 Sahil Adriouch1,2*
Sahil Adriouch1,2* Emilie Franck1,2 Laurent Drouot1,2
Emilie Franck1,2 Laurent Drouot1,2 Carole Bonneau1 Nelly Jolinon3 Anna Salvetti3
Carole Bonneau1 Nelly Jolinon3 Anna Salvetti3 Olivier Boyer1,2,4*
Olivier Boyer1,2,4*- 1 INSERM U905, Faculty of Medicine and Pharmacy, Rouen, France
- 2 IFRMP 23, Institute for Biomedical Research, University of Rouen, Rouen, France
- 3 INSERM U758 and École Normale Supérieure de Lyon, Lyon, France
- 4 Department of Immunology, Rouen University Hospital, Rouen, France
Initially thought as being non-immunogenic, recombinant AAVs have emerged as efficient vector candidates for treating monogenic diseases. It is now clear however that they induce potent immune responses against transgene products which can lead to destruction of transduced cells. Therefore, developing strategies to circumvent these immune responses and facilitate long-term expression of transgenic therapeutic proteins is a main challenge in gene therapy. We evaluated herein a strategy to inhibit the undesirable immune activation that follows muscle gene transfer by administration of CTLA-4/Ig to block the costimulatory signals required early during immune priming and by using gene transfer of PD-1 ligands to inhibit T cell functions at the tissue sites. We provide the proof of principle that this combination immunoregulatory therapy targeting two non-redundant checkpoints of the immune response, i.e., priming and effector functions, can improve persistence of transduced cells in experimental settings where cytotoxic T cells escape initial blockade. Therefore, CTLA-4/Ig plus PD-L1/2 combination therapy represents a candidate approach to circumvent the bottleneck of immune responses directed toward transgene products.
Introduction
Since the original reports describing the use of adeno-associated virus (AAV) vectors for transfer of β-galactosidase gene to muscle (Kessler et al., 1996; Xiao et al., 1996; Fisher et al., 1997), recombinant AAVs (rAAV) have emerged as very efficient and potentially non-immunogenic vector candidates for delivering therapeutic genes to a variety of tissues and treating monogenic diseases. As they poorly activate innate immunity and weakly transduce dendritic cells, rAAV appear as far less immunogenic than adenoviral vectors (Zhang et al., 2000; Zaiss et al., 2002; McCaffrey et al., 2008). Nevertheless, it has rapidly become clear that rAAV vectors carrying various transgenes can, under different conditions, induce potent immune responses that could ultimately lead to destruction of transduced cells in vivo and consecutive disappearance of transgene expression (Manning et al., 1997, 1998; Halbert et al., 1998; Brockstedt et al., 1999). Not surprisingly therefore, their use has even been proposed in genetic vaccination protocols aimed at eliciting cellular and humoral immune responses against different microorganisms (Kuck et al., 2006; Du et al., 2008). In primates and humans, rAAV administration has also been documented to elicit significant cytotoxic CD8+ T cell responses directed against the viral as well as the transgenic “exogenous” proteins, resulting in the destruction of transduced cells and complete loss of transgene expression (Manno et al., 2006; Mingozzi et al., 2007; Gao et al., 2009). Additionally, on the side of the humoral immunity, production of neutralizing antibodies targeting capsid proteins may also prevent vector readministration and accelerate the loss of the therapeutic protein through the formation of immune complexes. Such immune complexes may further sensitize the cellular immune response by enhancing cross-presentation of the transgenic protein by the antigen-presenting cells (APC). Therefore, developing strategies to circumvent immune responses and facilitate long-term expression of transgenic therapeutic proteins has been identified as one of today’s main challenges for the translation of rAAV vectors into the clinic (Mingozzi and High, 2011a,b; Nayak and Herzog, 2011).
Depending on the experimental situation, rAAV-mediated gene transfer can either lead to durable transgene expression or, conversely, to the rapid formation of neutralizing antibodies and/or destruction of transduced cells by cytotoxic cells. Several factors influencing the immune response against transgenic proteins encoded by the rAAV vectors have now been identified including host species, route of administration, vector dose, immunogenicity of the transgenic protein, inflammatory status of the host and capsid serotype (Mays and Wilson, 2011). These factors are thought to influence immunogenicity by triggering innate immunity, cytokine production, APC maturation, antigen presentation and, ultimately, priming of naïve T lymphocytes to functional effectors. Therefore, the idea to dampen immune activation by interfering with these very mechanisms has logically emerged with the aim to induce a short-term immunosuppression, avoid the early immune priming that follows vector administration and promote long-term tolerance (Zaiss and Muruve, 2008).
Here, we evaluated two different strategies to inhibit the undesirable immune activation that follows muscle gene transfer by acting at two different checkpoints of the immune response, i.e., on T cell priming or on the functions of activated T cells that may escape such priming blockade. We used the administration of CTLA-4/Ig to inhibit the substantial immune priming that immediately follow vector injection. Indeed, CTLA-4/Ig represent a potent immunosuppressive fusion protein that reversibly prevents T cell activation (Wallace et al., 1995) and is now used in the clinic to treat inflammatory diseases such as rheumatoid arthritis (Bluestone et al., 2006). Its immunomodulatory action depends on its competitive inhibitory effect on the CD28/B7 pathway thereby preventing the pivotal CD28-dependent costimulation required to fully activate T lymphocytes (Salomon and Bluestone, 2001). As a second strategy, we turned to immunomodulatory molecules that could protect transduced muscle fibers from immune attacks by activated T cells. For that, we aimed at stimulating the inhibitory PD-1 molecule expressed on T cells upon activation, by the gene transfer of its ligands PD-L1 or PD-L2 to muscle cells (Freeman et al., 2000; Latchman et al., 2001; Ishida et al., 2002). We show herein that acting on these two non-redundant mechanisms of tolerance provides synergistic effects that prolong transgene expression in muscle even in the presence of circulating cytotoxic T cells directed against the transgene product.
Materials and Methods
Animal Experiments
Female C57BL/6 (B6) mice were obtained from Centre d’Elevage Janvier (Le Genest Saint Isle, France). Mice were all between 7 and 10 weeks of age at beginning of experiments. For transduction with rAAV vectors, mice back legs were shaved under general anesthesia and titrated 1 × 1011 vector genomes (vg) rAAV2/1-Ova or rAAV2/8-Ova were injected (50 μl in PBS) in the gastrocnemius muscles. Where indicated, 1011 vg rAAV2/1-PD-L1 or rAAV2/1-PD-L2 were mixed with 1011 vg rAAV-Ova and co-injected using the same procedure. For costimulation blockade experiments, mice were injected i.p. with 200 μg CTLA-4/Ig (Chimerigen laboratories, MF-110A4) diluted in 200 μl of PBS.
In some experiments, lymphocytes from tolerized mice were transferred to conditioned recipients to further evaluate the presence of anti-Ova lymphocytes and to study their functionality in vivo. For that, 50 × 106 splenocytes harvested from individual mice that have be treated 80 days before with AAV-Ova, with or without immunomodulatory regimens, were injected i.v. into 5 Gy-irradiated syngenic C57BL/6 recipient. One day after, each individual mouse was shaved and injected subcutaneously with 1 × 106 syngenic EG7 tumor cells expressing the Ova antigen and known to be sensitive in vivo to CD8+ T cell cytotoxicity in primed animals (Moore et al., 1988). Tumor sizes were measured with a digital caliper three times a week during 26 days. Tumor volume was calculated as length × width × [(length + width)/2].
All animal experiments were approved by the local institutional ethic committee for animal experimentation (authorization #0211-22 “Comité Régional d’Éthique en Expérimentation Animale de Normandie”).
Plasmidic Constructs and Preparation of Recombinant AAV Vectors
The rAAV-Ova vector, a kind gift of Roland W. Herzog, was described previously (Wang et al., 2005). The cDNA encoding mouse PD-L1 (CD274) or PD-L2 (CD273) were cloned using standard molecular biology procedures and introduced in the SSV9–CAG plasmidic backbone after digestion with EcoRI. The resulting expression cassette, flanked by AAV serotype 2 inverted terminal repeats (ITRs), contains the CAG promoter combining the cytomegalovirus early enhancer and the chicken β-actin promoter, a chicken β-actin intron, and a rabbit β-globin polyadenylation signal. rAAV2/1 and rAAV2/8 vectors were generated using a standard helper-virus free three-plasmid transient transfection method and pseudotyped with either AAV1 or AAV8 capsid proteins. Vectors were purified by two cesium chloride gradient centrifugations and dialyzed against PBS as described (Salvetti et al., 1998). Genome titers of vector preparations were assayed by Dot-blot hybridization using a probe to detect the CAG or CMV promoter.
SOVA and Anti-Ova IgG ELISA
Quantification of soluble Ova (sOVA) concentration in serum was performed by Ova-specific ELISA. Microtiter plates were coated with polyclonal rabbit anti-Ova antibodies (1:3000 dilution, RayBiotech) and bound sOVA was detected using biotinylated rabbit polyclonal anti-Ova antibodies (1:5000 dilution, Abcam) and streptavidin–peroxidase (1:15000, Roche).
Detection of serum anti-Ova IgG antibodies was performed by ELISA using Ova-coated microtiter plates. Anti-Ova IgG antibodies were detected using biotinylated polyclonal goat anti-mouse IgG and revealed using the mouse ExtrAvidin kit (Sigma-Aldrich). IgG titers were defined as the dilution yielding the half maximum optical density obtained with control serums and was calculated using sigmoid curve fitting using GraphPad prism software.
Quantification of Ova CDNA and MRNA in Transduced Muscles
To analyze the quantities of Ova DNA and Ova mRNA present in transduced muscles of treated mice at indicated time points, a real-time PCR assay was developed. Muscles collected from each mouse were kept at −80°C before DNA and RNA extraction performed using the phenol/chloroform method and the RNeasy Fibrous Tissue Mini Kit (Qiagen) following the manufacturer’s instructions.
For quantification of Ova DNA, the primers used were Ova-F (5′-AAG CAG GCA GAG AGG TGG TA-3′), Ova-R (5′-GAA TGG ATG GTC AGC CCT AA-3′), CD8a-F (5′-GGT GCA TTC TCA CTC TGA GTT CC-3′), and CD8a-R (5′-GCA GAC AGA GCT GAT TTC CTA TGT G-3′). For all reaction mixtures, 10 μl of FastStart Universal SYBR green master mix (Roche) was used in a final volume of 20 μl. Ova primers were used at 500 nM and CD8a primers at 400 nM. Approximately 10 ng of DNA was added in a 5-μl volume and always set up in duplicate. Each qPCR was performed under the following conditions: 10 min hot-start denaturation at 95°C and 40 amplification cycles (10 s at 95°C, 30 s at 60°C). The melting temperatures of the final double-strand DNA products were determined by gradual heating from 60 to 95°C over 20 min. All qPCRs were performed with a StepOnePlus real-time PCR system (Applied Biosystems) and corresponding software. Absolute amounts of Ova and CD8a amplicons, in arbitrary units, were determined using serial dilutions of pAAV–CMV–OVA plasmid or pTOPO-CD8a plasmid as a standard. The data were expressed as Ova/CD8a ratios, fixed at 1 for PBS-injected control mice.
For quantification of Ova mRNA, 100 ng of total RNA were reverse transcribed using iScript DNA Synthesis Kit (Biorad). Then, 2 μl of cDNA were subjected to real-time PCR amplification using Ova primers, β-actin-F (5′-AAG ATC TGG CAC CAC ACC TTC T-3′) and β-actin-R (5′-TTT TCA CGG TTG GCC TTA GG-3′) primers. For all reaction mixtures, 10 μl of FastStart Universal SYBR green master mix (Roche) was used in a final volume of 20 μl, Ova primers were used at 500 nM and β-actin primers at 400 nM. The same qPCR program as above-described conditions were used. The absolute amount of Ova mRNA for each sample was then normalized against the β-actin mRNA amount (arbitrary units) and determined using serial dilutions of pAAV–CMV–OVA plasmid and β-actin purified PCR product.
Flow Cytometry and ELISpot Assays
Fluorescently labeled anti-CD4 (RM4-5), -CD8 (53-6.7), -CD44 (IM7), -CD62L (MEL-14), -PD-1 (J43), -PD-L1 (B7-H1), -PD-L2 (B7-DC) monoclonal antibodies (mAb), and unlabeled anti-CD16/CD32 antibodies were all purchased from eBioscience. PE-conjugated H-2Kb/Ova257–264 pentamers were used to detect the CD8+ cells that specifically recognize the immunodominant Ova-derived SIINFEKL peptide using the manufacturers’ protocol (Proimmune). Flow cytometry measurements of single cell suspensions derived from lymph nodes, spleen, or blood samples were performed using standard procedures and acquired on a FACSCanto (BD Biosciences) instrument. Flow cytometry analyses were performed using the FlowJo software (Tree Star).
Enzyme-linked immunospot (ELISpot) assays were performed following the instructions of the manufacturer (Diaclone). Briefly, PVDF membrane plates were coated with capture antibody against mouse IFNΓ and blocked with a 2% skimmed milk solution prepared in PBS. 2 × 105 Splenocytes per well were cultured in vitro for 36 h in the presence of 10 μg/ml SIINFEKL peptide. Plates were revealed after incubation with anti-mouse IFNΓ detection antibody coupled to biotin followed with a streptavidin–alkaline phosphatase conjugate and exposure to a ready-to-use solution of nitro-blue tetrazolium (NBT) and 5-bromo-4-chloro-3′-indolyphosphate (BCIP) for chromogenic development. Plates were analyzed with an ELISpot plate reader and a dedicated ImmunoSpots software (C.T.L.).
Data Representation and Statistical Analysis
Results were expressed as mean ± SEM. Significance was assessed by non-parametric one-way ANOVA (Kruskal–Wallis tests) using the GraphPad Prism software. Results were considered statistically significant when the p value was inferior to 0.05 (*), to 0.01(**) or to 0.001 (***).
Results
Characterization of the Anti-Ova Cellular and Humoral Immune Responses after RAAV2/1-Ova or RAAV2/8-Ova-Mediated Muscle Gene Transfer
We used rAAV2/1-Ova and rAAV2/8-Ova administration to model a gene therapy setting where the transgene encodes for a highly immunogenic secreted protein. We first evaluated the capacity of these vectors to transduce muscle cells upon direct i.m. injection. Injection of 1011 viral genome (vg) rAAV2/1-Ova or rAAV2/8-Ova similarly resulted in efficient transduction, as attested by the detection of Ova DNA and mRNA in the injected muscles quantified by qPCR and qRT-PCR respectively (Figure 1A). No significant difference was found when comparing the transduction efficiency of rAAV2/1-Ova and rAAV2/8-Ova in these conditions.
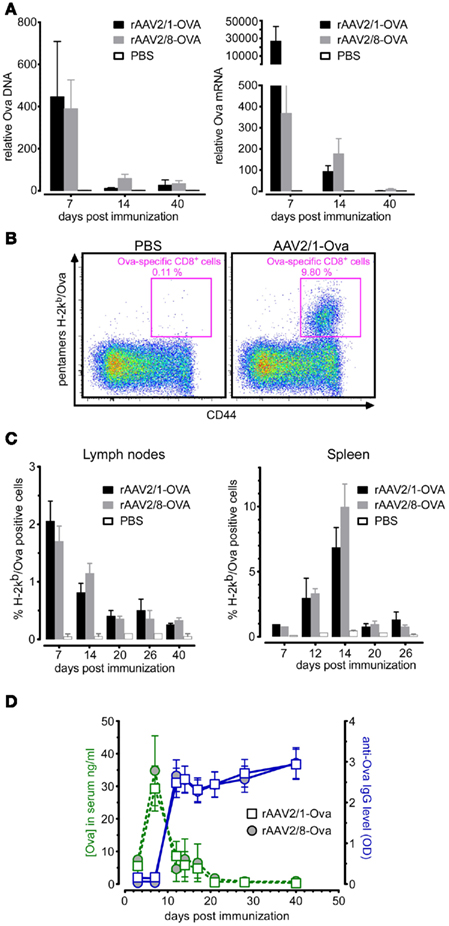
Figure 1. Characterization of anti-Ova immune responses following muscle gene transfer using rAAV2/1-Ova or rAAV2/8-Ova. Group of four to seven mice were injected with titrated 1 × 1011 viral genomes (vg) of either rAAV2/1-Ova or rAAV2/8-Ova in the gastrocnemius muscles at day 0 and then culled at different time points. (A) Gastrocnemius muscles were analyzed for the presence of Ova DNA and mRNA by qPCR and qRT-PCR, respectively, at 7, 14, and 40 days post-gene transfer. (B) Representative flow cytometry profile and (C) bar graphs showing the percentage of Ova-specific CD8+ T cells positively stained with H-2Kb/Ova pentamers in draining lymph nodes and spleen at different time points after gene transfer. (D) Sera were harvested from individual mice at different time points and analyzed by ELISA for the detection of sOva (green lines) and anti-Ova IgG (blue lines).
Transgene expression in muscle was found to be high early after transduction but rapidly declined over time, suggesting the occurrence of an immune response against transduced cells (Figure 1A). To investigate this point, we quantified the cellular and humoral immune responses directed against the xenogenic Ova transgene product by flow cytometry and ELISA. Staining with MHC-I pentamers presenting the immunodominant Ova-derived SIINFEKL peptide, designated hereafter as H-2Kb/Ova pentamers, allowed to monitor the expansion of CD8+ lymphocytes in spleen and draining lymph nodes of rAAV-Ova challenged animals. This analysis revealed that both rAAV2/1-Ova and rAAV2/8-Ova injected in the gastrocnemius muscles induced a robust expansion of anti-Ova CD8+ lymphocytes (Figures 1B,C). Interestingly, the kinetics and intensity of T cell priming was found to be different when comparing the cells harvested from the draining lymph nodes and from the spleen. In the lymph nodes draining the injected muscles, anti-Ova CD8+ T cells expanded early after AAV challenge to reach around 2% of the CD8+ T cell compartment at day 7 post-injection (Figure 1C, left panel). In contrast, when analyzing splenocytes, anti-Ova CD8+ T cells were found to reach up to 10% of the total CD8+ subset but only at day 14 after i.m. AAV injection (Figure 1C, right panel). These results are consistent with a model in which the local cellular immune response measured in the draining lymph nodes would be induced by an rAAV leakage from the injected muscle and the consequent transduction of non-muscle cells such as dendritic cells, as shown for AAV5 (Xin et al., 2006) or AAV1 (Lu and Song, 2009) for instance. The systemic response detected in the spleen would rather reflect systemic immunization directed against the circulating sOva protein after it has been produced by transduced muscle cells. This interpretation is supported by the kinetics of sOva apparition and antibodies production (Figure 1D). Indeed, in the serum of AAV-treated mice, sOva reached a maximum at day 7 post-injection and rapidly declined thereafter (Figure 1D, green curves). Consistently, anti-Ova IgG appeared between day 7 and 12 after AAV i.m. injection, correlating with the disappearance of sOva in the serum after day 7 (Figure 1D, blue curves). The latter probably reflects the in vivo clearance of sOva following the formation of antigen–antibody immune complexes, although we cannot completely rule out that anti-Ova IgG antibodies may mask Ova epitopes in the immunoassay. Together, these data reveal that i.m. injection of rAAV encoding sOva is strongly immunogenic and provides a mouse model that is suited to stringently evaluate the efficacy of tolerance induction protocols.
CTLA-4/Ig but not PD-L1 Gene Transfer Prevents the Priming of the Immune Responses Following RAAV2/1-Ova Administration
We investigated the possibility to block immune responses directed against the transgene product by two distinct immunomodulatory strategies, one blocking lymphocyte priming (i.e., CTLA-4/Ig) and the other inhibiting the function of lymphocytes targeting transduced muscle fibers (i.e., activation of the PD-1 pathway by muscle gene transfer of PD-1 ligands). Indeed, we have previously shown in transgenic mice that express Ova as a neo-autoantigen in muscle that tolerant transgenic anti-Ova CD8+ T cells up-regulated PD-1, suggesting that PD-1 plays a role in the induction of tolerance to skeletal muscle-expressed Ag (Calbo et al., 2008). As expected, the anti-Ova CD8+ T cell response that followed an i.m. injection of rAAV2/1-Ova (Figures 1B,C) was accompanied by an up-regulation of PD-1 on CD8+ T cells that exhibit an activated CD44hi phenotype (Figure 2A). As activated T cells express PD-1 and PD-1 ligands inhibit their activation and function, we generated rAAV2/1-PD-L1 and rAAV2/1-PD-L2 vectors and first verified that they were able to efficiently transduce HEK cells in vitro (Figure 2B). The expected consequence of injecting these vectors i.m. is that they should not prevent T cell priming but rather inhibit the cytotoxic activity of T cells directed against muscle-expressed transgenic proteins.
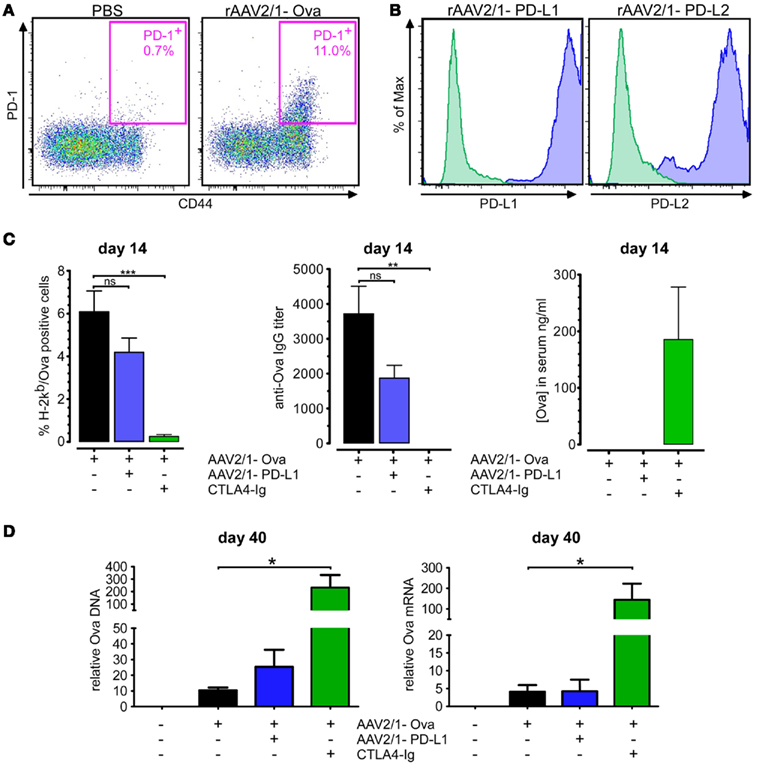
Figure 2. A single injection of CTLA-4/Ig dramatically inhibits immune responses and improves transgene persistence at early time points. (A) Mice were injected as in Figure 1 with 1 × 1011 vg rAAV2.1-Ova in the gastrocnemius muscles at day 0. Splenic CD8+ T cells were analyzed at day 14 by flow cytometry for expression of CD44 and PD-1. (B) rAAV2/1-PD-L1 and rAAV2/1-PD-L2 vectors were designed, produced, and tested for their capacity to transduce HEK-293 cells in vitro. For this, cells were analyzed by flow cytometry 3–5 days after their transduction with 1/10th dilution of the concentrated virus stocks. Controls correspond to unmanipulated HEK-293 parental cells stained with the same antibodies (green histograms). (C) The immunosuppressive potential of rAAV2/1-PD-L1 and CTLA-4/Ig were evaluated in vivo. Mice were injected with 1011 vg rAAV2/1-Ova in the gastrocnemius muscles and received or not a co-injection of 1011 vg rAAV2.1-PD-L1, or 200 μg of CTLA-4/Ig injected contemporaneously by the i.p. route. Blood samples were then collected 14 days later to analyze the percentage of anti-Ova CD8+ T cells, the level of anti-Ova IgG and the presence of sOva in the serum. (D) Gastrocnemius muscles were then collected at day 40, and Ova DNA and mRNA were quantified by qPCR and qRT-PCR.
To investigate the in vivo immunomodulatory potential of CTLA-4/Ig and rAAV2/1-PD-L1, we either administered 200 μg of CTLA-4/Ig i.p. or co-injected rAAV2/1-PD-L1 i.m. at the same time as rAAV2/1-Ova, and monitored the cellular and humoral immune response 14 day after. Results showed that even a single dose of CTLA-4/Ig was efficient to completely prevent the priming of anti-Ova CD8+ T cells (Figure 2C, left panel) and the apparition of anti-Ova IgG in the serum of treated animals (Figure 2C, middle panel). Accordingly, this immunosuppressive treatment allowed a sustained sOva production that could be assayed in the serum at day 14 whereas it was undetectable in controls (Figure 2C right panel). We concluded from these data that transient immunosuppression by a single dose of CTLA-4/Ig is very efficient to prevent the priming of the cellular and humoral response at early time points after AAV transduction.
In contrast, despite a slight but not statistically significant tendency to reduce immune responses, rAAV2/1-PD-L1 co-injected with rAAV2/1-Ova failed to provide the same effect on immune priming (Figure 2C, left and middle panels). Also, treatment with rAAV2/1-PD-L1 did not allow the persistence of sOva in the serum in agreement with the significant amount of anti-Ova IgG detected in the serum of these animals (Figure 2C). This expected result confirmed that local transduction of muscle cells with rAAV2/1-PD-L1 can not interfere with the systemic immune priming against sOva since activation of the PD-1/PD-L1 pathway was anticipated to regulate T cells functions rather than their priming.
We further analyzed the presence of Ova DNA and mRNA in muscles at day 40 in the different groups of treated mice by qPCR and qRT-PCR, respectively. This further confirmed that suppression of immune priming by CTLA-4/Ig promotes the persistence of transduced cells in vivo as measured by the detection of significantly higher levels of Ova DNA and mRNA in the muscles of treated animals (Figure 2D). By contrast, injection of rAAV2/1-PD-L1 did not significantly improve transgene persistence in these settings, in line with the prominent systemic immune response detected at day 14 (Figure 2D).
Combined Treatment with CTLA-4/Ig and rAAV2/1-PD-L1 Synergistically Acts to Improve Transgene Persistence and Expression
Whereas a single dose of CTLA-4/Ig was efficient to suppress the systemic immune response at day 14 (Figures 3A,B left panels), this was not the case when further monitoring the immune response at day 40. Indeed, anti-Ova CD8+ T cell expansion as well as anti-Ova IgG immune responses were readily detectable at day 40 in mice that had received rAAV2/1-Ova and CTLA-4/Ig at day 0 (Figures 3A,B). Therefore, early suppression of immune priming by a single dose of CTLA-4/Ig is not sufficient to permanently wipe out the immune responses against transgene product in these experimental conditions. Instead, continuous production of sOva by transduced muscle cells sensitizes the immune response at later time points when CTLA-4/Ig has been cleared from the circulation of treated mice.
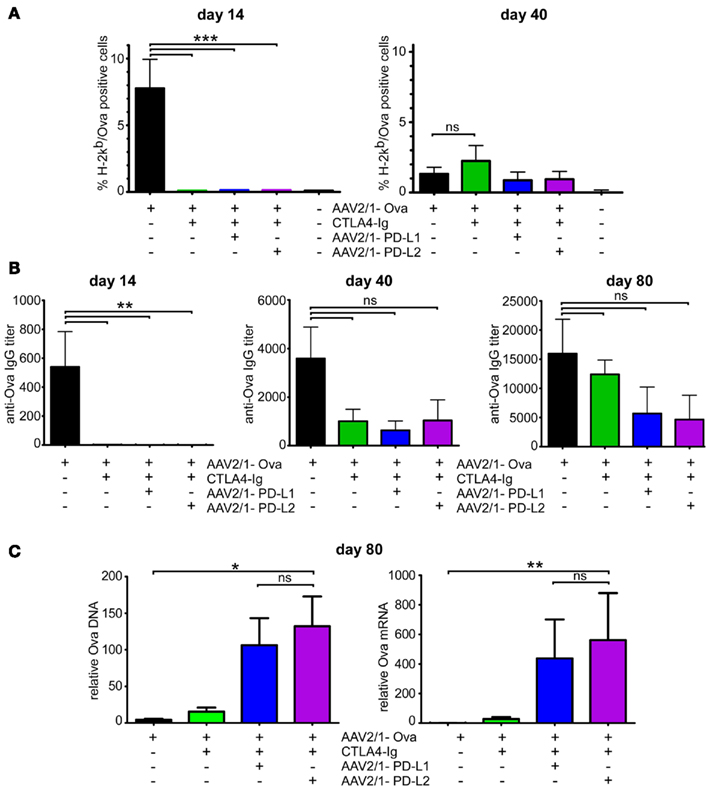
Figure 3. Combination therapies using CTLA-4/Ig and rAAV-PD-L1 or rAAV-PD-L2 significantly improve transgene persistence and transcription. Mice were injected with 1 × 1011 vg rAAV2/1-Ova in the gastrocnemius muscles at day 0 and received at the same time an immunomodulatory regimen consisting of either 200 μg of CTLA-4/Ig alone or the same amount of CTLA-4/Ig together with 1 × 1011 vg rAAV2/1-PD-L1 or rAAV2/1-PD-L2. (A) Blood samples were collected at day 14 and 40 to evaluate the percentage of CD8+ T cells specifically recognizing the immunodominant epitope derived from Ova by flow cytometric analyses. (B) Sera were collected at days 14, 40, and 80 to assay the level of anti-Ova IgG antibodies by ELISA. (C) Injected gastrocnemius muscles were collected at day 80 and analyzed for the presence of Ova DNA and mRNA by qPCR and qRT-PCR respectively.
We next tested whether rAAV2/1-PDL-L1 or rAAV2/1-PDL-L2 could synergize with CTLA-4/Ig to improve transgene tolerance at later time points when the initial CTLA-4/Ig monotherapy was evidently not efficient alone to completely block immune sensitization against sOva. This combined strategy may indeed target two non-redundant mechanisms of immunomodulation acting either on the APC side for CTLA-4/Ig or on the target tissue side for rAAV2/1-PD-L1 and rAAV2/1-PDL-L2. To test this possibility, groups of mice were transduced i.m. with rAAV2/1-Ova and also received at the same time either rAAV2/1-PD-L1 or rAAV2/1-PD-L2 i.m. and CTLA-4/Ig i.p. combination therapies. We then evaluated the cellular and humoral immune responses at different time points and finally evaluated the persistence of transduced muscle cells by quantification of Ova DNA and mRNA at day 80 (Figure 3).
Remarkably, whereas CTLA-4/Ig alone was inefficient, rAAV2/1-PD-L1 co-administered with CTLA-4/Ig at day 0 significantly improved transgene persistence, as measured by quantification of Ova DNA by qPCR (Figure 3C, left panel). Accordingly, mRNA analysis by qRT-PCR at day 80 revealed sustained transcription of the transgene in muscle (Figure 3C, right panel). Of note, rAAV2/1-PD-L2 appeared equally as effective as rAAV2/1-PD-L1 to provide this effect when combined with CTLA-4/Ig. Importantly, this protective effect occurred in a setting where neither CTLA-4/Ig alone nor the tested combination therapies could block immune response at days 40 and 80 (Figures 3A,B). To further attest the presence of functional anti-Ova cytotoxic T cells at day 80 (a time point when pentamer staining is not sensitive enough to detect anti-Ova T cells, not shown), we analyzed by ELISpot the capacity of CD8+ T cells to secrete IFNΓ when stimulated with the Ova-derived immunodominant SIINFEKL peptide. This assay revealed the presence of low numbers of functional anti-Ova CD8+ T cells in the three groups of animals that had received CTLA-4/Ig with or without rAAV-PD-L1 or rAAV-PD-L2, whereas the assay was negative for mice that did not receive rAAV-Ova and strongly positive for those that received rAAV-Ova in the absence of any immunoregulatory adjuvant therapy (Figure 4A). To definitively demonstrate that these IFNΓ-secreting T cells were functional in vivo, we performed adoptive transfer experiments to non-lethally irradiated syngenic recipients that were inoculated with Ova-bearing tumor cells. Whereas tumors rapidly developed in recipients that had received T cells from control mice that only received PBS, recipient mice receiving lymphocytes from rAAV2/1-Ova injected donors rapidly developed an anti-tumor immune response that resulted in complete tumor rejection (Figure 4B). In agreement with the lower numbers of anti-Ova CD8+ T cells detected by ELISpot, recipient mice that had received immune cells from donors treated with CTLA-4/Ig or the combined CTLA-4/Ig plus rAAV-PD-L1 or rAAV-PD-L2 therapy reacted with a slight delay but were also was also capable to efficiently control tumor growth (Figure 4B). Together, these results indicate that the cytotoxic activity of transgene product specific T cells that have escaped costimulation blockade can be functionally blocked by AAV-mediated gene transfer of PD-1 ligands in muscle tissues while they remain capable to reject tumor cells.
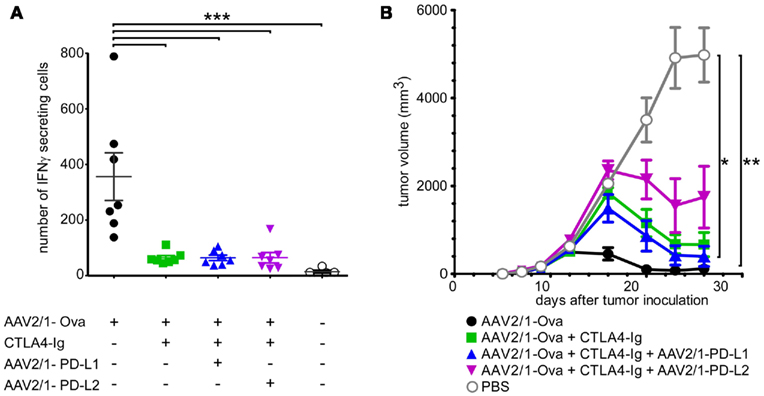
Figure 4. Detection of anti-Ova T cells in animals treated with combination therapies. Seven mice per group were injected with 1 × 1011 vg rAAV2/1-Ova in the gastrocnemius muscles at day 0 and received at the same time combination therapies as in Figure 3. Splenocytes were then harvested 80 days after and analyzed for the presence of lymphocytes capable to respond to Ova antigen. (A) Splenocytes were analyzed by ELISpot as described in the Section “Materials and Methods” for their capacity to secrete IFNΓ after in vitro restimulation with the Ova-derived immunodominant SIINFEKL peptide. (B) 5 × 107 Splenocytes from the same mice were also adoptively transferred to 5 Gy-irradiated syngenic mice inoculated 1 day after with Ova-bearing EG7 tumor cells. The capacity of transferred lymphocytes to reject the tumors was monitored three times per week by measuring the tumor size with a digital caliper.
Discussion
Gene therapy for muscular dystrophies and other monogenic diseases aims at achieving long-term expression of a functional form of an otherwise deficient gene in target tissues. In this context, the product of the therapeutic gene is structurally different from its abnormal or absent counterpart, and consequently viewed as non-self by the immune system. Additional signals from the viral vector may further strengthen adverse innate and adaptive immune reactions against the transgene product, while the vector itself can be targeted by antiviral pre-existing or acquired immunity. Although the presence of natural epitopes in the non-mutated regions of the protein or the occasional occurrence of mutation reversion in a minute population of patients’ cells (Klein et al., 1992) may yield some level of immunological tolerance, it remains that acquired immunity to the transgene product still represents one of the major obstacles to the success of gene therapy. Here, we provide the proof of principle that a combination immunoregulatory therapy targeting two non-redundant checkpoints of the immune response, i.e., priming and effector functions, can promote persistence of transduced target cells and transgene transcription thereof even when some cytotoxic T cells have escaped initial control.
A first strategy for the blockade of adaptive immunity is obviously to target costimulation pathways and therefore provide an early control on lymphocyte priming. Whereas several costimulatory pathways have now been identified, CD28-mediated T cell costimulation by APC expressing CD80/CD86 molecules from the B7 family is clearly prominent. Indeed, CD28 signaling strongly enhances T cell proliferation and survival, cytokine production and prevents induction of anergy after TCR-mediated activation (Boise et al., 1995). Consequently, anti-CD80 (B7.1) or anti-CD86 (B7.2) antibodies inhibit T cell priming and genetically engineered CD28-deficient T cells are strongly impaired in their capacity to expand (Green et al., 1994; Salomon and Bluestone, 2001). T cells activation also promotes up-regulation of the negative regulator CTLA-4, another ligand of CD80 and CD86 molecules, which transmits inhibitory signals that regulate activated T cell and provides a key homeostatic mechanism of the immune response (Fife and Bluestone, 2008). One of the early striking evidence of the key role of this molecule is that CTLA-4-deficient animal rapidly die from massive lymphoproliferative syndrome and dysregulation of immunity (Waterhouse et al., 1995).
One convenient way to pharmacologically block the CD28 costimulation pathway is to use the soluble CD80/CD86 ligand CTLA-4/Ig. This fusion protein prevents CD28 interaction with CD80 and CD86 molecules on naïve and activated T cells and therefore provides a strong and early inhibition on T cell priming (June et al., 1990; Lenschow et al., 1992; Larsen et al., 2005). CTLA-4/Ig may also induce a negative signalization on the APC side through the induction of indoleamine2,3-dioxygenase (IDO) which catalyses the production of inhibitory kynurenine from tryptophan (Grohmann et al., 2002). CTLA-4/Ig may also possibly provide some level of APC depletion in vivo, although this aspect has been poorly documented and should critically depend on the form of CTLA-4/Ig used. Importantly, CTLA-4/Ig is now clinically available for the treatment of diseases such as rheumatoid arthritis (Dumont, 2004) and it is thus reasonable to propose its evaluation in the field of gene therapy. Indeed, there has been experimental evidence that CTLA-4/Ig in combination with a monoclonal antibody to CD40L (MR1) in the context of gene therapy can block immune responses and allow vector readministration in the mouse (Halbert et al., 1998; Lorain et al., 2008). It is difficult to weight the relative roles of CTLA-4/Ig and MR1 in these experiments but a plausible hypothesis is that CTLA-4/Ig provides the strongest immunoregulatory contribution since MR1 alone has been reported to be less efficient in similar experimental conditions (Manning et al., 1998). The results reported herein are consistent with these earlier reports and provide definitive evidence that CTLA-4/Ig alone efficiently, but only transiently, blocks anti-transgene T cell priming.
One concern about using CTLA-4/Ig in gene therapy is that discontinuation of treatment exposes patients to immunization against the transgene product. The present results illustrate this point since initial CTLA-4/Ig efficiently blocks the anti-Ova cellular and humoral responses while sustained sOva production finally elicits this undesired response when CTLA-4/Ig is probably cleared from the circulation. Our experimental conditions have been purposely designed to model this very situation where the immune response escapes the initial immunoregulatory regimen in order to evaluate if a second strategy could provide additional protection of transduced cells, i.e., PD-L1/2 gene therapy.
The PD-1/PD-L1 pathway has recently been identified as a negative regulator of immunity (Sharpe et al., 2007). The PD-1 immunoreceptor is also a member of the CD28/CTLA-4 family which, upon interaction with either one of its two ligands PD-L1 or PD-L2, down-modulates TCR signaling, reduces cytokine production and affects T cell survival by recruiting the SHP-2 tyrosine phosphatase, thereby dampening the PI3K and Akt pathway (Francisco et al., 2010). In BALB/c mice, PD-1 deficiency leads to the spontaneous development of a lethal autoimmune cardiomyopathy (Nishimura and Honjo, 2001). This disease is mediated by autoantibodies directed against the cardiac muscle autoantigen troponin I (Okazaki et al., 2003). Hence, PD-1 controls the physiological tolerance to muscle autoantigens. Also, PD-L1 was recently shown to regulate CD8+ T cell-mediated muscle injury in a mouse model of myocarditis (Grabie et al., 2007). Several PD-1/PD-L1 dependant mechanisms may act synergistically to induce an effective effector T cell blockade and contribute to tolerance induction. As mentioned earlier, PD-1 engagement on the surface of T cell is known to significantly inhibit TCR signaling thereby preventing T cell activation and cytokine production. In addition to this mechanism, PD-L1 was recently shown to be a potent inducer of adaptive Tregs. Indeed, PD-L1 expressed on CD8+ dendritic cell subset plays an important role on the conversion of naive lymphocytes toward FoxP3+ regulatory T cells (Wang et al., 2008). Thus, engagement of PD-1 on T cells could induce cell-intrinsic tolerance (by blocking TCR signaling) as well as a dominant form of immunological tolerance by promoting the emergence of FoxP3+ adaptive Tregs. Of note, manipulation of the PD-1/PD-L1 axis is also used by some tumors cells to tolerize surrounding lymphocytes. Indeed, expression of PD-L1 on the surface of malignant cells has been suggested to represent a subversive strategy used by tumors to escape to immuno-surveillance (Iwai et al., 2002). Thus, we believe that up-regulation of the expression of PD-L1 on the surface of muscle cells by the means of gene transfer should be able to induce tolerization of the immune system by preventing cytotoxicity of PD-1-expressing activated lymphocytes. However, PD-L1 gene transfer alone was not sufficient in our model to prevent the immune response directed against transduced muscle fibers (Figure 2), suggesting that muscle expression of PD-L1, i.e., at distance from lymphoid compartments, is probably not capable of inducing sufficient adaptive FoxP3+ Tregs. Another explanation would be that PD-L1 expressed in muscle is not effective to block fully activated T cells that have received normal costimulatory signals (i.e., in the absence of CTLA-4/Ig co-administration). This interpretation would be in line with the finding that PD-1 inhibitory pathway could be overcome by CD28 costimulatory ligation in the presence of IL-2 (Freeman et al., 2000; Latchman et al., 2001; Carter et al., 2002; Ishida et al., 2002). Hence, the use of CTLA-4/Ig to block early CD28 costimulatory signaling may create favorable conditions where escaping T cells could become sensitive to PD-L1/2. This strengthens the notion that CTLA-4 and PD-1 signaling represent two distinct pathways acting synergistically to maintain tolerance (Fife and Bluestone, 2008). As shown herein, neither CTLA-4/Ig alone nor combination therapies could block immune response at days 40 and 80 (Figures 3 and 4). Our results are thus compatible with the notion that the use of rAAV2/1-PD-L1 or rAAV2/1-PD-L2 in combination with CTLA-4/Ig, does not completely inhibit the immune priming against sOva that most probably occurs at distance from the site of transduction (e.g., spleen and/or lymph nodes) but instead provides an additional protection by disarming lymphocytes within the target tissue.
It has been argued that the priming of T cells directed to the transgene product may be somehow defective after rAAV-mediated gene transfer (Lin et al., 2007a,b). However, in our experimental settings, transduced muscle fibers progressively disappear in the absence of immunoregulatory treatment (Figure 2D). Further, we show that even after CTLA-4/Ig alone or in combination therapies, an anti-Ova immune response is evidenced in ELISpot assays and in an in vivo model of tumor rejection upon adoptive transfer of T cells in recipient animals (Figure 4). Hence, forced up-regulation of PD-1 ligands in muscle cells yielded a level of protection of transduced cells from the cytotoxic assault of circulating anti-Ova lymphocytes and provided in combination therapy a means to prolong transgene persistence and transcription. Therefore, CTLA-4/Ig plus PD-L1/2 combination therapy represents a candidate approach to circumvent the bottleneck of immune responses directed toward the transgene product and may deserve further investigation for non-secreted transgenic protein models and in larger animals.
Contribution
Sahil Adriouch designed research, performed experiments, analyzed data, and co-wrote the manuscript; Anna Salvetti and Olivier Boyer designed research and co-wrote the manuscript; and Emilie Franck, Laurent Drouot, Carole Bonneau, and Nelly Jolinon performed experiments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Roland W. Herzog for kindly providing the rAAV-Ova vector, and Delphine Pannetier for excellent technical assistance. This work was supported by grants from the “Association Française contre les Myopathies (AFM).” Emilie Franck was recipient of a joint Ph.D. fellowship from INSERM and “Région Haute-Normandie.”
References
Bluestone, J. A., St Clair, E. W., and Turka, L. A. (2006). CTLA4Ig: bridging the basic immunology with clinical application. Immunity 24, 233–238.
Boise, L. H., Minn, A. J., Noel, P. J., June, C. H., Accavitti, M. A., Lindsten, T., and Thompson, C. B. (1995). CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3, 87–98.
Brockstedt, D. G., Podsakoff, G. M., Fong, L., Kurtzman, G., Mueller-Ruchholtz, W., and Engleman, E. G. (1999). Induction of immunity to antigens expressed by recombinant adeno-associated virus depends on the route of administration. Clin. Immunol. 92, 67–75.
Calbo, S., Delagreverie, H., Arnoult, C., Authier, F. J., Tron, F., and Boyer, O. (2008). Functional tolerance of CD8+ T cells induced by muscle-specific antigen expression. J. Immunol. 181, 408–417.
Carter, L., Fouser, L. A., Jussif, J., Fitz, L., Deng, B., Wood, C. R., Collins, M., Honjo, T., Freeman, G. J., and Carreno, B. M. (2002). PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur. J. Immunol. 32, 634–643.
Du, L., Zhao, G., Lin, Y., Sui, H., Chan, C., Ma, S., He, Y., Jiang, S., Wu, C., Yuen, K. Y., Jin, D. Y., Zhou, Y., and Zheng, B. J. (2008). Intranasal vaccination of recombinant adeno-associated virus encoding receptor-binding domain of severe acute respiratory syndrome coronavirus (SARS-CoV) spike protein induces strong mucosal immune responses and provides long-term protection against SARS-CoV infection. J. Immunol. 180, 948–956.
Dumont, F. J. (2004). Technology evaluation: abatacept, Bristol-Myers Squibb. Curr. Opin. Mol. Ther. 6, 318–330.
Fife, B. T., and Bluestone, J. A. (2008). Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 224, 166–182.
Fisher, K. J., Jooss, K., Alston, J., Yang, Y., Haecker, S. E., High, K., Pathak, R., Raper, S. E., and Wilson, J. M. (1997). Recombinant adeno-associated virus for muscle directed gene therapy. Nat. Med. 3, 306–312.
Francisco, L. M., Sage, P. T., and Sharpe, A. H. (2010). The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242.
Freeman, G. J., Long, A. J., Iwai, Y., Bourque, K., Chernova, T., Nishimura, H., Fitz, L. J., Malenkovich, N., Okazaki, T., Byrne, M. C., Horton, H. F., Fouser, L., Carter, L., Ling, V., Bowman, M. R., Carreno, B. M., Collins, M., Wood, C. R., and Honjo, T. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034.
Gao, G., Wang, Q., Calcedo, R., Mays, L., Bell, P., Wang, L., Vandenberghe, L. H., Grant, R., Sanmiguel, J., Furth, E. E., and Wilson, J. M. (2009). Adeno-associated virus-mediated gene transfer to nonhuman primate liver can elicit destructive transgene-specific T cell responses. Hum. Gene Ther. 20, 930–942.
Grabie, N., Gotsman, I., DaCosta, R., Pang, H., Stavrakis, G., Butte, M. J., Keir, M. E., Freeman, G. J., Sharpe, A. H., and Lichtman, A. H. (2007). Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 116, 2062–2071.
Green, J. M., Noel, P. J., Sperling, A. I., Walunas, T. L., Gray, G. S., Bluestone, J. A., and Thompson, C. B. (1994). Absence of B7-dependent responses in CD28-deficient mice. Immunity 1, 501–508.
Grohmann, U., Orabona, C., Fallarino, F., Vacca, C., Calcinaro, F., Falorni, A., Candeloro, P., Belladonna, M. L., Bianchi, R., Fioretti, M. C., and Puccetti, P. (2002). CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3, 1097–1101.
Halbert, C. L., Standaert, T. A., Wilson, C. B., and Miller, A. D. (1998). Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J. Virol. 72, 9795–9805.
Ishida, M., Iwai, Y., Tanaka, Y., Okazaki, T., Freeman, G. J., Minato, N., and Honjo, T. (2002). Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol. Lett. 84, 57–62.
Iwai, Y., Ishida, M., Tanaka, Y., Okazaki, T., Honjo, T., and Minato, N. (2002). Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. U.S.A. 99, 12293–12297.
June, C. H., Ledbetter, J. A., Linsley, P. S., and Thompson, C. B. (1990). Role of the CD28 receptor in T-cell activation. Immunol. Today 11, 211–216.
Kessler, P. D., Podsakoff, G. M., Chen, X., McQuiston, S. A., Colosi, P. C., Matelis, L. A., Kurtzman, G. J., and Byrne, B. J. (1996). Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc. Natl. Acad. Sci. U.S.A. 93, 14082–14087.
Klein, C. J., Coovert, D. D., Bulman, D. E., Ray, P. N., Mendell, J. R., and Burghes, A. H. (1992). Somatic reversion/suppression in duchenne muscular dystrophy (DMD): evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am. J. Hum. Genet. 50, 950–959.
Kuck, D., Lau, T., Leuchs, B., Kern, A., Muller, M., Gissmann, L., and Kleinschmidt, J. A. (2006). Intranasal vaccination with recombinant adeno-associated virus type 5 against human papillomavirus type 16 L1. J. Virol. 80, 2621–2630.
Larsen, C. P., Pearson, T. C., Adams, A. B., Tso, P., Shirasugi, N., Strobert, E., Anderson, D., Cowan, S., Price, K., Naemura, J., Emswiler, J., Greene, J., Turk, L. A., Bajorath, J., Townsend, R., Hagerty, D., Linsley, P. S., and Peach, R. J. (2005). Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am. J. Transplant. 5, 443–453.
Latchman, Y., Wood, C. R., Chernova, T., Chaudhary, D., Borde, M., Chernova, I., Iwai, Y., Long, A. J., Brown, J. A., Nunes, R., Greenfield, E. A., Bourque, K., Boussiotis, V. A., Carter, L. L., Carreno, B. M., Malenkovich, N., Nishimura, H., Okazaki, T., Honjo, T., Sharpe, A. H., and Freeman, G. J. (2001). PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268.
Lenschow, D. J., Zeng, Y., Thistlethwaite, J. R., Montag, A., Brady, W., Gibson, M. G., Linsley, P. S., and Bluestone, J. A. (1992). Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science 257, 789–792.
Lin, J., Zhi, Y., Mays, L., and Wilson, J. M. (2007a). Vaccines based on novel adeno-associated virus vectors elicit aberrant CD8+ T-cell responses in mice. J. Virol. 81, 11840–11849.
Lin, S. W., Hensley, S. E., Tatsis, N., Lasaro, M. O., and Ertl, H. C. (2007b). Recombinant adeno-associated virus vectors induce functionally impaired transgene product-specific CD8+ T cells in mice. J. Clin. Invest. 117, 3958–3970.
Lorain, S., Gross, D. A., Goyenvalle, A., Danos, O., Davoust, J., and Garcia, L. (2008). Transient immunomodulation allows repeated injections of AAV1 and correction of muscular dystrophy in multiple muscles. Mol. Ther. 16, 541–547.
Lu, Y., and Song, S. (2009). Distinct immune responses to transgene products from rAAV1 and rAAV8 vectors. Proc. Natl. Acad. Sci. U.S.A. 106, 17158–17162.
Manning, W. C., Paliard, X., Zhou, S., Pat Bland, M., Lee, A. Y., Hong, K., Walker, C. M., Escobedo, J. A., and Dwarki, V. (1997). Genetic immunization with adeno-associated virus vectors expressing herpes simplex virus type 2 glycoproteins B and D. J. Virol. 71, 7960–7962.
Manning, W. C., Zhou, S., Bland, M. P., Escobedo, J. A., and Dwarki, V. (1998). Transient immunosuppression allows transgene expression following readministration of adeno-associated viral vectors. Hum. Gene Ther. 9, 477–485.
Manno, C. S., Pierce, G. F., Arruda, V. R., Glader, B., Ragni, M., Rasko, J. J., Ozelo, M. C., Hoots, K., Blatt, P., Konkle, B., Dake, M., Kaye, R., Razavi, M., Zajko, A., Zehnder, J., Rustagi, P. K., Nakai, H., Chew, A., Leonard, D., Wright, J. F., Lessard, R. R., Sommer, J. M., Tigges, M., Sabatino, D., Luk, A., Jiang, H., Mingozzi, F., Couto, L., Ertl, H. C., High, K. A., and Kay, M. A. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347.
Mays, L. E., and Wilson, J. M. (2011). The complex and evolving story of T cell activation to AAV vector-encoded transgene products. Mol. Ther. 19, 16–27.
McCaffrey, A. P., Fawcett, P., Nakai, H., McCaffrey, R. L., Ehrhardt, A., Pham, T. T., Pandey, K., Xu, H., Feuss, S., Storm, T. A., and Kay, M. A. (2008). The host response to adenovirus, helper-dependent adenovirus, and adeno-associated virus in mouse liver. Mol. Ther. 16, 931–941.
Mingozzi, F., and High, K. A. (2011a). Immune responses to AAV in clinical trials. Curr. Gene Ther. 11, 321–330.
Mingozzi, F., and High, K. A. (2011b). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 12, 341–355.
Mingozzi, F., Maus, M. V., Hui, D. J., Sabatino, D. E., Murphy, S. L., Rasko, J. E., Ragni, M. V., Manno, C. S., Sommer, J., Jiang, H., Pierce, G. F., Ertl, H. C., and High, K. A. (2007). CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13, 419–422.
Moore, M. W., Carbone, F. R., and Bevan, M. J. (1988). Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 54, 777–785.
Nayak, S., and Herzog, R. W. (2011). Progress and prospects: immune responses to viral vectors. Gene Ther. 17, 295–304.
Nishimura, H., and Honjo, T. (2001). PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 22, 265–268.
Okazaki, T., Tanaka, Y., Nishio, R., Mitsuiye, T., Mizoguchi, A., Wang, J., Ishida, M., Hiai, H., Matsumori, A., Minato, N., and Honjo, T. (2003). Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 9, 1477–1483.
Salomon, B., and Bluestone, J. A. (2001). Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu. Rev. Immunol. 19, 225–252.
Salvetti, A., Oreve, S., Chadeuf, G., Favre, D., Cherel, Y., Champion-Arnaud, P., David-Ameline, J., and Moullier, P. (1998). Factors influencing recombinant adeno-associated virus production. Hum. Gene Ther. 9, 695–706.
Sharpe, A. H., Wherry, E. J., Ahmed, R., and Freeman, G. J. (2007). The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 8, 239–245.
Wallace, P. M., Rodgers, J. N., Leytze, G. M., Johnson, J. S., and Linsley, P. S. (1995). Induction and reversal of long-lived specific unresponsiveness to a T-dependent antigen following CTLA4Ig treatment. J. Immunol. 154, 5885–5895.
Wang, L., Dobrzynski, E., Schlachterman, A., Cao, O., and Herzog, R. W. (2005). Systemic protein delivery by muscle-gene transfer is limited by a local immune response. Blood 105, 4226–4234.
Wang, L., Pino-Lagos, K., de Vries, V. C., Guleria, I., Sayegh, M. H., and Noelle, R. J. (2008). Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 105, 9331–9336.
Waterhouse, P., Penninger, J. M., Timms, E., Wakeham, A., Shahinian, A., Lee, K. P., Thompson, C. B., Griesser, H., and Mak, T. W. (1995). Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988.
Xiao, X., Li, J., and Samulski, R. J. (1996). Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J. Virol. 70, 8098–8108.
Xin, K. Q., Mizukami, H., Urabe, M., Toda, Y., Shinoda, K., Yoshida, A., Oomura, K., Kojima, Y., Ichino, M., Klinman, D., Ozawa, K., and Okuda, K. (2006). Induction of robust immune responses against human immunodeficiency virus is supported by the inherent tropism of adeno-associated virus type 5 for dendritic cells. J. Virol. 80, 11899–11910.
Zaiss, A. K., Liu, Q., Bowen, G. P., Wong, N. C., Bartlett, J. S., and Muruve, D. A. (2002). Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 76, 4580–4590.
Zaiss, A. K., and Muruve, D. A. (2008). Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther. 15, 808–816.
Keywords: gene therapy, AAV vectors, costimulation, CTLA-4, PD-1, PD-L1, PD-L2, tolerance
Citation: Adriouch S, Franck E, Drouot L, Bonneau C, Jolinon N, Salvetti A and Boyer O (2011) Improved immunological tolerance following combination therapy with CTLA-4/Ig and AAV-mediated PD-L1/2 muscle gene transfer. Front. Microbio. 2:199. doi: 10.3389/fmicb.2011.00199
Received: 26 July 2011; Paper pending published: 15 August 2011;
Accepted: 06 September 2011; Published online: 29 September 2011.
Edited by:
Roland W. Herzog, University of Florida, USACopyright: © 2011 Adriouch, Franck, Drouot, Bonneau, Jolinon, Salvetti and Boyer. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Sahil Adriouch and Olivier Boyer, INSERM U905, Faculty of Medicine and Pharmacy, 22 boulevard Gambetta, F-76000 Rouen, France. e-mail:c2FoaWwuYWRyaW91Y2hAdW5pdi1yb3Vlbi5mcg==;b2xpdmllci5ib3llckBjaHUtcm91ZW4uZnI=