

 Kristie L. Rose2
Kristie L. Rose2
- 1 Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC, USA
- 2 Mass Spectrometry Research Center, Department of Biochemistry, Vanderbilt University, Nashville, TN, USA
- 3 Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, USA
- 4 Department of Craniofacial Biology, Medical University of South Carolina, Charleston, SC, USA
- 5 Division of Infectious Diseases, Medical University of South Carolina, Charleston, SC, USA
We recently established a key role for host sphingomyelin synthase (SMS) in regulating the killing activity of neutrophils against Cryptococcus neoformans. In this paper, we studied the effect of C. neoformans on the killing activity of neutrophils and whether SMS would still be a player against C. neoformans in immunocompromised mice lacking T and natural killer (NK) cells (Tgε26 mice). To this end, we analyzed whether C. neoformans would have any effect on neutrophil survival and killing in vitro and in vivo. We show that unlike Candida albicans, neither the presence nor the capsule size of C. neoformans cells have any effect on neutrophil viability. Interestingly, melanized C. neoformans cells totally abrogated the killing activity of neutrophils. We monitored how exposure of neutrophils to C. neoformans cells would interfere with any further killing activity of the conditioned medium and found that pre-incubation with live but not “heat-killed” fungal cells significantly inhibits further killing activity of the medium. We then studied whether activation of SMS at the site of C. neoformans infection is dependent on T and NK cells. Using matrix-assisted laser desorption–ionization tissue imaging in infected lung we found that similar to previous observations in the isogenic wild-type CBA/J mice, SM 16:0 levels are significantly elevated at the site of infection in mice lacking T and NK cells, but only at early time points. This study highlights that C. neoformans may negatively regulate the killing activity of neutrophils and that SMS activation in neutrophils appears to be partially independent of T and/or NK cells.
Introduction
The fact that an infection caused by Cryptococcus neoformans still cannot be fully contained by the host is starkly evident. Almost all reviews on host defense against the ubiquitous environmental fungus emphasize the role of cell-mediated immunity (CMI), which is critical for containment of fungal cells through the activation of macrophages and neutrophils, resulting in granuloma formation (Huffnagle et al., 1991a,b; Goldman et al., 1994). While there are several studies elucidating the role and mechanisms by which alveolar macrophages control a C. neoformans infection (Feldmesser et al., 2001; Luberto et al., 2003; Fan et al., 2005; Shao et al., 2005; Alvarez and Casadevall, 2006; Ma et al., 2006; Rittershaus et al., 2006; Garcia et al., 2008; Stano et al., 2009), very little is known on the mechanisms by which neutrophils neutralize C. neoformans. Neutrophils are more effective killers than macrophages against fungi (Diamond et al., 1972; Miller and Mitchell, 1991) and they possess the innate ability to kill microbes without a CMI-mediated activation. We have recently shown that host sphingomyelin synthase (SMS) plays a pivotal role in the regulation of the killing activity of neutrophils against C. neoformans through a diacylglycerol-protein kinase D (DAG-PKD) dependent mechanism (Qureshi et al., 2010). In order to further understand the cryptococcal–neutrophil interaction, we have undertaken additional studies utilizing the HL-60 cell line model system and a mouse model lacking T and natural killer (NK) cells (Tgε26) to gain a better understanding of this important relationship in protecting the host.
Materials and Methods
Materials, Strains, and Growing Media
Cryptococcus neoformans variety grubii serotype A strain H99 (WT), Candida albicans strain A39, and Saccharomyces cerevisiae JK9-3dα (WT) were routinely grown in yeast extract/peptone/2% dextrose-rich (YPD-rich) medium. C. neoformans Δgsc1 and Δisc1 mutant strains were created previously in our laboratory (Heung et al., 2005; Rittershaus et al., 2006; Shea et al., 2006). C. neoformans GAL7::IPC1 strain was created from M001, an ade2 isogenic strain derivative of H99, as described previously (Luberto et al., 2001). Melanized strain H99 was grown on BD BBL™ Stacker™ bird seed agar plates (cat. 297875). HL-60 cells (ATCC® CCL-240™) were cultured at 37°C, 5% CO2 in RPMI 1640, supplemented with L-glutamine, 20% heat-inactivated FBS, and 1% penicillin and streptomycin. RPMI 1640 medium, FBS, and penicillin–streptomycin were from Gibco/Invitrogen; pooled human serum, retinoic acid, and DMSO were from Sigma.
HL-60 Viability Assay
To quantify the effect of C. neoformans, C. albicans, and S. cerevisiae on neutrophils, HL-60 cells were differentiated by incubation in the presence of 1.3% (v/v) DMSO and 2.5 μM retinoic acid for 72 h in growth medium. Cells were then washed with RPMI serum-free medium and 8 × 104 granulocytes/ml re-suspended in fresh RPMI containing 10% pooled human serum. These were co-cultured with 4 × 103 C. neoformans, C. albicans, or S. cerevisiae (20:1 ratio HL-60:yeast)/ml for 24 h at 37°C. At time points specified in the Section “Results,” HL-60 cells were counted using trypan blue to determine their viability.
HL-60 Medium Killing Assay
To quantify the effect of HL-60 medium on C. neoformans, the killing assay described by Spellberg et al. (2007) was used. HL-60 cells were differentiated by incubation in the presence of 1.3% (v/v) DMSO and 2.5 μM retinoic acid for 72 h in growth medium. Cells were then washed with RPMI serum-free and 8 × 104 neutrophils/ml re-suspended in fresh RPMI containing 10% pooled human serum for 1, 2, 4, 6, 8, and 16 h. Conditioned media (900 μl) were then collected by centrifugation and 4 × 103 cells/ml C. neoformans (20:1 ratio HL-60:C. neoformans) incubated for 4 h at 37°C. At the end of the incubation, the medium was serially diluted and streaked onto YPD agar, and the plates incubated for 48 h at 30°C. CFUs were counted to assess killing of C. neoformans compared with control cultures of C. neoformans alone with no HL-60 medium. In order to check the effect of pre-incubating HL-60 cells with “heat-killed” C. neoformans, 4 × 103 C. neoformans cells/ml were boiled at 100°C for 10 min prior to addition to HL-60.
Effect of Pre-Incubation with C. neoformans on HL-60D Medium Killing Activity
In order to examine the effect of pre-incubating HL-60 cells with C. neoformans prior to removing the conditioned HL-60 medium for further experimentation, 8 × 104 neutrophils/ml were incubated with 4 × 103 C. neoformans H99 cells/ml for 1, 2, 4, and 6 h. At the specified time points, a portion of the cells were serially diluted and streaked onto YPD agar, and incubated for 48 h at 30°C. CFUs were counted to assess killing of C. neoformans at the various time points. At the same time, the remainder of the cells was centrifuged and the conditioned medium (800 μl) transferred to fresh wells. About 4 × 103 fresh C. neoformans cells/ml were then added and allowed to incubate for a further 4 h at 37°C. At the end of the incubation, the medium was serially diluted and streaked onto YPD agar, and incubated for 48 h at 30°C. CFUs were counted to assess killing of C. neoformans compared with control cultures of C. neoformans alone with no HL-60 medium.
Effect of Capsule on Killing of C. neoformans by Differentiated HL-60 (HL-60D)
Cryptococcus neoformans WT H99 and Δcap59 were used for capsule growth experiments. The yeast cells were grown overnight in 10 ml YPD at 30°C with shaking, collected in the logarithmic phase of growth by centrifugation (10 min at 3000 rpm at room temperature), washed twice with phosphate-buffered saline (PBS), and then incubated at a cell density of 5 × 106 cells/ml in the capsule growth inducing medium (DMEM buffered with 25 mM HEPES pH 7.2) overnight at 37°C, 5% CO2. HL-60D cells were washed with RPMI and 8 × 104 cells/ml re-suspended in fresh RPMI supplemented with 10% pooled human serum and incubated for 6 h at 37°C, 5% CO2. Conditioned media (900 μl) were then collected by centrifugation and 4 × 103 cells/ml C. neoformans H99 or Δcap59 added and allowed to incubate for 4 h at 37°C. At the end of the incubation, the medium was serially diluted and streaked onto YPD agar, and incubated for 48 h at 30°C. CFUs were counted to assess killing of C. neoformans compared with control cultures of C. neoformans alone with no HL-60 medium.
Effect of Melanin on Killing of C. neoformans by HL-60D Medium
Following differentiation, HL-60D cells were washed with RPMI and 8 × 104 cells/ml re-suspended in fresh RPMI supplemented with 10% pooled human serum and incubated for 6 h at 37°C, 5% CO2. Conditioned media (900 μl) were then collected by centrifugation and 4 × 103 melanized or non-melanized C. neoformans cells/ml added and allowed to incubate for 4 h at 37°C. At the end of the incubation, the medium was serially diluted and streaked onto YPD agar, and incubated for 48 h at 30°C. CFUs were counted to assess killing of melanized vs non-melanized C. neoformans.
Neutrophil Killing Assay
Approximately 1.6 × 105 freshly isolated murine neutrophils/ml were incubated in 124 ml sterile filtered PBS supplemented with 10% pooled mouse serum for 4 h in the presence of 9.92 × 104 C. neoformans cells/ml (10 μl used; 20:1 ratio murine neutrophils:C. neoformans). At the end of the incubation, the cultures were serially diluted and streaked onto YPD agar, and incubated for 48 h at 30°C. CFUs were counted to assess killing of C. neoformans compared with control cultures of C. neoformans alone with no neutrophils. Murine neutrophils were obtained from Tgε26 mice using the method of Shimizu et al. (2001) with the following modifications. Following harvest of the cells and a single wash with PBS, the collected cells were re-suspended in 4 ml PBS and neutrophils separated from mononuclear cells using 3 ml Ficoll-Paque™ PREMIUM (GE Healthcare, cat. 17-5442-02). The mixture was centrifuged at 400×g for 40 min at 20°C. The resulting pellet was collected and red blood cells were lysed with 5 ml ice-cold water in ice for 1 min. Cold PBS (10 ml) was then added and the mixture centrifuged at 400×g for 10 min at 4°C. The resulting pellet was then re-suspended in 500 μl PBS. Mouse sera were obtained from Tgε26 mice.
Animal Studies
Four- to six-week-old Tgε26 mice from the Animal Core Facility, Medical University of South Carolina, Charleston, were used for this study. Mice were anesthetized by intraperitoneal injection of 60 μl of a xylazine–ketamine mixture containing 5 mg xylazine and 95 mg ketamine per kg of body weight. The wild-type strain (H99) of C. neoformans was grown in YPD medium for 24 h at 30°C. The fungal cells were harvested, washed three times in PBS, and re-suspended in PBS at a concentration of 2.5 × 107 cells/ml. Mice were infected intranasally with 20 μl containing 5 × 105 cells. Mice were fed ad libitum and monitored by inspection twice a day. Mice that appeared moribund or in pain were sacrificed using CO2 inhalation followed by cervical dislocation. All animal procedures were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee and followed the guidelines of the American Veterinary Medical Association.
Tissue Sectioning and Sample Preparation
At 6, 12, and 15 days post-infection, mice were euthanized and lungs were harvested and flash-frozen in dry-ice/ethanol, then stored at −80°C until ready for use. The organ was attached to the cryostat sample stage using a small bead of optimal cutting temperature compound (OCT) at the base of the tissue only. The tissue was sectioned to a thickness of 30 μm at a temperature of −26°C using a cryotome (Microm HM 550, Walldorf, Germany). The sections were thaw-mounted onto conductive indium tin oxide (ITO) coated conductive glass slides for mass spectral analysis (Bruker Daltonics, Billerica, MA, USA). The resistance of the ITO coated microscope slide was 40 Ω over a distance of 1 cm. The tissue was allowed to warm on the microscope slides for 10 s before refreezing and storage at −80°C. For mass spectrometric analysis, the tissue sections were removed from the freezer and placed in a dessicator for 30 min prior to matrix deposition. A freshly prepared solution of 2,5-dihydroxybenzoic acid (DHB; Sigma; 40 mg/ml in 70% ethanol), was applied to the tissue by repeated cycles using a thin layer chromatography (TLC) sprayer. Each spray cycle was followed by 45–60 s of drying time, and the cycle repeated until an even coverage of matrix across the entire tissue was achieved.
MALDI mass spectrometry SM imaging
Matrix-assisted laser desorption–ionization (MALDI) mass spectral analysis was carried out using a reflector time-of-flight mass spectrometer (Bruker Autoflex III TOF-TOF, Bruker Daltonik, Bremen, Germany) operating in positive ion mode with a +20-kV accelerating potential. The laser beam size was set to medium, and operated at 200 Hz. Using Bruker Peptide Standard 1 (Bruker Daltonik, Bremen, Germany), a linear, external calibration was applied to the instrument before data collection. Mass spectral data sets were acquired over each whole mouse lung using flexImaging™ software (Bruker Daltonik, Bremen, Germany) in the mass range of m/z 500–1200 with a raster step size of 100 μm and 250 laser shots per spectrum. After data acquisition, molecular images were reconstituted using flexImaging™ software. Each data set consists of approximately 4000 individual sampling locations, each representing one pixel in the resultant image. Data was normalized using flexImaging™ software, and each m/z signal plotted ± 0.5 mass units. For display purposes, signals between sampling locations were interpolated and pixel intensities were scaled to utilize the entire dynamic range. Tandem mass spectrometry was used to identify signals detected in the MALDI imaging data sets. Lipids to be identified were extracted from mouse lung tissue by homogenization of the tissue in 70% ethanol. Samples were centrifuged at 45,000 rpm for 30 min at 4°C using a Beckman Optima TL Ultracentrifuge with a TLA45 rotor (Beckman Coulter, Inc., Fullerton, CA, USA), and the supernatant containing extracted lipids removed. Samples were concentrated using a speed-vac (Labconco, Kansas City, MO, USA), and spotted on a MALDI plate using 40 mg/ml DHB in 70% ethanol. Standard solutions of known lipids were also spotted in a similar manner. A timed ion gate was used for precursor ion selection and the fragments generated were further accelerated with 19 kV in the LIFT cell, and detected following passage through the reflectron. No CID gas was used for fragmentation of the precursor ions. Signals in the MALDI tissue imaging data set were identified based on matching fragmentation spectra of lipids extracted from the tissue and prepared lipid standard solutions.
SM Determination
For lipid analysis in lung tissues, lungs from two mice from each time point were homogenized in 5 ml homogenization buffer consisting of 0.25 M sucrose, 0.5 mM EDTA, 25 mM KCl, and 50 mM Tris–HCl at pH 7.4. Then, 1 mg of homogenate was used for the mass spectrometry analysis at the Medical University of South Carolina Lipidomics Facility whereas an aliquot was used for the quantitation of glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Qureshi et al., 2010) by Western blot using the LabWorks Image Acquisition and Analysis software from UVP BioImaging Systems, version 4.5. Lipids were extracted and analyzed using established protocols in the facility (Bielawski et al., 2006, 2009).
Statistics
All experiments were performed at least in triplicate. Statistical analyses of the data were performed using Student’s t-test, and P < 0.05 was considered statistically significant.
Results
Cryptococcus neoformans Does Not Affect the Viability of HL-60D Cells
To determine the fate of HL-60 granulocytes exposed to C. neoformans, the viability of the granulocytes was determined at 1, 2, 3, 4, 5, 6, and 24 h following co-incubation with C. neoformans cells at a ratio of 20:1 (HL-60D:C. neoformans). As a comparison, C. albicans, which is known to affect viability (Mullick et al., 2004), was used as a positive control. Figure 1 shows that C. albicans induced a significant level of mortality starting at 2 h in the HL-60 cells whereas C. neoformans did not. In contrast to C. albicans, but similar to C. neoformans, S. cerevisiae does not elicit a mortality response in HL-60.
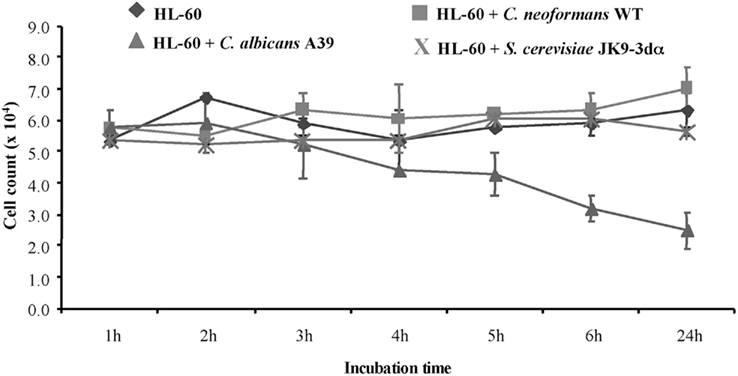
Figure 1. Cryptococcus neoformans does not affect HL-60D viability. To quantify the effect of co-incubation, differentiated HL-60 cells (HL-60D) were incubated either alone or with C. neoformans WT (H99), C. albicans A39, or S. cerevisiae JK9-3dα. C. albicans affects viability of HL-60 cells whereas S. cerevisiae or C. neoformans do not.
Killing Activity of Neutrophils is Solely due to Secreted Factors
In order to determine the optimal time point for incubation of differentiated HL-60 cells in fresh medium prior to exposure to C. neoformans, following differentiation 8 × 104 granulocytes/ml were incubated in fresh RPMI medium containing 10% human serum for 1, 2, 4, 6, 8, and 16 h. The medium was then collected by centrifugation at 3000 rpm for 10 min and 4 × 103 C. neoformans cells/ml added and incubated for 4 h at 37°C. Figure 2 shows that the percentage killing of C. neoformans was greatest at 6 h of incubation of differentiated HL-60 (HL-60D) in fresh medium and that the killing effect diminishes after this time. However the presence of heat-killed C. neoformans has no effect on the down-stream killing activity of the conditioned medium. Together with the results shown in Figure 1 in which viability of HL-60D at 6 h is not reduced, it appears that the killing of C. neoformans is solely due to secreted anti-cryptococcal factors (ACFs) by HL-60D.
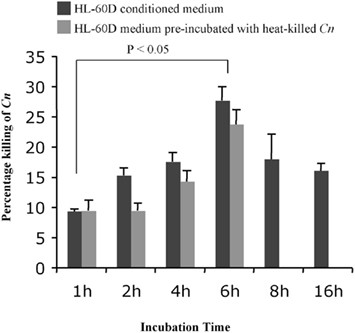
Figure 2. Time course of the killing activity of neutrophils against C. neoformans. Six hours is the peak for the killing of HL-60D in fresh medium (10% HS + RPMI) prior to exposure to live C. neoformans. Incubation with heat-killed C. neoformans (up to 6 h) has no effect on the down-stream killing activity of the conditioned medium.
Pre-Incubation of Differentiated HL-60 (HL-60D) Cells with Live C. neoformans Significantly Inhibits any Further Killing Activity of HL-60D Medium
We next wished to test the effect of pre-incubation with C. neoformans on the killing ability of HL-60D medium (Figure 3). To this aim, following a 72-h incubation with 1.3% (v/v) DMSO and 2.5 μM retinoic acid as described above, 8 × 104 cells were washed, re-suspended in 1 ml fresh RPMI containing 10% pooled human serum and incubated with 4 × 103 C. neoformans cells for 1, 2, 4, and 6 h. At each time point, the killing activity was determined by counting CFUs and comparing to control cultures of C. neoformans alone with no HL-60 cells. At the same time, 800 μl of conditioned medium containing ACFs was collected by centrifugation at each time point and incubated with 4 × 103 fresh C. neoformans cells/ml for a further 4 h at 37°C. It was found that pre-incubation of HL-60D cells with C. neoformans decreases further killing ability of the ACFs in HL-60D medium.
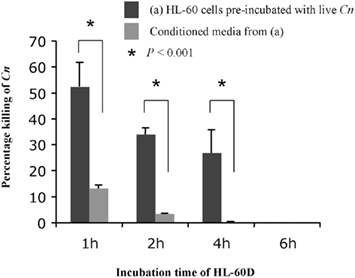
Figure 3. Pre-incubation of HL-60D cells with C. neoformans significantly inhibits further killing activity of HL-60D medium. Pre-incubation with C. neoformans decreases any further killing of anti-cryptococcal factors in HL-60D medium.
Extent of C. neoformans Killing by HL-60D is Not Related to Capsule Size but is Related to Melanin Production
Since C. neoformans factors are able to neutralize the killing activity of the medium we sought to examine the effect of capsule and melanin on the killing activity of the conditioned medium collected from HL-60D. C. neoformans produces a polysaccharide capsule, which consists of glucuronoxylomannan (GXM) and galactoxylomannan (GalXM), as well as mannoprotein (Bose et al., 2003). These components all occupy spatially separate and discrete regions in the capsule of C. neoformans (Jesus et al., 2010). Surprisingly the presence of capsule made no difference to the killing activity of HL-60D medium (Figure 4A), in line with previous findings suggesting that the extent to which different C. neoformans strains (capsular and acapsular) were killed by neutrophils was not consistently related to the size of the capsule or the entire cell (Miller and Mitchell, 1991). In order to determine the effect of HL-60D medium on the killing of melanized vs non-melanized C. neoformans, HL-60 granulocytes were incubated with melanized H99 for 4 h. Melanin has been shown to interfere with numerous host defense mechanisms, and it is well known that melanized C. neoformans cells are less susceptible to the toxic effects of microbicidal peptides than non-melanized cells (Doering et al., 1999). In addition, melanization protects C. neoformans against injury secondary to nitrogen or oxygen derived radical attack (Nosanchuk and Casadevall, 2003, 2006). In accordance with this, Figure 4B shows that HL-60D medium is unable to kill melanized C. neoformans, with no cell death observed suggesting that melanin totally protects C. neoformans from the killing activity of the conditioned medium collected from HL-60D.
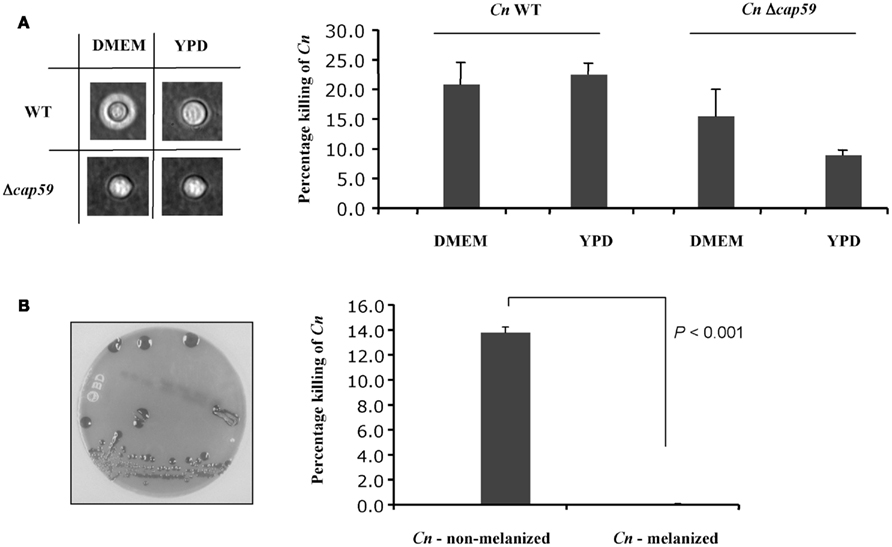
Figure 4. Effect of C. neoformans capsule and melanin on neutrophils killing activity. (A) Capsule size does not affect HL-60D killing ability. The acapsular strain Δcap59 has no capsule, but is more resistant to killing by HL-60D, presumably due to its thicker cell wall. (B) Non-melanized C. neoformans is killed by HL-60D whereas melanin confers resistance against anti-cryptococcal factors.
Fungal Inositol Sphingolipids and Glucosylceramide Content Affect C. neoformans Susceptibility to Neutrophils
Fungal inositol containing sphingolipids have been implicated in the regulation of the sensitivity of C. neoformans to oxidative and nitrosative stresses (Shea et al., 2006). At the same time, glucosylceramide is essential for fungal growth in host extracellular environments (Rittershaus et al., 2006). Interestingly, intracellular growth is regulated by a different sphingolipid (phytoceramide C26) which is mainly produce by Isc1. We therefore wished to investigate whether the HL-60D medium killing of C. neoformans correlated with the amount of complex sphingolipids present in the H99 strain. In order to test this, we used C. neoformans WT H99, Δgcs1, GAL7::IPC1, and Δisc1 mutant strains, each having a different “fitness” compared to the WT strain. The Δgcs1 mutant stain was included in the study as it shows no growth defect in the intracellular environment, compared to Δisc1 which grows poorly at low pH and is susceptible to intracellular compounds such as nitric oxide and hydrogen peroxide. Following incubation of these strains for 4 h with HL-60D medium, they were streaked onto YPD agar plates and CFUs counted after 48 h incubation at 30°C. We found that the GAL7::IPC1 cells, in which the level of inositol sphingolipids compared to the WT is decreased (Luberto et al., 2001), were significantly less susceptible to the killing activity of HL-60D medium compared to WT cells (Figure 5). Similarly, the glucosylceramide-lacking strain (Δgcs1) was slightly more resistant (although not significant) to the killing activity of the medium. Interestingly, the Δisc1 mutant which accumulates inositol sphingolipids (Henry et al., 2011) was as sensitive to the killing as the wild-type (Figure 5).
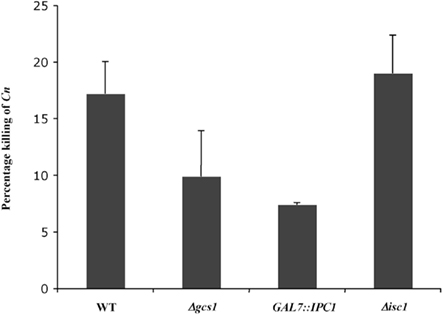
Figure 5. Fungal inositol sphingolipids and glucosylceramide content affect C. neoformans susceptibility to neutrophils. Decrease in inositol sphingolipids (GAL7::IPC1) compared to WT seems to confer resistance to killing. Lack of glucosylceramide (Δgcs1) is not significantly more resistant to killing. Δisc1 accumulates inositol sphingolipids and lacks phytoceramide C26 but it is as sensitive to killing as WT.
Neutrophils from Immunocompromised Tgε26 Mice Kill C. neoformans
Since cryptococcosis is an opportunistic fungal disease often seen in patients with impaired cellular immunity such as acquired immunodeficiency syndrome (AIDS), and since the host defense is regulated by CMI (Lim and Murphy, 1980) where CD4+ T cells play a central role in limiting infection (Hill and Harmsen, 1991; Huffnagle et al., 1991a; Kawakami, 2002; Uezu et al., 2004), we were interested in studying whether neutrophils from an immunocompromised host lacking NK and T cells would still be able to kill C. neoformans. We had previously observed that neutrophils obtained from immunocompetent mice having a fully functional immune system were able to kill C. neoformans (Qureshi et al., 2010). To this aim, we employed fresh murine neutrophils from Tgε26 mice, and carried out the killing assay as described in the Section “Materials and Methods”. It was found that these mouse neutrophils kill C. neoformans even though they lack NK and T cells (Figure 6).
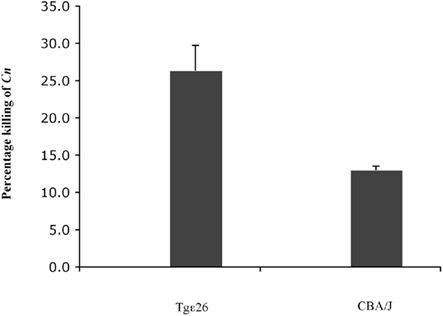
Figure 6. Effect of freshly isolated murine neutrophils from immunocompromised host on C. neoformans. Neutrophils from both immunocompromised Tgε26 mice as well as immunocompetant CBA/J mice display the ability to kill C. neoformans, even though Tgε26 mice lack T and NK cells.
Sphingomyelin 16:0 in Lungs of Mice Lacking NK and T Cells is Upregulated When Mice are Infected with C. neoformans
Matrix-assisted laser desorption/ionization-mass spectrometric imaging (MALDI–MSI) allows the visualization of the spatial distribution of specific molecules according to their m/z ratio within thin sections of tissue. The identification of the imaged lipid was accomplished by MALDI tandem mass spectrometry where fragmentation patterns were compared to those of sphingomyelin standards (see Materials and Methods). In our previous studies we showed that killing activity of neutrophils is mediated by SMS activity, and that there is a differential distribution of SM species as observed by MALDI–MSI. It is known that infection by C. neoformans mainly occurs in immunodeficient subjects, particularly those in which T cells count is drastically low (e.g., HIV positive subjects; Kovacs et al., 1985; King and Dewitt, 2010). Since we found that neutrophils isolated from the Tgε26 mice are able to kill C. neoformans very efficiently (Figure 6), and in previous studies we found that this killing ability is mediated by SMS activity (Qureshi et al., 2010), it was important to determine whether SMS activity would also have a role against C. neoformans under conditions of T cell deficiency. Thus, we examined the distribution of SM (as a read-out of SMS activity) species in C. neoformans-infected vs un-infected lung of Tgε26 mice. Overall, we found that SM 16:0 was distributed more homogenously in Tgε26 compared to CBA/J infected lungs (Figure 7). However, SM 16:0 was still clustered around the site of C. neoformans infection where an intense neutrophil infiltration was observed especially at day 15 of infection, although it was much less organized than what we observed in CBA/J lungs (Figure 7).

Figure 7. Effect of C. neoformans infection on the level of SM 16:0 in lungs of mice lacking NK and T cells. Lungs of Tgε26 mice un-infected and infected with C. neoformans wild-type H99 strain were processed for MALDI tissue imaging. Sphingomyelin (SM) 16:0 species ([M + Na]+ m/z 725) is distributed throughout the lungs at all timepoints. Min and Max, minimum and maximum intensity of SM 16:0 in the lung, respectively.
Lipid Analysis by HPLC-MS/MS
Since we found that SM 16:0 was elevated in the lungs of Tgε26 mice, we wondered whether any other subspecies of SM not observed by MALDI–MSI would also be elevated. To address this question, we examined the level of different species of SM, such as 16:0, 18:0, 18:1, 20:0, 20:1, 22:0, 22:1, 24:0, and 24:1 in lung of Tgε26 infected mice. As expected, we found that among the measured species, SM 16:0 and 24:1 were the most abundant. Indeed, these two species have been shown to be the most abundant in many cellular types and tissues (Fitzgerald et al., 1995; Venable et al., 1995; Bielawska et al., 2000; Merrill et al., 2005; Kono et al., 2006; Valsecchi et al., 2007; Jin et al., 2008a). Importantly, LC–MS results showed that SM 16:0 was significantly elevated at day 12 of infection compared to un-infected mice (Figure 7) but not at a later time point, as we observed in CBA/J mice. These results suggest that the production of this sphingolipid might be independent of T cell activation in immunocompromised mice but only at early time points of infection. In fact, the basal levels of SM 16:0 in Tgε26 un-infected lung (Figure 8) was similar to the levels found in CBA/J un-infected lung (Qureshi et al., 2010). We could not detect any SM 14:0, 26:0, or 26:1, suggesting that these species are either not produced by cells found in the mouse lung or that their levels are too low to be detected by MS.
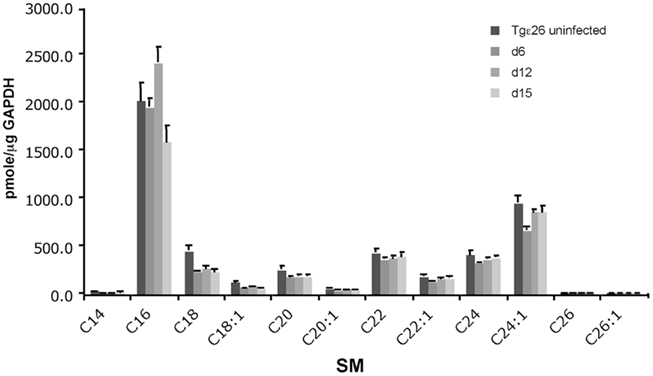
Figure 8. Quantitative analysis of the lung sphingolipids upon C. neoformans infection. Liquid Chromatography–Mass Spectrometry (LC–MS/MS) analysis of immunocompromised mouse lungs infected with wild-type C. neoformans show that SM 16:0 is the major species elevated at the site of infection. Other SM species remain constant or are not detected.
Discussion
In this investigation, the effect of pre-incubation of C. neoformans with HL-60 derived neutrophils has been studied. Our results show that pre-incubation of neutrophils with live but not heat-killed C. neoformans inhibits any further killing by secreted ACFs. Furthermore, activation of SMS in neutrophils, which is responsible for modulating their killing activity, appears to be partially independent of T and/or NK cell activation.
Neutrophils play important roles in host defense against all classes of infectious agents including C. neoformans. They constitute the second line of defense against pathogens after alveolar macrophages because, once an inflammatory response is initiated, neutrophils are the first cells to be recruited to the site of infection (Schleimer et al., 1989). The microbicidal arsenal of neutrophils includes the formation of reactive oxygen and nitrogen species, hydrolytic enzymes, and antimicrobial peptides, all of which target microbes (Smith, 1994). Neutrophils can also kill extracellular pathogens via the formation of neutrophil extracellular traps (NETs), which have activity against C. albicans and Aspergillus fumigatus (Urban et al., 2006a,b; Bruns et al., 2010). Notably, neutrophils are more effective killers of C. neoformans than macrophages, and they possess the innate ability to kill microbes without a cell-mediated activation. So while it has long been appreciated that neutrophils kill C. neoformans in part via generation of fungicidal oxidants, non-oxidative mechanisms also make a significant contribution (Mambula et al., 2000). Interestingly, transfusion of human neutrophils significantly improved the survival of mice challenged with fungal organisms (Spellberg et al., 2005, 2007), suggesting that these cells are able to control the infection through their antifungal activity.
To gain a better understanding of the neutrophil–C. neoformans interaction, and specifically the interaction of ACFs secreted by differentiated HL-60 (HL-60D) cells into the medium, we turned our attention to several in vitro systems. Before studying the response of neutrophils upon C. neoformans challenge however, we verified that the viability of HL-60 derived neutrophils used in this study is not reduced upon exposure to C. neoformans, as is the case with C. albicans where viability in the HL-60 granulocytic population is significantly affected (Mullick et al., 2004) upon phagocytosis of C. albicans by neutrophils, which in turn induces the HL-60 derived neutrophils to undergo apoptosis. In the case of C. neoformans, we have shown that killing is induced through the release of ACFs instead of phagocytosis (Qureshi et al., 2010). This suggests that, in contrast to C. albicans, C. neoformans cells may have developed other mechanisms to protect themselves against the killing activity of neutrophils. Thus, we sought to determine whether C. neoformans would be able to neutralize the antifungal action of the medium.
Interestingly, secretion of the ACFs by HL-60 derived neutrophils into the medium, and subsequent killing of C. neoformans, is maximal at 6 h. However, co-incubation of HL-60D cells with C. neoformans shows maximal killing at 1 h (Figure 3), followed by steady-state killing at 2 and 4 h, and no killing at 6 h, at which time it is known that C. neoformans cells start doubling in RPMI (Miller and Mitchell, 1991). Therefore 4 h was chosen as the time point for C. neoformans killing by HL-60D medium. Surprisingly, when the medium of HL-60D cells is used for further killing of C. neoformans, after the preliminary C. neoformans–HL-60D co-incubation, percent killing is severely reduced, implying that co-incubation of HL-60D cells with C. neoformans decreases the killing ability of the ACF in the medium, either because some C. neoformans binds such factors and thus neutralizes them and/or because C. neoformans degrades such factors. Thus C. neoformans appears to play a pivotal role in the killing-degradation cycle of the ACFs. The presence of the pathogen induces neutrophils to kill C. neoformans, but degradation of the ACFs which are secreted into the medium by neutrophils is also driven by the presence of C. neoformans. This observation is supported by studies showing that C. neoformans is able to secrete a variety of proteins including proteases (Eigenheer et al., 2007). In order to better understand this interaction, we sought to explore specifically the effect of C. neoformans virulence factors upon the ACFs.
The antiphagocytic polysaccharide capsule and melanin are important virulence components of C. neoformans. The regulation of these virulence factors has been reviewed recently in several excellent works (Li and Mody, 2010; Alspaugh et al., 2011; Fox et al., 2011; Idnurm et al., 2011; Nielsen and Kwon-Chung, 2011; Pfaller et al., 2011). Interestingly, the presence of capsule made little to no difference on killing of C. neoformans by the ACF in HL-60D medium. Notably, the acapsular strain C. neoformans Δcap59 was more resistant to killing by ACF, regardless of growth conditions. This is likely due to the fact that despite lacking a polysaccharide capsule and the protective architecture conferred upon it by the main capsular components, C. neoformans Δcap59 has a thicker cell wall which may provide some resistance to killing. Melanin on the other hand confers excellent resistance to C. neoformans against ACFs in the HL-60D medium.
Inositol phosphoryl ceramide synthase 1 (Ipc1) is a fungal enzyme that transfers inositol phosphate from phosphatidylinositol (PI) to phytoceramide, producing IPC and diacylglycerol (DAG; Kuroda et al., 1999; Heidler and Radding, 2000). Once produced, IPC is metabolized by Isc1 (Dickson and Lester, 1999; Henry et al., 2011), and, by doing so, it regulates the levels of complex sphingolipids and very long chain phytoceramides in C. neoformans (Shea et al., 2006; Garcia et al., 2008; Tommasino et al., 2008; Henry et al., 2011). Thus, we wished to examine the effect of the modulation of these sphingolipids on killing under conditions where Ipc1 was downregulated or Isc1 deleted. Loss of Isc1 leads to an accumulation of complex sphingolipids and a specific depletion of phytoceramide C26, whereas no changes are observed with phytoceramide C18 or other subspecies (Garcia et al., 2008). Downregulation of Ipc1 (GAL7::IPC1, grown in glucose) shows less complex sphingolipids, no change in phytoceramide C26 and accumulation of C18 phytoceramides compared to the WT strain.
The observation that the GAL7::IPC1 strain (which contains less inositol sphingolipids) is less susceptible to the killing activity of neutrophils would argue that the presence of complex sphingolipids may favor the action of the ACFs present in the medium. However, the fact that accumulation of complex sphingolipids (Δisc1) does not render the cells more susceptible than the wild-type does not support this hypothesis. On the other hand, the increased resistance to killing by neutrophils may be due to accumulation of phytoceramide C18 (observed in the GAL7::IPC1 strain). This hypothesis is supported by the results showing that the Δgcs1 strain is slightly more resistant and, in this strain, ceramide C18 accumulates (Rittershaus et al., 2006). Taken together with the results of the Δisc1 strain where no change in phytoceramide (or ceramide) C18 correlates with no change in susceptibility, this would support the hypothesis that phytoceramide C18 and ceramide C18 play a role in resistance.
In continuation of our work on the role of host SMS in the regulation of the killing activity of neutrophils against C. neoformans, we investigated whether neutrophils derived from immunocompromised mice would still kill C. neoformans. Neutrophils from transgenic epsilon 26 mice (Tgε26), deficient in NK cells and T cells, were used to explore the killing of C. neoformans WT. Macrophages, monocytes, and granulocytes are present and functional in these mice (Wang et al., 1994, 1997). NK cells constitute a major component of the innate immune system and are responsible for release of cytoplasmic granules that kill pathogen cells, whereas T cells play a major role in CMI. The fact that neutrophils from Tgε26 mice were able to kill C. neoformans despite lacking T cells implies that activation of SMS in the host is partially independent of T and/or NK cells.
Finally, using MALDI–MS imaging of lung tissue from Tgε26 mice infected with C. neoformans WT we observed that SM 16:0 is found in the immunocompromised mouse lung similarly as in the immunocompetent lung, suggesting that NK and/or T cells are not necessary to produce a basal level of SM 16:0. When the lungs were infected with C. neoformans though, we found that SM 16:0 was elevated only at day 12 of the infection and its overall distribution was much less organized in the immunocompromised compared to the immunocompetent host, suggesting that T and/or NK cells may have a role in activating SMS in the lung, and coordinating the SMS response of neutrophils against C. neoformans, especially when the infection progresses. It has been suggested that sphingolipid-rich microdomains form platforms for the regulation and transduction of T cell receptors which cluster at the center of the T cell-antigen presenting cell interface during T cell activation. T cell clustering was shown to be impaired in an SMS knockdown cell line (Jurkat-SMS1/kd), indicating that SM and hence SMS may be important for full T cell activation (Jin et al., 2008b), and in the case where T cells are lacking, SMS will be somewhat impaired as one of its regulatory functions will be curtailed.
In conclusion, we have shown that pre-incubation of live but not heat-killed C. neoformans inhibits further killing activity of ACFs. Neither the presence of C. neoformans nor the capsule size has any effect on neutrophil viability but melanin confers resistance to C. neoformans against neutrophil killing. Finally SMS activation in neutrophils may be independent of T cell and/or NK cells activation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the Lipidomics Core facility for sphingolipid analysis. This work was supported in part by the Burroughs Welcome Fund, in part by Grants AI56168, AI78493, AI87541, and AI71142 (to Maurizio Del Poeta) from the National Institute of Health, in part by RR17677 Project 2 (to Maurizio Del Poeta) from the Centers of Biomedical Research Excellence Program of the National Center for Research Resources, in part by NIH C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. Dr. Maurizio Del Poeta is a Burroughs Wellcome New Investigator in the Pathogenesis of Infectious Diseases.
References
Alspaugh, J. A., Nichols, C. B., Xue, C., Shen, W. C., and Wang, P. (2011). “G-protein signaling pathways: regulating morphogenesis and virulence of Cryptococcus,” in Cryptococcus: From Human Pathogen to Model Yeast, eds J. Heitman, T. R. Kozel, K. J. Kwon-Chung, J. R. Perfect, and A. Casadevall (Washington, DC: ASM Press), 153–165.
Alvarez, M., and Casadevall, A. (2006). Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr. Biol. 16, 2161–2165.
Bielawska, A., Szulc, Z., and Hannun, Y. A. (2000). Synthesis of key precursors of radiolabeled sphingolipids. Meth. Enzymol. 311, 518–535.
Bielawski, J., Pierce, J. S., Snider, J., Rembiesa, B., Szulc, Z. M., and Bielawska, A. (2009). Comprehensive quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods Mol. Biol. 579, 443–467.
Bielawski, J., Szulc, Z. M., Hannun, Y. A., and Bielawska, A. (2006). Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39, 82–91.
Bose, I., Reese, A. J., Ory, J. J., Janbon, G., and Doering, T. L. (2003). A yeast under cover: the capsule of Cryptococcus neoformans. Eukaryotic Cell 2, 655–663.
Bruns, S., Kniemeyer, O., Hasenberg, M., Aimanianda, V., Nietzsche, S., Thywissen, A., Jeron, A., Latge, J. P., Brakhage, A. A., and Gunzer, M. (2010). Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 6, e1000873. doi: 10.1371/journal.ppat.1000873
Diamond, R. D., Root, R. K., and Bennett, J. E. (1972). Factors influencing killing of Cryptococcus neoformans by human leukocytes in vitro. J. Infect. Dis. 125, 367–376.
Dickson, R. C., and Lester, R. L. (1999). Metabolism and selected functions of sphingolipids in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 1438, 305–321.
Doering, T. L., Nosanchuk, J. D., Roberts, W. K., and Casadevall, A. (1999). Melanin as a potential cryptococcal defence against microbicidal proteins. Med. Mycol. 37, 175–181.
Eigenheer, R. A., Jin Lee, Y., Blumwald, E., Phinney, B. S., and Gelli, A. (2007). Extracellular glycosylphosphatidylinositol-anchored mannoproteins and proteases of Cryptococcus neoformans. FEMS Yeast Res. 7, 499–510.
Fan, W., Kraus, P. R., Boily, M. J., and Heitman, J. (2005). Cryptococcus neoformans gene expression during murine macrophage infection. Eukaryotic Cell 4, 1420–1433.
Feldmesser, M., Tucker, S., and Casadevall, A. (2001). Intracellular parasitism of macrophages by Cryptococcus neoformans. Trends Microbiol. 9, 273–278.
Fitzgerald, V., Blank, M. L., and Snyder, F. (1995). Molecular species of sphingomyelin in sphingomyelinase-sensitive and sphingomyelinase-resistant pools of HL-60 cells. Lipids 30, 805–809.
Fox, D. S., Djordjevic, J. T., and Sorrell, T. (2011). “Signaling cascades and enzymes as Cryptococcus virulence factors,” in Cryptococcus: From Human Pathogen to Model Yeast, eds J. Heitman, T. R. Kozel, K. J. Kwon-Chung, J. R. Perfect, and A. Casadevall (Washington, DC: ASM Press), 217–234.
Garcia, J., Shea, J., Alvarez-Vasquez, F., Qureshi, A., Luberto, C., Voit, E. O., and Del Poeta, M. (2008). Mathematical modeling of pathogenicity of Cryptococcus neoformans. Mol. Syst. Biol. 4, 183.
Goldman, D., Lee, S. C., and Casadevall, A. (1994). Pathogenesis of pulmonary Cryptococcus neoformans infection in the rat. Infect. Immun. 62, 4755–4761.
Heidler, S. A., and Radding, J. A. (2000). Inositol phosphoryl transferases from human pathogenic fungi. Biochim. Biophys. Acta 1500, 147–152.
Henry, J., Guillotte, A., Luberto, C., and Del Poeta, M. (2011). Characterization of inositol phospho-sphingolipid-phospholipase C 1 (Isc1) in Cryptococcus neoformans reveals unique biochemical features. FEBS Lett. 585, 635–640.
Heung, L. J., Kaiser, A. E., Luberto, C., and Del Poeta, M. (2005). The role and mechanism of diacylglycerol-protein kinase C1 signaling in melanogenesis by Cryptococcus neoformans. J. Biol. Chem. 280, 28547–28555.
Hill, J. O., and Harmsen, A. G. (1991). Intrapulmonary growth and dissemination of an avirulent strain of Cryptococcus neoformans in mice depleted of CD4+ or CD8+ T cells. J. Exp. Med. 173, 755–758.
Huffnagle, G. B., Yates, J. L., and Lipscomb, M. F. (1991a). Immunity to a pulmonary Cryptococcus neoformans infection requires both CD4+ and CD8+ T cells. J. Exp. Med. 173, 793–800.
Huffnagle, G. B., Yates, J. L., and Lipscomb, M. F. (1991b). T cell-mediated immunity in the lung: a Cryptococcus neoformans pulmonary infection model using SCID and athymic nude mice. Infect. Immun. 59, 1423–1433.
Idnurm, A., Bahn, Y. S., Shen, W. C., Rutherford, J. C., and Muhlschlegel, F. A. (2011). “Sensing extracellular signals in Cryptococcus neoformans,” in Cryptococcus: From Human Pathogen to Model Yeast, eds J. Heitman, T. R. Kozel, K. J. Kwon-Chung, J. R. Perfect, and A. Casadevall (Washington, DC: ASM Press), 175–187.
Jesus, M. D., Nicola, A. M., Chow, S. K., Lee, I. R., Nong, S., Specht, C. A., Levitz, S. M., and Casadevall, A. (2010). Glucuronoxylomannan, galactoxylomannan, and mannoprotein occupy spatially separate and discrete regions in the capsule of Cryptococcus neoformans. Virulence 1, 500–508.
Jin, J., Hou, Q., Mullen, T. D., Zeidan, Y. H., Bielawski, J., Kraveka, J. M., Bielawska, A., Obeid, L. M., Hannun, Y. A., and Hsu, Y. T. (2008a). Ceramide generated by sphingomyelin hydrolysis and the salvage pathway is involved in hypoxia/reoxygenation-induced Bax redistribution to mitochondria in NT-2 cells. J. Biol. Chem. 283, 26509–26517.
Jin, Z. X., Huang, C. R., Dong, L., Goda, S., Kawanami, T., Sawaki, T., Sakai, T., Tong, X. P., Masaki, Y., Fukushima, T., Tanaka, M., Mimori, T., Tojo, H., Bloom, E. T., Okazaki, T., and Umehara, H. (2008b). Impaired TCR signaling through dysfunction of lipid rafts in sphingomyelin synthase 1 (SMS1)-knockdown T cells. Int. Immunol. 20, 1427–1437.
Kawakami, K. (2002). Role of natural killer T cells in host defense against cryptococcal infection. Rev Iberoam Micol 19, 149–154.
King, J. W., and Dewitt, M. L. (2010). Cryptococcosis. eMedicine. Available at http://emedicine.medscape.com/article/215354 [accessed December 14, 2010; Online].
Kono, M., Dreier, J. L., Ellis, J. M., Allende, M. L., Kalkofen, D. N., Sanders, K. M., Bielawski, J., Bielawska, A., Hannun, Y. A., and Proia, R. L. (2006). Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J. Biol. Chem. 281, 7324–7331.
Kovacs, J. A., Kovacs, A. A., Polis, M., Wright, W. C., Gill, V. J., Tuazon, C. U., Gelmann, E. P., Lane, H. C., Longfield, R., Overturf, G., Macher, A. M., Fauci, A. S., Parrillo, J., Bennett, J. E., and Masur, H. (1985). Cryptococcosis in the acquired immunodeficiency syndrome. Ann. Intern. Med. 103, 533–538.
Kuroda, M., Hashida-Okado, T., Yasumoto, R., Gomi, K., Kato, I., and Takesako, K. (1999). An aureobasidin A resistance gene isolated from Aspergillus is a homolog of yeast AUR1, a gene responsible for inositol phosphorylceramide (IPC) synthase activity. Mol. Gen. Genet. 261, 290–296.
Lim, T. S., and Murphy, J. W. (1980). Transfer of immunity to cryptococcosis by T-enriched splenic lymphocytes from Cryptococcus neoformans-sensitized mice. Infect. Immun. 30, 5–11.
Luberto, C., Martinez-Marino, B., Taraskiewicz, D., Bolanos, B., Chitano, P., Toffaletti, D. L., Cox, G. M., Perfect, J. R., Hannun, Y. A., Balish, E., and Del Poeta, M. (2003). Identification of App1 as a regulator of phagocytosis and virulence of Cryptococcus neoformans. J. Clin. Invest. 112, 1080–1094.
Luberto, C., Toffaletti, D. L., Wills, E. A., Tucker, S. C., Casadevall, A., Perfect, J. R., Hannun, Y. A., and Del Poeta, M. (2001). Roles for inositol-phosphoryl ceramide synthase 1 (IPC1) in pathogenesis of C. neoformans. Genes Dev. 15, 201–212.
Ma, H., Croudace, J. E., Lammas, D. A., and May, R. C. (2006). Expulsion of live pathogenic yeast by macrophages. Curr. Biol. 16, 2156–2160.
Mambula, S. S., Simons, E. R., Hastey, R., Selsted, M. E., and Levitz, S. M. (2000). Human neutrophil-mediated nonoxidative antifungal activity against Cryptococcus neoformans. Infect. Immun. 68, 6257–6264.
Merrill, A. H. Jr., Sullards, M. C., Allegood, J. C., Kelly, S., and Wang, E. (2005). Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 36, 207–224.
Miller, M. F., and Mitchell, T. G. (1991). Killing of Cryptococcus neoformans strains by human neutrophils and monocytes. Infect. Immun. 59, 24–28.
Mullick, A., Elias, M., Harakidas, P., Marcil, A., Whiteway, M., Ge, B., Hudson, T. J., Caron, A. W., Bourget, L., Picard, S., Jovcevski, O., Massie, B., and Thomas, D. Y. (2004). Gene expression in HL60 granulocytoids and human polymorphonuclear leukocytes exposed to Candida albicans. Infect. Immun. 72, 414–429.
Nielsen, K., and Kwon-Chung, K. J. (2011). “A role for mating in cryptococcal virulence,” in Cryptococcus: From Human Pathogen to Model Yeast, eds J. Heitman, T. R. Kozel, K. J. Kwon-Chung, J. R. Perfect, and A. Casadevall (Washington, DC: ASM Press), 167–174.
Nosanchuk, J. D., and Casadevall, A. (2003). The contribution of melanin to microbial pathogenesis. Cell. Microbiol. 5, 203–223.
Nosanchuk, J. D., and Casadevall, A. (2006). Impact of melanin on microbial virulence and clinical resistance to antimicrobial compounds. Antimicrob. Agents Chemother. 50, 3519–3528.
Pfaller, M. A., Lodge, J. K., and Ghannoum, M. A. (2011). “Drug resistance in Cryptococcus: epidemiology and molecular mechanisms,” in Cryptococcus: From Human Pathogen to Model Yeast, eds J. Heitman, T. R. Kozel, K. J. Kwon-Chung, J. R. Perfect, and A. Casadevall (Washington, DC: ASM Press), 203–216.
Qureshi, A., Subathra, M., Grey, A., Schey, K., Del Poeta, M., and Luberto, C. (2010). Role of sphingomyelin synthase in controlling the antimicrobial activity of neutrophils against Cryptococcus neoformans. PLoS ONE 5, e15587. doi: 10.1371/journal.pone.0015587
Rittershaus, P. C., Kechichian, T. B., Allegood, J. C., Merrill, A. H. Jr., Hennig, M., Luberto, C., and Del Poeta, M. (2006). Glucosylceramide synthase is an essential regulator of pathogenicity of Cryptococcus neoformans. J. Clin. Invest. 116, 1651–1659.
Schleimer, R. P., Freeland, H. S., Peters, S. P., Brown, K. E., and Derse, C. P. (1989). An assessment of the effects of glucocorticoids on degranulation, chemotaxis, binding to vascular endothelium and formation of leukotriene B4 by purified human neutrophils. J. Pharmacol. Exp. Ther. 250, 598–605.
Shao, X., Mednick, A., Alvarez, M., Van Rooijen, N., Casadevall, A., and Goldman, D. L. (2005). An innate immune system cell is a major determinant of species-related susceptibility differences to fungal pneumonia. J. Immunol. 175, 3244–3251.
Shea, J. M., Kechichian, T. B., Luberto, C., and Del Poeta, M. (2006). The cryptococcal enzyme inositol phosphosphingolipid-phospholipase C confers resistance to the antifungal effects of macrophages and promotes fungal dissemination to the central nervous system. Infect. Immun. 74, 5977–5988.
Shimizu, Y., Dobashi, K., Iizuka, K., Horie, T., Suzuki, K., Tukagoshi, H., Nakazawa, T., Nakazato, Y., and Mori, M. (2001). Contribution of small GTPase Rho and its target protein rock in a murine model of lung fibrosis. Am. J. Respir. Crit. Care Med. 163, 210–217.
Smith, J. A. (1994). Neutrophils, host defense, and inflammation: a double-edged sword. J. Leukoc. Biol. 56, 672–686.
Spellberg, B. J., Collins, M., Avanesian, V., Gomez, M., Edwards, J. E. Jr., Cogle, C., Applebaum, D., Fu, Y., and Ibrahim, A. S. (2007). Optimization of a myeloid cell transfusion strategy for infected neutropenic hosts. J. Leukoc. Biol. 81, 632–641.
Spellberg, B. J., Collins, M., French, S. W., Edwards, J. E. Jr., Fu, Y., and Ibrahim, A. S. (2005). A phagocytic cell line markedly improves survival of infected neutropenic mice. J. Leukoc. Biol. 78, 338–344.
Stano, P., Williams, V., Villani, M., Cymbalyuk, E. S., Qureshi, A., Huang, Y., Morace, G., Luberto, C., Tomlinson, S., and Del Poeta, M. (2009). App1: an antiphagocytic protein that binds to complement receptors 3 and 2. J. Immunol. 182, 84–91.
Tommasino, N., Villani, M., Qureshi, A., Henry, J., Luberto, C., and Del Poeta, M. (2008). Atf2 transcription factor binds to the APP1 promoter in Cryptococcus neoformans: stimulatory effect of diacylglycerol. Eukaryotic Cell 7, 294–301.
Uezu, K., Kawakami, K., Miyagi, K., Kinjo, Y., Kinjo, T., Ishikawa, H., and Saito, A. (2004). Accumulation of gammadelta T cells in the lungs and their regulatory roles in Th1 response and host defense against pulmonary infection with Cryptococcus neoformans. J. Immunol. 172, 7629–7634.
Urban, C. F., Lourido, S., and Zychlinsky, A. (2006a). How do microbes evade neutrophil killing? Cell. Microbiol. 8, 1687–1696.
Urban, C. F., Reichard, U., Brinkmann, V., and Zychlinsky, A. (2006b). Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 8, 668–676.
Valsecchi, M., Mauri, L., Casellato, R., Prioni, S., Loberto, N., Prinetti, A., Chigorno, V., and Sonnino, S. (2007). Ceramide and sphingomyelin species of fibroblasts and neurons in culture. J. Lipid Res. 48, 417–424.
Venable, M. E., Lee, J. Y., Smyth, M. J., Bielawska, A., and Obeid, L. M. (1995). Role of ceramide in cellular senescence. J. Biol. Chem. 270, 30701–30708.
Wang, B., Biron, C., She, J., Higgins, K., Sunshine, M. J., Lacy, E., Lonberg, N., and Terhorst, C. (1994). A block in both early T lymphocyte and natural killer cell development in transgenic mice with high-copy numbers of the human CD3E gene. Proc. Natl. Acad. Sci. U.S.A. 91, 9402–9406.
Keywords: neutrophils, sphingolipid, sphingomyelin, immunodeficient mice, fungal infection, Cryptococcus neoformans
Citation: Qureshi A, Grey A, Rose KL, Schey KL and Del Poeta M (2011) Cryptococcus neoformans modulates extracellular killing by neutrophils. Front. Microbio. 2:193. doi: 10.3389/fmicb.2011.00193
Received: 19 July 2011;
Paper pending published: 05 August 2011;
Accepted: 31 August 2011;
Published online: 21 September 2011.
Edited by:
Vishnu Chaturvedi, Wadsworth Center, USAReviewed by:
Oscar Zaragoza, Instituto de Salud Carlos III, SpainMarcio Rodrigues, Federal University of Rio de Janeiro, Brazil
Copyright: © 2011 Qureshi, Grey, Rose, Schey and Del Poeta. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Maurizio Del Poeta, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, 173 Ashley Avenue, BSB 512A, Charleston, SC 29425, USA. e-mail:ZGVscG9ldGFAbXVzYy5lZHU=