stabilizes the proto-oncogene pituitary tumor-transforming gene 1 (PTTG1) protein, which is known to be overexpressed in HCC (Molina-Jimenez et al., 2010) and which has previously been shown to bind to and inhibit p53 (Bernal et al., 2002). However, whether PTTG1 ubiquitination is directly or indirectly inhibited by HBx remains to be determined.
Poxvirus
Unlike the other viruses described in this section, the members of the poxvirus family are not well known for their ability to interfere with the host cell cycle. However, there have been several reports noted for vaccinia (Koziorowska et al., 1971; Puckett and Moss, 1983; Yoo et al., 2008). Poxviruses are presumed to manipulate the host ubiquitin system via (a) their ankyrin-like proteins, which have been found to interact with Cul1 ligase complexes, (b) their BTB-Kelch proteins, which interact with Cul3 ligase complexes (reviewed in Shchelkunov, 2010), and (c) their conserved p28 proteins that are E3 ligases (Huang et al., 2004). Amongst these, only the Myxoma M-T5 ankyrin repeat protein has been shown to affect the cell cycle, and it does so by overcoming the G0/G1 arrest that normally takes place in infected cells. However, a direct role for ubiquitin in this process has not been shown (Johnston et al., 2005). Interestingly, several poxviruses were recently found to encode a RING domain protein referred to as poxvirus APC/cyclosome activator (PACR), which inhibits the function of the APC E3 ligase complex (Mo et al., 2009). PACR is homologous to the RING domain protein APC11, which is the APC subunit responsible for binding to Ub-charged E2 ligases. That same group’s subsequent finding that PACR competes with APC11 for binding to the APC complex suggests that PACR is a dominant negative inhibitor of the APC complex (Mo et al., 2010). Because APC normally functions to maintain cells in a quiescent state, the authors suggest that PACR-dependent inhibition of APC may promote viral DNA replication by pushing host cells toward S-phase.
Human Immunodeficiency Virus-1
The small accessory proteins encoded by HIV-1 (and variably by HIV-2 and SIV) include Vif, Vpu, and Vpx/Vpr. Each of these proteins has been shown to function as adaptors for various Cul-based E3 ligase complexes, which, at least in the case of Vif and Vpu, have clear benefits for the virus with respect to replication and egress (see below). Among the numerous activities attributed to Vpr, only a subset has been shown to depend upon Vpr’s ability to associate with the DDB1-Cul4 associated factor 1 (DCAF1), which functions as the specificity determinant for a Cul4 E3 ligase complex (see references in Casey et al., 2010). In the mid-1990s, several laboratories observed that Vpr induces a G2 cell cycle arrest in dividing cells (Jowett et al., 1995; Rogel et al., 1995). While the benefit to the virus remains unclear, many other groups subsequently found that this activity is dependent upon Vpr’s engagement of the DCAF1/DDB1/Cul4 Ub ligase. However, due to the Vpr-induced change in the levels of a large number of host proteins, which includes transcription factors, cytokines, and chemokines (see references in Casey et al., 2010), some have argued that the G2 arrest is a mere byproduct of those changes. In support of this notion, one group has shown that Vif can also arrest cells in G2 (DeHart et al., 2008), and others have found that DCAF1 is itself required for normal S-phase progression (McCall et al., 2008). These findings suggest that the titration of host Ub ligases may result in G2 arrest, and perhaps other Vpr phenotypes as well.
Viral Budding
One of the more intriguing aspects of the viral lifecycle is the manner in which enveloped viruses acquire their membranes. After the assembly of viral proteins at host membranes, the subsequent formation of a mature viral particle requires the deformation of the membrane, gathering it around the viral particle, culminating in an energetically unfavorable “pinching-off” event that separates the viral envelope from the host membrane. Although there appear to be examples of viruses that rely solely on their own integral membrane proteins to accomplish this feat (reviewed in Chen and Lamb, 2008), many other viruses instead require both Ub and the host vacuolar protein sorting (VPS) machinery (reviewed in Martin-Serrano, 2007). The general story that has emerged is that viral budding proceeds in a manner analogous to that in which ubiquitinated membrane proteins destined for lysosomal degradation are recognized by the host VPS machinery, packaged into multivesicular bodies (MVB; reviewed in Davies et al., 2009), and then delivered to lysosomes. For those viruses whose budding is VPS-dependent, their viral Gag or matrix proteins are found to harbor so-called late budding domains (L-domains), which are short motifs that function as binding sites for various VPS components. L-domains are so named because mutations within these motifs lead to the formation of viral particles with membranes that have failed to complete the budding process. There appear to be three primary classes of L-domains; PT/SAP, which interacts with tumor susceptibility gene 101 (Tsg101), a component of the VPS complex endosomal sorting complex required for transport-I (ESCRT-I), PPXY, which interacts with members of the Nedd4 family of Ub ligases, and YPDL, which interacts with apoptosis-linked-gene-2 product, ALG-2–interacting protein X, (ALIX; reviewed in Chen and Lamb, 2008; Calistri et al., 2009). Remarkably, L-domains appear to be interchangeable, and in some cases, a single Gag/Matrix protein will encode more than one L-domain. Once an L-domain is recognized by its cognate VPS component(s), it is thought that the ESCRT machinery (including ESCRT-I, -II, and -III) is recruited to supply the force necessary for virus budding. A role for Ub in this process first came from studies showing that retroviral Gag proteins were ubiquitinated (Putterman et al., 1990; Ott et al., 1998). Likewise, proteasome inhibitors have been shown to prevent budding (Patnaik et al., 2000; Schubert et al., 2000; Strack et al., 2000). The finding that PPXY L-domains bind Nedd4 E3 ligases further supported a role for ubiquitination. In an effort to determine if L-domains are themselves ubiquitinated, several groups have mutated all lysines within the vicinity of L-domains, and while this inhibits budding in some cases (Spidel et al., 2004; Gottwein et al., 2006), in others, it has little effect (Zhadina et al., 2007). However, because Ser, Thr, and Cys residues are now recognized as alternate “ubiquitin acceptors,” many of the earlier studies that solely focused on lysine will perhaps need to be revisited.
Retroviruses
In an attempt to clarify the requirement for Ub in retroviral budding, Zhadina and Bieniasz (2010) have recently engineered a foamy virus Gag protein that generates VLPs when provided with a variety of L-domains. When Nedd4-dependent PPXY motifs were inserted, they found that budding occurred, and that Gag was not ubiquitinated in the process. Because catalytically active Nedd4 ligases were required, their data suggests that if Gag is not ubiquitinated, a cellular protein(s) is instead targeted. However, when they generated Gag variants without an L-domain, but that were directly fused to Ub, budding was efficient, and depended upon an intact Vps system. This somewhat perplexing result hints at the plasticity of the system, and indicates that as long as ubiquitination occurs in the vicinity of the L-domain, budding can take place. Similar results have also been obtained for HIV-1 by Weiss et al. (2010) who find that a number of Nedd4 ligases, including yeast Rsp5, will promote VLP formation when targeted to Gag, but that budding does not correlate with the ability of these ligases to ubiquitinate Gag. The identity of at least one cellular ubiquitination target is perhaps suggested by Sette et al. (2010) who have recently provided evidence that ALIX binds to HIV-1 Gag, recruits Nedd4.1, and is then itself ubiquitinated by Nedd4.1 to promote egress.
Filoviruses
Like the retroviral Gag proteins, the VP40 matrix proteins encoded by both Ebola (Harty et al., 2000) and Marburg (Urata et al., 2007) viruses can form VLPs in the absence of other viral proteins. Ebola VP40 harbors both PPXY and PT/SAP L-domains, and appears to engage both Nedd4 and Tsg101, respectively (Timmins et al., 2003). On the other hand, Marburg virus encodes only a PPXY motif. Interestingly, it has been shown that while deletion of the PPXY motif severely limits Marburg VLP formation, co-expression of the viral nucleoprotein (NP), or glycoprotein (GP) with the VP40 PPXY mutant rescues budding (Urata et al., 2007). In that same paper it was found that Tsg101 is recruited to VP40 in a PPXY-dependent manner, providing a unique example of non-PT/SAP-dependent Tsg101 interaction. This same group has more recently shown that the Marburg VP40 PPXY motif also interacts with Nedd4 (Urata and Yasuda, 2010). Coupled with their earlier data, this result indicates that the filoviruses require both Nedd4 and Tsg101 for VLP formation, and that at least in the case of Marburg, NP and/or GP may also play a role in recruiting those proteins.
Paramyxoviruses
The paramyxoviruses are another group of enveloped RNA viruses, which appear less consistent in their use of L-domains and the MVB apparatus. For example, parainfluenza virus 5 (PIV5) budding requires matrix (M), nucleocapsid (NP), and spike glycoproteins (Schmitt et al., 2002), none of which appear to encode obvious L-domain sequences. However, PIV5 budding has been shown to depend on the host VPS machinery, as well as to be sensitive to proteasome inhibitors, suggesting commonalities between the retro- and filoviral budding pathways (Schmitt et al., 2005). Interestingly, the PIV5 M protein was found to harbor a unique FPIV L-domain that was discovered via complementation of an HIV-1 PTAP Gag mutant (Schmitt et al., 2005). Mutation of that motif in a recombinant PIV5 virus led to drops in viral titers by five orders of magnitude compared to the wildtype, demonstrating the importance of this sequence to viral production. However, it remains to be seen if the PIV5 FPIV motif functions as a binding site for either ESCRT components or Nedd4 ligases. While present in some paramyxoviruses, the FPIV motif is not observed in the M proteins from measles, Sendai, and Nipah virus, suggesting variability in paramyxoviral budding pathways. Support for this notion comes from studies of Nipah virus, which encodes an M protein that is necessary and sufficient for VLP production (Ciancanelli and Basler, 2006). Wang et al. (2010) have recently shown that prior to budding from the host plasma membrane, the Nipah M protein must first pass through the nucleus. Treatment of infected cells with proteasome inhibitors trapped M protein within the nucleus, and like PIV5, lowered viral titers substantially, suggesting that ubiquitination is involved in M protein trafficking. The authors went on to provide evidence that M protein is monoubiquitinated on four separate residues, including a lysine within a putative bipartite nuclear localization signal, and that this same lysine is likely to be ubiquitinated in order for M protein to be exported from the nucleus. They finished by showing that Nipah virus release was sensitive to levels of the proteasome inhibitor bortezomib that are readily achievable in human patients. While the precise mechanisms are not known, it is clear that Ub plays a role in the Nipah virus lifecycle. However, because proteasome inhibitors appear to block upstream events (e.g., M protein release from the nucleus), a specific role for Ub in Nipah budding is less certain.
Rhabdoviruses
Similar to other Gag/Matrix proteins discussed above, the vesicular stomatitis virus (VSV) M protein can promote VLP formation in the absence of other viral proteins (Harty et al., 1999), and this has been shown to depend upon a PPXY motif within the M protein (Craven et al., 1999). As one might expect for PPXY L-domain proteins, VSV M protein was found to bind Nedd4 E3 ligases, and proteasome inhibitors limit viral release, suggesting that Ub is involved (Harty et al., 2001). However, unlike other viruses that utilize PPXY L-domains, VSV budding does not appear to depend upon either Tsg101 or downstream players in the ESCRT pathway (Irie et al., 2004), indicating that the combination of an L-domain and a Ub-dependent budding mechanism is not a guarantee of dependence upon the host VPS pathway.
Arenaviruses
The Lassa virus matrix protein (Z) alone results in the production of VLPs, and harbors both PTAP and PPXY L-domains (Perez et al., 2003; Eichler et al., 2004). Similar to other enveloped RNA viruses, it has been demonstrated that budding of Lassa virus Z-protein VLPs is dependent upon Tsg101 and other ESCRT components (Urata et al., 2006). However, while the New World Tacaribe virus Z protein also promotes VLP budding, it harbors no obvious L-domain sequences and is not dependent upon Tsg101 (Urata et al., 2009). Nevertheless, Tacaribe Z-protein budding was still shown to require downstream Vps components, and so it is likely that arenavirus budding will, in general, depend upon the host MVB pathway.
DNA Viruses
While not all enveloped viruses bud through the plasma membrane, they are still faced with the same mechanistic problems. HBV budding can be subcategorized into the formation of either non-infectious subviral particles (SVP, composed of only the three viral envelope proteins), or fully infectious particles (recently reviewed in Patient et al., 2009), each of which appears to bud through cytoplasmic membranes. Interestingly, release of infectious HBV from cells has been shown to depend on the host VPS pathway (Kian Chua et al., 2006; Lambert et al., 2007; Watanabe et al., 2007). Moreover, the HBV core protein harbors a PPXY motif that has been shown to interact with Nedd4 (Rost et al., 2006), implicating a role for ubiquitination. Mutation of the two Lys residues within the core protein had no effect on budding (Garcia et al., 2009), but, as described above, alternate Ub acceptor residues may instead be targeted. Members of the herpesviridae appear to sequentially bud first through the inner nuclear membrane and then again through the TGN membrane (reviewed in Mettenleiter et al., 2009). The release of both Herpes simplex virus 1 (HSV-1; Calistri et al., 2007; Crump et al., 2007; Pawliczek and Crump, 2009) and HCMV (Tandon et al., 2009) particles has been shown to require components of the MVB pathway, suggesting that this may be a common theme among the herpesviruses. However, the details regarding their use of Ub in these budding events awaits further experimentation.
Viral Release
The recent identification of Tetherin (BST-2/CD317/HM1.24) as an IFN-induced anti-viral factor that restricts the egress of HIV-1 and other enveloped viruses by tethering mature virions to the host cell surface (Neil et al., 2008; Jouvenet et al., 2009) has revealed additional viral countermeasures that require the assistance of the UPS. The ability of the HIV-1 accessory protein Vpu to enhance viral release has been known for quite some time, however the mechanism for this function was not clear. Viral egress appeared to be separable from Vpu’s prominent role in inducing the ER-associated proteasomal degradation of the HIV-1 receptor, CD4 (Schubert and Strebel, 1994; Schubert et al., 1996), although some viral release enhancement has been attributed to CD4 depletion (Bour et al., 1999). However, recent evidence suggests that, similar to the situation with CD4, Vpu acts as a substrate recognition factor for an SCF Ub ligase complex to target Tetherin for downregulation (Van Damme et al., 2008; Douglas et al., 2009; Goffinet et al., 2009; Iwabu et al., 2009; Mangeat et al., 2009; Mitchell et al., 2009). Several studies by our group and others have concluded that in contrast to CD4, Vpu promotes the degradation of Tetherin within the lysosome (Douglas et al., 2009; Iwabu et al., 2009; Mitchell et al., 2009), although under certain conditions proteasomal degradation has been observed (Goffinet et al., 2009; Mangeat et al., 2009; reviewed in Douglas et al., 2010). A recent paper suggests that Vpu-mediated ubiquitination of Tetherin can occur on all lysine and non-lysine Ub targets in the cytoplasmic tail (Tokarev et al., 2010). Before this tethering function was known, our group had identified BST-2 in a proteomics screen for targets of the viral Ub-ligase K5 of the gammaherpesvirus KSHV (Bartee et al., 2006). Later we showed that K5 induces ubiquitination of Tetherin’s cytosolic lysines, which leads to both its lysosomal degradation and the enhancement of KSHV egress (Mansouri et al., 2009). Other viruses, such as HIV-2 (Douglas et al., 2009; Le Tortorec and Neil, 2009), SIV (Jia et al., 2009; Zhang et al., 2009), and Ebola (Kaletsky et al., 2009) also express antagonists of Tetherin, but there is no evidence thus far to indicate that they utilize the UPS for this function.
As discussed previously, adenoviruses express the E4orf6 and E1B55k proteins that complex with cellular E3 Ub ligases to degrade cellular proteins involved in apoptosis and cell cycle regulation. A recent proteomics study looking for new substrates of E4orf6 and E1B55k/Cul5 found numerous host proteins that were both up- and down-regulated. One of the substrates identified was α3 integrin, which was degraded in the presence of the Adenoviral proteins (Dallaire et al., 2009a). This same group showed that α3 integrin degradation promotes cell detachment from the extracellular matrix and that this may contribute to viral release and spreading (Dallaire et al., 2009b).
Evading Innate Immune Mechanisms
The interface between viruses and the host innate immune responses has also provided numerous examples in which viruses harness the UPS. IFN is the host’s first line of defense against viral infection and there are numerous examples in which viruses employ the UPS to block either IFN production or its anti-viral effects. The following section is sub-divided into the main steps of the IFN pathway that are targeted by various viral classes, with an emphasis on the most recent evidence for viral misappropriation of the UPS to subvert anti-viral immunity (Table 4).
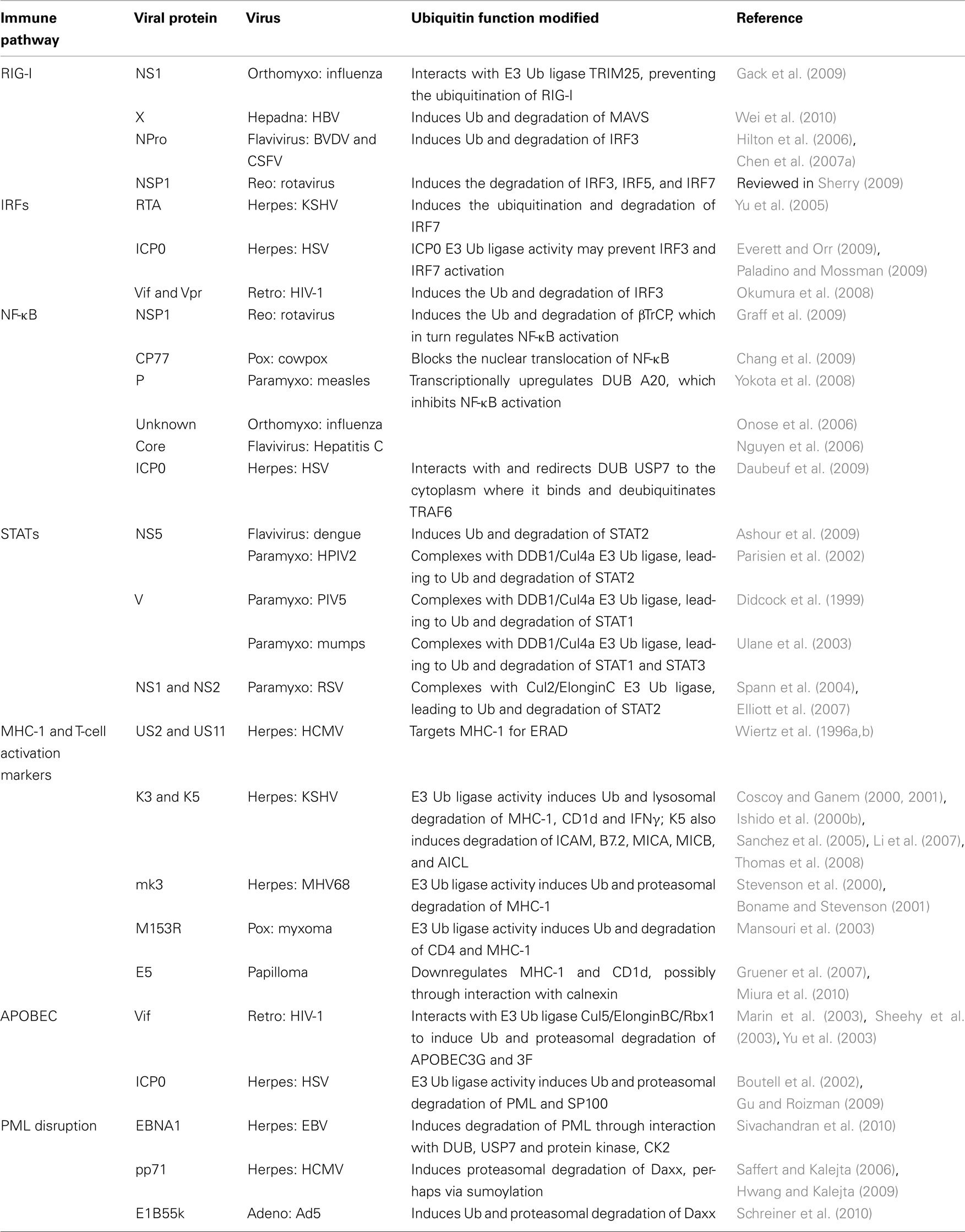
Table 4. Evasion of the host immune system.
Downregulation of Factors Leading to IFN Production
The host cell has a variety of viral sensing mechanisms that ultimately trigger IFN production (recently reviewed in Kanneganti, 2010). This signaling cascade begins with several classes of pattern-recognition receptors (PRRs) including the Toll-like receptors (TLRs), Retinoic acid inducible gene-I (RIG-I)-like receptors (RLRs), and the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs). Signaling through these molecules in turn leads to the activation of NF-κB, IRF3, and IRF7, which promote IFNα/β expression. Many viruses have evolved mechanisms that utilize the UPS to degrade specific players within these pathways, thereby limiting IFN production.
Retinoic acid inducible gene-I
In an effort to inhibit IFN activation, influenza and HBV utilize Ub-dependent mechanisms to target RIG-I either directly or through its downstream effector the mitochondrial anti-viral signaling protein (MAVS). In order to initiate the anti-viral signaling cascade, RIG-I must first be ubiquitinated by the E3 ligase tripartite motif (TRIM) protein 25. The influenza NS1 protein interacts with TRIM25, thereby preventing the ubiquitination of RIG-I. Gack et al. (2009) suggest that NS1 works by blocking the oligomerization of TRIM25, which is critical for its E3 ligase activity. The HBV X protein (HBx) was recently found to inhibit IFN-β production by interacting with MAVS, leading to its ubiquitination and proteasomal degradation (Wei et al., 2010). One particular MAVS lysine mutant exhibited increased IFN-β activation, suggesting that this is the residue targeted for HBx-dependent ubiquitination. However, the particular E3 Ub ligase involved in this process has not been identified.
IFN response factors
Other common viral targets degraded with the help of the UPS to limit IFN production are IRF3 and IRF7. Examples can be found among the flaviviruses, reoviruses, and herpesviruses. Bovine viral diarrhea virus (BVDV) and classical swine fever virus (CSFV) are flaviviruses that encode NPro, a papain-like protease that acts as an IFN antagonist by binding and inducing the polyubiquitination and subsequent degradation of IRF3 (Hilton et al., 2006; Chen et al., 2007a). However, NPro protease activity does not appear to be required for this function. Moreover, NPro from CSFV was itself degraded in a UPS-dependent manner in the absence of IRF3 (Seago et al., 2010). Whether NPro acts as a Ub ligase or recruits a cellular Ub ligase remains to be determined. The rotavirus NSP1 protein has also been shown to block IFN production by inducing the degradation of IRF3, IRF5, and IRF7 (reviewed in Sherry, 2009). Graff et al. (2007) showed that the NSP1 zinc-binding domain is important for IRF3 degradation, and their data suggests that NSP1 may be acting as an E3 Ub ligase. Herpesviruses have also been shown to inhibit IFN activation by interfering with IRFs. As discussed above, the KSHV RTA protein can function as both a transcriptional activator and an E3 ligase. In addition to its other targets (see above), RTA also induces the ubiquitination and degradation of IRF7 (Yu et al., 2005). HSV encodes the multifunctional protein ICP0, which has been shown to interfere with several stages of the IFN pathway, including the prevention of IRF3 and IRF7 activation (reviewed in Paladino and Mossman, 2009). ICP0 E3 Ub ligase activity has been implicated in this function. However, recent data indicates that inhibition of signal transducer and activator of transcription (STAT) or IRF3 pathways does not enhance the replication of ICP0-deleted viruses (Everett et al., 2008). These results suggest that ICP0’s role in subversion of the IFN response may be secondary to its involvement in promyelocytic leukemia (PML) and sp100 degradation (see below; Everett and Orr, 2009). Lastly, HIV-1 has been shown to downregulate IRF3 protein levels in infected cells, and Okumura et al. (2008) have provided evidence that this is due to the action of the HIV-1 E3 Ub adaptor proteins Vif and Vpr (Okumura et al., 2008). However, while ectopically expressed Vpr and Vif were shown to induce IRF3 ubiquitination and degradation in transfected 293T cells, IRF3 degradation was still observed in T-cells infected with an HIV-1 vpr/vif double mutant. Recent work from another group has confirmed IRF3 downregulation by HIV-1, and while their data supported a significant role for Vpr in that process, Vif did not appear to have an effect (Doehle et al., 2009).
NF-κB
NF-κB activation provides yet another route to IFN production and is thus a target for several viral antagonists. In addition to its ability to degrade IRF3 (see above), the rotavirus NSP1 protein also mediates the ubiquitination and degradation of the cellular E3 Ub ligase F-box protein beta-transducin repeating containing protein (βTrCP), which regulates NF-κB activation via the degradation of the NF-κB inhibitor IκB (Graff et al., 2009). This provides an interesting example of a single virally encoded Ub ligase targeting two separate mechanisms for IFN activation. The cowpox host-range protein CP77 has also been found to inhibit NF-κB activation. CP77 contains both an F-box motif that mediates binding to the SCF complex and a series of ankyrin repeats responsible for binding to NF-κB. While it was expected that CP77 would simply serve as an adaptor targeting NF-κB for ubiquitination by the SCF complex, no CP77-dependent ubiquitination or degradation of NF-κB was observed (Chang et al., 2009). Curiously, both the CP77 F-box and ankyrin repeats are required in order to prevent NF-κB nuclear translocation. Because NF-κB was not directly targeted for ubiquitination/degradation, the authors propose that CP77 somehow sequesters NF-κB to prevent its activation and nuclear translocation (Chang et al., 2009). Measles virus appears to exhibit a cell-type specific suppression of NF-κB activation, which is active in monocytes but not epithelial cells (Indoh et al., 2007). This activity is mediated by the viral P protein, which has been shown to transcriptionally upregulate the cellular DUB A20, thus leading to the inhibition of NF-κB-dependent IFN induction via the deubiquitination of TRAF6 (Yokota et al., 2008). As described in a previous section, A20 induction by EBV LMP1 was shown to inhibit IRF7, which consequently prevents IRF7-dependent IFN induction (Ning and Pagano, 2010). Influenza (Onose et al., 2006) and hepatitis C (Nguyen et al., 2006) have also been shown to inactivate NF-κB via A20 induction. Therefore, multiple viruses appear to utilize this cellular DUB as a means to inhibit two separate IFN-induction pathways. Finally, HSV appears to hijack a different cellular DUB to evade the immune system. The viral transcriptional activator ICP0 has been shown to interact with and redirect the cellular DUB USP7 to the cytoplasm where it deubiquitinates TRAF6 and IKKγ, which are required for activation of the canonical NF-κB pathway (Daubeuf et al., 2009).
Downregulation of IFN Signaling Molecules (STATs)
Once IFN is produced, it binds to the IFN-receptor on neighboring cells, thereby activating the STAT signaling cascade that ultimately results in the transcriptional activation of many anti-viral IFN-stimulated genes (ISGs). This makes targeting STATs another common means by which viruses ablate the IFN response. For example, both the flaviviruses and paramyxoviruses have evolved UPS-dependent methods to eliminate STATs. The Dengue virus NS5 polymerase interacts with STAT2, leading to STAT2 ubiquitination and proteasome-dependent degradation. NS5 is initially expressed as part of a single polyprotein that is cleaved by viral and host proteases into more than 10 individual proteins. Interestingly, NS5 alone can interact with STAT2, but does not induce degradation except in the context of the polyprotein (Ashour et al., 2009). It is not known whether a cellular Ub ligase is involved or if the proteolytic processing of the viral polyprotein somehow leads to the degradation of STAT2. NS5 has also been shown to prevent IFN-induced phosphorylation of STAT2, although this does not appear to involve the UPS (Mazzon et al., 2009). The antagonism of IFN by paramyxoviruses is more complicated due to the varied targeting of multiple STATs by different viral family members. For example, among the rubulavirus genus, the human parainfluenza type 2 (HPIV2) V protein directs the ubiquitination and degradation of STAT2 (Parisien et al., 2002), while the PIV5 V protein induces the degradation of STAT1 (Didcock et al., 1999). In addition, the Mumps V protein targets both STAT1 and STAT3 (Ulane et al., 2003). Although the targets are varied, it appears that the V proteins from these three viruses are all able to form similar E3 Ub ligase complexes with DDB1 and Cul4A, which are referred to as V protein-dependent degradation complexes (VDC; Ulane and Horvath, 2002). Respiratory syncytial virus (RSV), a paramyxovirus from the pneumovirus genus, does not encode a V protein homolog. However STAT2 is still degraded in a UPS-dependent manner that requires the viral NS1 and NS2 proteins (Spann et al., 2004). It has since been discovered that the RSV NS1 protein interacts with ElonginC and Cul2 to direct the ubiquitination and degradation of STAT2 (Elliott et al., 2007).
Downregulation of IFN-Induced Proteins
Interferon is responsible for activating many anti-viral genes. Therefore, those viruses that fail to avoid early IFN signaling still have many opportunities to circumvent downstream anti-viral events.
MHC-1, MHC-like molecules, and T-cell activation markers
The most well-characterized IFN-induced anti-viral target known to be downregulated by many virus classes is MHC-1. The host uses MHC-1 as a means to present viral antigens at the cell surface for recognition by cytotoxic T-cells. In response, viruses have developed numerous ways to evade this immune surveillance mechanism. Several members of the herpesvirus family have been shown to usurp the UPS to downregulate MHC-1 and in some cases T-cell activation markers from the cell surface. For example, the HCMV US2 and US11 gene products act in the ER to target MHC-1 for Ub-dependent ER-associated degradation (ERAD; Wiertz et al., 1996a,b). ERAD is normally used by the cell to eliminate misfolded proteins from the ER. US2 and US11 are not Ub ligases. Instead, they target MHC-1 for dislocation from the ER into the cytoplasm, where MHC-1 is then subjected to proteasomal degradation. Interestingly, although both viral proteins function similarly, they appear to interact with different Ub ligases and translocation machinery complexes. Studies of US11-mediated MHC-1 dislocation facilitated the initial identification of cellular components of the normal ERAD pathway, including Derlin 1, SeL1L, VIMP, p97 ATPase, AUP1, and UbxD8 (Lilley and Ploegh, 2004; Ye et al., 2004; Mueller et al., 2006, 2008). The E3 Ub ligases associated with Derlin1 are HRD1 and gp78 (Lilley and Ploegh, 2005), however it has not been confirmed whether these ligases are involved in the US11-mediated downregulation of MHC-1. In contrast, US2 mediates MHC-1 dislocation through interactions with the signal peptide peptidase (SPP; Loureiro et al., 2006) and the E3 ligase TRC8 (translocation in renal carcinoma; Stagg et al., 2009). The gammaherpesviruses also target MHC-1 for degradation, although they accomplish this task by encoding their own E3 Ub ligases. KSHV encodes the E3 Ub ligases K3 and K5, also known as modulators of immune recognition 1 and 2 (MIR1 and MIR2), respectively, which target MHC-1 for ubiquitination and subsequent lysosomal degradation (Coscoy and Ganem, 2000; Ishido et al., 2000b). Using a proteomics approach, several groups have recently demonstrated that K5 decorates surface-associated MHC-1 with a mixed-linkage ubiquitin chain, and that this stimulates the Epsin1-dependent endocytosis and subsequent lysosomal degradation of MHC-1 (Boname et al., 2010; Goto et al., 2010). Although they are 40% homologous to one another, K3 and K5 do not target the same molecules. K3 can downregulate all classes of MHC-1, HLA-A, B, C, and E, while K5 is specific for HLA-A and B (Ishido et al., 2000b). In contrast, K3 and K5 can each downregulate the IFNγ receptor (Li et al., 2007). While the removal of MHC-1 from the cell surface may be a good strategy to prevent cytotoxic T-cell killing, it can also increase the likelihood of being targeted by NK cells, which eliminate cells with low MHC-1 surface expression. The differential targeting of MHC-1 molecules by K3 and K5 may be one mechanism for the virus to block CTL killing without overtly activating NK cells. In addition, K5 has been shown to downregulate ICAM-1 and B7.2, which are costimulatory molecules for both T-cell and NK cell activation (Coscoy and Ganem, 2001), thus preventing NK cell killing (Ishido et al., 2000a). To further protect cells from NK cytotoxicity, K5 has also been shown to downregulate the NKG2D ligands MHC class I-related chain A (MICA), MICB, and the ligand for NKp80, activation-induced C-type lectin (AICL; Thomas et al., 2008). CD1d, another NK and T-cell activation marker, is downregulated by both K3 and K5, which decreases CD1d-restricted T-cell activation (Sanchez et al., 2005). MHV68, the mouse homolog of KSHV, only encodes mK3, which has also been shown to downregulate MHC-1 (Stevenson et al., 2000). However, instead of resulting in lysosomal MHC-1 degradation, mK3 appears to function in the ER via interaction with the transporter associated with antigen processing protein (TAP; Lybarger et al., 2003; Wang et al., 2005), thereby targeting MHC-1 for proteasomal degradation (Boname and Stevenson, 2001). Similarly, the retroperitoneal fibromatosis-associated herpesvirus (RFHV) also encodes a single K3/K5 homolog referred to as rfK3, which appears to downregulate MHC-I and ICAM, but not B7.2, in a Ub-dependent manner (Harris et al., 2010). Members of the Poxviridae also encode their own Ub ligases. The rabbit myxomavirus M153R protein contains a RING-CH domain and appears to downregulate both MHC-1 and CD4 in a manner reminiscent of MHC-1 downregulation by K3 and K5 (Mansouri et al., 2003), suggesting a common immune evasion strategy for the large DNA viruses.
The HPV E5 protein has recently been shown to downregulate the T-cell activation factor CD1d in a proteasome-dependent manner. E5 has been proposed to accomplish this by inhibiting the calnexin-dependent trafficking of CD1d (Miura et al., 2010). Interestingly, E5 has also been demonstrated to downregulate MHC-1 (Ashrafi et al., 2005). However, this has been attributed to E5’s interaction with the vacuolar ATPase, which leads to endosome acidification and retention of MHC-1 in the Golgi. This has also been one of the proposed models for E5’s retention of EGF-R as described above. However, like CD1d, MHC-1 also binds to E5 and calnexin in the ER, and E5 can only mediate MHC-1 downregulation in calnexin-expressing cells (Gruener et al., 2007), suggesting a potential role for the UPS in HPV’s immune evasion strategies.
An interesting caveat to emerge from the study of these viral Ub ligases is their ability to target non-lysine residues for ubiquitination. KSHV K3 ubiquitination of MHC-1 provided the first report of an E3 Ub ligase targeting a cysteine for ubiquitination via a thioester linkage (Cadwell and Coscoy, 2005). Subsequent studies have revealed that K5 also targets MHC-1 cysteine residues (Cadwell and Coscoy, 2008), and that mK3 conjugates ubiquitin to MHC-1 serine and threonine residues via ester bonds (Wang et al., 2007). Interestingly, data from a recent mK3 study suggests that while the RING-CH domain of these viral Ub ligases plays a role in targeting non-lysine residues, sequences outside the RING-CH domain are also required for this altered specificity (Herr et al., 2009).
Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3
The apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3 (APOBEC3) genes encode a family of cytidine deaminases, several of which have been shown to be part of an innate anti-retroviral defense that inhibits retroviral replication (Sheehy et al., 2002). Of the family members, APOBEC3G and 3F appear to have the highest anti-viral activity. These enzymes are incorporated into nascent virions where they subsequently deaminate cytosines in the minus DNA strand generated via reverse transcription. This action results in G to A transitions in the genomic retroviral RNA, which leads to potentially lethal nonsense and missense mutations. Aside from the “mutator” phenotype, some groups have shown that APOBEC3 might have other deleterious effects on the virus, as mutants without deaminase activity still show anti-retroviral activity (reviewed in Neil and Bieniasz, 2009). The lentiviral family of retroviruses express the Vif accessory protein that counteracts this restriction by inducing the ubiquitination and subsequent proteasomal degradation of APOBEC3G (Marin et al., 2003; Sheehy et al., 2003). Vif harbors a unique SOCS-box motif that allows Vif to complex with the Cul5/ElonginBC/Rbx1 E3 Ub ligase (Yu et al., 2003, 2004). There is currently some debate as to whether Vif induces the direct ubiquitination of APOBEC (Iwatani et al., 2009) or if Vif is itself ubiquitinated and acts as a suicide molecule that delivers APOBEC to the proteasome (Dang et al., 2008). Most recently, one group has demonstrated the Vif-dependent polyubiquitination of an APOBEC mutant devoid of lysine residues (Shao et al., 2010). They later showed that in the absence of lysines, the APOBEC N-terminal-MET residue is targeted for ubiquitination (Wang et al., 2011). This same group has shown that APOBEC degradation can occur without simultaneous Vif degradation, weighing against a Vif suicide model (Shao et al., 2010).
Disruption of the Promyelocytic Leukemia Nuclear Bodies
The PML protein was first identified and named for its role in acute promyelocytic leukemia (APL). The PML protein normally aggregates with other proteins (Daxx, ATRX, SP100, and SUMO) in the nucleus, forming distinct structures that have been shown to be involved in many cellular functions, including cell cycle regulation, DNA damage repair, apoptosis, transcriptional regulation, and IFN response to viral infection (see references in Everett and Chelbi-Alix, 2007). APL results from a chromosomal translocation at the PML locus. This gives rise to a pair of new PML–retinoic acid receptor fusion proteins, neither of which allow for the formation and assembly of functional PML NB (Melnick and Licht, 1999). Herpesviruses and adenoviruses express proteins that disrupt the PML NBs, thus preventing IFN-induced anti-viral responses and resulting in enhanced viral transcription and replication (reviewed in Everett and Chelbi-Alix, 2007). For simplicity, we discuss all the examples of PML NB disruption in this section, although the impact that these have on viral functions is varied. HSV-1 encodes ICP0, which induces the disruption of PML NBs via degradation of the complex proteins PML and SP100. This degradation is proteasome dependent and relies on the ICP0 RING domain E3 Ub ligase function (Boutell et al., 2002; Gu and Roizman, 2009). EBV disrupts PML NBs through the action of the viral transactivator EBNA1, in a process that leads to the development of nasopharyngeal carcinoma (Sivachandran et al., 2008). A recent study suggests that EBNA1’s disruption of PML NBs requires its interaction with host proteins including the USP7 DUB and the CK2 protein kinase (Sivachandran et al., 2010). The phosphorylation of PML by CK2 is important for PML’s subsequent polyubiquitination and proteasomal degradation (Scaglioni et al., 2008). Sivachandran et al. (2010) hypothesize that EBNA1 enhances CK2’s effect on PML, but also requires USP7 binding for complete PML NB disruption. The role that USP7 plays here is not clear, given that its catalytic domain is not required (Sivachandran et al., 2010). Daxx is a host-encoded transcription repressor that can inhibit viral gene expression, and is yet another PML NB component targeted for degradation by viruses. The HCMV tegument protein pp71 induces the proteasomal degradation of Daxx, thereby relieving the IFN-induced inhibition on viral immediate early (IE) gene expression (Saffert and Kalejta, 2006). While no cellular Ub ligase has been implicated, data has been put forth suggesting that the pp71-mediated degradation of Daxx (Hwang and Kalejta, 2007) may instead involve SUMOylation (Hwang and Kalejta, 2009). However, a Daxx mutant that is inefficiently SUMOylated was still degraded by pp71 and exhibited no difference in IE gene expression (Hwang and Kalejta, 2009). It has recently been shown that adenovirus E1B55K also targets Daxx for ubiquitination and proteasomal destruction (Schreiner et al., 2010). Schreiner et al. (2010) found that knocking down Daxx via siRNA enhanced Ad5 replication, and that unlike E1B55K’s other Ub-dependent functions, E4orf6 was not required.
Unknown Functions
There are many examples of virus-specific effects on the UPS that we have not described here in detail because their advantage to the virus has not yet been determined. However, we have included them along with references in Table 5. It is expected that as these “unknown functions” are resolved, they will contribute to the ever-increasing number of instances whereby viruses utilize the host ubiquitin system (Table 5).
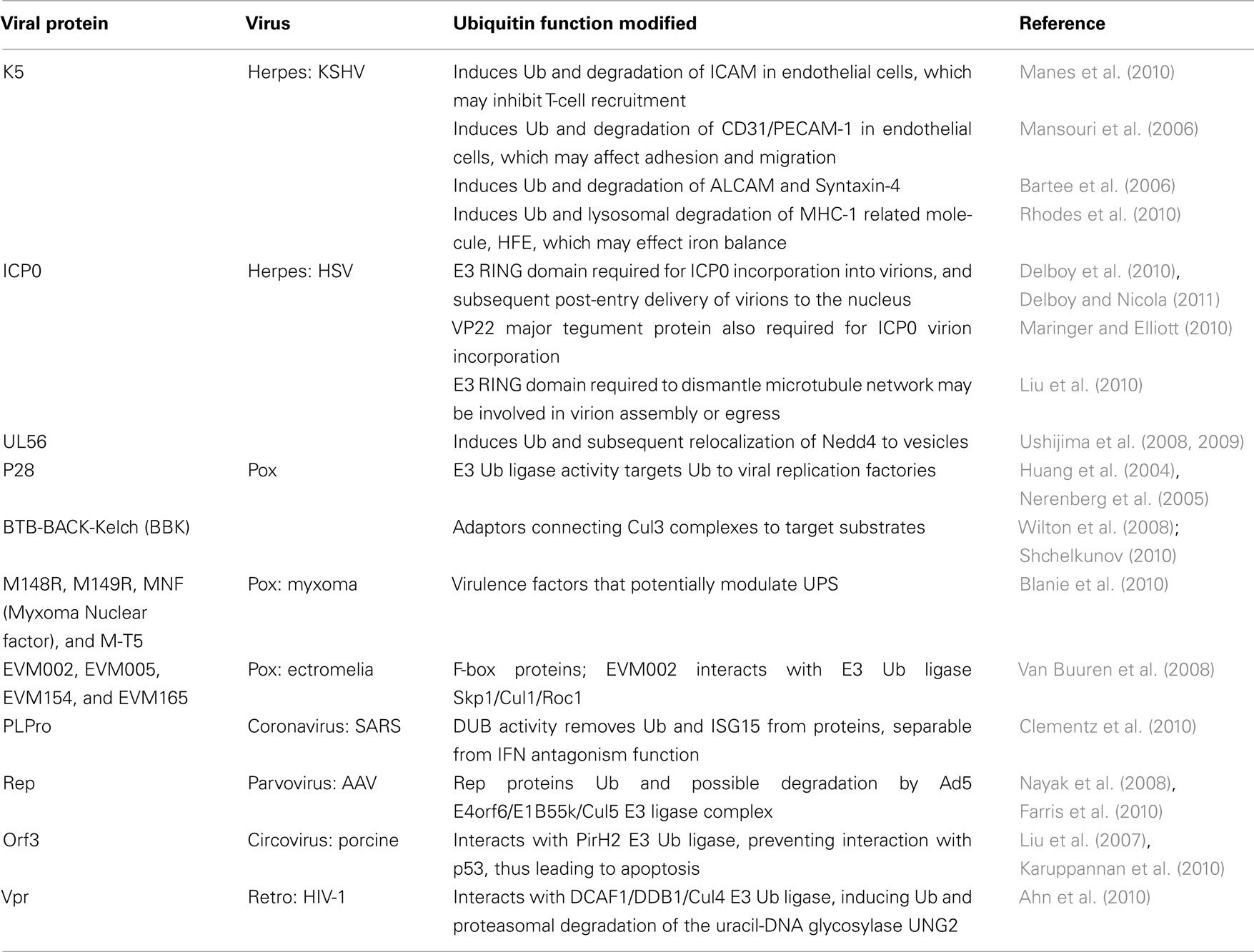
Table 5. Role of UPS in unknown viral functions.
Summary
As this review has hopefully made abundantly clear, viruses depend upon ubiquitin at virtually all points within the lifecycle (see Figure 1). The prevalence of ubiquitin in normal cellular processes makes this unsurprising; by simply co-evolving with their hosts, viruses have had to learn to “speak” the ubiquitin language fluently in order to maintain their high level of control in infected cells. This can be accomplished via more or less elegant means; viruses with limited coding capacity can utilize small adaptor proteins to redirect or modify the activity of cellular ligases, while more complex viruses can encode their own E3 ligases and DUBs. In many cases, viral manipulation of ubiquitin is required to specifically fend off host countermeasures, while in others ubiquitin must be harnessed for viral replication functions. Additionally, although we have focused on ubiquitin in this review, it is readily apparent that viruses can manipulate the other ubiquitin-like modifiers as well (see examples presented in Isaacson and Ploegh, 2009).
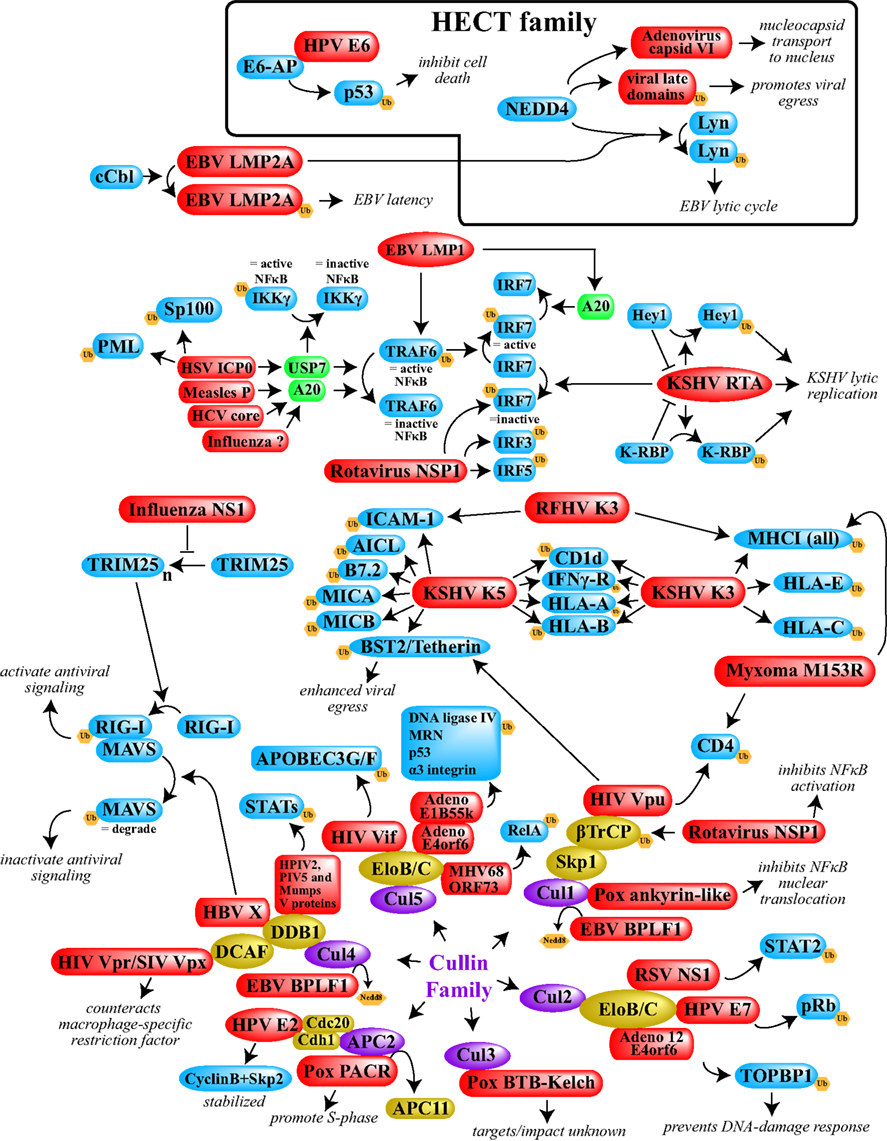
Figure 1. A schematic overview depicting examples of viral interference with the host ubiquitin system. Included here are cases in which either the E3 ligases or specific targets are known. With the exception of the cellular DUBs shown in green, the Cullin proteins shown in purple, and the various Cullin complex members shown in gold, cellular proteins are shown in blue. All viral proteins are shown in red. Ub, ubiquitin. The box at the top contains the relatively few cases in which HECT ligases are utilized. The remainder of the examples shown depend upon RING-family ligases. In the majority of cases shown, ubiquitination leads to degradation of the target protein. When degradation is not the outcome, or when ubiquitination of normal targets is prevented, the resulting phenotype has been annotated. See main text for specific references.
While much of the current ubiquitin literature has focused on events involving lysine-targeted polyubiquitination of targets and their subsequent degradation via the UPS, the recognition that (a) non-lysine residues can be targeted and (b) that ubiquitination itself comprises myriad different topological variants with a vast array of non-degradation outcomes is already leading the field to cast a wider net regarding ubiquitin-dependent phenotypes. The development of new proteomics tools (recently reviewed in Shi et al., 2011) has greatly enhanced our ability to detect and study these events, and such methods will in all likelihood continue to improve.
Many of the current examples of viral manipulation of the host ubiquitin system relate to aspects of innate immunity that must be overcome, but it is clear that many cellular process that are normally controlled by ubiquitin such as the cell cycle or the MVB pathway can be readily reprogrammed to benefit a virus. This unfortunately does not provide many new drug-able targets, as alterations made to any existing, ubiquitin-dependent host processes by small molecules will likely lead to other off-target pathologies. However, in those situations where a viral adaptor protein is used to link a host protein to a Ub-ligase complex, the physical interface between the viral protein and the host protein may provide us with an interaction that can safely be inhibited or prevented. In the near term, the catalytic sites of the virally encoded DUBs present the most attractive therapeutic targets and they are in fact the subject of intense investigation by numerous groups (Chen et al., 2007b, 2009; Ratia et al., 2008; Ghosh et al., 2010).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahn, J., Vu, T., Novince, Z., Guerrero-Santoro, J., Rapic-Otrin, V., and Gronenborn, A. M. (2010). HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-RING E3 ubiquitin ligase for proteasome-dependent degradation. J. Biol. Chem. 285, 37333–37341.
Aravind, L., and Koonin, E. V. (2000). The U box is a modified RING finger – a common domain in ubiquitination. Curr. Biol. 10, R132–R134.
Ashour, J., Laurent-Rolle, M., Shi, P. Y., and Garcia-Sastre, A. (2009). NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 83, 5408–5418.
Ashrafi, G. H., Haghshenas, M. R., Marchetti, B., O’Brien, P. M., and Campo, M. S. (2005). E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 113, 276–283.
Ayinde, D., Maudet, C., Transy, C., and Margottin-Goguet, F. (2010). Limelight on two HIV/SIV accessory proteins in macrophage infection: is Vpx overshadowing Vpr? Retrovirology 7, 35.
Bandi, P., Garcia, M. L., Booth, C. J., Chisari, F. V., and Robek, M. D. (2010). Bortezomib inhibits hepatitis B virus replication in transgenic mice. Antimicrob. Agents Chemother. 54, 749–756.
Banks, L., Pim, D., and Thomas, M. (2003). Viruses and the 26S proteasome: hacking into destruction. Trends Biochem. Sci. 28, 452–459.
Bartee, E., Mccormack, A., and Fruh, K. (2006). Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2, e107. doi: 10.1371/journal.ppat.0020107
Basler, M., Lauer, C., Beck, U., and Groettrup, M. (2009). The proteasome inhibitor bortezomib enhances the susceptibility to viral infection. J. Immunol. 183, 6145–6150.
Bellanger, S., Blachon, S., Mechali, F., Bonne-Andrea, C., and Thierry, F. (2005). High-risk but not low-risk HPV E2 proteins bind to the APC activators Cdh1 and Cdc20 and cause genomic instability. Cell Cycle 4, 1608–1615.
Bellanger, S., Tan, C. L., Nei, W., He, P. P., and Thierry, F. (2010). The human papillomavirus type 18 E2 protein is a cell cycle-dependent target of the SCFSkp2 ubiquitin ligase. J. Virol. 84, 437–444.
Bernal, J. A., Luna, R., Espina, A., Lazaro, I., Ramos-Morales, F., Romero, F., Arias, C., Silva, A., Tortolero, M., and Pintor-Toro, J. A. (2002). Human securin interacts with p53 and modulates p53-mediated transcriptional activity and apoptosis. Nat. Genet. 32, 306–311.
Bienz, M. (2006). The PHD finger, a nuclear protein-interaction domain. Trends Biochem. Sci. 31, 35–40.
Blackford, A. N., Patel, R. N., Forrester, N. A., Theil, K., Groitl, P., Stewart, G. S., Taylor, A. M., Morgan, I. M., Dobner, T., Grand, R. J., and Turnell, A. S. (2010). Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc. Natl. Acad. Sci. U.S.A. 107, 12251–12256.
Blanchette, P., and Branton, P. E. (2009). Manipulation of the ubiquitin-proteasome pathway by small DNA tumor viruses. Virology 384, 317–323.
Blanchette, P., Kindsmuller, K., Groitl, P., Dallaire, F., Speiseder, T., Branton, P. E., and Dobner, T. (2008). Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J. Virol. 82, 2642–2651.
Blanie, S., Gelfi, J., Bertagnoli, S., and Camus-Bouclainville, C. (2010). MNF, an ankyrin repeat protein of myxoma virus, is part of a native cellular SCF complex during viral infection. Virol. J. 7, 56.
Boname, J. M., and Stevenson, P. G. (2001). MHC class I ubiquitination by a viral PHD/LAP finger protein. Immunity 15, 627–636.
Boname, J. M., Thomas, M., Stagg, H. R., Xu, P., Peng, J., and Lehner, P. J. (2010). Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic 11, 210–220.
Bottcher, S., Maresch, C., Granzow, H., Klupp, B. G., Teifke, J. P., and Mettenleiter, T. C. (2008). Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J. Virol. 82, 6009–6016.
Bour, S., Perrin, C., and Strebel, K. (1999). Cell surface CD4 inhibits HIV-1 particle release by interfering with Vpu activity. J. Biol. Chem. 274, 33800–33806.
Boutell, C., Sadis, S., and Everett, R. D. (2002). Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76, 841–850.
Boyer, S. N., Wazer, D. E., and Band, V. (1996). E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 56, 4620–4624.
Breitschopf, K., Bengal, E., Ziv, T., Admon, A., and Ciechanover, A. (1998). A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 17, 5964–5973.
Bres, V., Kiernan, R. E., Linares, L. K., Chable-Bessia, C., Plechakova, O., Treand, C., Emiliani, S., Peloponese, J. M., Jeang, K. T., Coux, O., Scheffner, M., and Benkirane, M. (2003). A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 5, 754–761.
Bres, V., Yoshida, T., Pickle, L., and Jones, K. A. (2009). SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol. Cell 36, 75–87.
Burroughs, A. M., Balaji, S., Iyer, L. M., and Aravind, L. (2007). Small but versatile: the extraordinary functional and structural diversity of the beta-grasp fold. Biol. Direct 2, 18.
Cadwell, K., and Coscoy, L. (2005). Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science 309, 127–130.
Cadwell, K., and Coscoy, L. (2008). The specificities of Kaposi’s sarcoma-associated herpesvirus-encoded E3 ubiquitin ligases are determined by the positions of lysine or cysteine residues within the intracytoplasmic domains of their targets. J. Virol. 82, 4184–4189.
Calistri, A., Salata, C., Parolin, C., and Palu, G. (2009). Role of multivesicular bodies and their components in the egress of enveloped RNA viruses. Rev. Med. Virol. 19, 31–45.
Calistri, A., Sette, P., Salata, C., Cancellotti, E., Forghieri, C., Comin, A., Gottlinger, H., Campadelli-Fiume, G., Palu, G., and Parolin, C. (2007). Intracellular trafficking and maturation of Herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 81, 11468–11478.
Carson, C. T., Orazio, N. I., Lee, D. V., Suh, J., Bekker-Jensen, S., Araujo, F. D., Lakdawala, S. S., Lilley, C. E., Bartek, J., Lukas, J., and Weitzman, M. D. (2009). Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 28, 652–662.
Carson, C. T., Schwartz, R. A., Stracker, T. H., Lilley, C. E., Lee, D. V., and Weitzman, M. D. (2003). The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 22, 6610–6620.
Casey, L., Wen, X., and De Noronha, C. M. (2010). The functions of the HIV1 protein Vpr and its action through the DCAF1.DDB1.Cullin4 ubiquitin ligase. Cytokine 51, 1–9.
Chang, H. S., Lin, C. H., Yang, C. H., Liang, Y. J., and Yu, W. C. (2010). The human papillomavirus-16 (HPV-16) oncoprotein E7 conjugates with and mediates the role of the transforming growth factor-beta inducible early gene 1 (TIEG1) in apoptosis. Int. J. Biochem. Cell Biol. 42, 1831–1839.
Chang, S. J., Hsiao, J. C., Sonnberg, S., Chiang, C. T., Yang, M. H., Tzou, D. L., Mercer, A. A., and Chang, W. (2009). Poxvirus host range protein CP77 contains an F-box-like domain that is necessary to suppress NF-kappaB activation by tumor necrosis factor alpha but is independent of its host range function. J. Virol. 83, 4140–4152.
Chen, B. J., and Lamb, R. A. (2008). Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372, 221–232.
Chen, C., and Zhuang, X. (2008). Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc. Natl. Acad. Sci. U.S.A. 105, 11790–11795.
Chen, X., Chou, C. Y., and Chang, G. G. (2009). Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir. Chem. Chemother. 19, 151–156.
Chen, Z., Rijnbrand, R., Jangra, R. K., Devaraj, S. G., Qu, L., Ma, Y., Lemon, S. M., and Li, K. (2007a). Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366, 277–292.
Chen, Z., Wang, Y., Ratia, K., Mesecar, A. D., Wilkinson, K. D., and Baker, S. C. (2007b). Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. J. Virol. 81, 6007–6018.
Cheng, C. Y., Gilson, T., Dallaire, F., Ketner, G., Branton, P. E., and Blanchette, P. (2010). The E4orf6/E1B55K E3 ubiquitin ligase complexes of human adenoviruses exhibit heterogenity in composition and substrate specificity. J. Virol. 85, 765–775.
Chiari, E., Lamsoul, I., Lodewick, J., Chopin, C., Bex, F., and Pique, C. (2004). Stable ubiquitination of human T-cell leukemia virus type 1 tax is required for proteasome binding. J. Virol. 78, 11823–11832.
Ciancanelli, M. J., and Basler, C. F. (2006). Mutation of YMYL in the Nipah virus matrix protein abrogates budding and alters subcellular localization. J. Virol. 80, 12070–12078.
Clementz, M. A., Chen, Z., Banach, B. S., Wang, Y., Sun, L., Ratia, K., Baez-Santos, Y. M., Wang, J., Takayama, J., Ghosh, A. K., Li, K., Mesecar, A. D., and Baker, S. C. (2010). Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84, 4619–4629.
Coscoy, L., and Ganem, D. (2000). Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. U.S.A. 97, 8051–8056.
Coscoy, L., and Ganem, D. (2001). A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest. 107, 1599–1606.
Craven, R. C., Harty, R. N., Paragas, J., Palese, P., and Wills, J. W. (1999). Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J. Virol. 73, 3359–3365.
Crump, C. M., Yates, C., and Minson, T. (2007). Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 81, 7380–7387.
Dallaire, F., Blanchette, P., and Branton, P. E. (2009a). A proteomic approach to identify candidate substrates of human adenovirus E4orf6-E1B55K and other viral cullin-based E3 ubiquitin ligases. J. Virol. 83, 12172–12184.
Dallaire, F., Blanchette, P., Groitl, P., Dobner, T., and Branton, P. E. (2009b). Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1B 55-kilodalton E3 ubiquitin ligase complex. J. Virol. 83, 5329–5338.
Dang, Y., Siew, L. M., and Zheng, Y. H. (2008). APOBEC3G is degraded by the proteasomal pathway in a Vif-dependent manner without being polyubiquitylated. J. Biol. Chem. 283, 13124–13131.
Daubeuf, S., Singh, D., Tan, Y., Liu, H., Federoff, H. J., Bowers, W. J., and Tolba, K. (2009). HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 113, 3264–3275.
Davies, B. A., Lee, J. R., Oestreich, A. J., and Katzmann, D. J. (2009). Membrane protein targeting to the MVB/lysosome. Chem. Rev. 109, 1575–1586.
DeHart, J. L., Bosque, A., Harris, R. S., and Planelles, V. (2008). Human immunodeficiency virus type 1 Vif induces cell cycle delay via recruitment of the same E3 ubiquitin ligase complex that targets APOBEC3 proteins for degradation. J. Virol. 82, 9265–9272.
Delboy, M. G., and Nicola, A. V. (2011). A pre-immediate early role for tegument ICP0 in the proteasome-dependent entry of Herpes simplex virus. J. Virol. 85, 5910–5918.
Delboy, M. G., Roller, D. G., and Nicola, A. V. (2008). Cellular proteasome activity facilitates Herpes simplex virus entry at a postpenetration step. J. Virol. 82, 3381–3390.
Delboy, M. G., Siekavizza-Robles, C. R., and Nicola, A. V. (2010). Herpes simplex virus tegument ICP0 is capsid associated, and its E3 ubiquitin ligase domain is important for incorporation into virions. J. Virol. 84, 1637–1640.
Demers, G. W., Halbert, C. L., and Galloway, D. A. (1994). Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology 198, 169–174.
Deshaies, R. J., and Joazeiro, C. A. P. (2009). RING domain E3 ubiquitin ligases. Ann. Rev. Biochem. 78, 399–434.
Didcock, L., Young, D. F., Goodbourn, S., and Randall, R. E. (1999). The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 73, 9928–9933.
Dikic, I., Wakatsuki, S., and Walters, K. J. (2009). Ubiquitin-binding domains – from structures to functions. Nat. Rev. Mol. Cell Biol. 10, 659–671.
Doehle, B. P., Hladik, F., Mcnevin, J. P., Mcelrath, M. J., and Gale, M. Jr. (2009). Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 83, 10395–10405.
Douglas, J. L., Gustin, J. K., Viswanathan, K., Mansouri, M., Moses, A. V., and Fruh, K. (2010). The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog. 6, e1000913. doi: 10.1371/journal.ppat.1000913
Douglas, J. L., Viswanathan, K., Mccarroll, M. N., Gustin, J. K., Fruh, K., and Moses, A. V. (2009). Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a betaTrCP-dependent mechanism. J. Virol. 83, 7931–7947.
Downs, J. A., and Jackson, S. P. (2004). A means to a DNA end: the many roles of Ku. Nat. Rev. Mol. Cell Biol. 5, 367–378.
Dul, B. E., and Walworth, N. C. (2007). The plant homeodomain fingers of fission yeast Msc1 exhibit E3 ubiquitin ligase activity. J. Biol. Chem. 282, 18397–18406.
Eichler, R., Strecker, T., Kolesnikova, L., Ter Meulen, J., Weissenhorn, W., Becker, S., Klenk, H. D., Garten, W., and Lenz, O. (2004). Characterization of the Lassa virus matrix protein Z: electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP). Virus Res. 100, 249–255.
Elliott, J., Lynch, O. T., Suessmuth, Y., Qian, P., Boyd, C. R., Burrows, J. F., Buick, R., Stevenson, N. J., Touzelet, O., Gadina, M., Power, U. F., and Johnston, J. A. (2007). Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J. Virol. 81, 3428–3436.
Everett, R. D., and Chelbi-Alix, M. K. (2007). PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89, 819–830.
Everett, R. D., and Orr, A. (2009). Herpes simplex virus type 1 regulatory protein ICP0 aids infection in cells with a preinduced interferon response but does not impede interferon-induced gene induction. J. Virol. 83, 4978–4983.
Everett, R. D., Young, D. F., Randall, R. E., and Orr, A. (2008). STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant Herpes simplex virus type 1 in human fibroblasts. J. Virol. 82, 8871–8881.
Farris, K. D., Fasina, O., Sukhu, L., Li, L., and Pintel, D. J. (2010). Adeno-associated virus small rep proteins are modified with at least two types of polyubiquitination. J. Virol. 84, 1206–1211.
Gack, M. U., Albrecht, R. A., Urano, T., Inn, K. S., Huang, I. C., Carnero, E., Farzan, M., Inoue, S., Jung, J. U., and Garcia-Sastre, A. (2009). Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449.
Garcia, M. L., Byfield, R., and Robek, M. D. (2009). Hepatitis B virus replication and release are independent of core lysine ubiquitination. J. Virol. 83, 4923–4933.
Gastaldello, S., Hildebrand, S., Faridani, O., Callegari, S., Palmkvist, M., Di Guglielmo, C., and Masucci, M. G. (2010). A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat. Cell Biol. 12, 351–361.
Ghosh, A. K., Takayama, J., Rao, K. V., Ratia, K., Chaudhuri, R., Mulhearn, D. C., Lee, H., Nichols, D. B., Baliji, S., Baker, S. C., Johnson, M. E., and Mesecar, A. D. (2010). Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation. J. Med. Chem. 53, 4968–4979.
Goffinet, C., Allespach, I., Homann, S., Tervo, H. M., Habermann, A., Rupp, D., Oberbremer, L., Kern, C., Tibroni, N., Welsch, S., Krijnse-Locker, J., Banting, G., Krausslich, H. G., Fackler, O. T., and Keppler, O. T. (2009). HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5, 285–297.
Gonzalez, C. M., Wang, L., and Damania, B. (2009). Kaposi’s sarcoma-associated herpesvirus encodes a viral deubiquitinase. J. Virol. 83, 10224–10233.
Goto, E., Yamanaka, Y., Ishikawa, A., Aoki-Kawasumi, M., Mito-Yoshida, M., Ohmura-Hoshino, M., Matsuki, Y., Kajikawa, M., Hirano, H., and Ishido, S. (2010). Contribution of Lysine 11-linked Ubiquitination to MIR2-mediated major histocompatibility complex class I internalization. J. Biol. Chem. 285, 35311–35319.
Gottwein, E., Jager, S., Habermann, A., and Krausslich, H. G. (2006). Cumulative mutations of ubiquitin acceptor sites in human immunodeficiency virus type 1 gag cause a late budding defect. J. Virol. 80, 6267–6275.
Gould, F., Harrison, S. M., Hewitt, E. W., and Whitehouse, A. (2009). Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 83, 6727–6738.
Grabbe, C., and Dikic, I. (2009). Functional roles of ubiquitin-like domain (ULD) and ubiquitin-binding domain (UBD) containing proteins. Chem. Rev. 109, 1481–1494.
Graff, J. W., Ettayebi, K., and Hardy, M. E. (2009). Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 5, e1000280. doi: 10.1371/journal.ppat.1000280
Graff, J. W., Ewen, J., Ettayebi, K., and Hardy, M. E. (2007). Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1 stability. J. Gen. Virol. 88, 613–620.
Gredmark-Russ, S., Isaacson, M. K., Kattenhorn, L., Cheung, E. J., Watson, N., and Ploegh, H. L. (2009). A gammaherpesvirus ubiquitin-specific protease is involved in the establishment of murine gammaherpesvirus 68 infection. J. Virol. 83, 10644–10652.
Gruener, M., Bravo, I. G., Momburg, F., Alonso, A., and Tomakidi, P. (2007). The E5 protein of the human papillomavirus type 16 down-regulates HLA-I surface expression in calnexin-expressing but not in calnexin-deficient cells. Virol. J. 4, 116.
Gu, H., and Roizman, B. (2009). The two functions of Herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J. Virol. 83, 181–187.
Harhaj, N. S., Sun, S. C., and Harhaj, E. W. (2007). Activation of NF-kappa B by the human T cell leukemia virus type I Tax oncoprotein is associated with ubiquitin-dependent relocalization of I kappa B kinase. J. Biol. Chem. 282, 4185–4192.
Harris, S., Lang, S. M., and Means, R. E. (2010). Characterization of the rhesus fibromatosis herpesvirus MARCH family member rfK3. Virology 398, 214–223.
Harty, R. N., Brown, M. E., Mcgettigan, J. P., Wang, G., Jayakar, H. R., Huibregtse, J. M., Whitt, M. A., and Schnell, M. J. (2001). Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75, 10623–10629.
Harty, R. N., Brown, M. E., Wang, G., Huibregtse, J., and Hayes, F. P. (2000). A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. U.S.A. 97, 13871–13876.
Harty, R. N., Paragas, J., Sudol, M., and Palese, P. (1999). A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 73, 2921–2929.
Herr, R. A., Harris, J., Fang, S., Wang, X., and Hansen, T. H. (2009). Role of the RING-CH domain of viral ligase mK3 in ubiquitination of non-lysine and lysine MHC I residues. Traffic 10, 1301–1317.
Hicke, L., Schubert, H. L., and Hill, C. P. (2005). Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 6, 610–621.
Hilton, L., Moganeradj, K., Zhang, G., Chen, Y. H., Randall, R. E., Mccauley, J. W., and Goodbourn, S. (2006). The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 80, 11723–11732.
Hoppe, T. (2005). Multiubiquitylation by E4 enzymes: “one size” doesn’t fit all. Trends Biochem. Sci. 30, 183–187.
Howie, H. L., Katzenellenbogen, R. A., and Galloway, D. A. (2009). Papillomavirus E6 proteins. Virology 384, 324–334.
Huang, J., Huang, Q., Zhou, X., Shen, M. M., Yen, A., Yu, S. X., Dong, G., Qu, K., Huang, P., Anderson, E. M., Daniel-Issakani, S., Buller, R. M., Payan, D. G., and Lu, H. H. (2004). The poxvirus p28 virulence factor is an E3 ubiquitin ligase. J. Biol. Chem. 279, 54110–54116.
Huh, K., Zhou, X., Hayakawa, H., Cho, J. Y., Libermann, T. A., Jin, J., Harper, J. W., and Munger, K. (2007). Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 81, 9737–9747.
Hwang, J., and Kalejta, R. F. (2007). Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367, 334–338.
Hwang, J., and Kalejta, R. F. (2009). Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J. Virol. 83, 6591–6598.
Ikeda, M., Ikeda, A., and Longnecker, R. (2001). PY motifs of Epstein-Barr virus LMP2A regulate protein stability and phosphorylation of LMP2A-associated proteins. J. Virol. 75, 5711–5718.
Ikeda, M., and Longnecker, R. (2009). The c-Cbl proto-oncoprotein downregulates EBV LMP2A signaling. Virology 385, 183–191.
Indoh, T., Yokota, S., Okabayashi, T., Yokosawa, N., and Fujii, N. (2007). Suppression of NF-kappaB and AP-1 activation in monocytic cells persistently infected with measles virus. Virology 361, 294–303.
Irie, T., Licata, J. M., Mcgettigan, J. P., Schnell, M. J., and Harty, R. N. (2004). Budding of PPxY-containing rhabdoviruses is not dependent on host proteins TGS101 and VPS4A. J. Virol. 78, 2657–2665.
Isaacson, M. K., and Ploegh, H. L. (2009). Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 5, 559–570.
Ishido, S., Choi, J. K., Lee, B. S., Wang, C., Demaria, M., Johnson, R. P., Cohen, G. B., and Jung, J. U. (2000a). Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity 13, 365–374.
Ishido, S., Wang, C., Lee, B. S., Cohen, G. B., and Jung, J. U. (2000b). Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74, 5300–5309.
Isobe, T., Hattori, T., Kitagawa, K., Uchida, C., Kotake, Y., Kosugi, I., Oda, T., and Kitagawa, M. (2009). Adenovirus E1A inhibits SCF(Fbw7) ubiquitin ligase. J. Biol. Chem. 284, 27766–27779.
Ivanov, A. V., Peng, H., Yurchenko, V., Yap, K. L., Negorev, D. G., Schultz, D. C., Psulkowski, E., Fredericks, W. J., White, D. E., Maul, G. G., Sadofsky, M. J., Zhou, M. M., and Rauscher, F. J. III. (2007). PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 28, 823–837.
Iwabu, Y., Fujita, H., Kinomoto, M., Kaneko, K., Ishizaka, Y., Tanaka, Y., Sata, T., and Tokunaga, K. (2009). HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 284, 35060–35072.
Iwatani, Y., Chan, D. S., Liu, L., Yoshii, H., Shibata, J., Yamamoto, N., Levin, J. G., Gronenborn, A. M., and Sugiura, W. (2009). HIV-1 Vif-mediated ubiquitination/degradation of APOBEC3G involves four critical lysine residues in its C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 106, 19539–19544.
Jarosinski, K., Kattenhorn, L., Kaufer, B., Ploegh, H., and Osterrieder, N. (2007). A herpesvirus ubiquitin-specific protease is critical for efficient T cell lymphoma formation. Proc. Natl. Acad. Sci. U.S.A. 104, 20025–20030.
Jha, S., Vande Pol, S., Banerjee, N. S., Dutta, A. B., Chow, L. T., and Dutta, A. (2010). Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 38, 700–711.
Jia, B., Serra-Moreno, R., Neidermyer, W., Rahmberg, A., Mackey, J., Fofana, I. B., Johnson, W. E., Westmoreland, S., and Evans, D. T. (2009). Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5, e1000429. doi:10.1371/journal.ppat.1000429
Johnston, J. B., Wang, G., Barrett, J. W., Nazarian, S. H., Colwill, K., Moran, M., and Mcfadden, G. (2005). Myxoma virus M-T5 protects infected cells from the stress of cell cycle arrest through its interaction with host cell cullin-1. J. Virol. 79, 10750–10763.
Jouvenet, N., Neil, S. J., Zhadina, M., Zang, T., Kratovac, Z., Lee, Y., Mcnatt, M., Hatziioannou, T., and Bieniasz, P. D. (2009). Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 83, 1837–1844.
Jowett, J. B., Planelles, V., Poon, B., Shah, N. P., Chen, M. L., and Chen, I. S. (1995). The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 69, 6304–6313.
Kaletsky, R. L., Francica, J. R., Agrawal-Gamse, C., and Bates, P. (2009). Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 106, 2886–2891.
Kamio, M., Yoshida, T., Ogata, H., Douchi, T., Nagata, Y., Inoue, M., Hasegawa, M., Yonemitsu, Y., and Yoshimura, A. (2004). SOCS1 [corrected] inhibits HPV-E7-mediated transformation by inducing degradation of E7 protein. Oncogene 23, 3107–3115.
Kanneganti, T. D. (2010). Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10, 688–698.
Karuppannan, A. K., Liu, S., Jia, Q., Selvaraj, M., and Kwang, J. (2010). Porcine circovirus type 2 ORF3 protein competes with p53 in binding to Pirh2 and mediates the deregulation of p53 homeostasis. Virology 398, 1–11.
Kattenhorn, L. M., Korbel, G. A., Kessler, B. M., Spooner, E., and Ploegh, H. L. (2005). A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 19, 547–557.
Kew, M. C. (2011). Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 26(Suppl. 1), 144–152.
Kfoury, Y., Setterblad, N., El-Sabban, M., Zamborlini, A., Dassouki, Z., El Hajj, H., Hermine, O., Pique, C., De the, H., Saib, A., and Bazarbachi, A. (2010). Tax ubiquitylation and SUMOylation control the dynamic shuttling of Tax and NEMO between Ubc9 nuclear bodies and the centrosome. Blood 117, 190–199.
Kian Chua, P., Lin, M. H., and Shih, C. (2006). Potent inhibition of human Hepatitis B virus replication by a host factor Vps4. Virology 354, 1–6.
Knight, J. S., Sharma, N., and Robertson, E. S. (2005a). Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 102, 18562–18566.
Knight, J. S., Sharma, N., and Robertson, E. S. (2005b). SCFSkp2 complex targeted by Epstein-Barr virus essential nuclear antigen. Mol. Cell. Biol. 25, 1749–1763.
Koegl, M., Hoppe, T., Schlenker, S., Ulrich, H. D., Mayer, T. U., and Jentsch, S. (1999). A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644.
Komander, D. (2009). The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37, 937–953.
Kornitzer, D., Sharf, R., and Kleinberger, T. (2001). Adenovirus E4orf4 protein induces PP2A-dependent growth arrest in Saccharomyces cerevisiae and interacts with the anaphase-promoting complex/cyclosome. J. Cell Biol. 154, 331–344.
Koziorowska, J., Wlodarski, K., and Mazurowa, N. (1971). Transformation of mouse emnbryo cells by vaccinia virus. J. Natl. Cancer Inst. 46, 225–241.
Lagunas-Martinez, A., Madrid-Marina, V., and Gariglio, P. (2010). Modulation of apoptosis by early human papillomavirus proteins in cervical cancer. Biochim. Biophys. Acta 1805, 6–16.
Lambert, C., Doring, T., and Prange, R. (2007). Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and gamma 2-adaptin. J. Virol. 81, 9050–9060.
Le Tortorec, A., and Neil, S. J. (2009). Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 83, 11966–11978.
Lee, T. H., Elledge, S. J., and Butel, J. S. (1995). Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J. Virol. 69, 1107–1114.
Leechanachai, P., Banks, L., Moreau, F., and Matlashewski, G. (1992). The E5 gene from human papillomavirus type 16 is an oncogene which enhances growth factor-mediated signal transduction to the nucleus. Oncogene 7, 19–25.
Li, Q., Means, R., Lang, S., and Jung, J. U. (2007). Downregulation of gamma interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J. Virol. 81, 2117–2127.
Li, W., Bengtson, M. H., Ulbrich, A., Matsuda, A., Reddy, V. A., Orth, A., Chanda, S. K., Batalov, S., and Joazeiro, C. A. (2008). Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS ONE 3, e1487. doi: 10.1371/journal.pone.0001487
Lichtig, H., Gilboa, D. A., Jackman, A., Gonen, P., Levav-Cohen, Y., Haupt, Y., and Sherman, L. (2010). HPV16 E6 augments Wnt signaling in an E6AP-dependent manner. Virology 396, 47–58.
Lilley, B. N., and Ploegh, H. L. (2004). A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429, 834–840.
Lilley, B. N., and Ploegh, H. L. (2005). Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 14296–14301.
Lin, C. H., Chang, H. S., and Yu, W. C. (2008). USP11 stabilizes HPV-16E7 and further modulates the E7 biological activity. J. Biol. Chem. 283, 15681–15688.
Liu, J., Zhu, Y., Chen, I., Lau, J., He, F., Lau, A., Wang, Z., Karuppannan, A. K., and Kwang, J. (2007). The ORF3 protein of porcine circovirus type 2 interacts with porcine ubiquitin E3 ligase Pirh2 and facilitates p53 expression in viral infection. J. Virol. 81, 9560–9567.
Liu, M., Schmidt, E. E., and Halford, W. P. (2010). ICP0 dismantles microtubule networks in Herpes simplex virus-infected cells. PLoS ONE 5, e10975. doi:10.1371/journal.pone.0010975
Loureiro, J., Lilley, B. N., Spooner, E., Noriega, V., Tortorella, D., and Ploegh, H. L. (2006). Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature 441, 894–897.
Lupfer, C., and Pastey, M. K. (2010). Decreased replication of human respiratory syncytial virus treated with the proteasome inhibitor MG-132. Virus Res. 149, 36–41.
Lupfer, C., Patton, K. M., and Pastey, M. K. (2010). Treatment of human respiratory syncytial virus infected Balb/C mice with the proteasome inhibitor bortezomib (Velcade, PS-341) results in increased inflammation and mortality. Toxicology 268, 25–30.
Lybarger, L., Wang, X., Harris, M. R., Virgin, H. W. T., and Hansen, T. H. (2003). Virus subversion of the MHC class I peptide-loading complex. Immunity 18, 121–130.
Ma, X. Z., Bartczak, A., Zhang, J., Khattar, R., Chen, L., Liu, M. F., Edwards, A., Levy, G., and Mcgilvray, I. D. (2010). Proteasome inhibition in vivo promotes survival in a lethal murine model of severe acute respiratory syndrome. J. Virol. 84, 12419–12428.
Mammas, I. N., Sourvinos, G., Giannoudis, A., and Spandidos, D. A. (2008). Human papilloma virus (HPV) and host cellular interactions. Pathol. Oncol. Res. 14, 345–354.
Manes, T. D., Hoer, S., Muller, W. A., Lehner, P. J., and Pober, J. S. (2010). Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins block distinct steps in transendothelial migration of effector memory CD4+ T cells by targeting different endothelial proteins. J. Immunol. 184, 5186–5192.
Mangeat, B., Gers-Huber, G., Lehmann, M., Zufferey, M., Luban, J., and Piguet, V. (2009). HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 5, e1000574. doi: 10.1371/journal.ppat.1000574
Mansouri, M., Bartee, E., Gouveia, K., Hovey Nerenberg, B. T., Barrett, J., Thomas, L., Thomas, G., Mcfadden, G., and Fruh, K. (2003). The PHD/LAP-domain protein M153R of myxomavirus is a ubiquitin ligase that induces the rapid internalization and lysosomal destruction of CD4. J. Virol. 77, 1427–1440.
Mansouri, M., Douglas, J., Rose, P. P., Gouveia, K., Thomas, G., Means, R. E., Moses, A. V., and Fruh, K. (2006). Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood 108, 1932–1940.
Mansouri, M., Viswanathan, K., Douglas, J. L., Hines, J., Gustin, J., Moses, A. V., and Fruh, K. (2009). Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 83, 9672–9681.
Marin, I. (2010). Ancient origin of animal U-box ubiquitin ligases. BMC Evol. Biol. 10, 331. doi: 10.1186/1471-2148-10-331
Marin, M., Rose, K. M., Kozak, S. L., and Kabat, D. (2003). HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9, 1398–1403.
Maringer, K., and Elliott, G. (2010). Recruitment of Herpes simplex virus type 1 immediate-early protein ICP0 to the virus particle. J. Virol. 84, 4682–4696.
Martin-Lluesma, S., Schaeffer, C., Robert, E. I., Van Breugel, P. C., Leupin, O., Hantz, O., and Strubin, M. (2008). Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 48, 1467–1476.
Maufort, J. P., Shai, A., Pitot, H. C., and Lambert, P. F. (2010). A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 70, 2924–2931.
Mayer, R. J. (2000). The meteoric rise of regulated intracellular proteolysis. Nat. Rev. Mol. Cell Biol. 1, 145–148.
Mazzon, M., Jones, M., Davidson, A., Chain, B., and Jacobs, M. (2009). Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 200, 1261–1270.
McCall, C. M., Miliani De Marval, P. L., Chastain, P. D. II, Jackson, S. C., He, Y. J., Kotake, Y., Cook, J. G., and Xiong, Y. (2008). Human immunodeficiency virus type 1 Vpr-binding protein VprBP, a WD40 protein associated with the DDB1-CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol. Cell. Biol. 28, 5621–5633.
Melnick, A., and Licht, J. D. (1999). Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93, 3167–3215.
Mettenleiter, T. C., Klupp, B. G., and Granzow, H. (2009). Herpesvirus assembly: an update. Virus Res. 143, 222–234.
Mitchell, R. S., Katsura, C., Skasko, M. A., Fitzpatrick, K., Lau, D., Ruiz, A., Stephens, E. B., Margottin-Goguet, F., Benarous, R., and Guatelli, J. C. (2009). Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5, e1000450. doi:10.1371/journal.ppat.1000450
Miura, S., Kawana, K., Schust, D. J., Fujii, T., Yokoyama, T., Iwasawa, Y., Nagamatsu, T., Adachi, K., Tomio, A., Tomio, K., Kojima, S., Yasugi, T., Kozuma, S., and Taketani, Y. (2010). CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: a possible mechanism for immune evasion by HPV. J. Virol. 84, 11614–11623.
Mo, M., Fleming, S. B., and Mercer, A. A. (2009). Cell cycle deregulation by a poxvirus partial mimic of anaphase-promoting complex subunit 11. Proc. Natl. Acad. Sci. U.S.A. 106, 19527–19532.
Mo, M., Fleming, S. B., and Mercer, A. A. (2010). Orf virus cell cycle regulator, PACR, competes with subunit 11 of the anaphase promoting complex for incorporation into the complex. J. Gen. Virol. 91, 3010–3015.
Molina-Jimenez, F., Benedicto, I., Murata, M., Martin-Vilchez, S., Seki, T., Antonio Pintor-Toro, J., Tortolero, M., Moreno-Otero, R., Okazaki, K., Koike, K., Barbero, J. L., Matsuzaki, K., Majano, P. L., and Lopez-Cabrera, M. (2010). Expression of pituitary tumor-transforming gene 1 (PTTG1)/securin in hepatitis B virus (HBV)-associated liver diseases: evidence for an HBV X protein-mediated inhibition of PTTG1 ubiquitination and degradation. Hepatology 51, 777–787.
Moody, C. A., and Laimins, L. A. (2010). Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10, 550–560.
Mueller, B., Klemm, E. J., Spooner, E., Claessen, J. H., and Ploegh, H. L. (2008). SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 105, 12325–12330.
Mueller, B., Lilley, B. N., and Ploegh, H. L. (2006). SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 175, 261–270.
Mui, M. Z., Roopchand, D. E., Gentry, M. S., Hallberg, R. L., Vogel, J., and Branton, P. E. (2010). Adenovirus protein E4orf4 induces premature APCCdc20 activation in Saccharomyces cerevisiae by a protein phosphatase 2A-dependent mechanism. J. Virol. 84, 4798–4809.
Nasr, R., Chiari, E., El-Sabban, M., Mahieux, R., Kfoury, Y., Abdulhay, M., Yazbeck, V., Hermine, O., De the, H., Pique, C., and Bazarbachi, A. (2006). Tax ubiquitylation and sumoylation control critical cytoplasmic and nuclear steps of NF-kappaB activation. Blood 107, 4021–4029.
Nayak, R., Farris, K. D., and Pintel, D. J. (2008). E4Orf6-E1B-55k-dependent degradation of de novo-generated adeno-associated virus type 5 Rep52 and capsid proteins employs a cullin 5-containing E3 ligase complex. J. Virol. 82, 3803–3808.
Neil, S., and Bieniasz, P. (2009). Human immunodeficiency virus, restriction factors, and interferon. J. Interferon Cytokine Res. 29, 569–580.
Neil, S. J., Zang, T., and Bieniasz, P. D. (2008). Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430.
Nerenberg, B. T., Taylor, J., Bartee, E., Gouveia, K., Barry, M., and Fruh, K. (2005). The poxviral RING protein p28 is a ubiquitin ligase that targets ubiquitin to viral replication factories. J. Virol. 79, 597–601.
Nguyen, H., Sankaran, S., and Dandekar, S. (2006). Hepatitis C virus core protein induces expression of genes regulating immune evasion and anti-apoptosis in hepatocytes. Virology 354, 58–68.
Ning, S., Campos, A. D., Darnay, B. G., Bentz, G. L., and Pagano, J. S. (2008). TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 28, 6536–6546.
Ning, S., and Pagano, J. S. (2010). The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J. Virol. 84, 6130–6138.
Oh, J. M., Kim, S. H., Cho, E. A., Song, Y. S., Kim, W. H., and Juhnn, Y. S. (2010). Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 31, 402–410.
Oh, K. J., Kalinina, A., Wang, J., Nakayama, K., Nakayama, K. I., and Bagchi, S. (2004). The papillomavirus E7 oncoprotein is ubiquitinated by UbcH7 and Cullin 1- and Skp2-containing E3 ligase. J. Virol. 78, 5338–5346.
Ohi, M. D., Vander Kooi, C. W., Rosenberg, J. A., Chazin, W. J., and Gould, K. L. (2003). Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Biol. 10, 250–255.
Okumura, A., Alce, T., Lubyova, B., Ezelle, H., Strebel, K., and Pitha, P. M. (2008). HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology 373, 85–97.
Onose, A., Hashimoto, S., Hayashi, S., Maruoka, S., Kumasawa, F., Mizumura, K., Jibiki, I., Matsumoto, K., Gon, Y., Kobayashi, T., Takahashi, N., Shibata, Y., Abiko, Y., Shibata, T., Shimizu, K., and Horie, T. (2006). An inhibitory effect of A20 on NF-kappaB activation in airway epithelium upon influenza virus infection. Eur. J. Pharmacol. 541, 198–204.
Ott, D. E., Coren, L. V., Copeland, T. D., Kane, B. P., Johnson, D. G., Sowder, R. C. II, Yoshinaka, Y., Oroszlan, S., Arthur, L. O., and Henderson, L. E. (1998). Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of Moloney murine leukemia virus. J. Virol. 72, 2962–2968.
Paladino, P., and Mossman, K. L. (2009). Mechanisms employed by Herpes simplex virus 1 to inhibit the interferon response. J. Interferon Cytokine Res. 29, 599–607.
Parisien, J. P., Lau, J. F., Rodriguez, J. J., Ulane, C. M., and Horvath, C. M. (2002). Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J. Virol. 76, 4190–4198.
Patient, R., Hourioux, C., and Roingeard, P. (2009). Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol. 11, 1561–1570.
Patnaik, A., Chau, V., and Wills, J. W. (2000). Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. U.S.A. 97, 13069–13074.
Pawliczek, T., and Crump, C. M. (2009). Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J. Virol. 83, 11254–11264.
Perez, M., Craven, R. C., and De La Torre, J. C. (2003). The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc. Natl. Acad. Sci. U.S.A. 100, 12978–12983.
Portis, T., Ikeda, M., and Longnecker, R. (2004). Epstein-Barr virus LMP2A: regulating cellular ubiquitination processes for maintenance of viral latency? Trends Immunol. 25, 422–426.
Puckett, C., and Moss, B. (1983). Selective transcription of vaccinia virus genes in template dependent soluble extracts of infected cells. Cell 35, 441–448.
Putterman, D., Pepinsky, R. B., and Vogt, V. M. (1990). Ubiquitin in avian leukosis virus particles. Virology 176, 633–637.
Querido, E., Blanchette, P., Yan, Q., Kamura, T., Morrison, M., Boivin, D., Kaelin, W. G., Conaway, R. C., Conaway, J. W., and Branton, P. E. (2001). Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 15, 3104–3117.
Raaben, M., Grinwis, G. C., Rottier, P. J., and De Haan, C. A. (2010a). The proteasome inhibitor Velcade enhances rather than reduces disease in mouse hepatitis coronavirus-infected mice. J. Virol. 84, 7880–7885.
Raaben, M., Posthuma, C. C., Verheije, M. H., Te Lintelo, E. G., Kikkert, M., Drijfhout, J. W., Snijder, E. J., Rottier, P. J., and De Haan, C. A. (2010b). The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 84, 7869–7879.
Rampias, T., Boutati, E., Pectasides, E., Sasaki, C., Kountourakis, P., Weinberger, P., and Psyrri, A. (2010). Activation of Wnt signaling pathway by human papillomavirus E6 and E7 oncogenes in HPV16-positive oropharyngeal squamous carcinoma cells. Mol. Cancer Res. 8, 433–443.
Randow, F., and Lehner, P. J. (2009). Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 11, 527–534.
Ratia, K., Pegan, S., Takayama, J., Sleeman, K., Coughlin, M., Baliji, S., Chaudhuri, R., Fu, W., Prabhakar, B. S., Johnson, M. E., Baker, S. C., Ghosh, A. K., and Mesecar, A. D. (2008). A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U.S.A. 105, 16119–16124.
Reinstein, E., Scheffner, M., Oren, M., Ciechanover, A., and Schwartz, A. (2000). Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene 19, 5944–5950.
Rhodes, D. A., Boyle, L. H., Boname, J. M., Lehner, P. J., and Trowsdale, J. (2010). Ubiquitination of lysine-331 by Kaposi’s sarcoma-associated herpesvirus protein K5 targets HFE for lysosomal degradation. Proc. Natl. Acad. Sci. U.S.A. 107, 16240–16245.
Robert, A., Miron, M. J., Champagne, C., Gingras, M. C., Branton, P. E., and Lavoie, J. N. (2002). Distinct cell death pathways triggered by the adenovirus early region 4 ORF 4 protein. J. Cell Biol. 158, 519–528.
Rodrigues, L., Filipe, J., Seldon, M. P., Fonseca, L., Anrather, J., Soares, M. P., and Simas, J. P. (2009). Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 28, 1283–1295.
Rogel, M. E., Wu, L. I., and Emerman, M. (1995). The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 69, 882–888.
Ros, C., and Kempf, C. (2004). The ubiquitin-proteasome machinery is essential for nuclear translocation of incoming minute virus of mice. Virology 324, 350–360.
Rost, M., Mann, S., Lambert, C., Doring, T., Thome, N., and Prange, R. (2006). Gamma-adaptin, a novel ubiquitin-interacting adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus maturation. J. Biol. Chem. 281, 29297–29308.
Rotin, D., and Kumar, S. (2009). Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409.
Saffert, R. T., and Kalejta, R. F. (2006). Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80, 3863–3871.
Saha, A., Halder, S., Upadhyay, S. K., Lu, J., Kumar, P., Murakami, M., Cai, Q., and Robertson, E. S. (2011). Epstein-Barr virus nuclear antigen 3C facilitates G1-S transition by stabilizing and enhancing the function of cyclin D1. PLoS Pathog. 7, e1001275. doi: 10.1371/journal.ppat.1001275
Saha, A., Murakami, M., Kumar, P., Bajaj, B., Sims, K., and Robertson, E. S. (2009). Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J. Virol. 83, 4652–4669.
Sanchez, D. J., Gumperz, J. E., and Ganem, D. (2005). Regulation of CD1d expression and function by a herpesvirus infection. J. Clin. Invest. 115, 1369–1378.
Sarkari, F., Sanchez-Alcaraz, T., Wang, S., Holowaty, M. N., Sheng, Y., and Frappier, L. (2009). EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog. 5, e1000624. doi: 10.1371/journal.ppat.1000624
Satheshkumar, P. S., Anton, L. C., Sanz, P., and Moss, B. (2009). Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 83, 2469–2479.
Scaglioni, P. P., Yung, T. M., Choi, S., Baldini, C., Konstantinidou, G., and Pandolfi, P. P. (2008). CK2 mediates phosphorylation and ubiquitin-mediated degradation of the PML tumor suppressor. Mol. Cell. Biochem. 316, 149–154.
Scheffner, M., Werness, B. A., Huibregtse, J. M., Levine, A. J., and Howley, P. M. (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136.
Schmitt, A. P., Leser, G. P., Morita, E., Sundquist, W. I., and Lamb, R. A. (2005). Evidence for a new viral late-domain core sequence, FPIV, necessary for budding of a paramyxovirus. J. Virol. 79, 2988–2997.
Schmitt, A. P., Leser, G. P., Waning, D. L., and Lamb, R. A. (2002). Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J. Virol. 76, 3952–3964.
Schreiner, S., Wimmer, P., Sirma, H., Everett, R. D., Blanchette, P., Groitl, P., and Dobner, T. (2010). Proteasome-dependent degradation of Daxx by the viral E1B-55K protein in human adenovirus-infected cells. J. Virol. 84, 7029–7038.
Schubert, U., Bour, S., Ferrer-Montiel, A. V., Montal, M., Maldarell, F., and Strebel, K. (1996). The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 70, 809–819.
Schubert, U., Ott, D. E., Chertova, E. N., Welker, R., Tessmer, U., Princiotta, M. F., Bennink, J. R., Krausslich, H. G., and Yewdell, J. W. (2000). Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. U.S.A. 97, 13057–13062.
Schubert, U., and Strebel, K. (1994). Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J. Virol. 68, 2260–2271.
Schulman, B. A., and Harper, J. W. (2009). Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331.
Seago, J., Goodbourn, S., and Charleston, B. (2010). The classical swine fever virus Npro product is degraded by cellular proteasomes in a manner that does not require interaction with interferon regulatory factor 3. J. Gen. Virol. 91, 721–726.
Selvey, L. A., Dunn, L. A., Tindle, R. W., Park, D. S., and Frazer, I. H. (1994). Human papillomavirus (HPV) type 18 E7 protein is a short-lived steroid-inducible phosphoprotein in HPV-transformed cell lines. J. Gen. Virol. 75(Pt 7), 1647–1653.
Sette, P., Jadwin, J. A., Dussupt, V., Bello, N. F., and Bouamr, F. (2010). The ESCRT-associated protein Alix recruits the ubiquitin ligase Nedd4-1 to facilitate HIV-1 release through the LYPXnL L domain motif. J. Virol. 84, 8181–8192.
Shackelford, J., and Pagano, J. S. (2004). Tumor viruses and cell signaling pathways: deubiquitination versus ubiquitination. Mol. Cell. Biol. 24, 5089–5093.
Shai, A., Pitot, H. C., and Lambert, P. F. (2010). E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 70, 5064–5073.
Shao, Q., Wang, Y., Hildreth, J. E., and Liu, B. (2010). Polyubiquitination of APOBEC3G is essential for its degradation by HIV-1 Vif. J. Virol. 84, 4840–4844.
Sharova, N., Wu, Y., Zhu, X., Stranska, R., Kaushik, R., Sharkey, M., and Stevenson, M. (2008). Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 4, e1000057. doi: 10.1371/journal.ppat.1000057
Shchelkunov, S. N. (2010). Interaction of orthopoxviruses with the cellular ubiquitin-ligase system. Virus Genes 41, 309–318.
Sheehy, A. M., Gaddis, N. C., Choi, J. D., and Malim, M. H. (2002). Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650.
Sheehy, A. M., Gaddis, N. C., and Malim, M. H. (2003). The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9, 1404–1407.
Sherry, B. (2009). Rotavirus and reovirus modulation of the interferon response. J. Interferon Cytokine Res. 29, 559–567.
Shi, Y., Xu, P., and Qin, J. (2011). Ubiquitinated proteome: ready for global? Mol. Cell. Proteomics 10, R110.006882.
Shiyanov, P., Nag, A., and Raychaudhuri, P. (1999). Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J. Biol. Chem. 274, 35309–35312.
Sivachandran, N., Cao, J. Y., and Frappier, L. (2010). Epstein-Barr virus nuclear antigen 1 hijacks the host kinase CK2 to disrupt PML nuclear bodies. J. Virol. 84, 11113–11123.
Sivachandran, N., Sarkari, F., and Frappier, L. (2008). Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 4, e1000170. doi: 10.1371/journal.ppat.1000170
Skaug, B., Jiang, X., and Chen, Z. J. (2009). The role of ubiquitin in NF-kappaB regulatory pathways. Annu. Rev. Biochem. 78, 769–796.
Sompallae, R., Gastaldello, S., Hildebrand, S., Zinin, N., Hassink, G., Lindsten, K., Haas, J., Persson, B., and Masucci, M. G. (2008). Epstein-Barr virus encodes three bona fide ubiquitin-specific proteases. J. Virol. 82, 10477–10486.
Spann, K. M., Tran, K. C., Chi, B., Rabin, R. L., and Collins, P. L. (2004). Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected]. J. Virol. 78, 4363–4369.
Spidel, J. L., Craven, R. C., Wilson, C. B., Patnaik, A., Wang, H., Mansky, L. M., and Wills, J. W. (2004). Lysines close to the Rous sarcoma virus late domain critical for budding. J. Virol. 78, 10606–10616.
Stagg, H. R., Thomas, M., Van Den Boomen, D., Wiertz, E. J., Drabkin, H. A., Gemmill, R. M., and Lehner, P. J. (2009). The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 186, 685–692.
Stevenson, P. G., Efstathiou, S., Doherty, P. C., and Lehner, P. J. (2000). Inhibition of MHC class I-restricted antigen presentation by gamma 2-herpesviruses. Proc. Natl. Acad. Sci. U.S.A. 97, 8455–8460.
Strack, B., Calistri, A., Accola, M. A., Palu, G., and Gottlinger, H. G. (2000). A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. U.S.A. 97, 13063–13068.
Straight, S. W., Herman, B., and Mccance, D. J. (1995). The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol. 69, 3185–3192.
Studamire, B., and Goff, S. P. (2008). Host proteins interacting with the Moloney murine leukemia virus integrase: multiple transcriptional regulators and chromatin binding factors. Retrovirology 5, 48.
Tandon, R., Aucoin, D. P., and Mocarski, E. S. (2009). Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 83, 10797–10807.
Teale, A., Campbell, S., van Buuren, N., Magee, W. C., Watmough, K., Couturier, B., Shipclark, R., and Barry, M. (2009). Orthopoxviruses require a functional ubiquitin-proteasome system for productive replication. J. Virol. 83, 2099–2108.
Thomas, M., Boname, J. M., Field, S., Nejentsev, S., Salio, M., Cerundolo, V., Wills, M., and Lehner, P. J. (2008). Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 1656–1661.
Timmins, J., Schoehn, G., Ricard-Blum, S., Scianimanico, S., Vernet, T., Ruigrok, R. W., and Weissenhorn, W. (2003). Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J. Mol. Biol. 326, 493–502.
Tokarev, A. A., Munguia, J., and Guatelli, J. C. (2010). Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J. Virol. 85, 51–63.
Tran, K., Mahr, J. A., and Spector, D. H. (2010). Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J. Virol. 84, 3079–3093.
Ulane, C. M., and Horvath, C. M. (2002). Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 304, 160–166.
Ulane, C. M., Rodriguez, J. J., Parisien, J. P., and Horvath, C. M. (2003). STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J. Virol. 77, 6385–6393.
Urata, S., Noda, T., Kawaoka, Y., Morikawa, S., Yokosawa, H., and Yasuda, J. (2007). Interaction of Tsg101 with Marburg virus VP40 depends on the PPPY motif, but not the PT/SAP motif as in the case of Ebola virus, and Tsg101 plays a critical role in the budding of Marburg virus-like particles induced by VP40, NP, and GP. J. Virol. 81, 4895–4899.
Urata, S., Noda, T., Kawaoka, Y., Yokosawa, H., and Yasuda, J. (2006). Cellular factors required for Lassa virus budding. J. Virol. 80, 4191–4195.
Urata, S., and Yasuda, J. (2010). Regulation of Marburg virus (MARV) budding by Nedd4.1: a different WW domain of Nedd4.1 is critical for binding to MARV and Ebola virus VP40. J. Gen. Virol. 91, 228–234.
Urata, S., Yasuda, J., and De La Torre, J. C. (2009). The z protein of the new world arenavirus tacaribe virus has bona fide budding activity that does not depend on known late domain motifs. J. Virol. 83, 12651–12655.
Ushijima, Y., Goshima, F., Kimura, H., and Nishiyama, Y. (2009). Herpes simplex virus type 2 tegument protein UL56 relocalizes ubiquitin ligase Nedd4 and has a role in transport and/or release of virions. Virol. J. 6, 168.
Ushijima, Y., Koshizuka, T., Goshima, F., Kimura, H., and Nishiyama, Y. (2008). Herpes simplex virus type 2 UL56 interacts with the ubiquitin ligase Nedd4 and increases its ubiquitination. J. Virol. 82, 5220–5233.
Van Buuren, N., Couturier, B., Xiong, Y., and Barry, M. (2008). Ectromelia virus encodes a novel family of F-box proteins that interact with the SCF complex. J. Virol. 82, 9917–9927.
Van Damme, N., Goff, D., Katsura, C., Jorgenson, R. L., Mitchell, R., Johnson, M. C., Stephens, E. B., and Guatelli, J. (2008). The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3, 245–252.
Wang, J., Sampath, A., Raychaudhuri, P., and Bagchi, S. (2001). Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene 20, 4740–4749.
Wang, X., Connors, R., Harris, M. R., Hansen, T. H., and Lybarger, L. (2005). Requirements for the selective degradation of endoplasmic reticulum-resident major histocompatibility complex class I proteins by the viral immune evasion molecule mK3. J. Virol. 79, 4099–4108.
Wang, X., Herr, R. A., Chua, W. J., Lybarger, L., Wiertz, E. J., and Hansen, T. H. (2007). Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 177, 613–624.
Wang, Y., Shao, Q., Yu, X., Kong, W., Hildreth, J. E., and Liu, B. (2011). N-terminal hemagglutinin tag renders lysine-deficient APOBEC3G resistant to HIV-1 Vif-induced degradation by reduced polyubiquitination. J. Virol. 85, 4510–4519.
Wang, Y. E., Park, A., Lake, M., Pentecost, M., Torres, B., Yun, T. E., Wolf, M. C., Holbrook, M. R., Freiberg, A. N., and Lee, B. (2010). Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 6, e1001186. doi: 10.1371/journal.ppat.1001186
Watanabe, T., Sorensen, E. M., Naito, A., Schott, M., Kim, S., and Ahlquist, P. (2007). Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. U.S.A. 104, 10205–10210.
Wei, C., Ni, C., Song, T., Liu, Y., Yang, X., Zheng, Z., Jia, Y., Yuan, Y., Guan, K., Xu, Y., Cheng, X., Zhang, Y., Wang, Y., Wen, C., Wu, Q., Shi, W., and Zhong, H. (2010). The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 185, 1158–1168.
Weiss, E. R., Popova, E., Yamanaka, H., Kim, H. C., Huibregtse, J. M., and Gottlinger, H. (2010). Rescue of HIV-1 release by targeting widely divergent NEDD4-type ubiquitin ligases and isolated catalytic HECT domains to Gag. PLoS Pathog. 6, e1001107. doi: 10.1371/journal.ppat.1001107
Welcker, M., and Clurman, B. E. (2005). The SV40 large T antigen contains a decoy phosphodegron that mediates its interactions with Fbw7/hCdc4. J. Biol. Chem. 280, 7654–7658.
Widjaja, I., De Vries, E., Tscherne, D. M., Garcia-Sastre, A., Rottier, P. J., and De Haan, C. A. (2010). Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J. Virol. 84, 9625–9631.
Wiertz, E. J., Jones, T. R., Sun, L., Bogyo, M., Geuze, H. J., and Ploegh, H. L. (1996a). The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769–779.
Wiertz, E. J., Tortorella, D., Bogyo, M., Yu, J., Mothes, W., Jones, T. R., Rapoport, T. A., and Ploegh, H. L. (1996b). Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384, 432–438.
Williams, C., Van Den Berg, M., Sprenger, R. R., and Distel, B. (2007). A conserved cysteine is essential for Pex4p-dependent ubiquitination of the peroxisomal import receptor Pex5p. J. Biol. Chem. 282, 22534–22543.
Wilton, B. A., Campbell, S., Van Buuren, N., Garneau, R., Furukawa, M., Xiong, Y., and Barry, M. (2008). Ectromelia virus BTB/kelch proteins, EVM150 and EVM167, interact with cullin-3-based ubiquitin ligases. Virology 374, 82–99.
Wodrich, H., Henaff, D., Jammart, B., Segura-Morales, C., Seelmeir, S., Coux, O., Ruzsics, Z., Wiethoff, C. M., and Kremer, E. J. (2010). A capsid-encoded PPxY-motif facilitates adenovirus entry. PLoS Pathog. 6, e1000808. doi: 10.1371/journal.ppat.1000808
Wong, J., Zhang, J., Si, X., Gao, G., and Luo, H. (2007). Inhibition of the extracellular signal-regulated kinase signaling pathway is correlated with proteasome inhibitor suppression of coxsackievirus replication. Biochem. Biophys. Res. Commun. 358, 903–907.
Xu, P., Duong, D. M., Seyfried, N. T., Cheng, D., Xie, Y., Robert, J., Rush, J., Hochstrasser, M., Finley, D., and Peng, J. (2009). Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145.
Yan, Z., Zak, R., Luxton, G. W., Ritchie, T. C., Bantel-Schaal, U., and Engelhardt, J. F. (2002). Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J. Virol. 76, 2043–2053.
Yang, Z., Yan, Z., and Wood, C. (2008). Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 82, 3590–3603.
Ye, Y., Shibata, Y., Yun, C., Ron, D., and Rapoport, T. A. (2004). A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429, 841–847.
Yokota, S., Okabayashi, T., Yokosawa, N., and Fujii, N. (2008). Measles virus P protein suppresses Toll-like receptor signal through up-regulation of ubiquitin-modifying enzyme A20. FASEB J. 22, 74–83.
Yoo, N. K., Pyo, C. W., Kim, Y., Ahn, B. Y., and Choi, S. Y. (2008). Vaccinia virus-mediated cell cycle alteration involves inactivation of tumour suppressors associated with Brf1 and TBP. Cell. Microbiol. 10, 583–592.
Yu, G. Y., and Lai, M. M. (2005). The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 79, 644–648.
Yu, X., Yu, Y., Liu, B., Luo, K., Kong, W., Mao, P., and Yu, X. F. (2003). Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302, 1056–1060.
Yu, Y., Wang, S. E., and Hayward, G. S. (2005). The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22, 59–70.
Yu, Y., Xiao, Z., Ehrlich, E. S., Yu, X., and Yu, X. F. (2004). Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 18, 2867–2872.
Zhadina, M., and Bieniasz, P. D. (2010). Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 6, e1001153. doi: 10.1371/journal.ppat.1001153
Zhadina, M., Mcclure, M. O., Johnson, M. C., and Bieniasz, P. D. (2007). Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 104, 20031–20036.
Zhang, B., Srirangam, A., Potter, D. A., and Roman, A. (2005). HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 24, 2585–2588.
Zhang, F., Wilson, S. J., Landford, W. C., Virgen, B., Gregory, D., Johnson, M. C., Munch, J., Kirchhoff, F., Bieniasz, P. D., and Hatziioannou, T. (2009). Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6, 54–67.
Keywords: ubiquitin, proteasome, virus, viral lifecycle, ubiquitin proteasome system, ubiquitin ligase complex
Citation: Gustin JK, Moses AV, Früh K and Douglas JL (2011) Viral takeover of the host ubiquitin system. Front. Microbio. 2:161. doi: 10.3389/fmicb.2011.00161
Received: 12 May 2011; Accepted: 14 July 2011;
Published online: 28 July 2011.
Edited by:
Daoguo Zhou, Purdue University, USAReviewed by:
Tomoko Kubori, Osaka University, JapanChaoyang Xue, University of Medicine and Dentistry of New Jersey, USA
Copyright: © 2011 Gustin, Moses, Früh and Douglas. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jean K. Gustin, Vaccine and Gene Therapy Institute, Oregon Health & Science University, 505 NW 185th Avenue, Beaverton, OR, USA. e-mail:Z3VzdGluakBvaHN1LmVkdQ==