 Yoshihisa Inoue1
Yoshihisa Inoue1
- 1 Laboratory Animal Research Center, Institute of Medical Science, The University of Tokyo, Tokyo, Japan
- 2 Department of Experimental Phylaxiology, Faculty of Medical and Pharmaceutical Sciences, Kumamoto University, Kumamoto, Japan
Measles, caused by measles virus (MeV) infection, is the leading cause of death in children because of secondary infections attributable to MeV-induced immune suppression. Recently, we have shown that wild-type MeVs induce the suppression of protein synthesis in host cells (referred to as “shutoff”) and that viral mRNAs are preferentially translated under shutoff conditions in infected cells. To determine the mechanism behind the preferential translation of viral mRNA, we focused on the 5′ untranslated region (UTR) of nucleocapsid (N) mRNA. The La/SSB autoantigen (La) was found to specifically bind to an N-5′UTR probe. Recombinant La enhanced the translation of luciferase mRNA containing the N-5′UTR (N-fLuc), and RNA interference of La suppressed N-fLuc translation. Furthermore, recombinant MeV lacking the La-binding motif in the N-5′UTR displayed delayed viral protein synthesis and growth kinetics at an early phase of infection. These results suggest that La induced predominant translation of N mRNA via binding to its 5′UTR under shutoff conditions. This is the first report on a cellular factor that specifically regulates paramyxovirus mRNA translation.
Introduction
Measles virus (MeV) infection causes a maculopapular rash, fever, cough, and coryza (Griffin, 2007). Measles, caused by MeV, is the leading cause of death in children, particularly in developing countries, because of secondary bacterial or parasitic infections attributable to MeV-induced immune suppression (Moss and Griffine, 2006). MeV also causes neurological diseases such as subacute sclerosing panencephalitis (Griffin, 2007). Although many research studies have been conducted, many aspects of the pathogenesis of MeV remain unclear.
Measles virus (genus Morbillivirus, within the family Paramyxoviridae) possesses a single-stranded RNA genome with negative polarity. MeV has six genes on its genome [nucleocapsid (N), phospho (P), matrix (M), fusion (F), hemagglutinin (H), and large (L)] and produces eight proteins (V and C proteins are produced from the P gene). The L gene encodes the L protein, an RNA-dependent RNA polymerase that transcribes the template RNA into new genomic RNA and viral mRNAs, together with the N and P proteins (Griffin, 2007). MeV mRNAs are capped at the 5′ end by L protein (Yoshikawa et al., 1986; Hercyk et al., 1988) and are believed to be translated in a cap-dependent manner.
Recently, we have shown that wild-type MeVs induce the “shutoff” of host cellular protein synthesis in infected cells (Inoue et al., 2009). Furthermore, we clarified that the phosphorylation of eukaryotic initiation factor (eIF) 2α and the binding of MeV-N protein to eIF3-p40, which are cellular initiation factors required for cap-dependent translation, are involved in the induction of this shutoff (Sato et al., 2007; Inoue et al., 2009). Severe suppression of the cap-dependent translation of host proteins was observed in infected cells, however, translated MeV proteins were still clearly evident (Inoue et al., 2009). Thus, it is considered that MeV mRNAs might have other mechanism(s) that facilitate their specific translation under shutoff conditions.
Viruses have acquired various mechanisms for efficient mRNA translation. Specifically, picornaviruses inhibit mRNA translation of host cells at the initiation step, and viral mRNA undergoes selective translation via an internal ribosome entry site (IRES) in its 5′ untranslated region (UTR; Jang, 2006). Several cellular factors are reported to bind to the IRES and play roles in translation independent of the cap structure (Jang, 2006).
On the other hand, the 5′UTR of MeV mRNAs is short (20–60 nt), except for F mRNA (570 nt), and does not contain a functional structure similar to the IRES. However, based on the predicted secondary structure of N-5′UTR, a short hairpin may form at its 5′ end (Figure 1A). This implies that some specific cellular factors may bind to the N-5′UTR via its sequence or secondary structure and induce selective viral mRNA translation. To determine the mechanisms underlying the specific translation of MeV mRNAs, we analyzed the translational efficiency with the N-5′UTR and attempted to identify cellular factors mediating the specific translation of N mRNA.
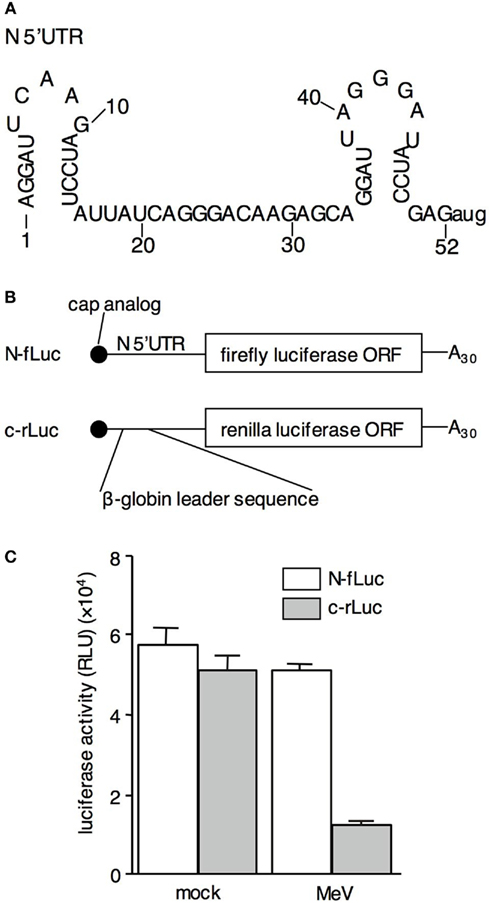
Figure 1. RNA containing the N-5′UTR escapes the shutoff effect induced by MeVHL infection. (A) Sequence and computer-predicted structure of the wild-type N-5′UTR. (B) Structure of N-fLuc reporter RNA and c-rLuc reporter RNA. The 3′UTR and poly A tail of both types of RNAs, and the 5′UTR of c-rLuc RNA were derived from the pTNT vector. (C) Luciferase activity in mock- or MeV-infected B95a cells co-transfected with N-fLuc (white bar) and c-rLuc RNA (gray bar).
Materials and Methods
Construction of Template Plasmid and in vitro Transcription of Reporter RNA
The DNA fragment containing the SP6 promoter followed by the N-5′UTR of the HL strain of MeV was amplified by PCR from the cDNA of the MeV genome, using primers 5′-ATTTAGGTGACACTATAGAGGATTCAAGATCCTATTATCA- GGGACAAGAGC-3′ and 5′-CCATGGCTCGGATATCCCTAATC- CTGCTCTTGTCCCTGAT-3′. The PCR fragment was subcloned into pCR2.1TOPO vector (Invitrogen, Carlsbad, CA, USA). After confirming the sequence, the SP6-N-5′UTR fragment was ligated to pGL3basic (Promega, Madison, WI, USA) at the KpnI and NcoI sites. The “CC” residues in front of the AUG codon of the luciferase open reading frame (ORF) at the NcoI site was deleted using a QuikChange mutagenesis kit (Stratagene, La Jolla, CA, USA; pGL3 + NΔCC). The N-fLucΔCC fragment was digested with XhoI and XbaI and inserted to the pTNT vector (Promega) at the same site (pTNT-N-fLuc). N-fLuc with the polyA tail fragment was inserted to pCR2.1TOPO at the XhoI and BamHI sites (N-fLuc template plasmid; pCR2.1-N-fLuc). Deletion of the N-5′UTR was performed with pGL3 + NΔCC using the QuikChange mutagenesis kit. After confirming the sequence, template plasmids were prepared in a similar way to pCR2.1-N-fLuc. The ORF of renilla luciferase, amplified from pRL-CMV (Promega) by PCR using primers containing the SalI site at the 5′ end and the EcoRI site at the 3′ end was inserted into pTNT at the SalI and EcoRI sites (pTNT-rLuc) as control RNA. The capped RNAs were transcribed from a linearized template with SP6 RNA polymerase using a RiboMAX Large Scale RNA production kit-SP6 (Promega) and cap analog (Promega). The capped RNA was purified by the method recommended by the manufacturer.
RNA Transfection
RNA was transfected into B95a cells (Kobune et al., 1990) derived from marmoset B cells which are highly susceptible to MeV infection, and grown in RPMI 1640 medium supplemented with 5% fetal calf serum (FCS) using Lipofectamine2000 (Invitrogen). After 4 h of incubation, the HL strain of MeV was infected into the B95a cells at a multiplicity of infection (moi) of 1. At 24 h post infection (hpi), the cells were harvested and luciferase activity was measured using a Dual Luciferase Assay kit (Promega).
Electrophoresis Mobility Shift Assay and Ultraviolet Cross-Link Assay
The fragment containing the SP6 promoter followed by the N-5′UTR containing the KpnI site at the 3′ end was amplified by PCR from the pGL3-NΔCC plasmid and subcloned into pCR2.1TOPO (pCR2.1-N). After confirming the sequence, pCR2.1-N was linearized using KpnI and blunted to delete a 3′ overhang at the 3′ end of 5′UTR using a blunting kit (TAKARA BIO INC., Shiga, Japan). The template of each deleted N-5′UTR probe was made from pCR2.1-N using a QuikChange mutagenesis kit. 32P-labeled RNA probes were transcribed from the templates with a riboprobe kit-SP6 (Promega) and [α-32P] UTP and purified by electrophoresis in an 8% polyacrylamide gel denatured by 8 M urea. The unlabeled probe was transcribed for competitor RNA. B95a extract was prepared from B95a cells that were frozen and thawed three times in buffer A [20 mM Tris-HCL (pH 7.5), 150 mM KCl, 1 mM DTT, and 1 mM EDTA]. His6-tagged recombinant La protein (rLa) was synthesized using the RTS 500 Wheat Germ CECF kit and pIVEX1.4WG (Roche Applied Science, Basel, Switzerland) in which the ORF of human La was inserted at the NcoI and SmaI sites. rLa was purified using a Protino Ni 1000 prepacked column kit (MACHEREY-NAGEL, Düren, Germany). EMSA and a UV cross-link assay were performed as previously described by Park and Katze (1995). After electrophoresis, the gels were dried, exposed to imaging plates (FUJI FILM, Tokyo, Japan) and visualized with FLA5100 (FUJI FILM). Competitor RNA was added to the B95a extract and preincubated at 30°C for 10 min before adding the probe. The antibody against La (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added to the B95a extract and preincubated on ice for 30 min prior to adding the probes.
In vitro Translation Assay
Cell-free translation S10 extracts from the suspension of COBL-a cells (derived from human umbilical cord cells and highly susceptible to MeV infection; Kobune et al., 2007) were prepared, and an in vitro translation assay was performed according to the method of Cuconati et al. (1998). After translation, luciferase activity was measured with PicaGene (Toyo Inki, Tokyo, Japan).
RNA Interference
The La-small interfering RNA (siRNA) target sequence (5′-GUUGAACCGUCUAACAACAGA-3′) was designed using siDirect software and purchased from RNAi Co., Ltd (Tokyo, Japan). Silencer Negative Control #1 siRNA was purchased from Ambion (Austin, TX, USA). Transfection experiments were performed using X-tremeGENE siRNA Transfection Reagent (Roche Applied Science) according to the manufacturer’s recommendation. After 48-h incubation, the cells were harvested and checked for the efficiency of RNAi by Western blotting with antibodies against La and β-actin (Santa Cruz Biotechnology). Reporter RNA was transfected into HepG2 cells after the 48-h incubation following siRNA transfection.
Reverse Genetics
To create recombinant MeV lacking the La-binding motif in the N-5′UTR, a previously established plasmid encoding the cDNA of the full-length genome of the HL strain of MeV was used (Terao-Muto et al., 2008). Deletion of six nucleotides was introduced into the N-5′UTR of the plasmid (genome position 66–71), and the resulting plasmid was designated pMeV-6del. The recombinant MeVHL-6del plasmid was generated from pMeV-6del. Briefly, 293 cells were placed in a six-well culture dish, inoculated with a recombinant vaccinia virus (MVA-T7) for 1 h, and then transfected with 1 μg of pMeV-6del, 1 μg of pKS-N1, 1 μg of pKS-P, and 0.3 μg of pGEM-L per well, which expressed N, P, and L proteins, respectively, under the control of a T7 promoter, using FuGENE6 (Roche Applied Science). After incubation for 3 days, the cells were co-cultivated with B95a cells at a concentration of 2 × 106 cells per well, and further incubated until extensive cytopathic effects were noted. The cells were collected and lysed by three cycles of freeze thawing. After sonication, samples were centrifuged at 1500 × g for 10 min to remove the cell debris and stored as a crude virus stock at −80°C.
Virus Growth
B95a cells (1 × 106 cells) were infected with wild-type MeVHL or MeVHL-6del at an moi of 0.001 for 1 h. The inoculum was removed and the cells were washed once with medium and then incubated in medium containing 2.5% FCS. Cells and supernatants were harvested at the indicated times, and three cycles of freeze thawing were carried out. Infectivity was determined by tissue culture infective dose 50 (TCID50) titration using the standard method. The experiment was repeated twice.
Results
Analysis of N-5′UTR Function in Translation
Measles virus mRNAs possess unknown mechanism(s) underlying their specific translation under shutoff conditions in MeV-infected cells. To determine whether the 5′UTR of N mRNA functions as the cis-element for translation in the MeV-infected cells, we constructed a reporter RNA (N-fLuc) that has the N-5′UTR in front of the ORF of the firefly luciferase gene (Figure 1B). As a control for cellular RNA translation, renilla luciferase RNA containing a leader sequence of β-globin mRNA in its 5′UTR was also constructed (c-rLuc). MeV was infected after the reporter RNAs were co-transfected into B95a cells. At 24 h postinfection, the cells were harvested, and luciferase activity was measured. The renilla luciferase activity derived from c-rLuc decreased (Figure 1C), confirming our previous results that MeV infection inhibited cap-dependent cellular mRNA translation (Inoue et al., 2009). In contrast, the firefly luciferase activity derived from N-fLuc was unaltered by MeV infection. This result suggests that N-5′UTR plays an important role in the specific translation of MeV-N mRNA in the shutoff conditions induced by MeV infection.
Identification of Cellular Proteins that Bind to N-5′UTR
To determine the presence of cellular proteins that specifically bind to the N-5′UTR, EMSA was performed using an N-5′UTR probe and B95a cell extracts (Figure 2A). The shift observed in the electrophoretic mobility of the N-5′UTR probe indicated the presence of a protein in the extract that interacted with the N-5′UTR probe (lane 1). Non-labeled RNA with the same sequence inhibited protein–RNA complex formation in a concentration-dependent manner (lanes 2–4). Since the protein–RNA interaction was not inhibited when yeast tRNA was used as the competitor RNA (lanes 5 and 6), the formation of this complex could be specific for N-5′UTR. A UV cross-link assay was performed to determine the molecular weight of proteins that bind to N-5′UTR. Several proteins that interacted with the N-5′UTR probe were observed (Figure 2B, lane 1). Among these, three proteins (of 52, 40, and 35 kDa) formed a specific complex with the N-5′UTR because their bands disappeared or the intensity of their bands decreased when non-labeled RNA was included in the reaction mixture (lane 2). The 52-kDa protein had the same molecular weight as that of the human La/SSB autoantigen (La). It has been reported that La binds to IRES in several viral-encoded RNAs, enhancing their translation (Jang, 2006). Although IRES is not present in MeV-N-5′UTR, we analyzed whether or not La bound to N-5′UTR. When an antibody against La was added to the EMSA reaction buffer prior to adding the probe, we observed that the specific complex (Figure 2C, lane 1) disappeared (lane 2). This was not observed with the antibody against the polypyrimidine tract binding protein (PTB; lane 3). To examine whether La specifically binds to N-5′UTR, rLa was constructed and subjected to EMSA along with N-5′UTR. When rLa was added, an rLa-probe complex was clearly observed (Figure 2D, lanes 2 and 3). Thus, several proteins in B95a cells specifically bind to N-5′UTR, one of which is La.
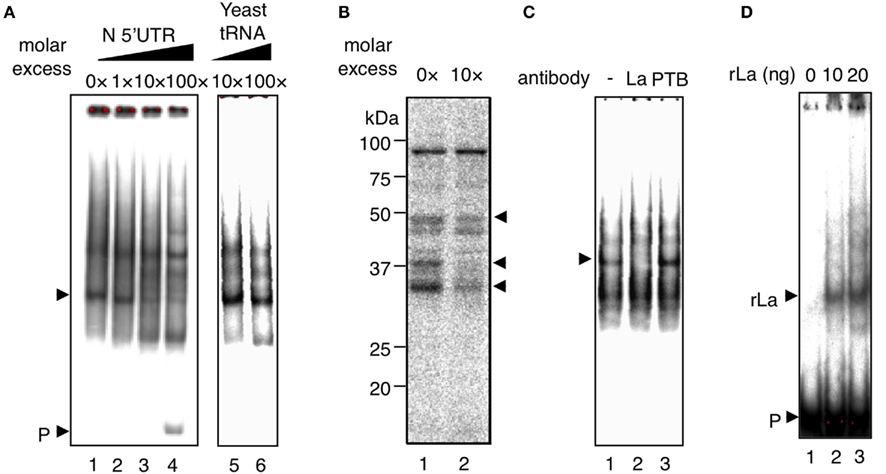
Figure 2. La specifically interacts with N-5′UTR. (A) EMSA was performed with the N-5′UTR probe (P) and B95a cell extract. A specific competitor was added at the indicated molar excess ratio (lanes 2–4). Yeast tRNA was added at the indicated molar excess ratio as a non-specific competitor (lanes 5 and 6). The specific complex is indicated by an arrow. (B) The UV cross-link assay revealed that the molecular weight of specific-binding protein (which disappeared with the addition of a specific competitor RNA, lane 2) was approximately 35, 40, or 50 kDa (indicated by arrows). (C) The antibody against La inhibited the interaction of N-5′UTR and specific-binding protein (arrow; lane 2). As a control experiment, antibody against PTB was added (lane 3). (D) Recombinant La binds to the N-5′UTR probe. The probe (P) without rLa is shown in lane 1. rLa at the indicated dose was added to the reaction (lanes 2 and 3). The arrow indicates the complex derived from the rLa-probe.
La-Binding Region of N-5′UTR
To determine the sequence in N-5′UTR that interacts with La, deleted N-5′UTR probes (probes d1–d5) were constructed (Figure 3A) and subjected to EMSA with rLa (Figure 3B). Each probe formed a complex with rLa, except the d2 probe that barely interacted with rLa (lane 3). Thus, nt 11–20 of N-5′UTR are required for the interaction with rLa. To evaluate the function of the deleted region in the N-5′UTR in translation, deletions similar to those in Figure 3A were introduced into N-fLuc (d1–d5-fLuc), and their luciferase activities were measured at 4 hpi in MeV-infected B95a cells co-transfected with mutant RNAs and c-rLuc. The luciferase activity from the translated d2-fLuc in the B95a cells was lower than that from other RNAs (Figure 3C). Because the observed decrease was by 30%, other region(s) than nt 11–20 may be involved in the interaction with La and the mRNA translation. However, this result clearly indicated that nt 11–20 of N-5′UTR was required for the interaction with La and the optimum translation of N-fLuc RNA.
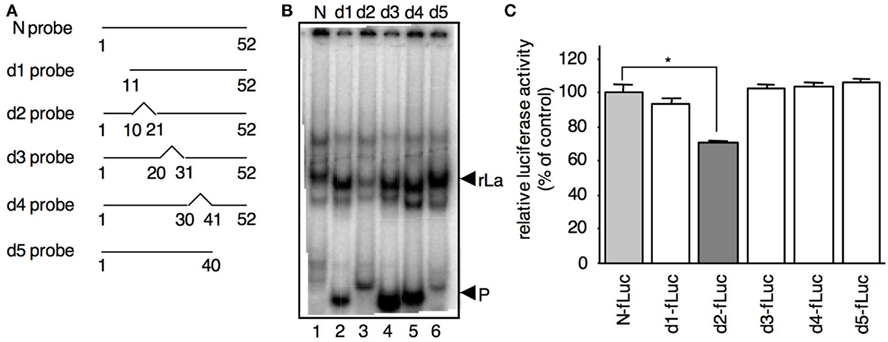
Figure 3. The interaction of La with N-5′UTR requires a specific nt sequence in N (nt 11–20). (A) Outline of the deleted N-5′UTR probes used in the EMSA. The broken lines indicate the deleted regions. (B) The binding activity of rLa to the d2 probe was lower than that of other probes. The interactions of rLa with the probes (La) and the free probe (P) are indicated by arrows. (C) Luciferase activity of B95a cells transfected with the reporter RNA containing various N-5′UTRs. The deletions indicated in (A) were introduced into N-fLuc (d1–d5-fLuc). c-rLuc was co-transfected as an internal control. The N-fLuc value was arbitrarily set at 100%. Statistical analysis was performed using the Student’s t-test; *P < 0.01.
La Enhances in vitro N-fLuc Reporter RNA Translation
To determine the role of La in translation via N-5′UTR, an in vitro translation assay was performed with a COBL-a S10 cell extract. N-fLuc or d2-fLuc RNA was translated in the COBL-a S10 extract containing rLa protein (Figure 4A). The luciferase activity translated from N-fLuc increased proportionally with the dose of rLa protein. The proportional increase was not observed for luciferase activity translated from d2-fLuc, indicating that La enhances N-fLuc translation via the N-5′UTR. The weak effect of the additional rLa on translation was probably due to the high amount of endogenous La in all extracts. La is a major mammalian cell component and its copy number reaches 2 × 107 copies/cell (Gottlieb and Steitz, 1989). In the case of the COBL-a S10 extract, a large amount of La was extracted (data not shown). To confirm the positive effect of La on translation, RNAi was performed to knock down La protein expression in HepG2 cells. Compared with the negative control siRNA-transfected cells, the La protein expression level decreased to 50% in La siRNA-transfected cells (Figure 4B). Under these conditions, N-fLuc or d2-fLuc RNA was transfected. Firefly luciferase activity decreased to 60% of the control cells following the knock down of La in the N-fLuc-transfected cells (Figure 4C). In contrast, the effect of La RNAi on the luciferase activity in d2-fLuc-transfected cells was less than that of N-fLuc (85% compared with the control cells). These results imply that La enhances N-fLuc translation by binding to the N-5′UTR in vivo.
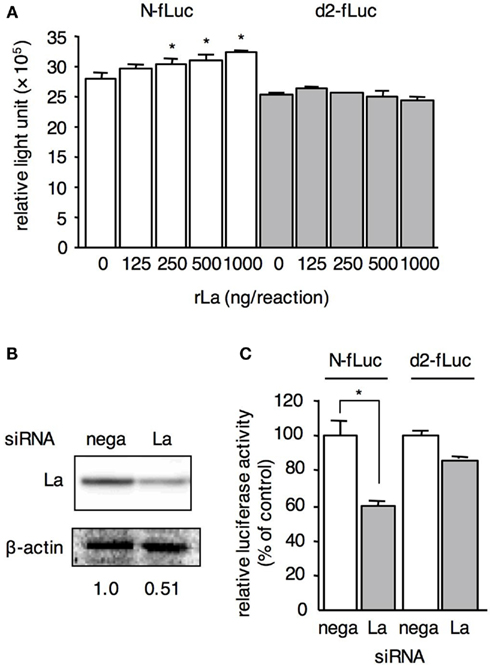
Figure 4. La enhances N-fLuc translation. (A) N-fLuc and d2-fLuc were translated in COBL-a S10 cell extracts with rLa at the indicated quantity. Statistical analysis was performed using the Student’s t-test; *P < 0.05. (B) Expression level of La in HepG2 cells transfected with La siRNA (La) or negative control siRNA (nega). After 48-h incubation, the expression levels of La and actin on the same membrane were detected by western blotting. The relative value of the La expression level was measured and is provided under the panels. (C) The translation of N-fLuc in La siRNA-transfected HepG2 cells decreased more than that in d2-fLuc. The luciferase activity in HepG2 cells transfected with negative control siRNA was arbitrarily set at 100%.
Interaction of La with N-5′UTR Affects MV Growth Efficiency at an Early Phase of Infection
We further investigated the effect of La on viral RNA in the replicating state using a recombinant MeV generated by reverse genetics (Terao-Muto et al., 2008). Because the genome length of Morbilliviruses, including MeV, is an integral of six (Kolakofsky et al., 1998), we synthesized a new deletion probe (6del-probe) by deleting nt 11–16 instead of nt 11–20 and tested the La interaction. La did not bind to the 6del-probe in EMSA, and a reporter RNA with the same deletion in the N-5′UTR showed low translation efficiency comparable to d2-fLuc (data not shown). Therefore, we performed reverse genetics using a plasmid encoding MeVHL full-length genome cDNA, which lacks 6 nt of the La-binding motif corresponding to the genome position 66–71, and generated the recombinant MeV (MeVHL-6del). Wild-type strain and MeVHL-6del were infected into B95a cells at an moi of 0.001 and harvested at each time point indicated. In cells infected with the 6del strain, the expression level of N protein lagged nearly 12 h behind that of wild-type infected cells (Figure 5A). Growth kinetics of the recombinant MeV indicated that production of infectious MeV particles in cells infected with MeVHL-6del was also delayed at an early phase of infection compared with the wild-type, and then recovered rapidly and reached similar levels to wild-type (Figure 5B). In MeV-infected cells, MeV-N is the first, most abundantly, and excessively synthesized among all viral proteins, and is necessary for viral genome replication and mRNA transcription, and therefore for virus growth. MeV-N synthesis was delayed in MeVHL-6del-infected cells approximately 12 h, and reached to the same amount as wild-type at 36 hpi (Figure 5A). Therefore, the virus growth kinetics was also delayed approximately 12 h, and increased rapidly between 24 and 36 hpi and reached the maximum titer at 48 hpi. Thus, the impaired La interaction with the N-5′UTR suppressed the translation efficiency of N mRNA and consequently virus replication at an early stage of infection. This result suggested that La contributes to synthesis of MeV proteins after infection.
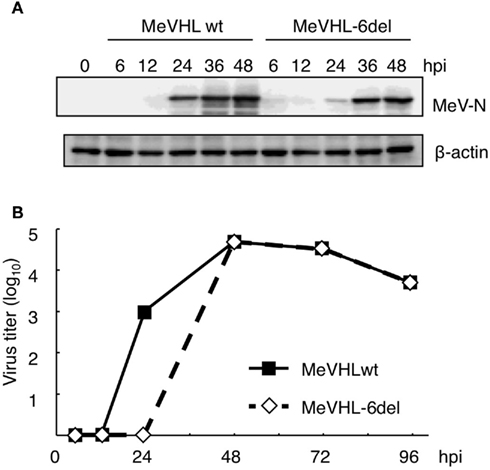
Figure 5. La interaction with N-5′UTR affects the replication of MeV at an early phase of infection. (A) The expression levels of N protein in B95a cells infected with wild-type MeV or MeVHL-6del were determined by western blotting. As an internal control, β-actin protein was measured. (B) The growth kinetics of MeVHL-6del. Wild-type MeV (straight line) and MeVHL-6del (dotted line) were inoculated to B95a cells at an moi of 0.001, and harvested at the indicated time point. The average of two independent measurements is shown.
Discussion
We demonstrated that the 5′UTR of MeV-N mRNA possesses a function for selective translation under the shutoff conditions induced by MeV infection (Figure 1). Moreover, La binds to the N-5′UTR and plays an important role in the induction of specific translation of N-5′UTR-driven mRNA in vitro and in vivo (Figures 2–4).
La was first described as an autoantigen in patients with the rheumatic diseases systemic lupus erythematosus and Sjogren’s syndrome (Mattioli and Reichlin, 1974; Alspaugh et al., 1976). To date, it has been revealed that La possesses at least two major functions. First, La associates with various nascent RNAs transcribed by RNA polymerase III, such as pre-tRNAs and pre-5S rRNA, and plays a role in the regulation of RNA processing and nuclear export and retention (reviewed in Ref. Wolin and Cedervall, 2002). In this process, the La-binding motif (UUU-OH 3′) and the underlying molecular mechanisms have been well characterized. Many studies have indicated that the other major function of La protein is in the translation of specific viral and cellular mRNAs. For example, La binds to IRES in the 5′UTR of viral RNAs such as poliovirus (Meerovitch et al., 1993) and hepatitis C virus (HCV) RNA (Jang, 2006), as well as some eukaryotic mRNAs, namely, human immunoglobulin heavy chain binding protein mRNA (Kim et al., 2001). La also interacts with the 5′ end of HIV mRNAs which contain no IRES but have a trans-activation response (TAR) element (Chang et al., 1994; Svitkin et al., 1994). These interactions are proposed to enhance RNA translation (Ali and Siddiqui, 1995; Pudi et al., 2004). In addition, La has a negative effect on the translation of mRNAs of ribosomal proteins and translation elongation factors which contain the 5′ terminal oligopyrimidine tract sequence (Zhu et al., 2001). These data suggest that La has various functions in translation which maintain the optimal activity of interacting RNA. In this study, we showed that La has a positive effect on the translation of MeV-N-5′UTR-driven mRNA, although it contains neither an IRES nor a TAR element. The precise mechanism of upregulation of mRNA translation by La has not yet been elucidated, however, one possible role has been discussed on the basis of the reported function of La. La protein has been reported to function as a helicase that unwinds double-stranded RNA (Xiao et al., 1994; Hühu et al., 1997). The TAR element of HIV RNA is a stem-loop structure and suppresses the translation of the following RNA (Parkin et al., 1988), and La-binding enhances the translation of TAR-driven RNA (Chang et al., 1994; Svitkin et al., 1994). These reports imply that La functions as a helicase on the 5′UTR of mRNA and serves as an RNA chaperone. The MeV-N-5′UTR has a small stem-loop structure at its 5′ end consisting of nt 1–15 (Figure 1A), and the region responsible for La-binding is found in the stem region (Figure 3). In Figure 3, d2 probe showed low interaction with La, while d1 probe, which also lacks a part of the stem-loop structure at 5′ end (Figure 1A), bound to La comparable to N probe. Interestingly, d1 probe is estimated to form an alternative short hairpin structure at the 5′ end, which consists of nt 12–26, from a secondary structure prediction (data not shown). It suggests that La bound to d1 probe via the alternative stem structure at 5′ end. Thus, unwinding of the stem structure by La may result in the efficient translation of MeV-N mRNA. Alternatively, previous reports indicated that the binding of La to the stem-loop IV region of HCV IRES facilitates the formation of the 48S ribosomal complex on IRES (Costa-Mattioli et al., 2004; Pudi et al., 2004). Therefore, the binding of La to the N-5′UTR might facilitate formation of the ribosomal complex at the 5′ end. Interestingly, secondary structure predictions have indicated that not only N but also other MeV mRNAs, except for F, possess similar small stem-loop structures on their 5′UTR, and our preliminary experiments suggest that each 5′UTR can bind to La in vitro (data not shown). These findings indicate that efficient translation of other MeV mRNAs under shutoff conditions is also upregulated by similar mechanisms.
Recently, an additional function of La has been proposed in negative strand viruses, in which La binds to the leader RNA of viruses such as the human parainfluenza virus, vesicular stomatitis virus, rinderpest virus, and respiratory syncytial virus (Wilstz et al., 1983; De et al., 1996; Raha et al., 2004; Bitko et al., 2008). In particular, the leader RNA of rinderpest virus, which is closely related to MeV, specifically binds to La and this interaction enhances the replication/transcription of viral RNA (Raha et al., 2004). In addition, a more recent report indicated that La binds to the leader RNA of respiratory syncytial virus and shields it from RIG-I followed by abrogation of the early viral activation of type I interferon (Bitko et al., 2008). These results imply that La contributes to virus replication in multiple systems.
A previous study suggested that sufficient intracellular accumulation of N protein is required for viral genome replication (Lamb and Kolakofsky, 2001). Moreover, a recent report showed that a siRNA that specifically targets MeV-N represses the synthesis of viral mRNAs and genomic RNA (Reuter et al., 2006). These findings indicate that the efficient expression of N protein is important for virus replication. N protein binds to the viral genome and forms a ribonucleoprotein complex with P and L proteins, which acts as a template for viral genome replication and transcription. Therefore, interference of N protein expression might result in the suppression of MeV transcription and replication. In this study, we demonstrated that the lack of La-binding to N-5′UTR, which elicits lower expression of N protein, caused a delay in N protein synthesis and virus replication at an early phase of infection (Figure 5). This suggests that the binding of La to N-5′UTR contributes to the rapid expression of N protein which in turn facilitates efficient virus growth.
In conclusion, our study revealed that a host protein, La autoantigen, enhances the translation of RNA containing N-5′UTR, and that La interaction can facilitate escape from the shutoff effect induced by MeV infection. This is the first report of a cellular factor that specifically regulates morbilliviral mRNA translation. Further studies are required to clarify the overall mechanism underlying the upregulation of mRNA translation and its implication in the pathogenesis of MeV.
Acknowledgments
We thank Dr. K. Ito (Institute of Medical Science, The University of Tokyo) for providing valuable insights and for critical reading of this manuscript. This work was supported by grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology, and the Bio-oriented Technology Research Advance Institution.
References
Ali, N., and Siddiqui, A. (1995). The La antigen binds 5′ noncoding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc. Natl. Acad. Sci. U.S.A. 94, 2249–2254.
Alspaugh, M. A., Talal, N., and Tan, E. M. (1976). Differentiation and characterization of autoantibodies and their antigens in Sjogren’s syndrome. Arthritis Rheum. 19, 216–222.
Bitko, V., Musiyenko, A., Bayfield, M. A., Maraia, R. J., and Barik, S. (2008). Cellular La protein shields nonsegmented negative-strand RNA viral leader RNA from RIG-I and enhances virus growth by diverse mechanisms. J. Virol. 82, 7977–7987.
Chang, Y. N., Kenan, D. J., Keene, J. D., Gatignol, A., and Jeang, K. T. (1994). Direct interactions between autoantigen La and human immunodeficiency virus leader RNA. J. Virol. 68, 7008–7020.
Costa-Mattioli, M., Svitkin, Y., and Sonenberg, N. (2004). La autoantigen is necessary for optimal function of the poliovirus and hepatitis C virus internal ribosome entry site in vivo and in vitro. Mol. Cell. Biol. 24, 6861–6870.
Cuconati, A., Molla, A., and Wimmer, E. (1998). Brefeldin A inhibits cell-free, de novo synthesis of poliovirus. J. Virol. 72, 6456–6464.
De, B. P., Gupta, S., Zhao, H., Drazba, J. A., and Banerjee, A. (1996). Specific interaction in vitro and in vivo of glyceraldehyde-3-phosphate dehydrogenase and LA protein with cis-acting RNAs of human parainfluenza virus type 3. J. Biol. Chem. 271, 24728–24735.
Gottlieb, E., and Steitz, J. A. (1989). The RNA binding protein La influences both the accuracy and the efficiency of RNA polymerase II transcription in vitro. EMBO J. 8, 841–850.
Griffin, D. E. (2007). “Measles virus,” in Fields Virology, 5th Edn, Vol. 2, eds D. M. Knipe, and P. M. Howley (Philadelphia, PA: Lippincott Williams & Wilkins), 1551–1586.
Hercyk, N., Horikami, S. M., and Moyer, S. A. (1988). The vesicular stomatitis virus L protein possesses the mRNA methyltransferase activities. Virology 163, 222–225.
Hühu, P., Prujin, G. J. M., Van Venrooji, W. J., and Bachmann, M. (1997). Characterization of the autoantigen La (SS-B) as a dsRNA unwinding enzyme. Nucleic Acids Res. 25, 410–416.
Inoue, Y., Tsukiyama-Kohara, K., Yoneda, M., Sato, H., and Kai, C. (2009). Inhibition of host protein synthesis in B95a cells infected with the HL strain of measles virus. Comp. Immunol. Microbiol. Infect. Dis. 32, 29–41.
Jang, S. K. (2006). Internal initiation: IRES elements of picornaviruses and hepatitis c virus. Virus Res. 119, 2–15.
Kim, A. K., Back, S. H., Rho, J., Lee, S. H., and Jang, S. K. (2001). La autoantigen enhances translation of BiP mRNA. Nucleic Acids Res. 29, 5009–5016.
Kobune, F., Ami, Y., Katayama, M., Takahashi, M., Tuul, R., Korukluoglu, G., Kiyohara, T., Miura, R., Sato, H., Yoneda, M., and Kai, C. (2007). A novel monolayer cell line derived from human umbilical cord blood cells shows high sensitivity to measles virus. J. Gen. Virol. 88, 1565–1567.
Kobune, F., Sakata, H., and Sugiura, A. (1990). Marmoset lymphoblastoid cells as a sensitive host for isolation of measles virus. J. Virol. 64, 700–705.
Kolakofsky, D., Pelet, T., Garcin, D., Hausmann, S., Curran, J., and Roux, L. (1998). Paramyxovirus RNA synthesis and the requirement for hexamer genome length: the rule of six revisited. J. Virol. 72, 891–899.
Lamb, R. A., and Kolakofsky, D. (2001). Fields virology, 4th Edn, Vol. 1. Philadelphia, PA: Lippincott Williams & Wilkins, 1305–1340.
Mattioli, M., and Reichlin, M. (1974). Heterogeneity of RNA protein antigen reactive with sera of patients with systemic lupus arythematosus. Description of a cytoplasmic nonribosomal antigen. Arthritis Rheum. 17, 421–429.
Meerovitch, K. S., Svitkin, Y. V., Lee, H. S., Lejbkowicz, F., Kenan, D. J., Chan, E. K., Agol, V. I., Keene, J. D., and Sonenberg, N. (1993). La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J. Virol. 67, 3798–3807.
Moss, W. J., and Griffine, D. E. (2006). Global measles elimination. Nat. Rev. Microbiol. 4, 900–908.
Park, Y. W., and Katze, M. G. (1995). Translational control by influenza virus. Identification of cis-acting sequence and trans-acting factors which may regulate selective viral mRNA translation. J. Biol. Chem. 270, 28433–28439.
Parkin, N. T., Cohen, E. A., Darveau, A., Rosen, C., Haseltine, W., and Sonenberg, N. (1988). Mutational analysis of the 5′non-coding region of human immunodeficiency virus type 1: effects of secondary structure on translation. EMBO J. 7, 2831–2837.
Pudi, R., Srivasan, P., and Das, S. (2004). La protein binding at the GCAC site near the initiator AUG facilitate the ribosomal assembly on the hepatitis c virus RNA to influence internal ribosomal entry site-mediated translation. J. Biol. Chem. 279, 29879–29888.
Raha, T., Pudi, R., Das, S., and Shaila, M. S. (2004). Leader RNA of rinderpest virus binds specifically with cellular La protein: a possible role in virus replication. Virus Res. 104, 101–109.
Reuter, T., Weissbrich, B., Schneider-Schaulies, S., and Schneider-Schaulies, J. (2006). RNA interference with measles virus N, P, and L mRNAs efficiently prevents and with matrix protein mRNA enhances viral transcription. J. Virol. 80, 5951–5957.
Sato, H., Masuda, M., Kanai, M., Tsukiyama-Kohara, K., Yoneda, M., and Kai, C. (2007). Measles virus N protein inhibits host translation by binding to eIF3-p40. J. Virol. 81, 11569–11576.
Svitkin, Y. V., Pause, A., and Sonenberg, N. (1994). Autoantigen alleviates translational repression by the 5′ leader sequence of the human immunodeficiency virus type 1 mRNA. J. Virol. 68, 7001–7007.
Terao-Muto, Y., Yoneda, M., Seki, T., Watanabe, A., Tsukiyama-Kohara, K., Fujita, K., and Kai, C. (2008). Heparin-like glycosaminoglycans prevent the infection of measles virus in SLAM-negative cell lines. Antiviral Res. 80, 370–376.
Wilstz, J., Kurilla, M. G., and Keene, J. D. (1983). A host protein (La) binds to a unique species of minus-sense leader RNA during replication of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A. 80, 5827–5831.
Xiao, Q., Sharp, T. V., Jeffrey, I. W., James, M. C., Prijin, G. J. M., Van Venrooji, W. J., and Clemens, M. J. (1994). The La antigen inhibits the activation of the interferon-inducible protein kinase PKR by sequestering and unwinding double-stranded RNA. Nucleic Acids Res. 22, 2512–2518.
Yoshikawa, Y., Mizumoto, K., and Yamanouchi, K. (1986). Characterization of messenger RNAs of measles virus. J. Gen. Virol. 67, 2807–2812.
Keywords: measles virus, 5′ untranslated region, La
Citation: Inoue Y, Sato H, Fujita K, Tsukiyama-Kohara K, Yoneda M and Kai C (2011) Selective translation of the measles virus nucleocapsid mRNA by La protein. Front. Microbio. 2:173. doi: 10.3389/fmicb.2011.00173
Received: 02 June 2011; Accepted: 02 August 2011;
Published online: 23 August 2011.
Edited by:
Akio Adachi, The University of Tokushima Graduate School, JapanReviewed by:
Kaoru Takeuchi, University of Tsukuba, JapanHak Hotta, Kobe University Graduate School of Medicine, Japan
Copyright: © 2011 Inoue, Sato, Fujita, Tsukiyama-Kohara, Yoneda and Kai. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Chieko Kai, Laboratory Animal Research Center, Institute of Medical Science, The University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8639, Japan. e-mail:Y2thaUBpbXMudS10b2t5by5hYy5qcA==