 Xiaopei Huang1 Yiping Yang1,2*
Xiaopei Huang1 Yiping Yang1,2*- 1 Department of Medicine, Duke University Medical Center, Durham, NC, USA
- 2 Department of Immunology, Duke University Medical Center, Durham, NC, USA
Gene therapy with recombinant viral vectors such as adenovirus and adenovirus-associated virus holds great promise in treating a wide range of diseases because of the high efficiency with which the viruses transfer their genomes into host cells in vivo. However, the activation of the host immune responses remains a major hurdle to successful gene therapy. Studies in the past two decades have elucidated the important role co-stimulation plays in the activation of both T and B cells. This review summarizes our current understanding of T cell co-stimulatory pathways, and strategies targeting these co-stimulatory pathways in gene therapy applications as well as potential future directions.
Introduction
Virus-based vectors such as adenovirus and adenovirus-associated virus (AAV) have been widely used in gene therapy applications due to their high efficiency of transduction into a variety of cells in vivo. Indeed, several impressive results using viral vector-based gene therapy have been reported in humans (Ashtari et al., 2011; Fischer et al., 2011). However, one major barrier to successful gene therapy with these viral vectors is the host immune responses to both viral vectors and the transgenes (Huang and Yang, 2009). Our understanding of the mechanisms of immune responses to viral vectors has greatly improved over the past two decades, and as a result, strategies have been developed to manipulate various immune pathways in order to regulate the immune responses to viral vectors. One promising therapeutic strategy is targeting the co-stimulatory pathways to modulate the function of T and B cells.
The original two-signal hypothesis (Lafferty et al., 1974, 1980) proposes that full T cell activation requires two signals: antigen-specific T cell recognition (signal 1) provided by the engagement of the TCR–CD3 complex with a processed antigenic peptide bound to the major histocompatibility complex (MHC) molecule on the surface of an antigen presenting cell (APC), and an antigen-non-specific co-stimulatory signal (signal 2) provided by the interactions between cell surface molecules on the APC and the T cell. Indeed, early work by Jenkins and Schwartz showed that in the absence of the co-stimulatory signal, T cells are rendered anergic, a state marked by the inability of T cells to respond to subsequent antigenic stimulation, leading to a model that co-stimulation plays a critical role in controlling the fate of T cell responses: activation or anergy (Schwartz et al., 1989; Jenkins et al., 1990). Since then, these seminal studies have stimulated tremendous growth of the co-stimulation field. In addition to signals 1 and 2, we now appreciate that other signals also participate in determining the fate of T cell activation: some of these signals are stimulatory and others are inhibitory (Sharpe, 2009). Furthermore, some other signals are required to promote the differentiation into different T helper cell subsets (Pipkin and Rao, 2009; Simpson et al., 2010).
Because of the importance of co-stimulation in determining the outcome of the immune response, manipulation of various co-stimulatory pathways to regulate host immune responses is of therapeutic interest. In this review, we first summarize the current knowledge of T cell co-stimulatory pathways. We then focus on strategies targeting co-stimulatory pathways and their potential implications in viral vector-mediated gene therapy.
T Cell Co-Stimulatory Pathways
Several co-stimulatory pathways have been characterized including both activating and inhibitory pathways (Figure 1). The positive activating signals are balanced by the negative inhibitory signals to achieve optimal control of T and B cell activation. The activating pathways such as B7 and CD28, CD40 ligand (CD40L) and CD40, inducible co-stimulatory molecule (ICOS) and ICOS-ligand (ICOS-L), and OX40 (CD134) and its ligand, OX40L, are critical for the activation of T and B cells, whereas inhibitory pathways such as B7 and cytotoxic T lymphocyte antigen-4 (CTLA-4), and programmed cell death-1 (PD-1) and PD ligand (PD-L) downregulate T cell activation. Here we mainly focus on the activating co-stimulatory pathways.
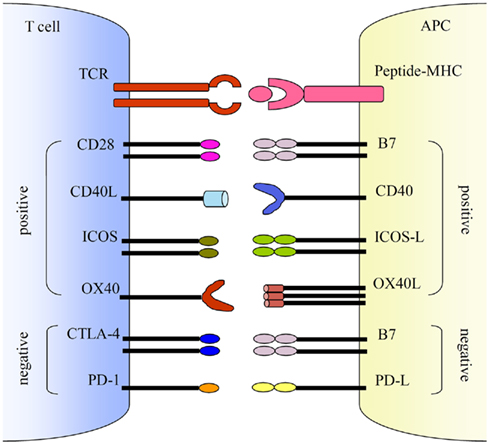
Figure 1. Co-stimulatory pathways. Co-stimulations either enhance or down-regulate T cell activation following the initial TCR and peptide-MHC ligation. Positive co-stimulatory pathways include B7–CD28, CD40L–CD40, ICOS–ICOS-L, and OX40–OX40L. Negative co-stimulatory pathways include B7–CTLA-4 and PD-1–PD-L.
B7-CD28/CTLA-4 Pathway
The CD28 glycoprotein, a member of the immunoglobulin superfamily, is expressed on the T cell membrane as disulfide-linked homodimers. It is expressed on the surface of 90% of human CD4+ T cells and 50% of human CD8+ T cells, and nearly 100% of murine T cells. The ligands for CD28 are B7-1 (CD80) and B7-2 (CD86), also members of the immunoglobulin superfamily, which are expressed by APCs such as DCs, macrophages, and activated B cells. Although B7-1 and B7-2 are co-expressed and share similar overall structure, they differ in their temporal responses to stimuli, with B7-2 induction occurring earlier and usually at much higher level than B7-1 in activated B cells and DCs (Hathcock et al., 1994). The expression of B7-1 and B7-2 is controlled by cytokines and cell–cell interactions. IL-4 is a potent inducer of B7-2 on B cells, whereas IFN-γ and IL-10 differentially regulate B7 expression, depending on cell types (Valle et al., 1991; Stack et al., 1994). Studies have shown that signals transmitted through the MHC class II cytoplasmic domain, which is required for effective antigen presentation, induce B7 expression on B cells (Nabavi et al., 1992).
Mice deficient in CD28 or treated with antagonists of CD28 exhibit substantially reduced proliferative responses (Green et al., 1994). Similarly, mice deficient in both B7-1 and B7-2 have compromised T cell-mediated responses (Borriello et al., 1997; Mcadam et al., 1998), whereas addition of B7 transfectants could augment T cell proliferation and IL-2 production induced with anti-CD3 or PMA in a CD28-dependent manner (Gimmi et al., 1991; Linsley et al., 1991). B7/CD28-mediated signaling enhances the production of IL-2 and IL-2 receptor in a nuclear factor (NF)-κB dependent manner, and upregulates the expression of other cytokines such as IL-1, IL-4, IL-5, TNF, and IFN-γ (reviewed in Lenschow et al., 1996; Kane et al., 2002); accelerates resting T cell entry into and progression through the cell cycle; plays a critical role in the development and differentiation of Th1 and Th2 T cell subsets (Van Der Pouw-Kraan et al., 1992; Seder et al., 1994; King et al., 1995); prevents anergy, and promotes T cell survival by enhancing the expression of the anti-apoptotic proteins such as BCL-xL. Recent studies have reported chromatin structural changes within minutes following T cell activation, and induction of DNA demethylation of IL-2 gene as early as 20 min after TCR/CD28 stimulation, suggesting a role for CD28 signaling in these early nuclear events, which are critical to the ensuing processes of cell proliferation and differentiation (Zhao et al., 1998; Acuto and Michel, 2003; Bruniquel and Schwartz, 2003).
Cytotoxic T lymphocyte antigen-4, also known as CD152, is expressed on the T cell membrane as disulfide-linked homodimers. Like CD28 and B7, they are also members of the immunoglobulin superfamily. The ligands for CTLA-4 are also B7-1 and B7-2, but its binding affinity for B7 molecules is about 20-fold greater than that of CD28. Although CTLA-4 and CD28 share sequence homology, CTLA-4 acts antagonistically of CD28, and delivers an inhibitory signal that down-regulates T cell activation. CTLA-4 knockout mice manifest lymphoproliferative disorder which is lethal by 3–4 weeks after birth, supporting the notion that CTLA-4 acts as a negative regulator of T cell activation and is vital for the control of lymphocyte homeostasis (Waterhouse et al., 1995). While CD28 is expressed by both resting and activated T cells, CTLA-4 is only expressed on activated T cells at much lower levels. CTLA-4 induction can be detected 24 h after T cell stimulation, with cell surface expression peaking around 2 days post activation, and returning to background levels by day 4 (Linsley et al., 1992; Walunas et al., 1994). Interestingly, CD28/B7-mediated signaling upregulates the expression of CTLA-4 which effectively competes with CD28 for B7-1/B7-2 at later stages of the immune response to counter T cell activation, suggesting a mechanism for achieving balance between positive and negative stimulation.
CD40L–CD40 Pathway
The CD40 glycoprotein is a member of the tumor necrosis factor-receptor (TNFR) superfamily. It is expressed on APCs such as B cells, DCs, activated macrophages as well as Langerhan cells, endothelial cells, and thymic epithelial cells (Buhlmann and Noelle, 1996). The ligand for CD40 (CD40L), a member of the TNF family, is preferentially expressed on the surface of activated CD4+ T cells in both humans and mice. Early studies have established an important role for CD40L–CD40 interactions in B cell activation and proliferation, as well as immunoglobulin (Ig) class switching (Allen et al., 1993; Disanto et al., 1993). For example, the underlying cause for a severe form of immunodeficiency, X-linked hyper IgM syndrome (HIGM) is a mutation in CD40L (Allen et al., 1993; Disanto et al., 1993). Patients with this disorder have elevated levels of IgM, but little to none IgG or IgA, and absent germinal centers.
In addition to the role in the humoral response, two seminal reports also demonstrate a critical role for the CD40L–CD40 pathway in the activation of T cell responses (Grewal et al., 1996; Yang and Wilson, 1996). In a murine model of experimental allergic encephalomyelitis (EAE), CD4+ T cell priming is defective in CD40L-deficient mice. These mice failed to develop EAE after priming with antigen and produced little to no IL-4 and IFN-γ (Grewal et al., 1996). Similarly, in a murine model of liver-directed gene transfer with adenoviral vectors, CD40L–CD40 signaling on APCs is essential for the development of CD8+ T cell response to adenoviral vector (Yang and Wilson, 1996). This is mediated by upregulating the expression of B7 molecules on APCs, which in turn promote the B7–CD28 pathway, leading to T cell activation (Yang and Wilson, 1996). Indeed, subsequent studies have further delineated that CD40–CD40L interactions between DCs and CD4+ T cells provide DCs with “licensing” in the activation of CD8+ T cells (Bennett et al., 1998; Ridge et al., 1998; Schoenberger et al., 1998).
The broad spectrum of cells that express CD40 indicates that CD40L–CD40 interactions may exert regulatory effects at multiple levels. CD40L–CD40 interactions also result in the production of IL-12, which promotes the differentiation of Th1 immunity (Trinchieri, 1994). Furthermore, the interactions between CD40L on activated T cells and CD40 on vascular endothelial cells can stimulate the vasculature at sites of inflammation and may play a role in inflammatory responses (Hollenbaugh et al., 1995).
ICOS Pathway
ICOS, a third member of the CD28/CTLA-4 family, is expressed on activated T cells, but very little on resting naïve T cells. ICOS binds specifically to its ligand (ICOS-L), also known as B7-related protein-1 (B7RP-1), which is expressed constitutively on B cells. In vivo, the interaction of ICOS with ICOS-L is critical for T cell-dependent B cell responses (Mcadam et al., 2001; Tafuri et al., 2001). In the absence of ICOS, germinal center formation is impaired and immunoglobulin class switching is defective.
In addition to providing help for B cells, ICOS plays an important role in the differentiation of unpolarized CD4+ T cells into Th1, Th2, Th17, and Treg lineages (Simpson et al., 2010). It has been shown that ICOS can promote both Th1 and Th2 responses. However, ICOS and ICOS-L interaction during the early stages of T cell differentiation favors Th2 response (Nurieva et al., 2003). Although ICOS is not required for Th17 differentiation during primary responses, it plays a pivotal role in promoting Th17 compartment by upregulating IL-23R during secondary responses (Bauquet et al., 2009). Recent studies also demonstrate a critical role of ICOS in maintaining the homeostasis of CD4 + Foxp3 + Treg (Burmeister et al., 2008; Ito et al., 2008).
PD-1 Pathway
Programmed cell death-1, an inhibitory receptor in the CD28 family, is expressed on CD4 and CD8 T cells, B cells, NKT cells, and some DC subsets upon activation (Keir et al., 2008). PD-1 has two ligands, PD-L1 and PD-L2. Upon ligation to either PD-L1 or PD-L2, PD-1 attenuates TCR signaling, leading to suppression of T cell responses. Indeed, in a model of chronic LCMV infection, PD-1 is overexpressed on the exhausted T cells and blockade of PD-1 and PD-L1 interaction led to the functional restoration of these exhausted T cells (Barber et al., 2006). Furthermore, the PD-1 pathway plays an important role in regulating immune responses to acute infections (Brown et al., 2010). In addition to providing inhibitory signals to T cells, a recent study has also revealed an essential role for PD-1 signaling in germinal center B cell survival and the formation and affinity of long-lived plasma cells (Good-Jacobson et al., 2010).
Targeting the B7–CD28/CTLA-4 Pathway
Early studies in transplant setting have demonstrated that blockade of the CD28/B7 co-stimulation with CTLA-4Ig, a soluble fusion protein consisting of the extracellular domains of CTLA-4 and the constant region of the IgG1 heavy chain, could cause host T cells directed against the grafted tissue anergic, therefore leading to the long-term survival of the graft (Lenschow et al., 1993). CTLA-4Ig acts through competition with CD28 for its ligands, B7-1 and B7-2, disrupting the co-stimulation required for complete T cell activation. In addition, studies using a combination of anti-B7-1 and anti-B7-2 monoclonal antibodies (mAbs) in an allogeneic pancreatic islet transplant setting showed this approach selectively delayed CD4+ T cell infiltration into the graft, leading to inhibition of transplant rejection (Lenschow et al., 1995). Similarly, CTLA-4Ig has been used successfully to suppress T cell responses in animal models of autoimmunity (Dall’era and Davis, 2004). These studies provide evidence that blocking co-stimulatory pathway is a viable strategy in inducing T cell anergy.
A number of studies have also investigated the effect of blocking the B7–CD28 co-stimulatory pathway in viral vector-mediated gene therapy. Administration of CTLA-4Ig blocked the formation of anti-β-galactosidase antibodies following retrovirus-mediated gene transfer to the liver (Puppi et al., 2004). Similarly, treatment with CTLA-4Ig resulted in more stable transgene expression after retrovirus-mediated gene therapy in mucopolysaccharidosis I mice (Ma et al., 2007). Furthermore, potent immunosuppression has been observed with a high-affinity variant of CTLA-4Ig, LEA29Y (belatacept; Larsen et al., 2005). In a mouse model of Duchenne muscular dystrophy (DMD), adenovirus vectors carrying murine CTLA-4Ig (AdmCTLA-4Ig) was coadministered with an adenoviral vector carrying a full-length murine dystrophin cDNA (AdmDys) into skeletal muscle (Jiang et al., 2004a). Stable expression of dystrophin in skeletal muscle up to 8 weeks was achieved, in contrast to the control group without CTLA-4Ig where dystrophin expression was significantly diminished within 8 weeks. CTL response to adenoviral vector was markedly suppressed in the group receiving co-administration of AdmCTLA-4Ig at 2-week time point, and cytokine production such as IFN-γ, IL-4, and IL-6 were much lower than the control group as well. No dystrophin-specific CTL response was observed in either group during the 2-week period. Neutralizing antibody against adenoviral vector was detected in both groups, with highest levels found in the control group, indicating that CTLA-4Ig partially blocked the humoral response against adenoviral vectors.
Similar strategies employing CTLA-4Ig to block the B7–CD28 co-stimulatory pathway have been investigated in a murine model of type II collagen-induced arthritis (Ijima et al., 2001). However, in other model systems, the effect of AdmCTLA-4Ig has been modest (Laumonier et al., 2003), suggesting other strategies in combination with CTLA-4Ig may be needed to achieve successful therapeutic effect.
Targeting the CD40/CD40 Ligand Pathway
The critical role of the CD40L–CD40 pathway in activation of both T and B cells suggests interfering with this pathway may prevent both cellular and humoral immune responses to virus, which is critical for stable transgene expression and the repeated administration of the viral vectors. Indeed, initial studies revealed that a transient blockade of CD40 signaling with an antibody to CD40L infused at the time of vector administration resulted in stable transgene expression and diminished production of neutralizing antibodies in a murine model of adenovirus-mediated gene transfer to the liver and the lungs (Yang et al., 1996). Later, anti-mouse CD40L mAb in combination with CTLA-4Ig has been shown to further prolong adenovirus-mediated transgene expression after both primary and secondary vector delivery (Kay et al., 1995). Similarly, the combined CD40L blockade and CTLA-4Ig approach also proved effective in a murine model of retrovirus-mediated gene therapy for mucopolysaccharidosis (Ma et al., 2007). More recently, the dual blockade strategy has been shown to act synergistically to prevent antigen-specific immune responses in non-human primates, leading to stable expression of transgene and diminished neutralizing antibody production, and in some cases allowing for repeated administration of adenovirus (Haegel-Kronenberger et al., 2004).
The effectiveness of the combined antibody blockade strategy led to the evaluation for the effect of systemic administration of AdmCTLA-4Ig and AdmCD40Ig (adenoviral vector carrying murine CD40Ig) in murine models of gene delivery to the muscle (Jiang et al., 2004b) and the liver (Schowalter et al., 1997). These studies showed that systemic delivery of both AdmCTLA-4Ig and AdmCD40Ig was required to inhibit the production of anti-adenoviral vector neutralizing antibody, and that AdmCTLA-4Ig alone was insufficient. This may be due to insufficient circulating level of mCTLA-4Ig to prevent anti-adenoviral capsid protein neutralizing antibody production, and that addition of circulating levels of mCD40Ig decreases B cell activation by T cells hence leading to diminished neutralizing antibody production.
Taken together, these data lend support to the effectiveness of the combined CD40 and CD28 blockade strategy in viral vector-mediated gene therapy in animal models. However, the efficacy of this approach in human gene therapy requires further exploration in clinical trials.
Targeting the ICOS/ICOS-L Pathway
T cell-dependent B cell responses are critically dependent on the interaction of ICOS with ICOS-L, which promotes germinal center formation and immunoglobulin class switching in vivo (Mcadam et al., 2001; Tafuri et al., 2001). It has been shown that anti-ICOS alone or in combination with CD40Ig or anti-CD40L can induce tolerance to islet allografts (Nanji et al., 2006) and inhibit allograft rejection in murine models of transplantation (Guillonneau et al., 2005; Taylor et al., 2005). In a murine model of non-viral mediated gene therapy for hemophilia A, blockade of the ICOS–ICOS-L pathway by anti-ICOS antibody alone was effective in inhibiting the formation of anti-factor VIII antibodies following plasmid DNA-mediated gene transfer (Peng et al., 2008). In this study, anti-ICOS treatment also resulted in elevation of Treg numbers and their suppressive activity (Peng et al., 2008). However, the efficacy of ICOS blockade has not been tested in animal models of viral vector-mediated gene transfer.
Novel Strategies for Targeting the Co-Stimulatory Pathway
Recent advances in our understanding of the role of the innate immunity in adaptive immune responses suggest potential novel strategies for targeting the co-stimulatory pathways. The innate immune system is the first line of defense against invading pathogens through recognition of pathogen-associated molecular patterns (PAMPs) by a set of receptors called pattern recognition receptors (PRRs; Huang and Yang, 2009). Among the PRRs identified to date, the best studied is the toll-like receptor (TLR) family. Thirteen TLRs have been identified in mammals, with each recognizing a unique set of PAMPs. Most TLRs signal through the myeloid differentiating factor 88 (MyD88)-dependent pathway, initiating the downstream signaling cascade leading to the induction of proinflammatory response characterized by the production of cytokines and chemokines (Akira et al., 2006). In addition, TLR pathways are critical for the ensuing adaptive immune responses by promoting DC maturation and function through upregulation of co-stimulatory molecules (Iwasaki and Medzhitov, 2004; Huang and Yang, 2010).
Recent studies have demonstrated that the innate immune response to AAV is mediated by TLR9 (Zhu et al., 2009; Martino et al., 2011). AAV mainly activates the TLR9–MyD88 pathway in pDCs, leading to the production of type I IFNs (Zhu et al., 2009). In vivo, the TLR9–MyD88 pathway is crucial to the activation of CD8 T cell response to both the transgene product and the AAV vector, leading to the loss of transgene expression and the generation of neutralizing antibodies to transgene product and AAV (Zhu et al., 2009). Thus, the TLR9–MyD88-type I IFN pathway plays a crucial role in promoting the adaptive immune responses to AAV.
Stimulation of TLR9 with its ligand, CpG-DNA leads to upregulation of surface expression of co-stimulatory molecules, such as CD40, B7-1/B7-2, and MHC class II on cDCs and macrophages (Hemmi et al., 2003). Therefore, DC maturation and Th1-like cytokine induced by TLR9 stimulation result in efficient and robust activation of Th1 and CD8 cytotoxic T lymphocytes (CTL; Lipford et al., 2000; Horkheimer et al., 2009). Type I IFNs, in addition to their direct anti-viral effects (Vaidya and Cheng, 2003), have also been shown to induce DC maturation by upregulating the expression of co-stimulatory molecules, which in turn promotes T cell responses (Hoebe et al., 2003). DCs matured by type I IFNs promote cross-priming of virus-specific CD8 T cells (Le Bon et al., 2003). Type I IFN can also promote effector function of virus-specific T cells (Cousens et al., 1999), and enhance the survival of activated T cells (Marrack et al., 1999). Furthermore, type I IFN signaling on DCs is important for the production of virus-specific IgM, whereas the generation of protective neutralizing antibodies to adenovirus critically depends on the type I IFN signaling on both CD4 T cells and B cells (Zhu et al., 2007).
A critical role for the TLR9 signaling pathway in the upregulation of co-stimulatory molecules and the induction of adaptive immune response to AAV suggests that strategies targeted to interfere with this signaling pathway may diminish T cell responses to the AAV vector and the transgene product, leading to prolonged transgene expression and reduction in inflammation, improving the safety and efficacy of AAV vectors for gene therapy in humans. Furthermore, blockade of the TLR9 signaling pathway will also likely inhibit antibody responses to the transgene product as well as the AAV vector.
One way to interfere with the TLR9–MyD88-type I IFN signaling pathway is to use antagonists specific to TLR9 such as inhibitory ODN to block the TLR9–MyD88 pathway (Zhu et al., 2009; Martino et al., 2011). Alternatively, blockade of type I IFNs by administering neutralizing antibodies to IFN-α and IFN-β may also prove effective as type I IFNs are required for adaptive immune responses to AAV (Zhu et al., 2009).
Future Perspectives
In this review, we have summarized the role of various T cell co-stimulatory pathways in the activation of both T and B cells and strategies targeting these pathways to improve the outcome of viral vector-mediated gene therapy. The majority of the studies so far are focused on targeting the B7–CD28 and the CD40L–CD40 pathways. It may be beneficial to investigate the effect of blocking other positive co-stimulatory pathways such as ICOS and ICOS-L, and OX40 and OX40L. In addition, strategies for stimulating the negative co-stimulatory pathways such as PD-1 and PD-L may prove effective. Given that the combined blockade of CD40 and CD28 pathways is more effective than the blockade of a single pathway alone, future studies should focus on targeting multiple pathways including both the positive and negative co-stimulatory pathways. In this regard, since the innate immunity is critical in regulating multiple co-stimulatory pathways, strategies targeting innate immune pathways may warrant further investigation. Ultimately, the utility of these strategies in improving the outcome of gene therapy with viral vectors will need to be tested in human clinical trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acuto, O., and Michel, F. (2003). CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat. Rev. Immunol. 3, 939–951.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Allen, R. C., Armitage, R. J., Conley, M. E., Rosenblatt, H., Jenkins, N. A., Copeland, N. G., Bedell, M. A., Edelhoff, S., Disteche, C. M., Simoneaux, D. K., Fanslow, W. C., Belmont, S., and Spriggs, M. K. (1993). CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science 259, 990–993.
Ashtari, M., Cyckowski, L. L., Monroe, J. F., Marshall, K. A., Chung, D. C., Auricchio, A., Simonelli, F., Leroy, B. P., Maguire, A. M., Shindler, K. S., and Bennett, J. (2011). The human visual cortex responds to gene therapy-mediated recovery of retinal function. J. Clin. Invest. 121, 2160–2168.
Barber, D. L., Wherry, E. J., Masopust, D., Zhu, B., Allison, J. P., Sharpe, A. H., Freeman, G. J., and Ahmed, R. (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687.
Bauquet, A. T., Jin, H., Paterson, A. M., Mitsdoerffer, M., Ho, I. C., Sharpe, A. H., and Kuchroo, V. K. (2009). The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 10, 167–175.
Bennett, S. R., Carbone, F. R., Karamalis, F., Flavell, R. A., Miller, J. F., and Heath, W. R. (1998). Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393, 478–480.
Borriello, F., Sethna, M. P., Boyd, S. D., Schweitzer, A. N., Tivol, E. A., Jacoby, D., Strom, T. B., Simpson, E. M., Freeman, G. J., and Sharpe, A. H. (1997). B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity 6, 303–313.
Brown, K. E., Freeman, G. J., Wherry, E. J., and Sharpe, A. H. (2010). Role of PD-1 in regulating acute infections. Curr. Opin. Immunol. 22, 397–401.
Bruniquel, D., and Schwartz, R. H. (2003). Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat. Immunol. 4, 235–240.
Buhlmann, J. E., and Noelle, R. J. (1996). Therapeutic potential for blockade of the CD40 ligand, gp39. J. Clin. Immunol. 16, 83–89.
Burmeister, Y., Lischke, T., Dahler, A. C., Mages, H. W., Lam, K. P., Coyle, A. J., Kroczek, R. A., and Hutloff, A. (2008). ICOS controls the pool size of effector-memory and regulatory T cells. J. Immunol. 180, 774–782.
Cousens, L. P., Peterson, R., Hsu, S., Dorner, A., Altman, J. D., Ahmed, R., and Biron, C. A. (1999). Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J. Exp. Med. 189, 1315–1328.
Disanto, J. P., Bonnefoy, J. Y., Gauchat, J. F., Fischer, A., and De Saint Basile, G. (1993). CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature 361, 541–543.
Fischer, A., Hacein-Bey-Abina, S., and Cavazzana-Calvo, M. (2011). Gene therapy for primary adaptive immune deficiencies. J. Allergy Clin. Immunol. 127, 1356–1359.
Gimmi, C. D., Freeman, G. J., Gribben, J. G., Sugita, K., Freedman, A. S., Morimoto, C., and Nadler, L. M. (1991). B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc. Natl. Acad. Sci. U.S.A. 88, 6575–6579.
Good-Jacobson, K. L., Szumilas, C. G., Chen, L., Sharpe, A. H., Tomayko, M. M., and Shlomchik, M. J. (2010). PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol. 11, 535–542.
Green, J. M., Noel, P. J., Sperling, A. I., Walunas, T. L., Gray, G. S., Bluestone, J. A., and Thompson, C. B. (1994). Absence of B7-dependent responses in CD28-deficient mice. Immunity 1, 501–508.
Grewal, I. S., Foellmer, H. G., Grewal, K. D., Xu, J., Hardardottir, F., Baron, J. L., Janeway, C. A. Jr., and Flavell, R. A. (1996). Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science 273, 1864–1867.
Guillonneau, C., Aubry, V., Renaudin, K., Seveno, C., Usal, C., Tezuka, K., and Anegon, I. (2005). Inhibition of chronic rejection and development of tolerogenic T cells after ICOS-ICOSL and CD40-CD40L co-stimulation blockade. Transplantation 80, 546–554.
Haegel-Kronenberger, H., Haanstra, K., Ziller-Remy, C., Ortiz Buijsse, A. P., Vermeiren, J., Stoeckel, F., Van Gool, S. W., Ceuppens, J. L., Mehtali, M., De Boer, M., Jonker, M., and Boon, L. (2004). Inhibition of costimulation allows for repeated systemic administration of adenoviral vector in rhesus monkeys. Gene Ther. 11, 241–252.
Hathcock, K. S., Laszlo, G., Pucillo, C., Linsley, P., and Hodes, R. J. (1994). Comparative analysis of B7-1 and B7-2 costimulatory ligands: expression and function. J. Exp. Med. 180, 631–640.
Hemmi, H., Kaisho, T., Takeda, K., and Akira, S. (2003). The roles of toll-like receptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell subsets. J. Immunol. 170, 3059–3064.
Hoebe, K., Janssen, E. M., Kim, S. O., Alexopoulou, L., Flavell, R. A., Han, J., and Beutler, B. (2003). Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat. Immunol. 4, 1223–1229.
Hollenbaugh, D., Mischel-Petty, N., Edwards, C. P., Simon, J. C., Denfeld, R. W., Kiener, P. A., and Aruffo, A. (1995). Expression of functional CD40 by vascular endothelial cells. J. Exp. Med. 182, 33–40.
Horkheimer, I., Quigley, M., Zhu, J., Huang, X., Chao, N. J., and Yang, Y. (2009). Induction of type I IFN is required for overcoming tumor-specific T-cell tolerance after stem cell transplantation. Blood 113, 5330–5339.
Huang, X., and Yang, Y. (2009). Innate immune recognition of viruses and viral vectors. Hum. Gene Ther. 20, 293–301.
Huang, X., and Yang, Y. (2010). Targeting the TLR9-MyD88 pathway in the regulation of adaptive immune responses. Expert Opin. Ther. Targets 14, 787–796.
Ijima, K., Murakami, M., Okamoto, H., Inobe, M., Chikuma, S., Saito, I., Kanegae, Y., Kawaguchi, Y., Kitabatake, A., and Uede, T. (2001). Successful gene therapy via intraarticular injection of adenovirus vector containing CTLA4IgG in a murine model of type II collagen-induced arthritis. Hum. Gene Ther. 12, 1063–1077.
Ito, T., Hanabuchi, S., Wang, Y. H., Park, W. R., Arima, K., Bover, L., Qin, F. X., Gilliet, M., and Liu, Y. J. (2008). Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity 28, 870–880.
Iwasaki, A., and Medzhitov, R. (2004). Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995.
Jenkins, M. K., Chen, C. A., Jung, G., Mueller, D. L., and Schwartz, R. H. (1990). Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J. Immunol. 144, 16–22.
Jiang, Z., Schiedner, G., Gilchrist, S. C., Kochanek, S., and Clemens, P. R. (2004a). CTLA4Ig delivered by high-capacity adenoviral vector induces stable expression of dystrophin in mdx mouse muscle. Gene Ther. 11, 1453–1461.
Jiang, Z., Schiedner, G., Van Rooijen, N., Liu, C. C., Kochanek, S., and Clemens, P. R. (2004b). Sustained muscle expression of dystrophin from a high-capacity adenoviral vector with systemic gene transfer of T cell costimulatory blockade. Mol. Ther. 10, 688–696.
Kane, L. P., Lin, J., and Weiss, A. (2002). It’s all relative: NF-kappaB and CD28 costimulation of T-cell activation. Trends Immunol. 23, 413–420.
Kay, M. A., Holterman, A. X., Meuse, L., Gown, A., Ochs, H. D., Linsley, P. S., and Wilson, C. B. (1995). Long-term hepatic adenovirus-mediated gene expression in mice following CTLA4Ig administration. Nat. Genet. 11, 191–197.
Keir, M. E., Butte, M. J., Freeman, G. J., and Sharpe, A. H. (2008). PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704.
King, C. L., Stupi, R. J., Craighead, N., June, C. H., and Thyphronitis, G. (1995). CD28 activation promotes Th2 subset differentiation by human CD4+ cells. Eur. J. Immunol. 25, 587–595.
Lafferty, K. J., Andrus, L., and Prowse, S. J. (1980). Role of lymphokine and antigen in the control of specific T cell responses. Immunol. Rev. 51, 279–314.
Lafferty, K. J., Misko, I. S., and Cooley, M. A. (1974). Allogeneic stimulation modulates the in vitro response of T cells to transplantation antigen. Nature 249, 275–276.
Larsen, C. P., Pearson, T. C., Adams, A. B., Tso, P., Shirasugi, N., Strobert, E., Anderson, D., Cowan, S., Price, K., Naemura, J., Emswiler, J., Greene, J., Turk, L. A., Bajorath, J., Townsend, R., Hagerty, D., Linsley, P. S., and Peach, R. J. (2005). Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am. J. Transplant. 5, 443–453.
Laumonier, T., Potiron, N., Boeffard, F., Chagneau, C., Brouard, S., Guillot, C., Soulillou, J. P., Anegon, I., and Le Mauff, B. (2003). CTLA4Ig adenoviral gene transfer induces long-term islet rat allograft survival, without tolerance, after systemic but not local intragraft expression. Hum. Gene Ther. 14, 561–575.
Le Bon, A., Etchart, N., Rossmann, C., Ashton, M., Hou, S., Gewert, D., Borrow, P., and Tough, D. F. (2003). Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 4, 1009–1015.
Lenschow, D. J., Su, G. H., Zuckerman, L. A., Nabavi, N., Jellis, C. L., Gray, G. S., Miller, J., and Bluestone, J. A. (1993). Expression and functional significance of an additional ligand for CTLA-4. Proc. Natl. Acad. Sci. U.S.A. 90, 11054–11058.
Lenschow, D. J., Walunas, T. L., and Bluestone, J. A. (1996). CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 14, 233–258.
Lenschow, D. J., Zeng, Y., Hathcock, K. S., Zuckerman, L. A., Freeman, G., Thistlethwaite, J. R., Gray, G. S., Hodes, R. J., and Bluestone, J. A. (1995). Inhibition of transplant rejection following treatment with anti-B7-2 and anti-B7-1 antibodies. Transplantation 60, 1171–1178.
Linsley, P. S., Brady, W., Grosmaire, L., Aruffo, A., Damle, N. K., and Ledbetter, J. A. (1991). Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp. Med. 173, 721–730.
Linsley, P. S., Greene, J. L., Tan, P., Bradshaw, J., Ledbetter, J. A., Anasetti, C., and Damle, N. K. (1992). Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 176, 1595–1604.
Lipford, G. B., Sparwasser, T., Zimmermann, S., Heeg, K., and Wagner, H. (2000). CpG-DNA-mediated transient lymphadenopathy is associated with a state of Th1 predisposition to antigen-driven responses. J. Immunol. 165, 1228–1235.
Ma, X., Liu, Y., Tittiger, M., Hennig, A., Kovacs, A., Popelka, S., Wang, B., Herati, R., Bigg, M., and Ponder, K. P. (2007). Improvements in mucopolysaccharidosis I mice after adult retroviral vector-mediated gene therapy with immunomodulation. Mol. Ther. 15, 889–902.
Marrack, P., Kappler, J., and Mitchell, T. (1999). Type I interferons keep activated T cells alive. J. Exp. Med. 189, 521–530.
Martino, A. T., Suzuki, M., Markusic, D. M., Zolotukhin, I., Ryals, R. C., Moghimi, B., Ertl, H. C., Muruve, D. A., Lee, B., and Herzog, R. W. (2011). The genome of self-complementary adeno-associated viral vectors increases toll-like receptor 9-dependent innate immune responses in the liver. Blood 117, 6459–6468.
Mcadam, A. J., Greenwald, R. J., Levin, M. A., Chernova, T., Malenkovich, N., Ling, V., Freeman, G. J., and Sharpe, A. H. (2001). ICOS is critical for CD40-mediated antibody class switching. Nature 409, 102–105.
Mcadam, A. J., Schweitzer, A. N., and Sharpe, A. H. (1998). The role of B7 co-stimulation in activation and differentiation of CD4+ and CD8+ T cells. Immunol. Rev. 165, 231–247.
Nabavi, N., Freeman, G. J., Gault, A., Godfrey, D., Nadler, L. M., and Glimcher, L. H. (1992). Signalling through the MHC class II cytoplasmic domain is required for antigen presentation and induces B7 expression. Nature 360, 266–268.
Nanji, S. A., Hancock, W. W., Luo, B., Schur, C. D., Pawlick, R. L., Zhu, L. F., Anderson, C. C., and Shapiro, A. M. (2006). Costimulation blockade of both inducible costimulator and CD40 ligand induces dominant tolerance to islet allografts and prevents spontaneous autoimmune diabetes in the NOD mouse. Diabetes 55, 27–33.
Nurieva, R. I., Duong, J., Kishikawa, H., Dianzani, U., Rojo, J. M., Ho, I., Flavell, R. A., and Dong, C. (2003). Transcriptional regulation of th2 differentiation by inducible costimulator. Immunity 18, 801–811.
Peng, B., Ye, P., Blazar, B. R., Freeman, G. J., Rawlings, D. J., Ochs, H. D., and Miao, C. H. (2008). Transient blockade of the inducible costimulator pathway generates long-term tolerance to factor VIII after nonviral gene transfer into hemophilia A mice. Blood 112, 1662–1672.
Pipkin, M. E., and Rao, A. (2009). Snapshot: effector and memory T cell differentiation. Cell 138, e601–e602.
Puppi, J., Guillonneau, C., Pichard, V., Bellodi-Privato, M., Cuturi, M. C., Anegon, I., and Ferry, N. (2004). Long term transgene expression by hepatocytes transduced with retroviral vectors requires induction of immune tolerance to the transgene. J. Hepatol. 41, 222–228.
Ridge, J. P., Di Rosa, F., and Matzinger, P. (1998). A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393, 474–478.
Schoenberger, S. P., Toes, R. E., Van Der Voort, E. I., Offringa, R., and Melief, C. J. (1998). T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393, 480–483.
Schowalter, D. B., Meuse, L., Wilson, C. B., Linsley, P. S., and Kay, M. A. (1997). Constitutive expression of murine CTLA4Ig from a recombinant adenovirus vector results in prolonged transgene expression. Gene Ther. 4, 853–860.
Schwartz, R. H., Mueller, D. L., Jenkins, M. K., and Quill, H. (1989). T-cell clonal anergy. Cold Spring Harb. Symp. Quant. Biol. 54(Pt 2), 605–610.
Seder, R. A., Germain, R. N., Linsley, P. S., and Paul, W. E. (1994). CD28-mediated costimulation of interleukin 2 (IL-2) production plays a critical role in T cell priming for IL-4 and interferon gamma production. J. Exp. Med. 179, 299–304.
Simpson, T. R., Quezada, S. A., and Allison, J. P. (2010). Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr. Opin. Immunol. 22, 326–332.
Stack, R. M., Lenschow, D. J., Gray, G. S., Bluestone, J. A., and Fitch, F. W. (1994). IL-4 treatment of small splenic B cells induces costimulatory molecules B7-1 and B7-2. J. Immunol. 152, 5723–5733.
Tafuri, A., Shahinian, A., Bladt, F., Yoshinaga, S. K., Jordana, M., Wakeham, A., Boucher, L. M., Bouchard, D., Chan, V. S., Duncan, G., Odermatt, B., Ho, A., Itie, A., Horan, T., Whoriskey, J. S., Pawson, T., Penninger, J. M., Ohashi, P. S., and Mak, T. W. (2001). ICOS is essential for effective T-helper-cell responses. Nature 409, 105–109.
Taylor, P. A., Panoskaltsis-Mortari, A., Freeman, G. J., Sharpe, A. H., Noelle, R. J., Rudensky, A. Y., Mak, T. W., Serody, J. S., and Blazar, B. R. (2005). Targeting of inducible costimulator (ICOS) expressed on alloreactive T cells down-regulates graft-versus-host disease (GVHD) and facilitates engraftment of allogeneic bone marrow (BM). Blood 105, 3372–3380.
Trinchieri, G. (1994). Interleukin-12: a cytokine produced by antigen-presenting cells with immunoregulatory functions in the generation of T-helper cells type 1 and cytotoxic lymphocytes. Blood 84, 4008–4027.
Vaidya, S. A., and Cheng, G. (2003). Toll-like receptors and innate antiviral responses. Curr. Opin. Immunol. 15, 402–407.
Valle, A., Aubry, J. P., Durand, I., and Banchereau, J. (1991). IL-4 and IL-2 upregulate the expression of antigen B7, the B cell counter structure to T cell CD28: an amplification mechanism for T-B cell interactions. Int. Immunol. 3, 229–235.
Van Der Pouw-Kraan, T., Van Kooten, C., Rensink, I., and Aarden, L. (1992). Interleukin (IL)-4 production by human T cells: differential regulation of IL-4 vs. IL-2 production. Eur. J. Immunol. 22, 1237–1241.
Walunas, T. L., Lenschow, D. J., Bakker, C. Y., Linsley, P. S., Freeman, G. J., Green, J. M., Thompson, C. B., and Bluestone, J. A. (1994). CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405–413.
Waterhouse, P., Penninger, J. M., Timms, E., Wakeham, A., Shahinian, A., Lee, K. P., Thompson, C. B., Griesser, H., and Mak, T. W. (1995). Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988.
Yang, Y., Su, Q., Grewal, I. S., Schilz, R., Flavell, R. A., and Wilson, J. M. (1996). Transient subversion of CD40 ligand function diminishes immune responses to adenovirus vectors in mouse liver and lung tissues. J. Virol. 70, 6370–6377.
Yang, Y., and Wilson, J. M. (1996). CD40 ligand-dependent T cell activation: requirement of B7-CD28 signaling through CD40. Science 273, 1862–1864.
Zhao, K., Wang, W., Rando, O. J., Xue, Y., Swiderek, K., Kuo, A., and Crabtree, G. R. (1998). Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell 95, 625–636.
Zhu, J., Huang, X., and Yang, Y. (2007). Innate immune response to adenoviral vectors is mediated by both toll-like receptor-dependent and -independent pathways. J. Virol. 81, 3170–3180.
Keywords: co-stimulation, viral vectors, gene therapy
Citation: Huang X and Yang Y (2011) Targeting co-stimulatory pathways in gene therapy. Front. Microbio. 2:202. doi: 10.3389/fmicb.2011.00202
Received: 28 June 2011;
Accepted: 07 September 2011;
Published online: 28 September 2011.
Edited by:
Roland W. Herzog, University of Florida, USACopyright: © 2011 Huang and Yang. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Yiping Yang, Departments of Medicine and Immunology, Duke University Medical Center, Box 103005, 106 Research Drive, Durham, NC 27710, USA. e-mail:eWFuZzAwMjlAbWMuZHVrZS5lZHU=