 Elizabeth M. Allwood1
Elizabeth M. Allwood1
- 1 Department of Microbiology, Monash University, Clayton, VIC, Australia
- 2 Department of Biochemistry and Molecular Biology, Monash University, Clayton, VIC, Australia
- 3 Australian Research Council Centre of Excellence in Structural and Functional Microbial Genomics, Monash University, Clayton, VIC, Australia
Burkholderia pseudomallei is the causative agent of melioidosis, a disease with high mortality that is prevalent in tropical regions of the world. A key component of the pathogenesis of melioidosis is the ability of B. pseudomallei to enter, survive, and replicate within mammalian host cells. For non-phagocytic cells, bacterial adhesins have been identified both on the bacterial surface and associated with Type 4 pili. Cell invasion involves components of one or more of the three Type 3 Secretion System clusters, which also mediate, at least in part, the escape of bacteria from the endosome into the cytoplasm, where bacteria move by actin-based motility. The mechanism of actin-based motility is not clearly understood, but appears to differ from characterized mechanisms in other bacterial species. A small proportion of intracellular bacteria is targeted by host cell autophagy, involving direct recruitment of LC3 to endosomes rather than through uptake by canonical autophagosomes. However, the majority of bacterial cells are able to circumvent autophagy and other intracellular defense mechanisms such as the induction of inducible nitric oxide synthase, and then replicate in the cytoplasm and spread to adjacent cells via membrane fusion, resulting in the formation of multi-nucleated giant cells. A potential role for host cell ubiquitin in the autophagic response to bacterial infection has recently been proposed.
Introduction
Burkholderia pseudomallei is a Gram-negative pathogen which is the causative agent of melioidosis, a serious invasive disease of humans and animals. Melioidosis is endemic to northern Australia, Papua New Guinea, Southeast Asia, most of the Indian subcontinent and southern China, Hong Kong, and Taiwan. It is well recognized that B. pseudomallei is “highly endemic” to northeast Thailand, northern Australia, Singapore, and Malaysia where many cases of melioidosis are diagnosed each year (Currie et al., 2008). Sporadic cases have also been reported in other regions including Brazil, Puerto Rico, and New Caledonia (Dorman et al., 1998; Aardema et al., 2005; Le Hello et al., 2005; Barth et al., 2007). B. pseudomallei is a natural inhabitant of rice paddies, still or stagnant waters, and moist tropical soils (Brett and Woods, 2000). Despite several decades of research on prevention and treatment, mortality from melioidosis remains very high (50% in northeast Thailand and 19% in Australia; Peacock 2006). Alarmingly, the mortality associated with septic shock remains close to 90% (Stone, 2007). Melioidosis accounts for 20% of all community-acquired septicemias in northeast Thailand (Chaowagul et al., 1989) and 32% of community-acquired bacteremic pneumonia and 6% of all bacteremias in northern Australia (Douglas et al., 2004).
Since the 1990s, it has been well accepted that a key component of the pathogenesis of B. pseudomallei is its ability to survive intracellularly in both phagocytic and non-phagocytic cells (Jones et al., 1996). The importance of intracellular survival is paramount, as this remarkable feature allows the bacterium to establish an infection while avoiding host immune responses. Indeed, the ability of B. pseudomallei to survive intracellularly explains numerous features of melioidosis, including latency, recrudescence, and treatment difficulty. Latency periods of more than 60 years have been documented (Ngauy et al., 2005). Activation of a latent infection is usually due to a decrease in the individual’s immunocompetence and can result in an acute, fulminant, and fatal infection (Koponen et al., 1991). Relapse is common in melioidosis patients; recrudescence of melioidosis following apparent clinical resolution of the primary infection usually results from reactivation rather than reinfection (Haase et al., 1995; Maharjan et al., 2005).
Burkholderia pseudomallei utilizes numerous strategies that enable it to survive in such a specialized niche as the intracellular environment (Figure 1). This review will focus on these strategies for intracellular survival, from adhesion, invasion, and endosome escape to actin-based motility, formation of multi-nucleated giant cells (MNGC), evasion of host cell autophagy, and interaction of B. pseudomallei with host cell ubiquitination mechanisms.
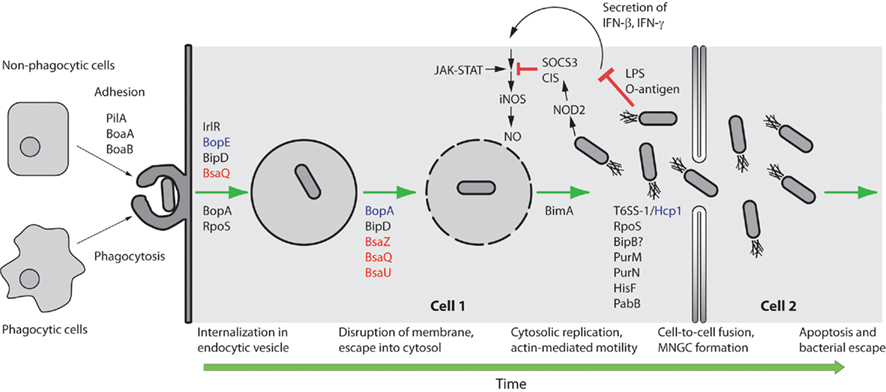
Figure 1. Schematic representation of selected steps in the intracellular lifestyle of B. pseudomallei. B. pseudomallei can be internalized by either phagocytic or non-phagocytic cells. In non-phagocytic cells PilA and the adhesins BoaA and BoaB are critical for uptake. For the internalization step in non-phagocytic cells the Bsa T3SS structural proteins BipD (translocator), BsaQ (structural component), and BopE (effector) are required. The IrlRS two-component signal transduction system is predicted to regulate the expression of other key gene(s) involved in internalization. For internalization in phagocytic cells the Bsa T3SS putative effector BopA and the alternate sigma factor RpoS play minor roles. The Bsa T3SS structural proteins BsaZ, BsaQ, and BsaU, the translocator protein BipD and the putative effector BopA are critical for membrane disruption and escape from the endocytic vesicle. Once in the cytoplasm B. pseudomallei can replicate and move by actin-based motility in a BimA-dependent fashion. B. pseudomallei LPS only weakly stimulates IFN secretion, which results in reduced iNOS expression and NO production. Furthermore, B. pseudomallei induces expression of suppressor of cytokine signaling 3 (SOCS3) and cytokine-inducible Src homology 2-containing (CIS) proteins which inhibit the Janus kinases – signal transducers and activators of transcription (JAK–STAT) signaling pathway and concomitant iNOS activation. Purine (PurM, PurN), histidine (HisF), and para-aminobenzoate (PabB) biosynthetic pathways are important for intracellular replication and survival, while the B. pseudomallei T6SS-1 Hcp protein, the alternate sigma factor RpoS, and the Bsa T3SS protein BipB are involved in stimulating cell-to-cell fusion and the formation of MNGC. NOD2 refers to nucleotide-binding oligomerization domain-containing protein 2. Green arrows are used to indicate progression of time. Red T-bar arrows indicate inhibitory interactions. Secretion system components are color coded with effectors in purple, translocators in black, and structural components in red.
The Adhesion of B. pseudomallei to Host Cells: An Early Step in Pathogenesis
It is a sine qua non that the internalization of pathogenic bacteria by mammalian cells is preceded by adhesion of the bacteria to the cell surface. It is therefore somewhat surprising that a detailed knowledge at the molecular level of the interaction of B. pseudomallei with the eukaryotic cell surface is less developed than that for many other bacterial pathogens. Comparisons between different studies using B. pseudomallei are difficult because of the use of different cell lines, bacterial strains, multiplicities of infection, and experimental conditions by different research groups. Nevertheless, the ability to adhere to the cell has been demonstrated to correlate with virulence in a number of studies. Five B. pseudomallei strains all adhered more strongly to human A549 lung epithelial cells than did five strains of the non-pathogenic species B. thailandensis (Kespichayawattana et al., 2004). The increased adhesion correlated with subsequent increased invasion and cytopathic effect, but, as in many other studies, the molecular basis for the difference was not determined. Likewise, a study which inferred, through inhibition studies, the attachment of B. pseudomallei to the GM1–GM2 glycosphingolipid ganglioside on undefined primary human pharyngeal epithelial cells did not investigate the bacterial component(s) involved (Gori et al., 1999).
Based on limited evidence derived from electron microscopy (EM), Ahmed et al. (1999 suggested that the adhesion of B. pseudomallei to an undefined population of primary human “pharyngeal” cells involved a bacterial surface structure which was designated, without supporting evidence, as capsular polysaccharide. Paradoxically, a more recent study with a genetically defined acapsular mutant (Reckseidler et al., 2001) found that the lack of capsule resulted in enhanced internalization into A549 and HeLa cells (Phewkliang et al., 2010); there was no effect on intracellular replication or cytotoxicity. Thus, while capsule clearly plays a key role in the ability of B. pseudomallei to cause disease (Reckseidler et al., 2001; Lazar Adler et al., 2009), there is no clear evidence for a role in cell adhesion or invasion (Table 1).
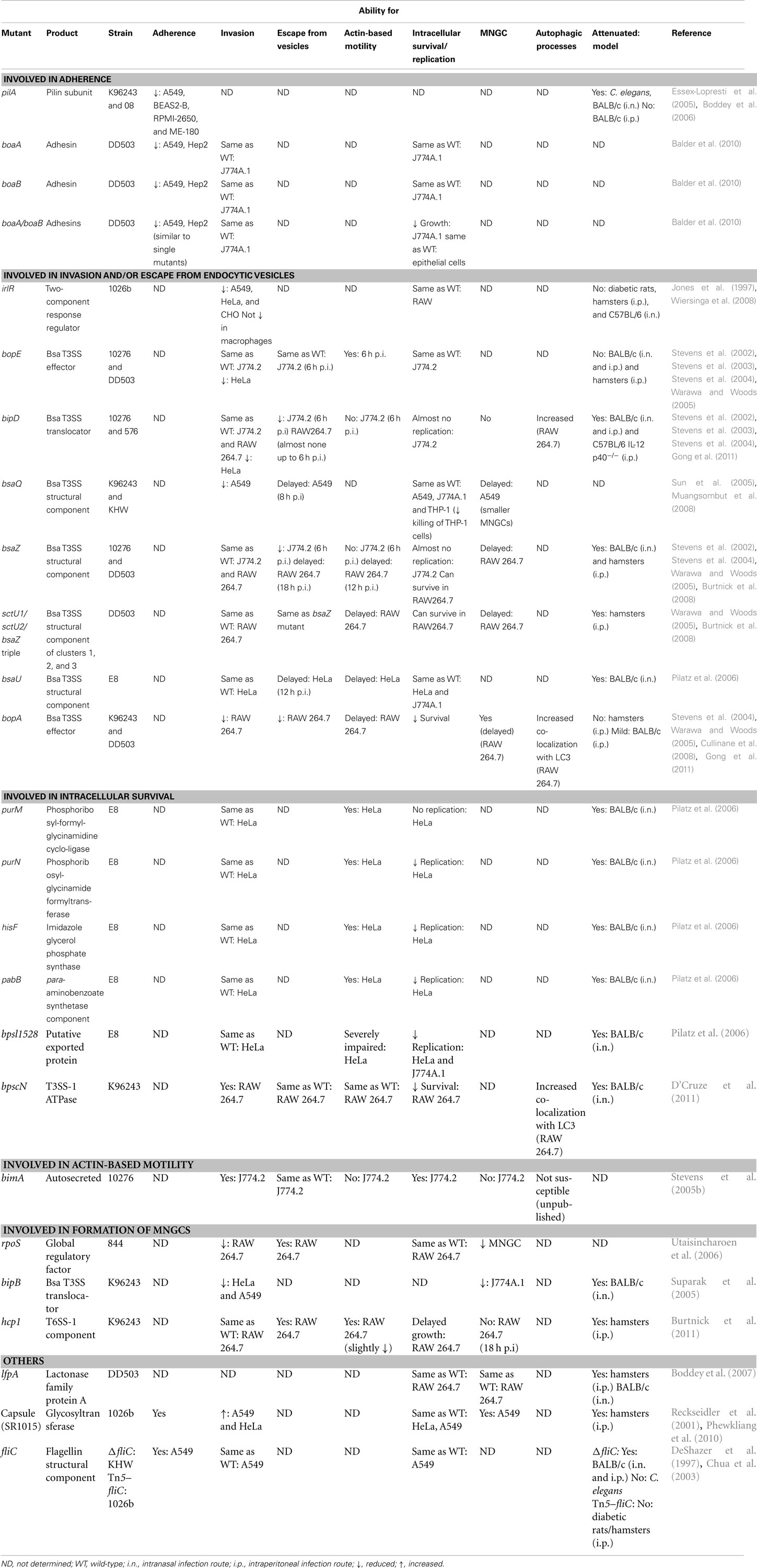
Table 1. Characterization of B. pseudomallei mutants with defects in intracellular life cycle.
Type 4 fimbriae (pili) are known to be involved in the adhesion of a large number of bacterial species to a range of cell types, tissues, and other surfaces (Pelicic, 2008). Consequently, these hair-like filaments, or their individual protein components, frequently constitute key adhesins that may be necessary for virulence. In common with many Gram-negative species, B. pseudomallei, produces Type 4 pili. Inactivation of the pilA gene, which encodes the pilin subunit protein, abrogated the production of pili and resulted in reduced adhesion of B. pseudomallei to three different human epithelial cell lines, including A549 cells (Essex-Lopresti et al., 2005). Interestingly, adhesion to A549 and BEAS2-B cells was almost completely abolished, consistent with pili being a major bacterial adhesin, whereas adhesion of the pilA mutant to RPMI-2650 cells was approximately 30% that of the wild-type, suggesting the presence of additional adhesins (Table 1). The basis for these differences remains unknown. The pilA gene can be regulated in response to temperature, with up-regulation at lower temperatures (27–30°C). However, a pilA B. pseudomallei mutant showed reduced adhesion to ME-180 cells derived from human cervical tissue (Table 1). Interestingly, PilA does not mediate direct adherence of B. pseudomallei strain 08 to eukaryotic cells. Rather, up-regulation of pilA at lower temperatures is responsible for the formation of B. pseudomallei microcolonies. These microcolonies in turn display a higher level of adherence to eukaryotic cells (Boddey et al., 2006). However, temperature regulation was not observed in strain K96243, the first strain to have its genome sequenced. Adhesion to six different human epithelial cell lines by strain 08 was also enhanced at 30°C, but the role of pili was not investigated (Brown et al., 2002). These studies again highlight the need for caution in extrapolating data derived with different B. pseudomallei strains and different eukaryotic cell lines.
Flagellum-mediated adhesion was a prerequisite for invasion of the free-living ameba Acanthamoeba astronyxis (Inglis et al., 2003). However, a defined aflagellar fliC mutant was markedly attenuated in BALB/c mice, but was unimpaired in its ability to invade A549 cells (Chua et al., 2003; Table 1). These findings suggest that flagellae are required for virulence in mice, but probably do not constitute a major adhesin for mammalian cells.
A recent study identified two novel adhesins in B. pseudomallei (Balder et al., 2010). The BoaA protein shares similarity with the YadA autotransporter adhesin of Yersinia enterocolitica and is present also in B. mallei. Recombinant BoaA expressed in E. coli enhanced the adhesion of E. coli to A549 and Hep2 cells, while inactivation of the boaA gene reduced, but did not abolish, adhesion of B. pseudomallei to both cell lines (Table 1; Figure 1). A second related gene, boaB, appears to be unique to B. pseudomallei. Expression of BoaB in E. coli also increased adhesion to A549 and Hep2 cells and mutation of boaB reduced adhesion to epithelial cell lines (Table 1; Figure 1). Significantly, a similar phenotype was observed with cultured normal human bronchial epithelium. A double boaA/boaB mutant exhibited levels of adhesion similar to those of either single mutant, with some residual binding remaining (Table 1). These findings suggest that multiple adhesins are involved in the adhesion of B. pseudomallei to eukaryotic cells. Moreover, different adhesins may be involved in the interaction of bacteria with different cell types.
Intracellular Invasion, Escape from Endocytic Vesicles, and Survival within the Cytoplasm
It is likewise a widely accepted notion that initial adherence of pathogenic bacteria to the external surface of host cells is followed immediately by intracellular invasion. Over the last decade, the pace of research addressing how B. pseudomallei gains access to the intracellular environment and how it subsequently escapes from endocytic vesicles into the cytoplasm has accelerated, providing much needed insight into this critical step of pathogenesis. Despite this knowledge, a comprehensive understanding of the precise mechanism of intracellular invasion and subsequent endosomal escape is still lacking.
Entry into Host Cells
Burkholderia pseudomallei is taken up by phagocytic cells such as polymorphonuclear leukocytes (Pruksachartvuthi et al., 1990; Egan and Gordon, 1996; Jones et al., 1996) and macrophages (Pruksachartvuthi et al., 1990; Harley et al., 1994, 1998b; Jones et al., 1996; Kespichayawattana et al., 2000; Utaisincharoen et al., 2001) and can invade non-phagocytic cell lines such as HeLa, CHO, A549, and Vero cultured epithelial cell lines (Jones et al., 1996; Harley et al., 1998a; Kespichayawattana et al., 2000). The invasiveness of B. pseudomallei and its avirulent counterpart, B. thailandensis, was compared in the human respiratory epithelial cell line A549; the invasive capacity of B. pseudomallei was significantly greater than that of B. thailandensis (Kespichayawattana et al., 2004).
A B. pseudomallei 1026b transposon mutagenesis screen identified a gene encoding a putative two-component response regulator that is involved in the invasion of the epithelial cell line A549, HeLa, and CHO cells, but not macrophages (Jones et al., 1997; Figure 1). The transposon mutant exhibited an invasion level only 10% that of the parental strain, 1026b, in epithelial cells. The disrupted two-component response regulator was therefore named irlR for invasion-related locus (Table 1). A putative two-component sensor, irlS, was located downstream of the response regulator. This irlRS two-component regulatory system is not involved in virulence, as the mutant remained virulent in diabetic rats, hamsters (Jones et al., 1997), and C57BL/6 mice (Wiersinga et al., 2008), suggesting that the tissue culture invasion defect observed in vitro was not related to pathogenesis in vivo.
Many diverse Gram-negative pathogenic bacteria use Type 3 Secretion Systems (T3SS) as a conserved, but highly adapted, virulence mechanism. The T3SS apparatus comprises approximately 20 different proteins assembled into a syringe-like structure that spans the bacterial inner and outer membranes, enabling the bacteria to secrete and inject effector molecules into the cytosol of eukaryotic host cells for the subversion of cellular functions (Hueck, 1998; Zhang et al., 2006; Sun and Gan, 2010). Although the components of the T3SS apparatus are conserved between species, the secreted proteins are highly divergent (Hueck, 1998). B. pseudomallei encodes three T3SS clusters. Clusters 1 and 2 are similar to the T3SS gene clusters encoding the Hrp secretons of the plant pathogens Ralstonia (formerly Burkholderia) solanacearum and Xanthomonas spp. (Winstanley et al., 1999; Rainbow et al., 2002). However, cluster 3 shares homology to the inv/spa/prg T3SS of Salmonella Typhimurium and the ipa/mxi/spa T3SS of Shigella flexneri (Attree and Attree, 2001; Stevens et al., 2001; Rainbow et al., 2002) and has been designated the Burkholderia secretion apparatus (bsa) T3SS (Stevens et al., 2002).
Numerous studies have demonstrated the importance of the Bsa T3SS in the intracellular survival, replication, and virulence of B. pseudomallei. Indeed, numerous structural components (BsaQ, BsaU, and BsaZ), translocator (BipD), and effector proteins (BopA and BopE) of the Bsa T3SS have been implicated in various aspects of the intracellular lifecycle of B. pseudomallei, including invasion of host cells, escape from endocytic vesicles, and intracellular survival. Studies have shown that the invasion event requires the proteins BopE, BipD (Stevens et al., 2003), and BsaQ (Muangsombut et al., 2008; Table 1; Figure 1). BopE shares similarity with the Salmonella SopE/SopE2 effector proteins which act as guanine nucleotide exchange factors (GEFs) for RhoGTPases that regulate the actin network (Hardt et al., 1998; Rudolph et al., 1999; Stender et al., 2000) and in turn facilitate bacterial invasion (Wood et al., 1996; Bakshi et al., 2000). Likewise, BopE facilitates bacterial invasion of non-phagocytic cells, as a B. pseudomallei 10276 bopE mutant displayed a significant reduction in invasion of HeLa cells when compared to the wild-type strain (Stevens et al., 2003). In turn, BopE induces rearrangements in the subcortical actin cytoskeleton and also exhibits GEF activity. It was concluded that in epithelial cells, BopE is likely to be translocated into the host cell cytosol, where it may facilitate membrane ruffling by acting as a GEF for Cdc42 and Rac1 (Stevens et al., 2003). This notion is supported by a subsequent study by Upadhyay et al. (2008) which showed that BopE and SopE/SopE2 catalytic domains adopt similar three-dimensional folds, but that BopE has a more compact conformation. An interaction of the catalytic domain of BopE78–261 with Cdc42 was also demonstrated. Interestingly, the SopE residues involved in contacting Cdc42 and the residues that were shown by mutation to be functionally important in SopE are conserved or conservatively substituted in BopE. Taken together, these data suggest that BopE uses the same mechanism as SopE and other Rho GEFs in catalyzing guanine nucleotide exchange in Rho GTPases, despite the fact that BopE possesses a more closed conformation (Upadhyay et al., 2008). The study performed by Stevens et al. (2003) also provided evidence that BipD, a translocator protein located on the tip of the needle (Johnson et al., 2007) and similar to Salmonella SipD, is also involved in invasion of non-phagocytic cells, as a 10276 bipD mutant was impaired in invasion of HeLa cells and notably, to an even greater extent than the bopE mutant described above, implying that several Bsa proteins may act in concert with BopE to facilitate bacterial invasion (Stevens et al., 2003; Stevens and Galyov, 2004).
BsaQ shares a high degree of identity with Salmonella Typhimurium InvA (56%; Ginocchio and Galán, 1995) and therefore by analogy, BsaQ is most likely to be a conserved inner-membrane channel protein, located at the base of the T3SS apparatus (Muangsombut et al., 2008). A B. pseudomallei K96243 bsaQ mutant exhibited a 30% reduction in invasion of A549 cells when compared to wild-type B. pseudomallei (Muangsombut et al., 2008). In turn, mutation of the bsaQ (Bsa structural component) gene prevented the secretion of at least two T3SS proteins, BopE and BipD (Muangsombut et al., 2008). Taken together, as it has been shown that BopE and BipD are required for invasion of non-phagocytic cells (Stevens et al., 2003), it can be speculated that the impaired invasion efficiency exhibited by the bsaQ mutant is a consequence of its inability to secrete the effector protein BopE and the translocator BipD. Interestingly, BopE and BipD do not appear to play a role in invasion of phagocytic cells, namely J774.2 murine macrophage-like cells, as bopE and bipD mutants were taken up by J774.2 cells in comparable numbers to the wild-type strain B. pseudomallei 10276 (Stevens et al., 2002). Likewise, we have shown that BipD is not necessary for invasion of RAW 264.7 macrophage-like cells (Gong et al., 2011; Table 1). These data indicate the importance of different T3SS mechanisms for the invasion of non-phagocytic and phagocytic cells.
Escape from Endocytic Vesicles
Once a bacterium gains access to the intracellular environment, it must rapidly invoke strategies to survive within this niche. Accordingly, within 15 min of internalization, B. pseudomallei strain 708a can escape from endocytic vesicles into the cytoplasm of RAW 264.7, HeLa, and human macrophage-like U937 cells (MOI of either 1000:1 or 100:1) by lysing endosome membranes thus avoiding degradation through the lysosomal system (Harley et al., 1998b). Several Bsa T3SS proteins play a role in escape of B. pseudomallei from endocytic vesicles.
We have recently shown that the putative Bsa T3SS effector protein BopA plays an important role in escape of B. pseudomallei from phagosomes (Cullinane et al., 2008; Gong et al., 2011); a bopA mutant displayed reduced intracellular survival and significantly increased co-localization with GFP–LC3 in RAW 264.7 cells when compared to cells infected with wild-type K96243 (Table 1), indicating the decreased ability of the mutant to escape from the phagosome (Cullinane et al., 2008). The same mutant showed reduced phagosomal escape compared to wild-type when analyzed by transmission electron microscopy (TEM); mutant bacteria that were free in the cytoplasm however, could form actin-tails and stimulate MNGC formation (Gong et al., 2011). These data indicate that BopA is important for efficient escape from phagosomes (Figure 1). However, the mechanism by which BopA modulates phagosomal escape is not known. Two possible functional domains have been identified in BopA that may contribute to its function in phagosomal escape. The first is a carboxy-terminal Rho GTPase inactivation domain (RID) which may function as a protease or acyltransferase acting on host molecules (Pei and Grishin, 2009). The second is a cholesterol binding domain (CBD; Kayath et al., 2010). In macrophages infected with Mycobacterium avium, cholesterol depletion triggered phagolysosomal or autophagolysosomal formation with consequent bacterial degradation (de Chastellier and Thilo, 2006). Similarly, the binding of cholesterol by BopA may lead to the accumulation of cholesterol on phagosome membranes, thereby limiting lysosomal recognition and/or fusion. The function of both domains is amenable to testing. Notably, BopA from B. mallei is important for invasion and intracellular survival within J774A.1 macrophage cells; a B. mallei bopA mutant showed reduced phagocytic uptake and reduced intracellular survival (Whitlock et al., 2008). In murine alveolar macrophage (MH-S) cells, a B. mallei bopA mutant was recovered at higher levels from intracellular compartments compared to wild-type B. mallei, suggesting that the bopA mutant remains within intracellular compartments longer than wild-type, resulting in decreased escape (Whitlock et al., 2009).
BsaZ, similar to Salmonella SpaS, is the last gene in the bsa gene cluster that encodes a structural component of the T3SS apparatus (Stevens et al., 2002). By examining the association of intracellular bacteria with lysosome-associated membrane glycoprotein-1 (LAMP-1) containing vacuoles, it has been shown that bsaZ and bipD mutants are highly delayed in their ability to escape from endocytic vesicles; 95.1 and 95.9% of bsaZ and bipD mutant bacteria respectively were LAMP-1 associated in J774.2 cells at 6 h post-infection (p.i.; Stevens et al., 2002). Similarly, in analyses using TEM, the bsaZ mutant was consistently observed only within vesicles with fully intact membranes (Stevens et al., 2002) and only 2% of bipD mutants were able to escape into the cytoplasm of RAW 264.7 cells by 6 h p.i. (Gong et al., 2011; Table 1). A similar study performed by Burtnick et al. (2008) also showed a phagosome escape defect for a bsaZ mutant in activated RAW 264.7 murine macrophage-like cells. Using TEM analyses they demonstrated that in concordance with results outlined by Stevens et al. (2002) at 6 h p.i., the bsaZ mutant remained in endocytic vesicles; however, by 12 h p.i. bacteria had begun to escape and by 18 h p.i. the majority of mutant bacteria were located in the cytoplasm (Burtnick et al., 2008). Thus, the bsaZ mutant does retain the ability to escape from endocytic vesicles, but this process is delayed by 6 to 12 h compared to the parent strain, DD503 (Burtnick et al., 2008; Table 1; Figure 1).
In non-phagocytic A549 cells, a bsaQ mutant also showed delayed escape from endocytic vesicles with 59.4% of mutant bacteria co-localizing with LAMP-1 positive vacuoles at 6 h p.i.; at 8 h p.i. further escape from vacuoles was observed (Muangsombut et al., 2008; Table 1; Figure 1). Likewise, a transposon mutant which disrupted the gene bsaU (Bsa structural component), which is thought to control needle length and whose product shares ∼20% similarity with Spa32 (Sun and Gan, 2010), was unable to escape from endocytic vesicles 6 h after infection of HeLa cells (Pilatz et al., 2006). The mutant however, retained the ability to multiply within the vacuoles, resulting in the formation of large vesicles containing many bacteria; at 12 h p.i. mutant bacteria began to escape, indicating a delayed escape phenotype for this mutant (Pilatz et al., 2006; Table 1; Figure 1).
The delayed escape phenotype observed for several Bsa T3SS mutants suggests that mechanisms other than the Bsa T3SS may be involved in escape from endocytic vesicles. It is noteworthy to consider the possibility of “cross-talk” taking place between the three T3SS clusters in B. pseudomallei. Interestingly, a triple mutant with stable deletions in the genes sctU1, sctU2, and bsaZ, which all encode the putative major inner-membrane subunits of T3SS clusters 1, 2, and 3 of B. pseudomallei, respectively, exhibited the same delayed escape phenotype as the bsaZ mutant described above (Burtnick et al., 2008; Table 1). This therefore provides evidence that components of the T3SS-1 and T3SS-2 are not responsible for the delayed escape phenotype observed for the bsaZ mutant and that other factors are involved in delayed phagosomal escape in RAW 264.7 cells (Burtnick et al., 2008). However, we have recently shown that the T3SS cluster 1 predicted ATPase encoded by bpscN is essential for full virulence of B. pseudomallei in a BALB/c mouse infection model (D’Cruze et al., 2011). A bpscN mutant was highly attenuated for virulence in BALB/c mice, displayed reduced intracellular replication in RAW 264.7 macrophage-like cells and increased co-localization with the autophagy marker protein LC3, but was unimpaired for escape from phagosomes. Thus, the T3SS cluster 1 does play a role in the intracellular survival of B. pseudomallei.
Intracellular Survival
Subsequent to invasion and escape from endocytic vesicles, the ability of B. pseudomallei to survive intracellularly is important for progression of infection. Screening of transposon mutants by Pilatz et al. (2006) identified nine mutants with reduced ability to form plaques on PtK2 cell monolayers, indicating a reduced capacity for intercellular spreading. Inactivation of bsaU and its role in escape from endocytic vesicles has been discussed earlier in this review. In addition to bsaU, Pilatz et al. (2006) showed that purine (purM and purN), histidine (hisF), and para-aminobenzoate (pabB) biosynthetic pathway genes and bpsl1528 are all important for intracellular replication and survival.
Neither purM, encoding a putative phosphoribosyl-formyl-glycinamidine cyclo-ligase, nor purN, encoding a putative phosphoribosylglycinamide formyltransferase, were required for invasion of HeLa cells; both mutants displayed wild-type levels of cellular invasion. However, both purine biosynthesis pathway mutants were defective for intracellular growth. Interestingly, the purM mutant showed no intracellular replication during the course of the experiment. Similarly, hisF (encoding a putative imidazole glycerol phosphate synthase) and pabB (encoding a putative para-aminobenzoate synthetase component) mutants also retained the ability to invade HeLa cells, but were impaired for intracellular growth. Although intracellular growth was impaired for purM, purN, hisF, and pabB mutants, actin-tail formation, and the ability to form membrane protrusions were not affected. These findings contrast with those for a bpsl1528 mutant (encoding a putative exported protein) that not only displayed severely restricted intracellular growth in HeLa and J774A.1 macrophages, but was also defective in formation of actin-tails and membrane protrusions.
Relevance for in vivo Pathogenesis
While studies focusing on intracellular invasion, escape from endocytic vesicles and subsequent intracellular replication and survival using various defined mutants have provided insight into the pathogenesis of B. pseudomallei in an in vitro setting, it is critical to understand the role of the corresponding gene products for in vivo pathogenesis. Numerous virulence studies have been carried out and it appears that, with the exception of the bopE (effector) mutant, a correlation exists between the in vitro invasion and escape defects observed for various Bsa T3SS structural component and translocator mutants, and their reduced ability to cause disease. The bsaU transposon mutant, bsaZ and bipD mutants described above were all attenuated in BALB/c mice following intranasal infection (Stevens et al., 2004; Pilatz et al., 2006). The bipD mutant was also attenuated following intraperitoneal infection and this correlated with reduced bacterial replication in the liver and spleen (Stevens et al., 2004), suggesting a role for BipD during both systemic and mucosal infection. A bsaZ triple mutant (with deletions in the bsaZ homologs, sctU, in clusters 1 and 2 as described above) was significantly attenuated in hamsters; however, double and single mutants in sctU of clusters 1 and 2 that did not share the cluster 3 bsaZ mutation were not attenuated (Warawa and Woods, 2005). However, a T3SS cluster 1 bpscN mutant was attenuated in BALB/c mice (D’Cruze et al., 2011). Taken together, these virulence data indicate that a functional Bsa T3SS apparatus is required for full virulence of B. pseudomallei. Further, BipD and therefore the Bsa T3SS, is functionally expressed during infection in vivo, as antibodies against BipD have been detected in convalescent melioidosis patient sera (Stevens et al., 2002; Allwood et al., 2008).
Interestingly, the bopE mutant was not attenuated in BALB/c mice following either intraperitoneal or intranasal infection (Stevens et al., 2004), indicating that the effector protein BopE, on its own, is not essential for virulence and that other effectors must play a role in subversion of normal cellular processes during both systemic and mucosal infection. In turn, Warawa and Woods (2005) also demonstrated that a bopE mutant and other putative effector mutants (bopA, bapA, and bapC) were not attenuated for virulence in the acute hamster model. However, a bopA mutant showed a mild attenuation in the BALB/c mouse model (Stevens et al., 2004). It has been theorized that the B. pseudomallei Bsa T3SS functions by delivering a set of effector proteins that target specific host cell pathways and that each individual effector alone may actually contribute little to virulence; however, the contribution of multiple effector proteins working in a cooperative manner may have a significant pathogenic impact (Galyov et al., 2010). As such, the generation of double and/or triple effector mutants would address this issue.
Similarly, a correlation also exists between the ability to replicate intracellularly and the ability to cause disease. All mutants identified by Pilatz et al. (2006) that were defective for intracellular growth (purM, purN, hisF, pabB, and bpsl1528) were also attenuated for virulence in BALB/c mice following intranasal infection. Interestingly, in the case of the purM, pabB, and bpsl1528 mutants, all animals not only survived following challenge with 107 CFU, but also showed no signs of disease (Pilatz et al., 2006). This study therefore highlights the important role that the purine, histidine and para-aminobenzoate biosynthetic pathways and bpsl1528 play in pathogenesis.
Taken together, these data indicate the importance of numerous genes and proteins involved in the intracellular life cycle of B. pseudomallei, including those involved in T3SS and various biosynthetic pathways. Importantly, the requirement for a functional T3SS with the ability to secrete effector proteins is vital for pathogenesis. Clearly, the secretion of numerous effector proteins that function in a collaborative manner, rather than individually, is critical for effective subversion of host cellular functions and therefore for pathogenesis.
Actin-Based Motility: A Crucial Mechanism for Cell-to-Cell Spread
Members of several genera, including Burkholderia, Listeria, Shigella, Rickettsia, and Mycobacterium, have the remarkable ability to propel themselves through the cytoplasm by polymerization of the eukaryotic cytoskeletal protein actin at the surface of one pole of the bacterium. The force generated by this actin-based motility propels the bacterium through the cytoplasm, at speeds that range from 3 to 87 μm/min, depending on the pathogen and the cell type (Stevens et al., 2006). This motility facilitates the formation of membrane protrusions into neighboring cells and concomitant cell-to-cell spread (Gouin et al., 2005; Stevens et al., 2006). Since the late 1980s, an abundance of research has furthered the understanding of how such intracellular pathogens manipulate the host actin polymerization machinery for their benefit. Ultimately, actin-based motility facilitates the avoidance of host innate and adaptive immune responses as the pathogen spreads directly from cell-to-cell.
The Arp2/3 Complex
The majority of pathogens capable of actin-based motility do so by exploiting the Arp2/3 complex. This conserved host protein complex consists of seven polypeptides and is involved in both actin filament nucleation and organization (Cossart, 2000). On its own, the Arp2/3 complex possesses weak biochemical activity. The complex becomes activated by nucleation promoting factors (NPFs), which include members of the Wiskott–Aldrich syndrome protein (WASP) family; this activation leads to a conformational change in the complex (Welch and Mullins, 2002; Goley et al., 2004; Gouin et al., 2005; Goley and Welch, 2006). This change initiates the formation of a new filament that emerges from an existing filament, generating a y-branched actin network (Mullins et al., 1998; Amann and Pollard, 2001; Goley and Welch, 2006). Intracellular bacteria have developed a number of different mechanisms for activating the Arp2/3 complex, thereby exploiting the complex for their own benefit. Research to date has identified two main strategies. These mechanisms include either the expression of mimics of the WASP family NPFs (e.g., Listeria monocytogenes ActA/Listeria ivanovii IactA, or Rickettsia RickA), or surface proteins that function by recruiting host family WASP (N-WASP) proteins (e.g., S. flexneri IcsA; Welch et al., 1997, 1998; Egile et al., 1999; May et al., 1999; Boujemaa-Paterski et al., 2001; Gouin et al., 2004, 2005; Jeng et al., 2004). The expression of the bacterial protein, predominately at one pole of the cell, therefore initiates actin polymerization by direct or indirect activation of the Arp2/3 complex at the surface of the pathogen (Stevens et al., 2005b).
BimA and its Role in Actin-Based Motility
Interestingly, it seems that B. pseudomallei employs a unique mechanism for activation of actin-based motility. Initial evidence that demonstrated the ability of B. pseudomallei to induce cellular actin rearrangement and membrane protrusions in both phagocytic and non-phagocytic cells was provided by Kespichayawattana et al. (2000). Actin rearrangement in a “comet” tail formation, typical of actin-based motility, was observed at one pole of the bacterium after staining B. pseudomallei-infected J774A.1 and HeLa cells with rhodamine-conjugated phalloidin. It was later discovered that actin-based motility of B. pseudomallei is dependent upon the autosecreted protein, BimA (Stevens et al., 2005b). Mutation of bimA abolished actin-based motility of B. pseudomallei; importantly however, the mutation did not influence the activity of the Bsa T3SS and the mutant retained the ability to escape from endosomes (Stevens et al., 2005b; Table 1; Figure 1). Evidence supporting the notion that actin-based motility is a prerequisite for intercellular spread was provided by Stevens et al. (2005b) and Sitthidet et al. (2011). A bimA mutant did not form membrane protrusions in J774.2 cells (Stevens et al., 2005b) and did not form plaques in A549 monolayers (Sitthidet et al., 2011), indicating the bimA mutant could not spread from cell-to-cell. Reminiscent of ActA and IcsA, BimA was found to localize at one pole of the bacterial cell where actin assembly takes place (Stevens et al., 2005b). Interestingly, BimA homologs in B. mallei and B. thailandensis (which also possess the ability to induce actin-based motility in J774.2 cells) can restore the actin-based motility defect of a B. pseudomallei bimA mutant (Stevens et al., 2005a).
A survey of the prevalence and sequence diversity of BimA in clinical and environmental isolates from endemic areas demonstrated intraspecies conservation of BimA in natural populations of B. pseudomallei, B. mallei, and B. thailandensis (Sitthidet et al., 2008). In addition, tandem WASP homology 2 (WH2) domains, which are predicted to mediate the binding of actin monomers, were conserved in sequenced B. pseudomallei strains (Sitthidet et al., 2008). These domains were later found to be required for actin binding, actin assembly, and therefore, actin-based motility (Sitthidet et al., 2011). A proline-rich motif (IP7, also known as PRM1) which is known to interact with profilin and functions to funnel actin monomers to the tip of actin filaments, was also conserved in sequenced B. pseudomallei strains. In addition, a tandem repeat of 13 amino acids adjacent to the IP7 motif on the amino-terminal side (NIPVPPPMPGGGA) was conserved in most sequenced B. pseudomallei strains (Sitthidet et al., 2008). However, later studies indicated that the 13 amino acid repeat and the IP7 proline-rich motif are not essential for actin-based motility in vitro (Sitthidet et al., 2011). The number of PDASX repeats that are adjacent to the WH2 domains on the carboxyl-terminal side was also found to vary within sequenced B. pseudomallei strains, with the number of repeats ranging from 2, 5 to 7 copies (Sitthidet et al., 2008). It was demonstrated that a tract of five PDASX direct repeats was required for actin-based motility and intercellular spread; however, they were not involved in actin binding. Interestingly, the rate of polymerization increased in a stepwise manner as the number of PDASX repeats increased from zero to two to five to seven, indicating that these repeats act in an additive manner to stimulate polymerization of pyrene-actin monomers in vitro (Sitthidet et al., 2011).
Burkholderia pseudomallei Employs a Unique Mechanism for Activation of Actin-Based Motility
Burkholderia pseudomallei does not activate actin-based motility via a Shigella-like mechanism, as N-WASP is not found on either the surface of B. pseudomallei or within actin-tails (Breitbach et al., 2003). This, and the fact that F-actin clustering results from transient expression of BimA, a characteristic that is seen with WASP overexpression (Stevens et al., 2005b), indicate that BimA may function as an NPF mimic to induce actin-based motility in infected cells (Stevens et al., 2006). However, the mechanism appears distinct from that of Listeria, as VASP proteins were not detected on the surface of B. pseudomallei and only very weakly throughout actin-tails (Breitbach et al., 2003); this is in contrast to Listeria where Ena/VASP proteins interact directly with ActA (Laurent et al., 1999). Confirmatory to these results, infection of N-WASP- or Ena/VASP-defective cells with B. pseudomallei demonstrated that these proteins are not essential for actin-based motility by B. pseudomallei (Breitbach et al., 2003).
Interestingly, while the Arp2/3 complex is strongly incorporated into B. pseudomallei actin-tails (Breitbach et al., 2003), its precise function in actin polymerization is not clear. BimA can stimulate actin polymerization in vitro in an Arp2/3 independent manner (Stevens et al., 2005b). In addition, overexpression of an Arp2/3 binding WCA (WH2, central, and acidic) domain of Scar1 (NPF), which leads to cytoplasmic sequestration and inhibition of the Arp2/3 complex, and therefore inhibition of motility of Listeria, Shigella, and Rickettsia conorii, had no effect on actin-based motility of B. pseudomallei (Breitbach et al., 2003). Based on these data, it appears that the mechanism of actin-based motility of B. pseudomallei is distinct from that identified for all other intracellular pathogens studied to date. It has been suggested that BimA alone may be sufficient for intracellular motility of B. pseudomallei; however, the requirement for other bacterial co-factors or different post-translational modifications of BimA cannot be ruled out (Stevens et al., 2006).
A Potential Role for Type VI Secretion System Cluster 1 and Other Gene Products in Actin-Based Motility
The B. pseudomallei genome encodes six functional Type 6 Secretion Systems originally designated T6SS-1-6 (Schell et al., 2007). A subsequent paper by Shalom et al. (2007) however, assigned the names tss-5, tss-4, tss-6, tss-3, tss-2, and tss-1, respectively, to these same gene clusters. As such, we will refer to the nomenclature used by Schell et al. (2007).
T6SS play a role in virulence in many bacterial pathogens where they function to inject effector proteins into host cells (Pukatzki et al., 2009). New evidence has demonstrated that in B. mallei, the T6SS-1 plays an important role in actin-based motility in RAW 264.7 cells; tssE mutants were able to undergo vacuolar escape, but once in the cytoplasm the mutants exhibited defects in actin polymerization and intra- and intercellular spread (Burtnick et al., 2010). Importantly, a homologous T6SS expressed in B. pseudomallei is similarly induced following uptake by RAW 264.7 cells. This system (T6SS-1; locus tags BPSS1493–BPSS1511) is flanked by bimA on one side and the Bsa T3SS locus on the other (Shalom et al., 2007). By analogy with B. mallei, it could be speculated that mutation of T6SS-1 in B. pseudomallei would also cause actin polymerization defects and raises the question as to whether secreted effectors of the T6SS, such as Hcp and/or VgrG, could interact with BimA. Importantly however, a T6SS-1 tssH mutant showed no difference in invasion and survival within RAW 264.7 cells compared to wild-type B. pseudomallei; its ability to polymerize actin and undergo actin-based motility was not investigated (Shalom et al., 2007). A subsequent study by Burtnick et al. (2011) has now shown that while a hcp1 (from T6SS-1) mutant did demonstrate a slightly reduced level of actin-based motility compared to wild-type B. pseudomallei, this difference was not as significant as that observed in the B. mallei tssE T6SS-1 mutant described above (Burtnick et al., 2010). tssE is predicted to encode a component of the T6SS-1 apparatus and appears to be required for Hcp1 secretion (Schell et al., 2007). Therefore, it appears likely that tssE is required for secretion of other effector(s) that may play a role in actin polymerization, as mutation of hcp1 on its own had little effect on actin-based motility. Interestingly however, mutation of hcp1 resulted in the absence of MNGC formation (discussed elsewhere in this review), delayed intracellular growth, and reduced cytotoxicity in RAW 264.7 cells (Burtnick et al., 2011).
Another gene which has been implicated for its potential role in actin-tail formation is bpsl1528 which encodes a putative exported protein (Pilatz et al., 2006). As described above, a bpsl1528 mutant was defective for intracellular growth; immunofluorescence microscopy revealed that actin-tail formation was severely impaired as only rudimentary actin-tails were observed and no protrusions could be detected in HeLa cells. However, the mutant also demonstrated reduced swimming motility. Pilatz et al. (2006) therefore speculated that the pleiotropic phenotype of the bpsl1528 mutant may be due to a regulatory function of the encoded protein on numerous virulence factors rather than to a single effector molecule.
In summary, the role of BimA in actin-based motility is widely accepted. However, further evidence has indicated that the process of actin polymerization leading to actin-based motility may occur as a result of a concerted effort involving other bacterial factor(s) that could involve the T6SS cluster 1 and other putative exported proteins. Interestingly, the process of activation of actin-based motility by B. pseudomallei is distinct from that observed for other pathogens and further studies are therefore required to address this issue.
Cell Fusion and the Formation of Multi-Nucleated Giant Cells: Important Mechanisms for the Progression of Infection
As outlined above, several species of intracellular pathogens are able to move throughout the cell cytoplasm by manipulating actin polymerization. For Shigella, actin-based motility is also sufficient for the movement of bacteria from cell-to-cell via membrane protrusions (Monack and Theriot, 2001). Indeed, recombinant E. coli cells expressing S. flexneri IcsA (required for actin polymerization) were able to produce membrane protrusions in HeLa cells and move into the cytoplasm of adjacent cells (Monack and Theriot, 2001). While actin-based motility is an essential prerequisite for the spread of B. pseudomallei from cell-to-cell (Stevens et al., 2005b; Sitthidet et al., 2011), cell fusion, and the production of MNGC also appears to be required. Infection of phagocytic or non-phagocytic cell lines with B. pseudomallei, B. mallei, or B. thailandensis results in cell-to-cell fusion and the formation of MNGC (Harley et al., 1998a). Furthermore, MNGC have also been observed following histopathological assessment of human tissue samples recovered from melioidosis patients (Wong et al., 1995), suggesting that MNGC formation occurs during B. pseudomallei infection. Other infectious agents such as human immunodeficiency virus (HIV; Ciborowski and Gendelman, 2006), cytomegalovirus (CMV; Kinzler and Compton, 2005), herpes simplex virus (HSV; Cole and Grose, 2003), and mycobacterial species (Utermöhlen et al., 2008) also drive the formation of MNGC.
Induction of cell fusion following B. pseudomallei infection was confirmed by analysis of co-cultures of J774A.1 cells that had been labeled either with cell tracker green CMFDA or cell tracker red CMTMR prior to mixing (Kespichayawattana et al., 2000). Orange–yellow MNGC were observed within 4–6 h after infection, indicating that some of the eukaryotic cell membranes had fused; no fused cells were observed in mock infected cultures. Similar results were obtained with HeLa and L929 cells, although the rate of cell fusion and MNGC formation was slower. B. pseudomallei infection leads to apoptosis of both fused and unfused cells. At 2 h p.i. approximately 3% of cells showed evidence of apoptotic changes and this increased to 43% by 6 h. Interestingly, within MNGC, both normal nuclei and those showing apoptotic changes could be observed (Kespichayawattana et al., 2000).
The molecular mechanisms leading to cell fusion and formation of MNGC following B. pseudomallei infection are poorly characterized. Macrophage cell fusion may be associated with chronic inflammation and can be induced in vitro by treatment of macrophages with certain cytokines, including macrophage colony-stimulating factor, granulocyte–macrophage colony-stimulating factor, interleukin (IL)-4 and IFN-γ (Kondo et al., 2009). Cell fusion in response to infections with the viruses CMV and HSV is associated with the production of viral glycoproteins (Cole and Grose, 2003; Kinzler and Compton, 2005). In B. pseudomallei, the ability to form MNGC has been associated with RpoS, BipB, T6SS-1, and the putative toxin-encoding genes bpsl0590 and bpsl0591 (Figure 1).
A B. pseudomallei rpoS mutant could replicate intracellularly almost indistinguishably from the wild-type strain, but was significantly impaired in its ability to induce MNGC formation; only 3% of cells infected with the rpoS mutant formed MNGC compared with 17% for cells infected with the wild-type strain (Utaisincharoen et al., 2006). However, the rpoS mutant was not tested for its ability to move intracellularly by actin-based motility (Table 1). The RpoS regulon in B. pseudomallei has been elucidated by comparing protein expression in wild-type and rpoS mutant strains; 70 proteins (derived from 58 unique genes) showed differential expression of >3-fold (p < 0.05) between the two strains (Osiriphun et al., 2009). The proteins encoded by these differentially expressed genes included bacterial stress response and cell envelope biogenesis components as well as some putative virulence factors, including a T3SS-1 component. Thus, it is likely that RpoS regulates the expression of a B. pseudomallei gene(s) that is directly involved in the stimulation of cell-to-cell fusion, but the exact gene(s) has not yet been identified.
The Bsa T3SS and the effector protein BipB have also been implicated in MNGC formation; a bipB mutant was unable to induce MNGC formation in J774A.1 macrophage-like cells at 6 h p.i. and showed 6 to 10-fold reduced MNGC formation at later time-points (Suparak et al., 2005). The ability to form MNGC was restored by complementation with intact bipB. However, it is unclear if BipB is specifically involved in cell fusion and MNGC formation, as the bipB mutant was also significantly impaired for invasion of HeLa cells and was not tested for its ability to move intracellularly by actin-based motility (Table 1).
Hcp proteins are critical components of T6SS. B. pseudomallei mutants with deletion of hcp genes from each of the six T6SS loci were tested for virulence in a hamster infection model; only the Δhcp1 mutant was attenuated for virulence (Burtnick et al., 2011). The Δhcp1 mutant was taken up by RAW 264.7 macrophages at a similar rate to the wild-type strain and could escape from phagosomes into the cytosol and move intracellularly by actin-based motility. Although the Δhcp1 mutant showed a reduced level of actin-based motility and a delayed intracellular growth phenotype, by 18 h p.i. the levels of intracellular wild-type and Δhcp1 mutant bacteria were indistinguishable. Importantly, no MNGC formation was observed in RAW 264.7 cells following infection with the Δhcp1 mutant despite high levels of intracellular bacteria (Table 1). These data suggest that a secreted effector(s) of the T6SS is critical for the cell fusion of macrophages and for the formation of MNGC. Indeed, Burtnick et al. 2011) proposed that the inability of the Δhcp1 mutant to induce MNGC formation may lead directly to its reduced intracellular growth defect, as the fusion with nutrient rich, uninfected macrophages may be critical for the intracellular replication of B. pseudomallei.
A genome-wide screen for B. pseudomallei products which could damage macrophage cells identified a wide range of B. pseudomallei genes, including the putative toxin-encoding genes bpsl0590 and bpsl0591 (Dowling et al., 2010). In this analysis, the supernatants from cultures of E. coli containing B. pseudomallei genomic fragments cloned in BAC or fosmid vectors, were tested for activity against J774-2 macrophage cells. More than 110 genomic regions were identified as conferring some level of anti-macrophage activity; however, the cloned genomic fragments contained multiple genes and the anti-macrophage activity was never localized to single genes. Importantly, treatment of J774-2 cells with supernatant from E. coli clones containing an approximately 15 kb region containing the genes bpsl0590 and bpsl0591, directly caused apoptosis and the formation of MNGC, suggesting that the putative toxins encoded by these genes may have a direct role in MNGC formation.
The MNGC formed following B. pseudomallei infection of macrophage-like cells show striking similarities to osteoclast cells, which are also MNGC. MNGC resulting from B. pseudomallei infection of RAW 264.7 cells express increased levels of mRNA encoding each of the osteoclast markers calcitonin receptor (CTR), cathepsin K (CTSK), and tartrate-resistant acid phosphatase (TRAP) compared to uninfected cells (Boddey et al., 2007). Interestingly, while B. thailandensis also induces the formation of MNGC, these cells do not express increased levels of the osteoclast markers CTR and CTSK, and show only a marginal increase in TRAP expression (Boddey et al., 2007). The B. pseudomallei gene lfpA (lactonase family protein A) was found to be required for maximal stimulation of expression of osteoclast markers in RAW 264.7 cells following B. pseudomallei infection; this gene is not present in B. thailandensis. Expression of lfpA itself was up-regulated approximately 107-fold in bacteria that were in contact with RAW 264.7 cells as compared to those growing in vitro. A B. pseudomallei lfpA mutant was able to replicate at levels indistinguishable from the wild-type strain in RAW 264.7 macrophage-like cells and form actin-tails, but showed reduced virulence in both Syrian hamsters and BALB/c mice (Boddey et al., 2007; Table 1). Taken together these data suggest that B. pseudomallei specifically induces the formation of MNGC through a number of mechanisms and the induction of these host cell changes may have evolved to benefit the bacteria as a niche for optimal replication and spread.
Inhibition of Macrophage Activation Facilitates Intracellular Survival
The ability of B. pseudomallei to survive and replicate in macrophages is at least in part due to the suppression of macrophage killing mechanisms, including the production of the free radical species nitric oxide (NO), although it should be noted that human macrophages produce less NO than rodent macrophages (for review see Carsillo et al., 2009). NO is one of the key antimicrobial molecules produced within macrophages and is important for the clearance of many intracellular pathogens, including Leishmania, Salmonella, and Mycobacterium (for review see Fang, 1997). In macrophages, NO production results from the activity of the inducible nitric oxide synthase (iNOS). Expression of iNOS is stimulated by the cytokines IFN-γ, IFN-β, TNFα, IL-1, and IL-2, and by pathogen-associated products including lipopolysaccharide (LPS) and lipoteichoic acid (Fang, 1997; Jacobs and Ignarro, 2001). However, macrophages infected with B. pseudomallei do not activate iNOS expression. This is due, at least in part, to the inability of B. pseudomallei-infected macrophages to produce IFN-γ and IFN-β, as stimulation of infected macrophages with either of these interferons results in increased iNOS expression and reduced intracellular survival of B. pseudomallei (Utaisincharoen et al., 2003, 2004).
The secretion of IFN-β by macrophages is up-regulated in response to LPS and also infection with Gram-negative, but not Gram-positive, bacteria, suggesting that LPS is critical for production of IFN-β (Utaisincharoen et al., 2003). The inability of macrophages infected with B. pseudomallei to produce IFN-β may be a direct consequence of the LPS structure of B. pseudomallei (Utaisincharoen et al., 2003), as purified B. pseudomallei LPS stimulated reduced NO expression in comparison to macrophages stimulated with E. coli or Salmonella enterica serovar Typhi LPS (Utaisincharoen et al., 2000). Furthermore, a B. pseudomallei wbiI mutant, which is predicted to express LPS lacking O-antigen, stimulated significant levels of iNOS and IFN-β production (Arjcharoen et al., 2007; Figure 1).
Both IFN-γ and IFN-β produce a transcriptional response through the Janus kinases-signal transducers and activators of transcription (JAK–STAT) signal transduction pathway. The receptor complexes for each IFN type are related but unique (Decker et al., 2002) and binding of IFN to the appropriate receptor results in STAT phosphorylation and dimerization. IFN-γ binding results in STAT-1 homodimerization, whereas IFN-β results in production of phosphorylated STAT-1 homodimers and STAT-1 and STAT-2 heterodimers. These phosphorylated STAT dimers are then translocated into the nucleus where they mediate changes in expression of a range of IFN-regulated genes including iNOS. However, the suppressor of cytokine signaling (SOCS) and cytokine-inducible Src homology 2-containing (CIS) proteins can inhibit STAT phosphorylation and therefore IFN responses (Ekchariyawat et al., 2005; Figure 1). RAW 264.7 macrophages exposed to either live or killed B. pseudomallei showed increased expression of SOCS3 and CIS but not SOCS1 and 2 (Ekchariyawat et al., 2005). This activation was specifically due to the presence of intracellular B. pseudomallei, rather than a secondary response to macrophage cytokine production, as increased SOCS3 and CIS mRNA was observed even following pre-treatment of macrophages with the protein inhibitor cyclohexamide (Ekchariyawat et al., 2005, 2007). Furthermore, up-regulation of SOCS3 mRNA was not observed in a macrophage cell line with reduced nucleotide-binding oligomerization domain-containing protein 2 (NOD2) expression, suggesting that the cytoplasmic pattern recognition receptor NOD2 is critical for SOCS3-mediated inhibition of iNOS expression (Pudla et al., 2010).
Evasion of Host Cell Autophagic Mechanisms by B. pseudomallei
Autophagy occurs in all eukaryotic cells and is critical for the regulated degradation of cellular components. During autophagy a portion of cytoplasm becomes enclosed within an isolation membrane (phagophore), generating a double-membrane vesicle, the autophagosome, which then, usually following initial fusion with an endosome, fuses with a lysosome. The material sequestered within the autophagosome is then degraded and the products exported to the cytosol for re-use. Autophagy is important in the context of cellular homeostasis for the removal and recycling of damaged or non-functional organelles and the destruction of certain long-lived proteins and other macromolecules (Ravikumar et al., 2010).
Recently, autophagy has become recognized as an important component of the eukaryotic innate immune system (Schmid and Munz, 2007; Ravikumar et al., 2010). In this context, autophagy can function as a defense mechanism against bacterial pathogens, resulting in their clearance. While some pathogens succumb to autophagic clearance and others modify autophagy to benefit their intracellular survival, it is clear that still other intracellular bacterial pathogens can evade autophagy to aid their survival (Hussey et al., 2009; Shahnazari and Brumell, 2011). Notably, two bacterial species, L. monocytogenes and S. flexneri, which can disrupt the phagosome membrane to allow escape into the cytosol, can be targeted by autophagy (Ogawa et al., 2005; Birmingham et al., 2007) when free in the cytosol.
Autophagy and B. pseudomallei
Bacterial escape from phagosomes, and the prospect of the escaped bacteria being subject to autophagy, led us to carry out the first analyses of the interaction of B. pseudomallei with host cell autophagic processes. In response to infection of macrophage RAW 264.7 cells, only a subset of bacteria co-localized with the autophagy marker protein LC3 (Cullinane et al., 2008). When cells were treated with rapamycin, a pharmacological inducer of autophagy, bacterial co-localization with LC3 was increased significantly and bacterial survival was reduced. Thus, autophagy was implicated as part of the host defense system against B. pseudomallei infection, although the mechanism by which most invading bacteria avoided host autophagic attack remained obscure. Moreover, we showed that the predicted bacterial Bsa T3SS effector protein, BopA, is involved in modulating the host cell response, as bopA mutant bacteria showed increased co-localization with LC3 and reduced intracellular survival (Cullinane et al., 2008).
Recently it was shown that LC3 can be recruited directly to bacteria-containing phagosomes (Sanjuan et al., 2007) via a process designated LC3-associated phagocytosis (LAP; Sanjuan et al., 2009). In RAW 264.7 cells infected with E. coli, LAP was induced by LPS treatment via toll-like receptors (TLRs), and involved the rapid translocation of the autophagic proteins Beclin1 and LC3 to the bacteria-containing phagosomes, leading to an increased level of phagocytosis and bacterial killing (Sanjuan et al., 2007). These reports led us to assess the nature of the compartment in which intracellular B. pseudomallei is sequestered. Through an extensive analysis of EM images of infected cells, we demonstrated that intracellular bacteria are either free in the cytosol or sequestered in single-membrane phagosomes, but only extremely rarely contained in double-membrane autophagosomes, suggesting that LC3 is recruited to B. pseudomallei-containing phagosomes (Gong et al., 2011; Figure 2).
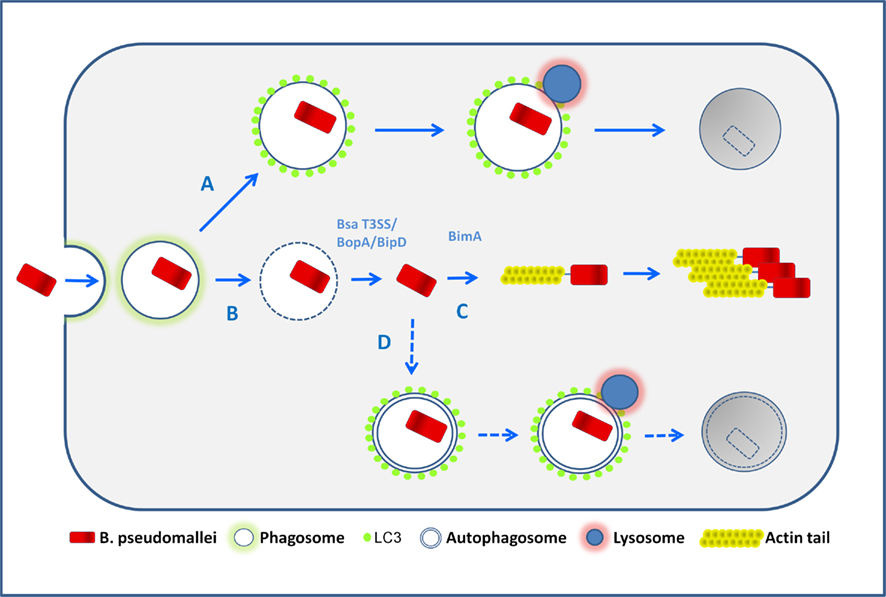
Figure 2. Possible fates of B. pseudomallei in infected macrophages. Following phagocytic uptake by macrophages, bacteria are first located within phagosomes. The majority of wild-type bacteria can escape from the phagosome into the cytosol (pathway B) in a process which is largely uncharacterized but involves the Bsa T3SS. Once free in the cytosol bacteria activate BimA-mediated actin-based motility and replicate (pathway C). Potentially some cytosolic bacteria may be sequestered in canonical autophagosomes (pathway D), but the available evidence suggests that this occurs very infrequently. The autophagy marker protein LC3 can be recruited to bacteria-containing phagosomes, a process designated LC3-associated phagocytosis (LAP) which stimulates further phagosomal maturation via recruitment of other proteins, and the subsequent fusion of phagosomes with lysosomes, leading to bacterial killing (pathway A). It is possible that bacteria may escape from LAP, but definitive evidence that this occurs is presently lacking.
Quantitative analysis of EM images of a bipD mutant, lacking a functional Bsa T3SS, indicated that it displayed no escape from phagosomes at 2 and 4 h p.i. However, bipD mutant bacteria displayed a high level of co-localization with LC3. Furthermore, bopA mutant bacteria that display increased co-localization with LC3 (Cullinane et al., 2008) also showed reduced phagosome escape (Gong et al., 2011). These observations indicate that B. pseudomallei co-localization with LC3 occurs as a result of direct recruitment of LC3 to phagosomes that contain bacteria and is not due to engulfment of bacteria by canonical autophagosomes (Figure 2).
Recruitment of LC3 to phagosomes in LAP led to their rapid acidification and enhanced killing of ingested E. coli in RAW 264.7 cells (Sanjuan et al., 2007, 2009). In our experiments, both bipD and bopA mutant bacteria showed decreased intracellular survival, enhanced co-localization with LC3 and entrapment in phagosomes (Gong et al., 2011). To confirm that these observations were consistent with LAP, we determined the dual co-localization of bacteria with LC3 and the lysosome marker LAMP-1. Such dual co-localization serves as a measure of the maturation of bacteria-containing LC3+ phagosomes by fusion with lysosomes. The percentage of bacteria co-localized with both LC3 and LAMP-1 was significantly higher for both mutants, suggesting that LC3 recruitment is associated with enhanced levels of phagolysosome maturation. Consistent with this proposal was the observation that only a very low percentage of LC3− bacteria-containing phagosomes were LAMP-1+. On the basis of these data we propose that LC3 recruitment to B. pseudomallei-containing phagosomes stimulates fusion of phagosomes with lysosomes, leading to enhanced killing of the internalized bacteria. However, this hypothesis remains to be demonstrated directly.
Collectively these observations (Gong et al., 2011) show that LAP, rather than canonical autophagy, is the mechanism which macrophage cells use in defense against B. pseudomallei infection. However LAP is relatively ineffective and most bacteria escape to the cytosol where they efficiently evade capture by canonical autophagy (Figure 2).
Escape from Autophagy Occurs by a Mechanism Different from that Used by Shigella
Shigella flexneri induces membrane damage to mediate bacterial escape from the phagosome into the cytosol, where it induces actin polymerization, mediated by the bacterial IcsA (VirG) protein (Makino et al., 1986; Goldberg and Theriot, 1995). In some cell types IcsA is specifically targeted by the autophagic membrane protein Atg5, thereby inducing autophagy (Ogawa et al., 2005). However, S. flexneri secretes a T3SS effector protein, IcsB, which binds to IcsA and competitively inhibits its interaction with Atg5, thus abrogating the induction of autophagy (Ogawa et al., 2005). Interestingly, B. pseudomallei encodes the proteins BimA and BopA which have 11 and 23% amino acid identity respectively to their S. flexneri homologs, IcsA and IcsB.
Although BimA mediates actin-based intracellular motility of B. pseudomallei (Stevens et al., 2005b), it lacks the IcsA-binding domain targeted by Atg5 (Ogawa et al., 2005). Thus, it would seem that BimA and IcsA are not functional counterparts in the context of autophagy evasion. Furthermore, bacterial two-hybrid analysis (our unpublished data) failed to establish an interaction between BopA and BimA, suggesting that the B. pseudomallei proteins do not interact in a manner comparable to their S. flexneri counterparts. Thus, we propose that B. pseudomallei evades autophagy by an alternative mechanism which currently remains unknown.
Ubiquitination-Mediated Autophagy
The specific modification of cellular proteins with polyubiquitin is a fundamental post-translational modification that occurs in all eukaryotic cells and is central to many cellular processes. Depending on the nature of the modification (K48- or K63-linked chains) polyubiquitination can result in altered functional properties, in targeting the protein for degradation by the 26S proteasome, or as shown recently, targeting of the material for lysosomal degradation by the autophagic pathway (for review see Komatsu and Ichimura, 2010).
Ubiquitination involves the conjugation of ubiquitin (Ub) moieties to target proteins. In mammalian cells, ubiquitination involves three enzymatic steps. Ub-activating enzymes (E1) transfer Ub to E2-conjugating enzymes, which then transfer the activated Ub to a lysine of the target protein through E3 ligases (Shabek and Ciechanover, 2010). Deubiquitinases are responsible for removing Ub chains from proteins (Shabek and Ciechanover, 2010).
The ubiquitination pathway does not operate in prokaryotes. Nevertheless, polyubiquitination has been shown in many cases to be important for regulating the host response to pathogens. Exploitation of host ubiquitination signaling pathways by pathogen effectors appears to be a common theme among bacteria (for review see Angot et al., 2007). A number of pathogen tactics has been described; they include effectors secreted by Type III and Type VI systems that (i) use the host ubiquitin proteasome system (UPS) to assure their own destruction; (ii) interfere with the ubiquitination level of key cellular proteins involved in innate immunity, or (iii) mimic E3 ligase subunits to manipulate the UPS to its own advantage.
A recent report suggests that, like a number of other pathogens, B. pseudomallei can modulate host ubiquitination to its own advantage. TssM, a secreted product of the T6SS cluster 5 encoded by the bpss1512 gene, was identified as a virulence factor for B. pseudomallei (Tan et al., 2010). In an acute BALB/c mouse infection model, the absence of TssM in B. pseudomallei increased the rate of death and inflammation in infected mice. TssM was alone found to be responsible for down-regulating host inflammatory responses by interfering with the ubiquitination of critical signaling intermediates and, in turn, the activation of NF-κB. Overexpression of TssM in HEK293T cells resulted in diminished ubiquitinated forms of TNFR-associated factor-3, TNFR-associated factor-6 and IκBα. The attenuation of NF-κB activity observed in macrophages infected with wild-type B. pseudomallei was restored in macrophages infected with a tssM mutant.
BPSS1512 shares 99% identity with TssM of B. mallei. In an earlier study it was shown that tssM encodes a broad-based deubiquitinase (Shanks et al., 2009). Cleavage of both K48- and K63-linked ubiquitinated substrates was demonstrated in enzymatic assays. In this study no cellular substrates or phenotype in animal models could be identified. Nevertheless, collectively these studies of TssM indicate that B. pseudomallei is able to modulate the host ubiquitination system through the action of a secreted deubiquitinase. It remains to be seen whether other host or bacterial substrates exist for the deubiquitinase activity of TssM, or whether B. pseudomallei can secrete other effectors capable of modulating the host Ub-pathway. Since several target cytosolic proteins are subject to the deubiquitinase activity of TssM, it seems likely that other cytosolic host proteins may well be susceptible.
It has been suggested that Ub-mediated targeting is a general and major pathway of selective autophagy in mammals. One possible set of targets for the deubiquitinase activity of TssM, yet to be investigated, are ubiquitinated proteins associated with bacteria and recognized by the autophagic pathway. There are several reports describing the co-localization with polyubiquitin of bacteria that have escaped from the phagosome. Species include Streptococcus pyogenes, L. monocytogenes, and Mycobacterium marinum (for review see Shahnazari and Brumell, 2011). A model has been proposed in which host or bacterial proteins associated with the bacterial surface are directly ubiquitinated by the host system. However, the identity of the ubiquitinated proteins has yet to be established. An alternate model has been proposed, based on observations of S. flexneri infections. When S. flexneri disrupts the phagosome membrane to escape into the cytosol, proteins associated with membrane remnants, including TRAF6 which is involved in NF-κB signaling, are polyubiquitinated, recruit the autophagy marker LC3 and adaptor p62, and are targeted to degradation through autophagy (Dupont et al., 2009). The presence of both P62 and other IKK components trapped in the membrane remnants led to the suggestion that the autophagy pathway is used to regulate the activation of inflammatory responses at early time-points in infection (Dupont et al., 2009). Notably, in autophagy deficient (Atg5−/−) cells and in cells in which autophagosome maturation was blocked (ATG4B dominant negative mutant), polyubiquitinated proteins and P62 accumulated on membrane remnants, and the early inflammatory and cytokine response was exacerbated (Dupont et al., 2009). Our preliminary data show that B. pseudomallei in macrophages do not show a high degree of co-localization with poly-Ub, suggesting that the host ubiquitination pathway might be modulated at a number of levels. One intriguing possibility is that the deubiquitinase activity of the TssM effector may play a role in removing Ub from proteins associated with the bacterial surface, thereby allowing the bacteria to avoid detection and elimination from the host cell by autophagy.
Burkholderia pseudomallei K96243 encodes a cyclomodulin (BPSS1385) that displays 26% identity with the cycle inhibiting factor (Cif) cyclomodulin produced by enteropathogenic and enterohemorrhagic E. coli (Jubelin et al., 2009; Yao et al., 2009). Cif from pathogenic E. coli is an effector which is injected into host cells via the T3SS machinery and provokes an irreversible cytopathic effect which is characterized by G1 and G2 cell cycle arrests and reorganization of the actin network (Nougayrède et al., 2001; Marchès et al., 2003; Taieb et al., 2006; Samba-Louaka et al., 2008; Jubelin et al., 2009). The B. pseudomallei cyclomodulin, termed CHBP (for Cif homolog in Burkholderia pseudomallei), reveals a papain-like fold with a conserved Cys-His-Gln catalytic triad and has been shown to be a functional homolog of Cif from E. coli (Jubelin et al., 2009; Yao et al., 2009). Interestingly, it was recently shown that CHBP is a potent inhibitor of the eukaryotic ubiquitination pathway and specifically deamidates Gln40 in ubiquitin and NEDD8 (a ubiquitin-like protein) and in turn attenuates ubiquitination both in vitro and during Burkholderia infection (Cui et al., 2010). Notably, CHBP was recognized by melioidosis patient serum (Felgner et al., 2009). At present there is no evidence to connect these observations concerning CHBP with the function of host autophagic pathways.
In summary, there is a number of intriguing observations that suggest a role for ubiquitin signaling and the avoidance of host autophagy in response to infection by B. pseudomallei. However, a more integrated understanding must await further experimentation.
Concluding Remarks
Burkholderia pseudomallei has evolved a large array of mechanisms to survive and replicate within eukaryotic cells, including mammalian phagocytic and non-phagocytic cells. As we have outlined in this review, the last two decades have seen significant advances in the elucidation of these mechanisms. However, much remains to be uncovered about the detailed interaction of bacterial and host cell factors at the molecular level. The apparently unique mechanism of actin-based motility in B. pseudomallei remains obscure. Likewise, many aspects of the roles of the three T3SS clusters in the interaction of bacterial and host cell molecules and their potential cross-talk remain to be defined. The possibility exists that the different clusters are involved in the invasion of, and replication within, different types of mammalian cells. The precise function of the six T6SS clusters also requires further investigation. While it is clear that most B. pseudomallei in cells are able to escape autophagic attack, the mechanisms are presently unknown. The possibility of host ubiquitination of the bacterial cell surface and whether B. pseudomallei possesses strategies to combat this process are currently under investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Original work in the authors’ laboratories was supported by the Australian Research Council and the National Health and Medical Research Council, Australia.
References
Aardema, H., Luijnenburg, E. M., Salm, E. F., Bijlmer, H. A., Visser, C. E., and Van’t Wout, J. W. (2005). Changing epidemiology of melioidosis? A case of acute pulmonary melioidosis with fatal outcome imported from Brazil. Epidemiol. Infect. 133, 871–875.
Ahmed, K., Enciso, H. D., Masaki, H., Tao, M., Omori, A., Tharavichikul, P., and Nagatake, T. (1999). Attachment of Burkholderia pseudomallei to pharyngeal epithelial cells: a highly pathogenic bacteria with low attachment ability. Am. J. Trop. Med. Hyg. 60, 90–93.
Allwood, E. M., Logue, C. A., Hafner, G. J., Ketheesan, N., Norton, R. E., Peak, I. R., and Beacham, I. R. (2008). Evaluation of recombinant antigens for diagnosis of melioidosis. FEMS Immunol. Med. Microbiol. 54, 144–153.
Amann, K. J., and Pollard, T. D. (2001). Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 98, 15009–15013.
Angot, A., Vergunst, A., Genin, S., and Peeters, N. (2007). Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathog. 3, e3. doi: 10.1371/journal.ppat.0030003
Arjcharoen, S., Wikraiphat, C., Pudla, M., Limposuwan, K., Woods, D. E., Sirisinha, S., and Utaisincharoen, P. (2007). Fate of a Burkholderia pseudomallei lipopolysaccharide mutant in the mouse macrophage cell line RAW 264.7: possible role for the O-antigenic polysaccharide moiety of lipopolysaccharide in internalization and intracellular survival. Infect. Immun. 75, 4298–4304.
Attree, O., and Attree, I. (2001). A second type III secretion system in Burkholderia pseudomallei: who is the real culprit? Microbiology 147, 3197–3199.
Bakshi, C. S., Singh, V. P., Wood, M. W., Jones, P. W., Wallis, T. S., and Galyov, E. E. (2000). Identification of SopE2, a Salmonella secreted protein which is highly homologous to SopE and involved in bacterial invasion of epithelial cells. J. Bacteriol. 182, 2341–2344.
Balder, R., Lipski, S., Lazarus, J. J., Grose, W., Wooten, R. M., Hogan, R. J., Woods, D. E., and Lafontaine, E. R. (2010). Identification of Burkholderia mallei and Burkholderia pseudomallei adhesins for human respiratory epithelial cells. BMC Microbiol. 10, 250. doi: 10.1186/1471-2180-10-250
Barth, A. L., de Abreu, E., Silva, F. A., Hoffmann, A., Vieira, M. I., Zavascki, A. P., Ferreira, A. G., da Cunha, L. G. J., Albano, R. M., and de Andrade Marques, E. (2007). Cystic fibrosis patient with Burkholderia pseudomallei infection acquired in Brazil. J. Clin. Microbiol. 45, 4077–4080.
Birmingham, C. L., Canadien, V., Gouin, E., Troy, E. B., Yoshimori, T., Cossart, P., Higgins, D. E., and Brumell, J. H. (2007). Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3, 442–451.
Boddey, J. A., Day, C. J., Flegg, C. P., Ulrich, R. L., Stephens, S. R., Beacham, I. R., Morrison, N. A., and Peak, I. R. (2007). The bacterial gene lfpA influences the potent induction of calcitonin receptor and osteoclast-related genes in Burkholderia pseudomallei-induced TRAP-positive multinucleated giant cells. Cell. Microbiol. 9, 514–531.
Boddey, J. A., Flegg, C. P., Day, C. J., Beacham, I. R., and Peak, I. R. (2006). Temperature-regulated microcolony formation by Burkholderia pseudomallei requires pilA and enhances association with cultured human cells. Infect. Immun. 74, 5374–5381.
Boujemaa-Paterski, R., Gouin, E., Hansen, G., Samarin, S., Le Clainche, C., Didry, D., Dehoux, P., Cossart, P., Kocks, C., Carlier, M. F., and Pantaloni, D. (2001). Listeria protein ActA mimics WASP family proteins: it activates filament barbed end branching by Arp2/3 complex. Biochemistry 40, 11390–11404.
Breitbach, K., Rottner, K., Klocke, S., Rohde, M., Jenzora, A., Wehland, J., and Steinmetz, I. (2003). Actin-based motility of Burkholderia pseudomallei involves the Arp 2/3 complex, but not N-WASP and Ena/VASP proteins. Cell. Microbiol. 5, 385–393.
Brett, P. J., and Woods, D. E. (2000). Pathogenesis of and immunity to melioidosis. Acta Trop. 74, 201–210.
Brown, N. F., Boddey, J. A., Flegg, C. P., and Beacham, I. R. (2002). Adherence of Burkholderia pseudomallei cells to cultured human epithelial cell lines is regulated by growth temperature. Infect. Immun. 70, 974–980.
Burtnick, M. N., Brett, P. J., Harding, S. V., Ngugi, S. A., Ribot, W. J., Chantratita, N., Scorpio, A., Milne, T. S., Dean, R. E., Fritz, D. L., Peacock, S. J., Prior, J. L., Atkins, T. P., and Deshazer, D. (2011). The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei. Infect. Immun. 79, 1512–1525.
Burtnick, M. N., Brett, P. J., Nair, V., Warawa, J. M., Woods, D. E., and Gherardini, F. C. (2008). Burkholderia pseudomallei type III secretion system mutants exhibit delayed vacuolar escape phenotypes in RAW 264.7 murine macrophages. Infect. Immun. 76, 2991–3000.
Burtnick, M. N., DeShazer, D., Nair, V., Gherardini, F. C., and Brett, P. J. (2010). Burkholderia mallei cluster 1 type VI secretion mutants exhibit growth and actin polymerization defects in RAW 264.7 murine macrophages. Infect. Immun. 78, 88–99.
Carsillo, M., Kutala, V. K., Puschel, K., Blanco, J., Kuppusamy, P., and Niewiesk, S. (2009). Nitric oxide production and nitric oxide synthase type 2 expression by cotton rat (Sigmodon hispidus) macrophages reflect the same pattern as human macrophages. Dev. Comp. Immunol. 33, 718–724.
Chaowagul, W., White, N. J., Dance, D. A., Wattanagoon, Y., Naigowit, P., Davis, T. M., Looareesuwan, S., and Pitakwatchara, N. (1989). Melioidosis: a major cause of community-acquired septicemia in northeastern Thailand. J. Infect. Dis. 159, 890–899.
Chua, K. L., Chan, Y. Y., and Gan, Y. H. (2003). Flagella are virulence determinants of Burkholderia pseudomallei. Infect. Immun. 71, 1622–1629.
Ciborowski, P., and Gendelman, H. E. (2006). Human immunodeficiency virus-mononuclear phagocyte interactions: emerging avenues of biomarker discovery, modes of viral persistence and disease pathogenesis. Curr. HIV Res. 4, 279–291.
Cole, N. L., and Grose, C. (2003). Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev. Med. Virol. 13, 207–222.
Cossart, P. (2000). Actin-based motility of pathogens: the Arp2/3 complex is a central player. Cell. Microbiol. 2, 195–205.
Cui, J., Yao, Q., Li, S., Ding, X., Lu, Q., Mao, H., Liu, L., Zheng, N., Chen, S., and Shao, F. (2010). Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science 329, 1215–1218.
Cullinane, M., Gong, L., Li, X., Lazar-Adler, N., Tra, T., Wolvetang, E., Prescott, M., Boyce, J. D., Devenish, R. J., and Adler, B. (2008). Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy 4, 744–753.
Currie, B. J., Dance, D. A., and Cheng, A. C. (2008). The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans. R. Soc. Trop. Med. Hyg. 102, S1–S4.
D’Cruze, T., Gong, L., Treerat, P., Ramm, G., Boyce, J. D., Prescott, M., Adler, B., and Devenish, R. J. (2011). A role for the Burkholderia pseudomallei type three secretion system cluster 1 bpscN gene in virulence. Infect. Immun. doi: 10.1128/IAI.01351-10. [Epub ahead of print].
de Chastellier, C., and Thilo, L. (2006). Cholesterol depletion in Mycobacterium avium-infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable Mycobacteria in phagolysosome-derived autophagic vacuoles. Cell. Microbiol. 8, 242–256.
Decker, T., Stockinger, S., Karaghiosoff, M., Müller, M., and Kovarik, P. (2002). IFNs and STATs in innate immunity to microorganisms. J. Clin. Invest. 109, 1271–1277.
DeShazer, D., Brett, P. J., Carlyon, R., and Woods, D. E. (1997). Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J. Bacteriol. 179, 2116–2125.
Dorman, S. E., Gill, V. J., Gallin, J. I., and Holland, S. M. (1998). Burkholderia pseudomallei infection in a Puerto Rican patient with chronic granulomatous disease: case report and review of occurrences in the Americas. Clin. Infect. Dis. 26, 889–894.
Douglas, M. W., Lum, G., Roy, J., Fisher, D. A., Anstey, N. M., and Currie, B. J. (2004). Epidemiology of community-acquired and nosocomial bloodstream infections in tropical Australia: a 12-month prospective study. Trop. Med. Int. Health 9, 795–804.
Dowling, A. J., Wilkinson, P. A., Holden, M. T., Quail, M. A., Bentley, S. D., Reger, J., Waterfield, N. R., Titball, R. W., and Ffrench-Constant, R. H. (2010). Genome-wide analysis reveals loci encoding anti-macrophage factors in the human pathogen Burkholderia pseudomallei K96243. PLoS ONE 5, e15693. doi: 10.1371/journal.pone.0015693
Dupont, N., Lacas-Gervais, S., Bertout, J., Paz, I., Freche, B., Van Nhieu, G. T., van der Goot, F. G., Sansonetti, P. J., and Lafont, F. (2009). Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6, 137–149.
Egan, A. M., and Gordon, D. L. (1996). Burkholderia pseudomallei activates complement and is ingested but not killed by polymorphonuclear leukocytes. Infect. Immun. 64, 4952–4959.
Egile, C., Loisel, T. P., Laurent, V., Li, R., Pantaloni, D., Sansonetti, P. J., and Carlier, M. F. (1999). Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 146, 1319–1332.
Ekchariyawat, P., Pudla, S., Limposuwan, K., Arjcharoen, S., Sirisinha, S., and Utaisincharoen, P. (2005). Burkholderia pseudomallei-induced expression of suppressor of cytokine signaling 3 and cytokine-inducible src homology 2-containing protein in mouse macrophages: a possible mechanism for suppression of the response to gamma interferon stimulation. Infect. Immun. 73, 7332–7339.
Ekchariyawat, P., Pudla, S., Limposuwan, K., Arjcharoen, S., Sirisinha, S., and Utaisincharoen, P. (2007). Expression of suppressor of cytokine signaling 3 (SOCS3) and cytokine-inducible Src homology 2-containing protein (CIS) induced in Burkholderia pseudomallei-infected mouse macrophages requires bacterial internalization. Microb. Pathog. 42, 104–110.
Essex-Lopresti, A. E., Boddey, J. A., Thomas, R., Smith, M. P., Hartley, M. G., Atkins, T., Brown, N. F., Tsang, C. H., Peak, I. R., Hill, J., Beacham, I. R., and Titball, R. W. (2005). A type IV pilin, PilA, contributes to adherence of Burkholderia pseudomallei and virulence in vivo. Infect. Immun. 73, 1260–1264.
Fang, F. C. (1997). Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J. Clin. Invest. 99, 2818–2825.
Felgner, P. L., Kayala, M. A., Vigil, A., Burk, C., Nakajima-Sasaki, R., Pablo, J., Molina, D. M., Hirst, S., Chew, J. S., Wang, D., Tan, G., Duffield, M., Yang, R., Neel, J., Chantratita, N., Bancroft, G., Lertmemongkolchai, G., Davies, D. H., Baldi, P., Peacock, S., and Titball, R. W. (2009). A Burkholderia pseudomallei protein microarray reveals serodiagnostic and cross-reactive antigens. Proc. Natl. Acad. Sci. U.S.A. 106, 13499–13504.
Galyov, E. E., Brett, P. J., and DeShazer, D. (2010). Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu. Rev. Microbiol. 64, 495–517.
Ginocchio, C. C., and Galán, J. E. (1995). Functional conservation among members of the Salmonella typhimurium InvA family of proteins. Infect. Immun. 63, 729–732.
Goldberg, M. B., and Theriot, J. A. (1995). Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc. Natl. Acad. Sci. U.S.A. 92, 6572–6576.
Goley, E. D., Rodenbusch, S. E., Martin, A. C., and Welch, M. D. (2004). Critical conformational changes in the Arp2/3 complex are induced by nucleotide and nucleation promoting factor. Mol. Cell 16, 269–279.
Goley, E. D., and Welch, M. D. (2006). The ARP2/3 complex: an actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 7, 713–726.
Gong, L., Cullinane, M., Treerat, P., Ramm, G., Prescott, M., Adler, B., Boyce, J. D., and Devenish, R. J. (2011). The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS ONE 6, e17852. doi: 10.1371/journal.pone.0017852
Gori, A. H., Ahmed, K., Martinez, G., Masaki, H., Watanabe, K., and Nagatake, T. (1999). Mediation of attachment of Burkholderia pseudomallei to human pharyngeal epithelial cells by the asialoganglioside GM1-GM2 receptor complex. Am. J. Trop. Med. Hyg. 61, 473–475.
Gouin, E., Egile, C., Dehoux, P., Villiers, V., Adams, J., Gertler, F., Li, R., and Cossart, P. (2004). The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature 427, 457–461.
Gouin, E., Welch, M. D., and Cossart, P. (2005). Actin-based motility of intracellular pathogens. Curr. Opin. Microbiol. 8, 35–45.
Haase, A., Melder, A., Smith-Vaughan, H., Kemp, D., and Currie, B. (1995). RAPD analysis of isolates of Burkholderia pseudomallei from patients with recurrent melioidosis. Epidemiol. Infect. 115, 115–121.
Hardt, W. D., Chen, L. M., Schuebel, K. E., Bustelo, X. R., and Galán, J. E. (1998). S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93, 815–826.
Harley, V. S., Dance, D. A., Drasar, B. S., and Tovey, G. (1998a). Effects of Burkholderia pseudomallei and other Burkholderia species on eukaryotic cells in tissue culture. Microbios 96, 71–93.
Harley, V. S., Dance, D. A., Tovey, G., McCrossan, M. V., and Drasar, B. S. (1998b). An ultrastructural study of the phagocytosis of Burkholderia pseudomallei. Microbios 94, 35–45.
Harley, V. S., Dance, D. A., Tovey, G., and Drasar, B. S. (1994). Interaction of Pseudomonas pseudomallei with macrophages. Biochem. Soc. Trans. 22, 88S.
Hueck, C. J. (1998). Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62, 379–433.
Hussey, S., Travassos, L. H., and Jones, N. L. (2009). Autophagy as an emerging dimension to adaptive and innate immunity. Semin. Immunol. 21, 233–241.
Inglis, T. J., Robertson, T., Woods, D. E., Dutton, N., and Chang, B. J. (2003). Flagellum-mediated adhesion by Burkholderia pseudomallei precedes invasion of Acanthamoeba astronyxis. Infect. Immun. 71, 2280–2282.
Jacobs, A. T., and Ignarro, L. J. (2001). Lipopolysaccharide-induced expression of interferon-beta mediates the timing of inducible nitric-oxide synthase induction in RAW 264.7 macrophages. J. Biol. Chem. 276, 47950–47957.
Jeng, R. L., Goley, E. D., D’Alessio, J. A., Chaga, O. Y., Svitkina, T. M., Borisy, G. G., Heinzen, R. A., and Welch, M. D. (2004). A Rickettsia WASP-like protein activates the Arp2/3 complex and mediates actin-based motility. Cell. Microbiol. 6, 761–769.
Johnson, S., Roversi, P., Espina, M., Olive, A., Deane, J. E., Birket, S., Field, T., Picking, W. D., Blocker, A. J., Galyov, E. E., Picking, W. L., and Lea, S. M. (2007). Self-chaperoning of the type III secretion system needle tip proteins IpaD and BipD. J. Biol. Chem. 282, 4035–4044.
Jones, A. L., Beveridge, T. J., and Woods, D. E. (1996). Intracellular survival of Burkholderia pseudomallei. Infect. Immun. 64, 782–790.
Jones, A. L., DeShazer, D., and Woods, D. E. (1997). Identification and characterization of a two-component regulatory system involved in invasion of eukaryotic cells and heavy-metal resistance in Burkholderia pseudomallei. Infect. Immun. 65, 4972–4977.
Jubelin, G., Chavez, C. V., Taieb, F., Banfield, M. J., Samba-Louaka, A., Nobe, R., Nougayrède, J. P., Zumbihl, R., Givaudan, A., Escoubas, J. M., and Oswald, E. (2009). Cycle inhibiting factors (CIFs) are a growing family of functional cyclomodulins present in invertebrate and mammal bacterial pathogens. PLoS ONE 4, e4855. doi: 10.1371/journal.pone.0004855
Kayath, C. A., Hussey, S., El hajjami, N., Nagra, K., Philpott, D., and Allaoui, A. (2010). Escape of intracellular Shigella from autophagy requires binding to cholesterol through the type III effector, IcsB. Microbes Infect. 12, 956–966.
Kespichayawattana, W., Intachote, P., Utaisincharoen, P., and Sirisinha, S. (2004). Virulent Burkholderia pseudomallei is more efficient than avirulent Burkholderia thailandensis in invasion of and adherence to cultured human epithelial cells. Microb. Pathog. 36, 287–292.
Kespichayawattana, W., Rattanachetkul, S., Wanun, T., Utaisincharoen, P., and Sirisinha, S. (2000). Burkholderia pseudomallei induces cell fusion and actin-associated membrane protrusion: a possible mechanism for cell-to-cell spreading. Infect. Immun. 68, 5377–5384.
Kinzler, E. R., and Compton, T. (2005). Characterization of human cytomegalovirus glycoprotein-induced cell-cell fusion. J. Virol. 79, 7827–7837.
Komatsu, M., and Ichimura, Y. (2010). Selective autophagy regulates various cellular functions. Genes Cells 15, 923–933.
Kondo, Y., Yasui, K., Yashiro, M., Tsuge, M., Kotani, N., and Morishima, T. (2009). Multi-nucleated giant cell formation from human cord blood monocytes in vitro, in comparison with adult peripheral blood monocytes. Clin. Exp. Immunol. 158, 84–90.
Koponen, M. A., Zlock, D., Palmer, D. L., and Merlin, T. L. (1991). Melioidosis. Forgotten, but not gone! Arch. Intern. Med. 151, 605–608.
Laurent, V., Loisel, T. P., Harbeck, B., Wehman, A., Gröbe, L., Jockusch, B. M., Wehland, J., Gertler, F. B., and Carlier, M. F. (1999). Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J. Cell Biol. 144, 1245–1258.
Lazar Adler, N. R., Govan, B., Cullinane, M., Harper, M., Adler, B., and Boyce, J. D. (2009). The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease? FEMS Microbiol. Rev. 33, 1079–1099.
Le Hello, S., Currie, B. J., Godoy, D., Spratt, B. G., Mikulski, M., Lacassin, F., and Garin, B. (2005). Melioidosis in New Caledonia. Emerging Infect. Dis. 11, 1607–1609.
Maharjan, B., Chantratita, N., Vesaratchavest, M., Cheng, A., Wuthiekanun, V., Chierakul, W., Chaowagul, W., Day, N. P., and Peacock, S. J. (2005). Recurrent melioidosis in patients in northeast Thailand is frequently due to reinfection rather than relapse. J. Clin. Microbiol. 43, 6032–6034.
Makino, S., Sasakawa, C., Kamata, K., Kurata, T., and Yoshikawa, M. (1986). A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell 46, 551–555.
Marchès, O., Ledger, T. N., Boury, M., Ohara, M., Tu, X., Goffaux, F., Mainil, J., Rosenshine, I., Sugai, M., De Rycke, J., and Oswald, E. (2003). Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol. Microbiol. 50, 1553–1567.
May, R. C., Hall, M. E., Higgs, H. N., Pollard, T. D., Chakraborty, T., Wehland, J., Machesky, L. M., and Sechi, A. S. (1999). The Arp2/3 complex is essential for the actin-based motility of Listeria monocytogenes. Curr. Biol. 9, 759–762.
Monack, D. M., and Theriot, J. A. (2001). Actin-based motility is sufficient for bacterial membrane protrusion formation and host cell uptake. Cell. Microbiol. 3, 633–647.
Muangsombut, V., Suparak, S., Pumirat, P., Damnin, S., Vattanaviboon, P., Thongboonkerd, V., and Korbsrisate, S. (2008). Inactivation of Burkholderia pseudomallei bsaQ results in decreased invasion efficiency and delayed escape of bacteria from endocytic vesicles. Arch. Microbiol. 190, 623–631.
Mullins, R. D., Heuser, J. A., and Pollard, T. D. (1998). The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. U.S.A. 95, 6181–6186.
Ngauy, V., Lemeshev, Y., Sadkowski, L., and Crawford, G. (2005). Cutaneous melioidosis in a man who was taken as a prisoner of war by the Japanese during World War II. J. Clin. Microbiol. 43, 970–972.
Nougayrède, J. P., Boury, M., Tasca, C., Marchès, O., Milon, A., Oswald, E., and De Rycke, J. (2001). Type III secretion-dependent cell cycle block caused in HeLa cells by enteropathogenic Escherichia coli O103. Infect. Immun. 69, 6785–6795.
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–731.
Osiriphun, Y., Wongtrakoongate, P., Sanongkiet, S., Suriyaphol, P., Thongboonkerd, V., and Tungpradabkul, S. (2009). Identification and characterization of RpoS regulon and RpoS-dependent promoters in Burkholderia pseudomallei. J. Proteome Res. 8, 3118–3131.
Pei, J., and Grishin, N. V. (2009). The Rho GTPase inactivation domain in Vibrio cholerae MARTX toxin has a circularly permuted papain-like thiol protease fold. Proteins 77, 413–419.
Phewkliang, A., Wongratanacheewin, S., and Chareonsudjai, S. (2010). Role of Burkholderia pseudomallei in the invasion, replication and induction of apoptosis in human epithelial cell lines. Southeast Asian J. Trop. Med. Public Health 41, 1164–1176.
Pilatz, S., Breitbach, K., Hein, N., Fehlhaber, B., Schulze, J., Brenneke, B., Eberl, L., and Steinmetz, I. (2006). Identification of Burkholderia pseudomallei genes required for the intracellular life cycle and in vivo virulence. Infect. Immun. 74, 3576–3586.
Pruksachartvuthi, S., Aswapokee, N., and Thankerngpol, K. (1990). Survival of Pseudomonas pseudomallei in human phagocytes. J. Med. Microbiol. 31, 109–114.
Pudla, M., Kananuruk, A., Limposuwan, K., Sirisinha, S., and Utaisincharoen, P. (2010). Nucleotide-binding oligomerization domain-containing protein 2 regulates suppressor of cytokine signaling 3 expression in Burkholderia pseudomallei- infected mouse macrophage cell line RAW 264.7. Innate Immun. doi: 10.1177/1753425910385484. [Epub ahead of print].
Pukatzki, S., McAuley, S. B., and Miyata, S. T. (2009). The type VI secretion system: translocation of effectors and effector-domains. Curr. Opin. Microbiol. 12, 11–17.
Rainbow, L., Hart, C. A., and Winstanley, C. (2002). Distribution of type III secretion gene clusters in Burkholderia pseudomallei, B. thailandensis and B. mallei. J. Med. Microbiol. 51, 374–384.
Ravikumar, B., Sarkar, S., Davies, J. E., Futter, M., Garcia-Arencibia, M., Green-Thompson, Z. W., Jimenez-Sanchez, M., Korolchuk, V. I., Lichtenberg, M., Luo, S., Massey, D. C., Menzies, F. M., Moreau, K., Narayanan, U., Renna, M., Siddiqi, F. H., Underwood, B. R., Winslow, A. R., and Rubinsztein, D. C. (2010). Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90, 1383–1435.
Reckseidler, S. L., DeShazer, D., Sokol, P. A., and Woods, D. E. (2001). Detection of bacterial virulence genes by subtractive hybridization: identification of capsular polysaccharide of Burkholderia pseudomallei as a major virulence determinant. Infect. Immun. 69, 34–44.
Rudolph, M. G., Weise, C., Mirold, S., Hillenbrand, B., Bader, B., Wittinghofer, A., and Hardt, W. D. (1999). Biochemical analysis of SopE from Salmonella typhimurium, a highly efficient guanosine nucleotide exchange factor for RhoGTPases. J. Biol. Chem. 274, 30501–30509.
Samba-Louaka, A., Nougayrède, J. P., Watrin, C., Jubelin, G., Oswald, E., and Taieb, F. (2008). Bacterial cyclomodulin Cif blocks the host cell cycle by stabilizing the cyclin-dependent kinase inhibitors p21 and p27. Cell. Microbiol. 10, 2496–2508.
Sanjuan, M. A., Dillon, C. P., Tait, S. W., Moshiach, S., Dorsey, F., Connell, S., Komatsu, M., Tanaka, K., Cleveland, J. L., Withoff, S., and Green, D. R. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257.
Sanjuan, M. A., Milasta, S., and Green, D. R. (2009). Toll-like receptor signaling in the lysosomal pathways. Immunol. Rev. 227, 203–220.
Schell, M. A., Ulrich, R. L., Ribot, W. J., Brueggemann, E. E., Hines, H. B., Chen, D., Lipscomb, L., Kim, H. S., Mrazek, J., Nierman, W. C., and DeShazer, D. (2007). Type VI secretion is a major virulence determinant in Burkholderia mallei. Mol. Microbiol. 64, 1466–1485.
Schmid, D., and Munz, C. (2007). Innate and adaptive immunity through autophagy. Immunity 27, 11–21.
Shabek, N., and Ciechanover, A. (2010). Degradation of ubiquitin: the fate of the cellular reaper. Cell Cycle 9, 523–530.
Shahnazari, S., and Brumell, J. H. (2011). Mechanisms and consequences of bacterial targeting by the autophagy pathway. Curr. Opin. Microbiol. 14, 68–75.
Shalom, G., Shaw, J. G., and Thomas, M. S. (2007). In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiology 153, 2689–2699.
Shanks, J., Burtnick, M. N., Brett, P. J., Waag, D. M., Spurgers, K. B., Ribot, W. J., Schell, M. A., Panchal, R. G., Gherardini, F. C., Wilkinson, K. D., and Deshazer, D. (2009). Burkholderia mallei tssM encodes a putative deubiquitinase that is secreted and expressed inside infected RAW 264.7 murine macrophages. Infect. Immun. 77, 1636–1648.
Sitthidet, C., Korbsrisate, S., Layton, A. N., Field, T. R., Stevens, M. P., and Stevens, J. M. (2011). Identification of motifs of Burkholderia pseudomallei BimA required for intracellular motility, actin binding, and actin polymerization. J. Bacteriol. 193, 1901–1910.
Sitthidet, C., Stevens, J. M., Chantratita, N., Currie, B. J., Peacock, S. J., Korbsrisate, S., and Stevens, M. P. (2008). Prevalence and sequence diversity of a factor required for actin-based motility in natural populations of Burkholderia species. J. Clin. Microbiol. 46, 2418–2422.
Stender, S., Friebel, A., Linder, S., Rohde, M., Mirold, S., and Hardt, W. D. (2000). Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol. Microbiol. 36, 1206–1221.
Stevens, J. M., Galyov, E. E., and Stevens, M. P. (2006). Actin-dependent movement of bacterial pathogens. Nat. Rev. Microbiol. 4, 91–101.
Stevens, J. M., Ulrich, R. L., Taylor, L. A., Wood, M. W., Deshazer, D., Stevens, M. P., and Galyov, E. E. (2005a). Actin-binding proteins from Burkholderia mallei and Burkholderia thailandensis can functionally compensate for the actin-based motility defect of a Burkholderia pseudomallei bimA mutant. J. Bacteriol. 187, 7857–7862.
Stevens, M. P., Stevens, J. M., Jeng, R. L., Taylor, L. A., Wood, M. W., Hawes, P., Monaghan, P., Welch, M. D., and Galyov, E. E. (2005b). Identification of a bacterial factor required for actin-based motility of Burkholderia pseudomallei. Mol. Microbiol. 56, 40–53.
Stevens, M. P., Brown, P. J., Wood, M. W., Wallis, T. S., and Galyov, E. E. (2001). “Cognate virulence gene clusters in Salmonella and B. pseudomallei,” in Proceedings of the World Melioidosis Congress, Perth.
Stevens, M. P., Friebel, A., Taylor, L. A., Wood, M. W., Brown, P. J., Hardt, W. D., and Galyov, E. E. (2003). A Burkholderia pseudomallei type III secreted protein, BopE, facilitates bacterial invasion of epithelial cells and exhibits guanine nucleotide exchange factor activity. J. Bacteriol. 185, 4992–4996.
Stevens, M. P., and Galyov, E. E. (2004). Exploitation of host cells by Burkholderia pseudomallei. Int. J. Med. Microbiol. 293, 549–555.
Stevens, M. P., Haque, A., Atkins, T., Hill, J., Wood, M. W., Easton, A., Nelson, M., Underwood-Fowler, C., Titball, R. W., Bancroft, G. J., and Galyov, E. E. (2004). Attenuated virulence and protective efficacy of a Burkholderia pseudomallei bsa type III secretion mutant in murine models of melioidosis. Microbiology 150, 2669–2676.
Stevens, M. P., Wood, M. W., Taylor, L. A., Monaghan, P., Hawes, P., Jones, P. W., Wallis, T. S., and Galyov, E. E. (2002). An Inv/Mxi-Spa-like type III protein secretion system in Burkholderia pseudomallei modulates intracellular behaviour of the pathogen. Mol. Microbiol. 46, 649–659.
Stone, R. (2007). Infectious disease. Racing to defuse a bacterial time bomb. Science 317, 1022–1024.
Sun, G. W., and Gan, Y. H. (2010). Unraveling type III secretion systems in the highly versatile Burkholderia pseudomallei. Trends Microbiol. 18, 561–568.
Sun, G. W., Lu, J., Pervaiz, S., Cao, W. P., and Gan, Y. H. (2005). Caspase-1 dependent macrophage death induced by Burkholderia pseudomallei. Cell. Microbiol. 7, 1447–1458.
Suparak, S., Kespichayawattana, W., Haque, A., Easton, A., Damnin, S., Lertmemongkolchai, G., Bancroft, G. J., and Korbsrisate, S. (2005). Multinucleated giant cell formation and apoptosis in infected host cells is mediated by Burkholderia pseudomallei type III secretion protein BipB. J. Bacteriol. 187, 6556–6560.
Taieb, F., Nougayrède, J. P., Watrin, C., Samba-Louaka, A., and Oswald, E. (2006). Escherichia coli cyclomodulin Cif induces G2 arrest of the host cell cycle without activation of the DNA-damage checkpoint-signalling pathway. Cell. Microbiol. 8, 1910–1921.
Tan, K. S., Chen, Y., Lim, Y. C., Tan, G. Y., Liu, Y., Lim, Y. T., Macary, P., and Gan, Y. H. (2010). Suppression of host innate immune response by Burkholderia pseudomallei through the virulence factor TssM. J. Immunol. 184, 5160–5171.
Upadhyay, A., Wu, H. L., Williams, C., Field, T., Galyov, E. E., van den Elsen, J. M., and Bagby, S. (2008). The guanine-nucleotide-exchange factor BopE from Burkholderia pseudomallei adopts a compact version of the Salmonella SopE/SopE2 fold and undergoes a closed-to-open conformational change upon interaction with Cdc42. Biochem. J. 411, 485–493.
Utaisincharoen, P., Anuntagool, N., Arjcharoen, S., Limposuwan, K., Chaisuriya, P., and Sirisinha, S. (2004). Induction of iNOS expression and antimicrobial activity by interferon (IFN)-beta is distinct from IFN-gamma in Burkholderia pseudomallei-infected mouse macrophages. Clin. Exp. Immunol. 136, 277–283.
Utaisincharoen, P., Anuntagool, N., Limposuwan, K., Chaisuriya, P., and Sirisinha, S. (2003). Involvement of beta interferon in enhancing inducible nitric oxide synthase production and antimicrobial activity of Burkholderia pseudomallei-infected macrophages. Infect. Immun. 71, 3053–3057.
Utaisincharoen, P., Arjcharoen, S., Limposuwan, K., Tungpradabkul, S., and Sirisinha, S. (2006). Burkholderia pseudomallei RpoS regulates multinucleated giant cell formation and inducible nitric oxide synthase expression in mouse macrophage cell line (RAW 264.7). Microb. Pathog. 40, 184–189.
Utaisincharoen, P., Tangthawornchaikul, N., Kespichayawattana, W., Anuntagool, N., Chaisuriya, P., and Sirisinha, S. (2000). Kinetic studies of the production of nitric oxide (NO) and tumour necrosis factor-alpha (TNF-alpha) in macrophages stimulated with Burkholderia pseudomallei endotoxin. Clin. Exp. Immunol. 122, 324–329.
Utaisincharoen, P., Tangthawornchaikul, N., Kespichayawattana, W., Chaisuriya, P., and Sirisinha, S. (2001). Burkholderia pseudomallei interferes with inducible nitric oxide synthase (iNOS) production: a possible mechanism of evading macrophage killing. Microbiol. Immunol. 45, 307–313.
Utermöhlen, O., Herz, J., Schramm, M., and Krönke, M. (2008). Fusogenicity of membranes: the impact of acid sphingomyelinase on innate immune responses. Immunobiology 213, 307–314.
Warawa, J., and Woods, D. E. (2005). Type III secretion system cluster 3 is required for maximal virulence of Burkholderia pseudomallei in a hamster infection model. FEMS Microbiol. Lett. 242, 101–108.
Welch, M. D., Iwamatsu, A., and Mitchison, T. J. (1997). Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 385, 265–269.
Welch, M. D., and Mullins, R. D. (2002). Cellular control of actin nucleation. Annu. Rev. Cell Dev. Biol. 18, 247–288.
Welch, M. D., Rosenblatt, J., Skoble, J., Portnoy, D. A., and Mitchison, T. J. (1998). Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science 281, 105–108.
Whitlock, G. C., Estes, D. M., Young, G. M., Young, B., and Torres, A. G. (2008). Construction of a reporter system to study Burkholderia mallei type III secretion and identification of the BopA effector protein function in intracellular survival. Trans. R. Soc. Trop. Med. Hyg. 102, S127–S133.
Whitlock, G. C., Valbuena, G. A., Popov, V. L., Judy, B. M., Estes, D. M., and Torres, A. G. (2009). Burkholderia mallei cellular interactions in a respiratory cell model. J. Med. Microbiol. 58, 554–562.
Wiersinga, W. J., de Vos, A. F., de Beer, R., Wieland, C. W., Roelofs, J. J., Woods, D. E., and van der Poll, T. (2008). Inflammation patterns induced by different Burkholderia species in mice. Cell. Microbiol. 10, 81–87.
Winstanley, C., Hales, B. A., and Hart, C. A. (1999). Evidence for the presence in Burkholderia pseudomallei of a type III secretion system-associated gene cluster. J. Med. Microbiol. 48, 649–656.
Wong, K. T., Puthucheary, S. D., and Vadivelu, J. (1995). The histopathology of human melioidosis. Histopathology 26, 51–55.
Wood, M. W., Rosqvist, R., Mullan, P. B., Edwards, M. H., and Galyov, E. E. (1996). SopE, a secreted protein of Salmonella dublin, is translocated into the target eukaryotic cell via a sip-dependent mechanism and promotes bacterial entry. Mol. Microbiol. 22, 327–338.
Yao, Q., Cui, J., Zhu, Y., Wang, G., Hu, L., Long, C., Cao, R., Liu, X., Huang, N., Chen, S., Liu, L., and Shao, F. (2009). A bacterial type III effector family uses the papain-like hydrolytic activity to arrest the host cell cycle. Proc. Natl. Acad. Sci. U.S.A. 106, 3716–3721.
Keywords: melioidosis, Burkholderia, pathogenesis, adhesion, intracellular survival, autophagy, ubiquitination
Citation: Allwood EM, Devenish RJ, Prescott M, Adler B and Boyce JD (2011) Strategies for intracellular survival of Burkholderia pseudomallei. Front. Microbio. 2:170. doi: 10.3389/fmicb.2011.00170
Received: 18 May 2011;
Paper pending published: 28 May 2011;
Accepted: 26 July 2011;
Published online: 22 August 2011.
Edited by:
Alfredo G. Torres, University of Texas Medical Branch, USAReviewed by:
Jonathan Mark Warawa, University of Louisville, USAJo Stevens, The University of Edinburgh, UK
Copyright: © 2011 Allwood, Devenish, Prescott, Adler and Boyce. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Ben Adler, Department of Microbiology, Monash University, Wellington Road, Clayton, VIC 3800, Australia. e-mail:YmVuLmFkbGVyQG1vbmFzaC5lZHU=