 Andrea L. H. Arnett1,2
Andrea L. H. Arnett1,2 Dilip Garikipati2†
Dilip Garikipati2† Zejing Wang3 Stephen Tapscott3 and Jeffrey S. Chamberlain2,4,5*
Zejing Wang3 Stephen Tapscott3 and Jeffrey S. Chamberlain2,4,5*- 1 Medical Scientist Training Program, University of Washington School of Medicine, Seattle, WA, USA
- 2 Department of Neurology, University of Washington School of Medicine, Seattle, WA, USA
- 3 Human Biology Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA
- 4 Department of Medicine, University of Washington School of Medicine, Seattle, WA, USA
- 5 Department of Biochemistry, University of Washington School of Medicine, Seattle, WA, USA
Recombinant adeno-associated viral (rAAV) vectors promote long-term gene transfer in many animal species. Significant effort has focused on the evaluation of rAAV delivery and the immune response in both murine and canine models of neuromuscular disease. However, canines provided for research purposes are routinely vaccinated against canine parvovirus (CPV). rAAV and CPV possess significant homology and are both parvoviruses. Thus, any immune response generated to CPV vaccination has the potential to cross-react with rAAV vectors. In this study, we investigated the immune response to rAAV6 delivery in a cohort of CPV-vaccinated canines and evaluated multiple vaccination regimens in a mouse model of CPV-vaccination. We show that CPV-vaccination stimulates production of neutralizing antibodies with minimal cross-reactivity to rAAV6. In addition, no significant differences were observed in the magnitude of the rAAV6-directed immune response between CPV-vaccinated animals and controls. Moreover, CPV-vaccination did not inhibit rAAV6-mediated transduction. We also evaluated the immune response to early rAAV6-vaccination in neonatal mice. The influence of maternal hormones and cytokines leads to a relatively permissive state in the neonate. We hypothesized that immaturity of the immune system would permit induction of tolerance to rAAV6 when delivered during the neonatal period. Mice were vaccinated with rAAV6 at 1 or 5 days of age, and subsequently challenged with rAAV6 exposure during adulthood via two sequential IM injections, 1 month apart. All vaccinated animals generated a significant neutralizing antibody response to rAAV6-vaccination that was enhanced following IM injection in adulthood. Taken together, these data demonstrate that the immune response raised against rAAV6 is distinct from that which is elicited by the standard parvoviral vaccines and is sufficient to prevent stable tolerization in neonatal mice.
Introduction
Adeno-associated virus (AAV) is a non-enveloped, single-stranded DNA virus that is a member of the Parvovirus family. The AAV genome is approximately 5 kb in size and is packaged within an icosahedral capsid that facilitates viral entry into susceptible cells (Schultz and Chamberlain, 2008). AAV-mediated gene transfer has been successfully demonstrated in numerous large and small animal models of human disease (Arnett et al., 2009; Wang et al., 2009), and recombinant AAV (rAAV) vectors are thus considered a prime candidate for use in the development of gene replacement strategies. rAAV vectors are limited by their small carrying capacity, but possess several attractive features that are advantageous for use as therapeutic reagents, including a broad range of tissue tropism and lack of pathogenicity (Schultz and Chamberlain, 2008). Over 12 serotypes and numerous variants of AAV have been identified. Each serotype has demonstrated a unique profile of tissue tropism that can be utilized to develop targeted therapies with enhanced tissue specificity (Zincarelli et al., 2008; Vandenberghe et al., 2009). For example, rAAV2 exhibits a high tropism for liver and has been used to treat hemophilia B via expression of Factor IX (Manno et al., 2006). rAAV6 has been shown to achieve a high level of transduction in both lung (Halbert et al., 2001, 2007) and striated muscle (Blankinship et al., 2004; Gregorevic et al., 2004, 2006), and is thus being studied to develop treatments for diseases such as cystic fibrosis (Flotte et al., 2007; Halbert et al., 2007), α1-antitrypsin deficiency (Halbert et al., 2010), and the muscular dystrophies (Arnett et al., 2009; Wang et al., 2009).
Stable transgene expression in transduced cells can be harnessed to treat diseases resulting from genetic deficiencies. However, a significant obstacle to the use of viral vectors is the development of host immune responses to both the transgene and the vector (Zaiss and Muruve, 2005, 2008; Nayak and Herzog, 2010). The rAAV genome is one of the simplest of viral gene therapy vectors, containing only the transgene expression cassette flanked by non-coding viral inverted terminal repeats that facilitate packaging and capsid assembly (Samulski et al., 1982). No viral genes are encoded within the engineered genome, which significantly reduces the risk of viral protein synthesis within the host and limits the potential immunogenicity of rAAV vectors. Initial studies regarding rAAV delivery have demonstrated the relative lack of a cell-mediated immune response to rAAV infection in naïve animals (Athanasopoulos et al., 2004; Warrington and Herzog, 2006). In contrast to adenovirus, rAAV does not efficiently trigger a strong, acute inflammatory response, resulting in inefficient activation of dendritic cells and other antigen presenting cells that influence a cytotoxic immune response (Zaiss and Muruve, 2005). These and other factors are thought to contribute to sustained transgene expression in targeted tissues. However, despite promising results in animal studies, clinical trials of rAAV gene delivery in humans have failed to demonstrate the same level of success. Results from multiple studies indicate that capsid-specific humoral and cell-mediated immunity limit tissue transduction and lead to gradual clearance of transduced cells (Manno et al., 2006; Mingozzi and High, 2007; Mingozzi et al., 2009). Thus, renewed effort is being made to understand and modulate factors governing the immune response to rAAV across multiple routes of delivery.
Evaluation of therapeutic constructs and delivery strategies in large animal models is a necessary step in the assessment of potential gene replacement therapies destined for clinical trials. In this regard, dogs can suitably model the physics and challenges of vector delivery to large volumes of tissue and model potential adverse reactions of an evolved mammalian immune system (Wang et al., 2009). The immune response to rAAV in canines has been investigated in several studies (Mount et al., 2002; Wang et al., 2007a, 2010; Yuasa et al., 2007; Ohshima et al., 2009; Halbert et al., 2010; Haurigot et al., 2010). We and others have previously evaluated rAAV delivery to striated muscle (Yuasa et al., 2007; Gregorevic et al., 2009; Ohshima et al., 2009) and have observed significant humoral and cell-mediated immunity utilizing a variety of rAAV serotypes (Wang et al., 2007a, 2010; Yuasa et al., 2007; Ohshima et al., 2009). However, canines used for research purposes are routinely vaccinated against several potential pathogens, one of which is canine parvovirus (CPV). CPV infection is associated with high mortality in young puppies and is very contagious (Patel and Heldens, 2009). High risk of CPV infection necessitates early vaccination in the majority of kennels. Like AAV, CPV is a member of the Parvovirus family and shares significant sequence identity with AAV and other family members. As follows, any immune response generated against CPV vaccination has the potential to cross-react with other members of the Parvovirus family, including AAV. Thus, it is important to consider the influence of CPV vaccine-related immunity on the rAAV-directed immune response in canines, as well as the influence of early exposure to virus and vaccine constituents in the maturing mammalian immune system. In this study, we investigate the immune response to rAAV6 delivery in CPV-vaccinated canines and evaluate multiple vaccination regimens in a mouse model of CPV-vaccination. In addition, we explore the influence of early rAAV6-vaccination on the immune response to repeat rAAV6-infection in adulthood.
Results
CPV Vaccination and Anti-AAV Activity in Dogs
Canine parvovirus-vaccination has the potential to stimulate production of antibodies that cross-react with rAAV, and may contribute to the rAAV-directed immune response that has been previously observed in canines (Wang et al., 2007a, 2010; Yuasa et al., 2007; Ohshima et al., 2009). To address this concern, we tested serum from a cohort of wild-type (wt) beagles that had been vaccinated following a standard vaccination regimen that included three separate intramuscular CPV-vaccinations between the ages of 3 and 7 weeks. Serum was collected at 8 weeks of age and evaluated for rAAV6 cross-reactive neutralizing antibodies (Figure 1). Assay results demonstrate that the neutralization efficiency of serum from CPV-vaccinated animals is minimal. At the strongest serum dilution tested (1:50), less than 10% inhibition was observed. Following the initial serum collection, the dogs were treated with rAAV6 via intravascular injection (5 × 1012 vector genomes/kg), and serum was again sampled for analysis 5 weeks later. As expected, exposure to rAAV6 capsids elicited a strong neutralizing antibody response (Figure 1), but prior CPV-vaccination did not prevent transduction of skeletal muscle (published in Gregorevic et al., 2009; Wang et al., 2010).
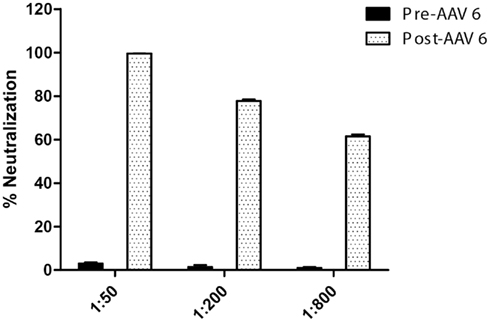
Figure 1. rAAV6 neutralization activity in CPV-vaccinated canines, pre- and post-rAAV6 injection. Beagle pups (n = 3) were vaccinated against CPV prior to 7 weeks of age. Serum neutralization activity was minimal prior to rAAV6 injection.
We also performed a series of Westerns to further analyze whether or not CPV vaccination generated antibodies that cross react with AAV capsids. Serum samples were collected from two groups of dogs (Figure 2). For Group 1, sera were collected from five CPV-vaccinated dogs between the ages of 8–12 months old as post-CPV but pre-rAAV samples, and then collected once more at 4 weeks after intramuscular (IM) injection of rAAV6 vectors. For Group 2, sera were collected from 5-week-old pups (n = 3) prior to CPV vaccination, followed by collection at 3 months after CPV-vaccination, and then 4 weeks after IM injection of rAAV6. Figure 2 shows representative data generated from the two groups, and essentially identical results were seen with all dogs. Pre-CPV serum isolated from the three dogs in Group 2 showed no obvious reactivity to rAAV2, rAAV6, or parvoviral capsids by western analysis. While parvovirus capsids were detected when post-parvo but pre-rAAV serum was used, the serum was not able to detect rAAV6 or rAAV2 above background (BSA was used as negative control). As expected, serum collected after rAAV6 treatment showed strong reactivity to both rAAV2 and 6, whereas the parvoviral signal remained similar in strength to what was observed prior rAAV injection. Together, these data suggested that CPV vaccination did not generate antibodies with detectable cross-reactivity to rAAV.
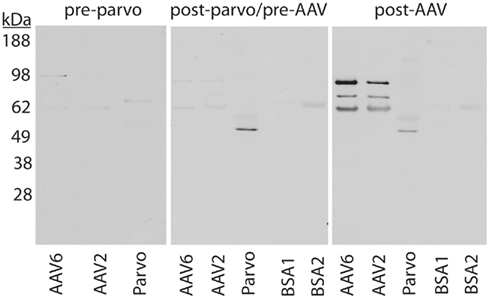
Figure 2. Western analysis of CPV vaccination and serum anti-AAV activity in canines. Serum isolated from CPV-vaccinated animals does not exhibit enhanced anti-rAAV6 activity in comparison to unvaccinated controls. Serum from dogs before CPV vaccination (pre-parvo), after CPV vaccination but before rAAV6 administration (post-parvo/pre-AAV), or after rAAV6 administration (post-AAV) were used as probes. kDa, kilodalton molecular marker; BSA, bovine serum albumin, used as negative control.
CPV-Vaccination and rAAV6 Transduction in Mice
CPV-vaccines have been developed and marketed by multiple agencies. Thus, both vaccination reagents and the timing of administration can vary from kennel to kennel. It is important to recognize that variation in vaccination regimens could influence the nature of the immune response, and it may not be appropriate to generalize results obtained from a single vaccination regimen in a relatively small cohort of animals. The above results from vaccinated beagles provide some insight regarding the immune response to CPV-vaccination and cross-reactivity to rAAV6. However, the study is limited by issues inherent to large animal models. Both ethical considerations and high cost necessitate the use of a small number of animals, and thus it is impractical to effectively assess multiple vaccination regimens in a canine model. In consideration of these factors, we continued the remainder of our vaccination studies in the mouse, which is more amenable to larger sample sizes. In addition, their smaller mass facilitates administration of a higher dose (per kilogram) of rAAV6 that can achieve body-wide transduction of skeletal muscle (Blankinship et al., 2004) and is more relevant to therapeutic dosing levels. While significant differences exist between canine and murine immune systems, the mouse has been utilized extensively as a model system for the study of vaccination responses, autoimmunity, and other aspects of mammalian immunology. Thus, commonalities between the immune systems of both animal models predict that the results regarding CPV-vaccination and generation of any potential rAAV6-directed immune response in the mouse may also be applicable to the canine model.
Mice were vaccinated following two different regimens that represent those commonly employed in kennels (Figure 3A). Regimen 1 utilized reagents that were equivalent to the vaccines administered to the above cohort of beagles. It consisted of a dose of Galaxy® PV, a modified live CPV vaccine, coupled with Intra trac® 3, an upper respiratory vaccine that is directed against adenovirus type-2, parainfluenza, and Bordetella bronchiseptica. Regimen 2 consisted of a combination of two commercial vaccines, Vanguard® Plus and Duramune® Max, both of which contain attenuated CPV, parainfluenza, adenovirus type-2, canine coronavirus, and canine distemper. The two vaccines are given together for the initial dose, followed by a single, additional dose of Vanguard® Plus 1 week later. Eight weeks post-vaccination, serum was collected from both cohorts and analyzed for cross-reactivity with rAAV6 capsids. Vaccinated animals exhibited a slightly elevated neutralizing antibody response compared to unvaccinated mice (Figure 3B). However, this difference was statistically significant only at the highest serum concentration.
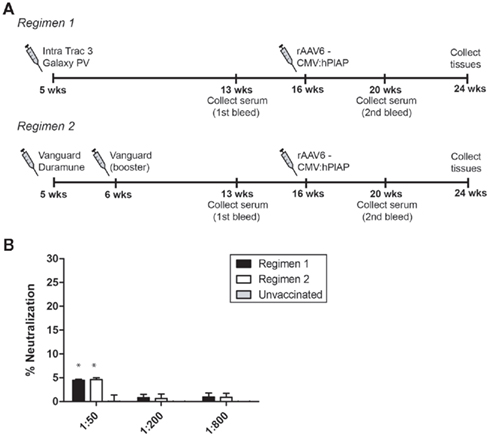
Figure 3. AAV6 neutralization activity and CPV vaccination in mice. (A) Schematic representing the timeline of CPV vaccination and rAAV6 injection. Serum was collected 4 weeks prior to rAAV6 injection and neutralization activity was quantified (B). Both vaccination schedules elicited a slight neutralizing antibody response that was detectable at 1:50 serum dilution (* indicates statistical significance compared to unvaccinated control, p < 0.05).
These three cohorts of mice were then injected with rAAV6 carrying the human placental alkaline phosphatase (hPlAP) reporter gene. Vector was administered via IV injection (2 × 1012 vg) and IM injection (1 × 1010 vg). Dual injection methods were employed to ensure maximum presentation of rAAV6 capsids to monitoring immune cells in both interstitial and intravascular compartments. One month post-rAAV injection, serum samples, and striated muscle tissues were collected for analysis (Figure 4). Transgene expression and vector genomes were quantified across different groups, but no significant difference in hPlAP enzymatic activity or genome copy number was found between either of the two vaccination regimens or the unvaccinated control mice (Figures 4C,D). In addition, no significant cellular inflammatory response was observed in transduced muscle isolated from vaccinated or unvaccinated mice (Figure 4B), suggesting that CPV-vaccination does not contribute to chronic inflammatory infiltration in rAAV-transduced muscle. As expected, neutralizing antibodies were generated following rAAV6 injection, but the response in CPV-vaccinated mice was not significantly different compared to control animals.
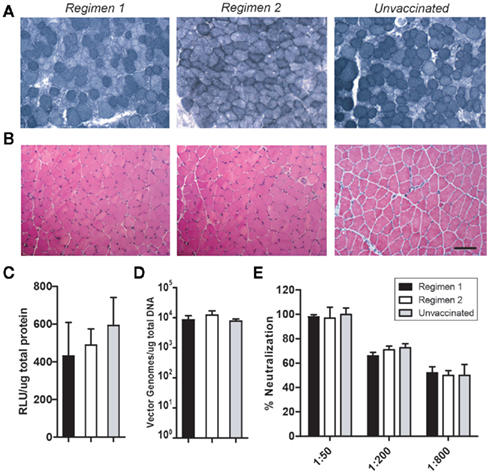
Figure 4. rAAV6 transduction is not impaired in CPV-vaccinated mice. Vaccinated animals (n = 6) received IM and IV injections of rAAV-hPlAP. Serum and tissues were collected 1 month post-injection. Histochemical staining of muscle cross-sections demonstrates robust hPlAP expression (A) and no significant inflammatory infiltrate (B). Quantification of hPlAP expression (C) and vector genome number (D) in muscle lysates revealed no significant difference between vaccinated and unvaccinated animals. CPV-vaccination did not significantly influence the neutralizing antibody response to rAAV6 injection (E). Scale bar 50 μm.
rAAV6 Vaccination in Neonatal Mice
The immune system in young mammals is immature, and neonatal exposure to antigen does not always elicit the same type of immune response as in the adult (reviewed in Jaspan et al., 2006). A diminished cytotoxic T cell response and the presence of circulating maternal antibodies can result in impaired induction of memory cells and persistent infection in newborns (Adkins et al., 2001). In addition, a tolerogenic response to antigens can be initiated in neonates (Morein et al., 2007; Verhasselt, 2010a), which could theoretically be harnessed to facilitate repeat administration of therapeutic vectors. Thus, it is possible that very early vaccination with rAAV6 could modulate the immune response to rAAV infection in a permissive manner. To investigate this prospect, we vaccinated mice with rAAV6 at either 1 or 5 days of age. Vector was delivered intraperitoneally at a dose of 1 × 1011 vg per mouse (Figure 5A). Serum was collected 4 weeks post-vaccination and rAAV6 neutralization activity was quantified prior to IM injection of rAAV6-hPlAP (1 × 1010 vg) in the left tibialis anterior (TA). Both cohorts of mice generated a significant neutralizing antibody response to neonatal rAAV6 vaccination (Figure 5B). However, neutralization activity in the Day 1 vaccination cohort was significantly lower compared to the Day 5 cohort. In addition, animals in the Day 1 cohort demonstrated a greater degree of individual variability in neutralization activity, which may be related to differences in immune system maturity and development between newborn and 5-day old mice. Interestingly, transduction of skeletal muscle was not significantly impacted by early vaccination in either cohort, despite the presence of circulating anti-rAAV6 antibodies (Figure 5C). These results suggest that the rAAV6-directed immune response initiated in vaccinated, neonatal mice may represent either a weak, ineffective primary immune response against re-infection or may instead be indicative of a tolerant response with the potential to remain permissive to viral infection upon rAAV6 re-exposure during adulthood.
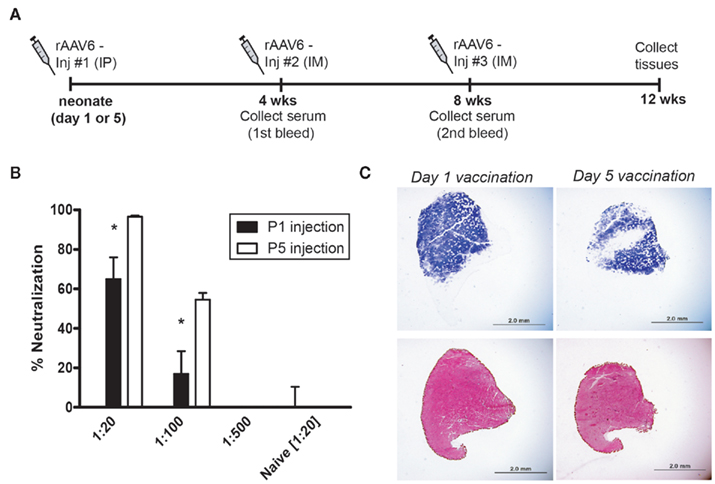
Figure 5. rAAV6 vaccination and neutralizing antibody response in neonatal mice. (A) rAAV6 injection and serum collection timeline. Mice were vaccinated (IP) at P1 or P5, and received two sequential injections (IM) of rAAV6 at 4 and 8 weeks. (B) Quantification of neutralizing antibody response from the first bleed, 4 weeks post-vaccination. Animals vaccinated on Day 1 generated a weaker response than those injected Day 5 (* indicates significant difference compared to P5 cohort, p < 0.05). (C) Cross-sections of tibialis anterior injected with rAAV6-hPlAP (Inj #2) at 4 weeks post-vaccination. Neutralizing antibody response did not prevent rAAV6 transduction in either cohort. hPLAP staining (top) and hematoxylin and eosin staining (bottom). Scale bar 2 mm.
Permissive Immunity Does not Persist in rAAV6-Vaccinated Mice
It has been previously shown that IM injection of rAAV stimulates a robust humoral immune response that is sufficient to prevent skeletal muscle transduction upon subsequent exposure to the same vector serotype (Burger et al., 2004; Riviere et al., 2006; Sabatino et al., 2007). As shown above, rAAV6-vaccinated mice responded positively to the first IM injection of rAAV6, but it was important to determine whether this permissive state would persist through repeated exposure to rAAV6 during adulthood. To evaluate whether mice had developed a functional tolerance for rAAV6, both Day 1 and Day 5 cohorts were given an additional IM injection of 1 × 1010 vg into the contralateral TA muscle. Serum was collected immediately prior to injection, and neutralization activity was quantified (Figure 6A). Both cohorts showed a strong neutralizing antibody response that was significantly enhanced when compared to the response obtained from the first bleed (Figure 5B). No significant difference in neutralization activity was observed between vaccinated animals and un-vaccinated controls. In addition, neutralization activity remained high even at more dilute serum concentrations, suggesting that the 4 week IM injection enhanced the rAAV6-directed immune response. Consistent with these observations, transduction in the contralateral TA was dramatically reduced and was limited to a small number of sparsely distributed muscle fibers (Figure 6B). These data indicate that rAAV6-vaccinated mice did not remain permissive to repeat infection and suggest that the success of the initial IM injection may be related to a weak primary immune response to rAAV6-vaccination, rather than induction of tolerance.
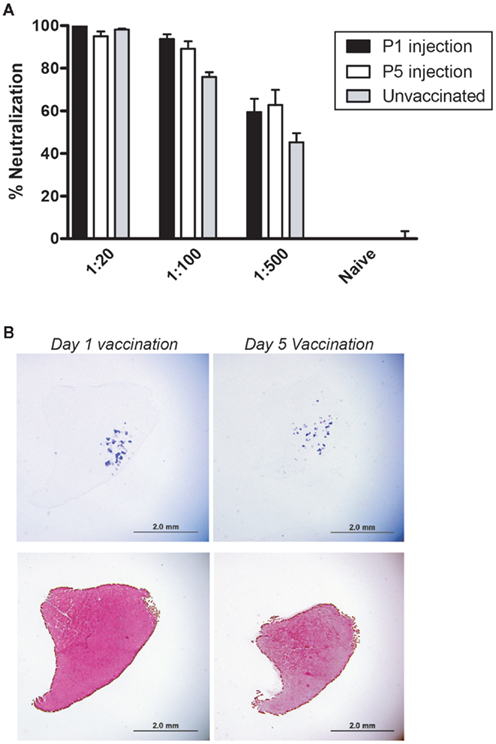
Figure 6. Permissive immunity does not persist in rAAV6-vaccinated mice. Animals received a third injection of rAAV6-hPlAP at 8 weeks post-vaccination. Serum collected immediately prior to injection (Inj #3) demonstrated high neutralization activity (A). rAAV6 transduction was dramatically reduced (B). hPLAP staining (top) and hematoxylin and eosin staining (bottom). Scale bar 2 mm.
Discussion
The immune response to rAAV vectors has emerged as a prominent issue in the development of gene replacement strategies. Initial studies suggested a relative lack of immunogenicity of rAAV vectors, but the success achieved in animal models has not been mirrored in human trials. Significant immune responses to the vector have been observed following intravascular delivery of rAAV to liver and direct injection into skeletal muscle. Exposure to wild-type AAV is common within the human population, and the frequency of sero-positivity for AAV-directed neutralizing antibodies approaches nearly 30% for AAV2 (Mingozzi and High, 2007). Pre-existing neutralizing antibodies to AAV have been shown to significantly inhibit transduction of hepatocytes in a clinical trial evaluating delivery of rAAV2 to hemophilia patients (Manno et al., 2006). A similar humoral response has been observed in cystic fibrosis gene therapy trials involving repeat administration of rAAV2 (Flotte et al., 2007). Disappointingly, the majority of clinical trials have resulted in the gradual loss of transgene expression. rAAV2-mediated delivery of human Factor IX (hFIX) to either skeletal muscle (Kay et al., 2000) or liver (Manno et al., 2006) resulted in therapeutic levels of hFIX in a subset of patients, but hFIX expression was eventually eliminated or reduced to non-therapeutic levels in all participants. In another clinical trial, rAAV1 was used to deliver lipoprotein lipase (LPL) via IM injection to patients with LPL-deficiency (Mingozzi et al., 2009). Once again, transgene expression rose to effective levels for a relatively short period before gradually dropping below therapeutic threshold. In a clinical trial of rAAV1-mediated delivery of α-sarcoglycan, two out of three patients responded well to the treatment, whereas the third patient failed to demonstrate successful gene transfer (Mendell et al., 2010). Extensive analysis of data generated from these clinical trials has indicated that both humoral and cell-mediated anti-capsid immune responses likely play a significant role in the elimination of transduced tissues (Manno et al., 2006; Mingozzi et al., 2009; Mendell et al., 2010).
A significant immune response has been observed in several studies of rAAV delivery in canines. Aspects of the canine immune response have mirrored findings in human clinical trials, including detection of a capsid-directed T cell response and gradual loss of transgene expression in non-immunosuppressed animals. Ohshima et al. (2009) observed a strong inflammatory response and T cell infiltration following delivery of rAAV2 or rAAV8 to skeletal muscle, though the magnitude of the inflammatory response was reduced in rAAV8-injected muscles. In addition, we have previously shown that injection of rAAV6 or rAAV1 in immunocompetent animals results in a similar sequence of events. An anti-capsid immune response led to local inflammation and clearance of the majority, but not all, transduced cells over a period of several weeks (Wang et al., 2007a, 2010). Loss of transgene expression was prevented by a short course of immunosuppression (Wang et al., 2007b). An rAAV-directed immune response has also limited transgene expression in a canine model of α1-antitrypsin deficiency (Halbert et al., 2010). These studies raise concerns regarding the immunogenicity of rAAV vectors and emphasize the need for careful evaluation of the potential immune response to rAAV delivery, especially in disorders that may require very high vector doses for systemic delivery, such as the muscular dystrophies.
Further study of the canine immune response to rAAV may provide insight toward addressing immunogenicity in clinical trials, but it is important to rule out the influence of CPV-vaccination regarding the generation of an rAAV-directed immune response. Antibody cross-reactivity has been demonstrated with closely related variants of AAV and between other related members of the Parvovirus family (Patel and Heldens, 2009). In addition, it has been shown that T cell receptors are reactive to epitopes that are conserved between different serotypes (Mingozzi and High, 2007; Mingozzi et al., 2009; Wang et al., 2010), suggesting that a cell-mediated response may be more broadly reactive across a related group of viral vectors. Here, we observed a low level of neutralization activity in both mouse and canine models of CPV-vaccination at serum dilutions of 1:50 (Figures 1 and 3). This activity was not measurable at more dilute serum concentrations, and Western analysis of serum from parvo-vaccinated and unvaccinated canines did not exhibit enhanced binding to rAAV particles (Figure 2). These results may indicate the presence of anti-CPV antibodies that possess low, cross-reactive affinity for rAAV6. Cross-reactive antibodies with weak affinity for rAAV6 would require a higher concentration of neutralizing antibodies to achieve significant neutralization. At more dilute serum concentrations, inefficient binding of cross-reactive antibodies would significantly limit neutralization activity. Further characterization of the antibody response would be necessary to quantify affinity strength and identify common epitope targets before cross-reactivity can be confirmed. However, it is important to emphasize that CPV-vaccination did not correlate with any significant inhibition of rAAV6 transduction (Figure 4), suggesting that a low level of neutralization activity may not play a role in generating a functionally significant rAAV6-directed immune response. In addition, we did not detect any significant inflammatory infiltration, nor did we observe any changes in muscle architecture that would be indicative of a cell-mediated, cytotoxic response (Figures 4A,B).
The CPV vaccination regimen utilized in these studies was chosen to reflect the vaccination regimens routinely employed in large kennels within the United States (Bioresources, 2006). A variety of CPV vaccines are available from different manufacturers, and availability in different regions is dependent on local laws, international licensing, and market preferences (Patel and Heldens, 2009). The regimens evaluated in this study are representative of the majority of vaccine components that are used by major manufacturers, and thus, our results are likely applicable to a wide selection of vaccines. However, it is possible that subtle differences in vaccines and vaccination schedules could influence the immune response. Additional studies evaluating vaccines from a larger number of manufacturers would be necessary to rule out this possibility.
In addition to CPV-vaccination, we explored the response to rAAV6 vaccination in neonatal mice. The immune system in newborn mammals is under-developed, and significant differences exist in the pattern of T and B cell activation between newborns and adults. Newborns exhibit a generalized deficiency in adaptive cellular responses, with a bias toward prolonged Th2-type immunity (Adkins et al., 2001). This observation has been attributed to the influence of maternal cytokines and hormones that promote maternal tolerance to fetal antigens. These cytokines persist in the fetal circulation and influence the neonatal immune response for a significant period after birth, leading to a relatively tolerogenic, and vulnerable state in the neonate (Morein et al., 2002). In this regard, the newborn period represents a time when the immune system is learning to balance induction of tolerance to self antigens with appropriate reactive immunity to foreign pathogens. Thus, viral vectors delivered during this developmental window have the potential to initiate a tolerant immune response.
The ability to achieve successful transduction following repeat administration of viral vectors would be advantageous, both in the clinic and in the realm of basic research. Unfortunately, repetitive administration of identical serotypes has been ineffective in the majority of cases due to the development of a significant humoral immune response to rAAV vectors (Burger et al., 2004; Riviere et al., 2006; Sabatino et al., 2007; Petry et al., 2008). It has been speculated that early vaccination with rAAV could stimulate a tolerant response to vector in the immature immune system and thus permit re-administration in adulthood. In this regard, previous studies have investigated the response to in utero adenoviral (Lipshutz et al., 2000; Bouchard et al., 2003) or AAV (Jerebtsova et al., 2002; Bouchard et al., 2003; Sabatino et al., 2007) vaccination in mice. These studies were limited to IM injection of rAAV serotypes 1, 2, and 5, and did not evaluate rAAV6. Considering that serotype-specific transduction profiles, dosage, and the route of administration can significantly influence the immune response to viral vectors (Mingozzi and High, 2007; Petry et al., 2008; Zaiss and Muruve, 2008), it is important to empirically determine the immune response rAAV6 vaccination.
Unfortunately, we were unable to induce persistent tolerance to rAAV6 utilizing neonatal vaccination. The neutralizing antibody response to the initial IP vaccination did not inhibit transduction during the first IM injection at 4 weeks of age. However, animals did not remain permissive to rAAV6 transduction. On the contrary, neutralization activity was significantly enhanced following the first IM injection, resulting in near-complete inhibition of transduction during the second IM injection. These results suggest that the initial vaccination triggered a weak primary immune response, which led to an enhanced secondary reaction to the first IM injection. However, without a more detailed characterization of the antibody response, we cannot rule out the existence of partial tolerance to specific viral epitopes. These findings are consistent with those previously observed following in utero delivery of rAAV1 and rAAV2 (Jerebtsova et al., 2002; Sabatino et al., 2007) and indicate that neonatal IP injection of rAAV6 does not facilitate repetitive administration of vector beyond a single, repeat injection.
In summary, we have evaluated the effects of CPV and rAAV vaccination on rAAV6-mediated transduction. The neutralizing antibody response to CPV-vaccinated animals is minimal and does not appear to significantly enhance either the humoral or cellular response to rAAV6 transduction. These data suggest that CPV-immunity is not a significant component of the rAAV6-directed immune response in canines, and support the use of canines as a valid model for further characterization of the immune response to rAAV6. In contrast, vaccination with rAAV6 in neonatal mice leads to a significant immune response that prevents repetitive administration of rAAV6. However, additional methods of tolerance induction may warrant further consideration, including oral delivery (Verhasselt, 2010b) and thymic expression of viral proteins (Chu et al., 2010).
Materials and Methods
AAV Production and Characterization
rAAV6 vector was generated as previously described (Grimm et al., 2003). Cells were co-transfected with an rAAV6 packaging plasmid pDGM6 and plasmid containing the expression cassette flanked by viral ITRs. Cellular pellets and supernatants were collected and processed through a 110S microfluidizer (Microfluidics, Newton, MA, USA), followed by clarification of the homogenate by filtration through a 0.22-μm filter. Additionally, an Amersham AKTA10 HPLC machine (Amersham, Piscataway, NJ, USA) was used for affinity purification on a HiTrap heparin column (Amersham). The column was then washed and vector was eluted and dialyzed against physiological Ringer’s solution. Vector was titered using HT-1080 cells as transduction targets, and Southern analysis was utilized to determine the number of genome-containing particles in the vector preparation.
Animal Experiments
Animal studies were performed in accordance with the guidelines set forth by the institutional Review office of University of Washington. C57BL/6 mice were bred in our animal facility. Mice were given vaccines or AAV injections according to the indicated schedule (see Results). Vaccinations were administered using either Intra Trac® 3 and Galaxy PV® (both supplied by Intervet/Schering-Plough Animal Health, Millsboro, DE, USA), Vanguard® Plus (Pfizer Animal Health, Exton, PA, USA) and Duramune® Max (Boehringer-Ingelheim Vetmedica, Inc., St. Joseph, MO, USA), or intraperitoneal administration of rAAV6-CMV-cre. rAAV6-CMV-hPlAP was delivered via either retro-orbital injection or IM injection (into the left TA muscle), as indicated. Blood samples were collected via retro-orbital route, under isofluorane-induced anesthesia. At the indicated timepoints, mice were euthanized according to approved protocol and tissue samples were collected for analysis.
Western Analysis
3 × 109 vector genome/well of AAV6 and AAV2 and 10 μl/well of parvovirus vaccine at 1:10 dilution were loaded onto a 4–12% NuPAGE Bis–Tris gel (Bio-Rad, USA). The gel was transferred onto nitrocellulose membrane (Bio-Rad, USA). Membranes were blocked with 5% non-fat milk, 0.1% Tween–PBS (w/v) overnight at 4°C and then incubated with serum at 1:200 dilution. Horseradish peroxidase (HRP)-labeled rabbit anti-dog Ig was used as secondary antibody at 1:25,000 (Jackson ImmunoResearch, USA). Immunoreactive proteins were visualized using the ECL system (Amersham, USA).
Cell Culture
The 293 human embryonic kidney cells and HT-1080 human fibrosarcoma cells were maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum supplemented with penicillin and streptomycin. Cells were cultured at 37°C in an atmosphere of 5% CO2.
Virus Neutralization Assay
Serum was prepared by centrifugation at 3000 rpm for 5 min, followed by heat inactivation at 56°C for 30 min. Virus neutralization assays were done as previously described (Halbert et al., 2000; Calcedo et al., 2009). Briefly, rAAV6-CMV-GFP or rAAV6-CMV-hPlAP was diluted to 1 × 109 genome-containing particles per ml. Serum was added to 100 μl of diluted virus to achieve the desired final serum dilution (1:20, 1:50, 1:200, 1:500, or 1:800). The virus and serum were incubated for 1 h at 37°C, and 80 μl were added to HT-1080 cells plated at 2 × 104 cells per well (12 well plates) the previous day. Two days following infection, plates transduced with rAAV6-CMV-GFP were counted with fluorescence activated cell sorting (FACS). Cells transduced with rAAV6-CMV-hPlAP were stained for hPlAP expression and the number of positive cells per field was quantified.
Histological Analysis
Muscle tissue was frozen in liquid nitrogen-cooled isopentane embedded in Tissue-Tek OCT medium (Sakura Finetek USA, Torrance, CA, USA) and sectioned transversely in cryostat at 10 μm. For hPLAP staining, sections were fixed with ice cold 4% paraformaldehyde, washed three times in cold phosphate-buffered saline, placed in 65°C phosphate-buffered saline for 90 min, rinsed in room temperature phosphate-buffered saline, and washed in alkaline phosphatase buffer (0.1 mol/l Tris–HCl pH 9.5, 0.1 mol/l NaCl, 0.01 mol/l MgCl2) for 10 min. Excess liquid was removed from the sections, and Sigma FAST BCIP/NBT substrate solution (Sigma, St Louis, MO, USA) was applied to each section for 30 min at room temperature, in the dark. Slides were rinsed three times in room temperature phosphate-buffered saline, dehydrated in 70% EtOH for 5 min, 2× (95% EtOH for 2 min), 2× (100% EtOH for 2 min), 2× (xylene for 3 min), and coverslipped with Permount mounting media (Fisher Scientific, Fair Lawn, NJ, USA). Images were captured using QIcam or Olympus digital cameras and processed using QCapture Pro (QImaging, BC, Canada). For hematoxylin and eosin m thickness were briefly fixed in methanol and staining, cryosections of 10 stained with Gill’s hematoxylin and eosin–phyloxine. The sections were washed, dehydrated, and cleared in xylene before mounting with Permount.
Luminometry Assay
After sacrifice of CPV-vaccinated mice, the gastrocnemius muscle was rapidly excised and flash frozen in liquid nitrogen. Frozen muscles were then powdered using a mortar and pestle and the protein was extracted with a protease-inhibiting buffer containing 137 mM NaCl, 20 mM Tris–HCl, pH 7.6, 2 mM MgCl2, 1 mM 2-mercaptoethanol, 0.2% Tween 20 (Amersham), and 1× Complete protease inhibitor (Roche, Indianapolis, IN, USA). Protein was quantified via spectrophotometric absorption using Bradford reagent (Peirce, Rockford, IL, USA). The extract was then analyzed for hPlAP expression using a commercial luminometry kit (Applied Biosystems, Carlsbad, CA, USA)
Vector Genome Quantification
Muscles were snap frozen in liquid nitrogen and then pulverized with a mortal and pestle. Pulverized muscle tissue was resuspended in tissue lysis buffer [0.5% NaDOC, 50 mM Tris, 150 mM NaCl, 1% Triton, 0.8% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA)]. DNA was isolated from cell and tissue lysates using a DNeasy blood and tissue kit (Qiagen, Valencia, CA, USA) according the manufacturer’s guidelines. Genome quantification was performed utilizing a SV40 polyA-specific probe and quantitative-PCR, as previously described (Gregorevic et al., 2004).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank James Allen and Eric Finn for assistance with vector production. Andrea L. H. Arnett was supported by the Medical Scientist Training Program, and Achievement Rewards for College Scientists, as well as a National Research Service Award (NIH F30NS068005). Supported by NIH grants AR40864 and AG33610, and a grant from the Muscular Dystrophy Association, USA (to Jeffrey S. Chamberlain).
References
Adkins, B., Bu, Y., and Guevara, P. (2001). The generation of Th memory in neonates versus adults: prolonged primary Th2 effector function and impaired development of Th1 memory effector function in murine neonates. J. Immunol. 166, 918–925.
Arnett, A. L., Chamberlain, J. R., and Chamberlain, J. S. (2009). Therapy for neuromuscular disorders. Curr. Opin. Genet. Dev. 19, 290–297.
Athanasopoulos, T., Graham, I. R., Foster, H., and Dickson, G. (2004). Recombinant adeno-associated viral (rAAV) vectors as therapeutic tools for Duchenne muscular dystrophy (DMD). Gene Ther. 11(Suppl. 1), S109–S121.
Bioresources, M. (2006). Beagle: Routine Vaccination and Treatment Procedures. North Rose: Marshall Bioresources.
Blankinship, M. J., Gregorevic, P., Allen, J. M., Harper, S. Q., Harper, H., Halbert, C. L., Miller, A. D., and Chamberlain, J. S. (2004). Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol. Ther. 10, 671–678.
Bouchard, S., MacKenzie, T. C., Radu, A. P., Hayashi, S., Peranteau, W. H., Chirmule, N., and Flake, A. W. (2003). Long-term transgene expression in cardiac and skeletal muscle following fetal administration of adenoviral or adeno-associated viral vectors in mice. J. Gene Med. 5, 941–950.
Burger, C., Gorbatyuk, O. S., Velardo, M. J., Peden, C. S., Williams, P., Zolotukhin, S., Reier, P. J., Mandel, R. J., and Muzyczka, N. (2004). Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. 10, 302–317.
Calcedo, R., Vandenberghe, L. H., Gao, G., Lin, J., and Wilson, J. M. (2009). Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 199, 381–390.
Chu, Q., Moreland, R. J., Gao, L., Taylor, K. M., Meyers, E., Cheng, S. H., and Scheule, R. K. (2010). Induction of immune tolerance to a therapeutic protein by intrathymic gene delivery. Mol. Ther. 18, 2146–2154.
Flotte, T. R., Ng, P., Dylla, D. E., McCray, P. B. Jr., Wang, G., Kolls, J. K., and Hu, J. (2007). Viral vector-mediated and cell-based therapies for treatment of cystic fibrosis. Mol. Ther. 15, 229–241.
Gregorevic, P., Allen, J. M., Minami, E., Blankinship, M. J., Haraguchi, M., Meuse, L., Finn, E., Adams, M. E., Froehner, S. C., Murry, C. E., and Chamberlain, J. S. (2006). rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 12, 787–789.
Gregorevic, P., Blankinship, M. J., Allen, J. M., Crawford, R. W., Meuse, L., Miller, D. G., Russell, D. W., and Chamberlain, J. S. (2004). Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 10, 828–834.
Gregorevic, P., Schultz, B. R., Allen, J. M., Halldorson, J. B., Blankinship, M. J., Meznarich, N. A., Kuhr, C. S., Doremus, C., Finn, E., Liggitt, D., and Chamberlain, J. S. (2009). Evaluation of vascular delivery methodologies to enhance rAAV6-mediated gene transfer to canine striated musculature. Mol. Ther. 17, 1427–1433.
Grimm, D., Kay, M. A., and Kleinschmidt, J. A. (2003). Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther. 7, 839–850.
Halbert, C. L., Allen, J. M., and Miller, A. D. (2001). Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J. Virol. 75, 6615–6624.
Halbert, C. L., Lam, S. L., and Miller, A. D. (2007). High-efficiency promoter-dependent transduction by adeno-associated virus type 6 vectors in mouse lung. Hum. Gene Ther. 18, 344–354.
Halbert, C. L., Madtes, D. K., Vaughan, A. E., Wang, Z., Storb, R., Tapscott, S. J., and Miller, A. D. (2010). Expression of human alpha1-antitrypsin in mice and dogs following AAV6 vector-mediated gene transfer to the lungs. Mol. Ther. 18, 1165–1172.
Halbert, C. L., Rutledge, E. A., Allen, J. M., Russell, D. W., and Miller, A. D. (2000). Repeat transduction in the mouse lung by using adeno-associated virus vectors with different serotypes. J. Virol. 74, 1524–1532.
Haurigot, V., Mingozzi, F., Buchlis, G., Hui, D. J., Chen, Y., Basner-Tschakarjan, E., Arruda, V. R., Radu, A., Franck, H. G., Wright, J. F., Zhou, S., Stedman, H. H., Bellinger, D. A., Nichols, T. C., and High, K. A. (2010). Safety of AAV factor IX peripheral transvenular gene delivery to muscle in hemophilia B dogs. Mol. Ther. 18, 1318–1329.
Jaspan, H. B., Lawn, S. D., Safrit, J. T., and Bekker, L. G. (2006). The maturing immune system: implications for development and testing HIV-1 vaccines for children and adolescents. AIDS 20, 483–494.
Jerebtsova, M., Batshaw, M. L., and Ye, X. (2002). Humoral immune response to recombinant adenovirus and adeno-associated virus after in utero administration of viral vectors in mice. Pediatr. Res. 52, 95–104.
Kay, M. A., Manno, C. S., Ragni, M. V., Larson, P. J., Couto, L. B., McClelland, A., Glader, B., Chew, A. J., Tai, S. J., Herzog, R. W., Arruda, V., Johnson, F., Scallan, C., Skarsgard, E., Flake, A. W., and High, K. A. (2000). Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat. Genet. 24, 257–261.
Lipshutz, G. S., Flebbe-Rehwaldt, L., and Gaensler, K. M. (2000). Reexpression following readministration of an adenoviral vector in adult mice after initial in utero adenoviral administration. Mol. Ther. 2, 374–380.
Manno, C. S., Pierce, G. F., Arruda, V. R., Glader, B., Ragni, M., Rasko, J. J., Ozelo, M. C., Hoots, K., Blatt, P., Konkle, B., Dake, M., Kaye, R., Razavi, M., Zajko, A., Zehnder, J., Rustagi, P. K., Nakai, H., Chew, A., Leonard, D., Wright, J. F., Lessard, R. R., Sommer, J. M., Tigges, M., Sabatino, D., Luk, A., Jiang, H., Mingozzi, F., Couto, L., Ertl, H. C., High, K. A., and Kay, M. A. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347.
Mendell, J. R., Rodino-Klapac, L. R., Rosales, X. Q., Coley, B. D., Galloway, G., Lewis, S., Malik, V., Shilling, C., Byrne, B. J., Conlon, T., Campbell, K. J., Bremer, W. G., Taylor, L. E., Flanigan, K. M., Gastier-Foster, J. M., Astbury, C., Kota, J., Sahenk, Z., Walker, C. M., and Clark, K. R. (2010). Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann. Neurol. 68, 629–638.
Mingozzi, F., and High, K. A. (2007). Immune responses to AAV in clinical trials. Curr. Gene Ther. 7, 316–324.
Mingozzi, F., Meulenberg, J. J., Hui, D. J., Basner-Tschakarjan, E., Hasbrouck, N. C., Edmonson, S. A., Hutnick, N. A., Betts, M. R., Kastelein, J. J., Stroes, E. S., and High, K. A. (2009). AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood 114, 2077–2086.
Morein, B., Abusugra, I., and Blomqvist, G. (2002). Immunity in neonates. Vet. Immunol. Immunopathol. 87, 207–213.
Morein, B., Blomqvist, G., and Hu, K. (2007). Immune responsiveness in the neonatal period. J. Comp. Pathol. 137(Suppl. 1), S27–S31.
Mount, J. D., Herzog, R. W., Tillson, D. M., Goodman, S. A., Robinson, N., McCleland, M. L., Bellinger, D., Nichols, T. C., Arruda, V. R., Lothrop, C. D. Jr., and High, K. A. (2002). Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood 99, 2670–2676.
Nayak, S., and Herzog, R. W. (2010). Progress and prospects: immune responses to viral vectors. Gene Ther. 17, 295–304.
Ohshima, S., Shin, J. H., Yuasa, K., Nishiyama, A., Kira, J., Okada, T., and Takeda, S. (2009). Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol. Ther. 17, 73–80.
Patel, J. R., and Heldens, J. G. (2009). Review of companion animal viral diseases and immunoprophylaxis. Vaccine 27, 491–504.
Petry, H., Brooks, A., Orme, A., Wang, P., Liu, P., Xie, J., Kretschmer, P., Qian, H. S., Hermiston, T. W., and Harkins, R. N. (2008). Effect of viral dose on neutralizing antibody response and transgene expression after AAV1 vector re-administration in mice. Gene Ther. 15, 54–60.
Riviere, C., Danos, O., and Douar, A. M. (2006). Long-term expression and repeated administration of AAV type 1, 2 and 5 vectors in skeletal muscle of immunocompetent adult mice. Gene Ther. 13, 1300–1308.
Sabatino, D. E., Mackenzie, T. C., Peranteau, W., Edmonson, S., Campagnoli, C., Liu, Y. L., Flake, A. W., and High, K. A. (2007). Persistent expression of hF.IX After tolerance induction by in utero or neonatal administration of AAV-1-F.IX in hemophilia B mice. Mol. Ther. 15, 1677–1685.
Samulski, R. J., Berns, K. I., Tan, M., and Muzyczka, N. (1982). Cloning of adeno-associated virus into pBR322: rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. U.S.A. 79, 2077–2081.
Schultz, B. R., and Chamberlain, J. S. (2008). Recombinant adeno-associated virus transduction and integration. Mol. Ther. 16, 1189–1199.
Vandenberghe, L. H., Wilson, J. M., and Gao, G. (2009). Tailoring the AAV vector capsid for gene therapy. Gene Ther. 16, 311–319.
Verhasselt, V. (2010a). Neonatal tolerance under breastfeeding influence. Curr. Opin. Immunol. 22, 623–630.
Verhasselt, V. (2010b). Oral tolerance in neonates: from basics to potential prevention of allergic disease. Mucosal Immunol. 3, 326–333.
Wang, Z., Allen, J. M., Riddell, S. R., Gregorevic, P., Storb, R., Tapscott, S. J., Chamberlain, J. S., and Kuhr, C. S. (2007a). Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum. Gene Ther. 18, 18–26.
Wang, Z., Kuhr, C. S., Allen, J. M., Blankinship, M., Gregorevic, P., Chamberlain, J. S., Tapscott, S. J., and Storb, R. (2007b). Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 15, 1160–1166.
Wang, Z., Chamberlain, J. S., Tapscott, S. J., and Storb, R. (2009). Gene therapy in large animal models of muscular dystrophy. ILAR J. 50, 187–198.
Wang, Z., Storb, R., Lee, D., Kushmerick, M. J., Chu, B., Berger, C., Arnett, A., Allen, J., Chamberlain, J. S., Riddell, S. R., and Tapscott, S. J. (2010). Immune Responses to AAV in Canine Muscle Monitored by Cellular Assays and Noninvasive Imaging. Mol. Ther. 18, 617–624.
Warrington, K. H. Jr., and Herzog, R. W. (2006). Treatment of human disease by adeno-associated viral gene transfer. Hum. Genet. 119, 571–603.
Yuasa, K., Yoshimura, M., Urasawa, N., Ohshima, S., Howell, J. M., Nakamura, A., Hijikata, T., Miyagoe-Suzuki, Y., and Takeda, S. (2007). Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther. 14, 1249–1260.
Zaiss, A. K., and Muruve, D. A. (2005). Immune responses to adeno-associated virus vectors. Curr. Gene Ther. 5, 323–331.
Zaiss, A. K., and Muruve, D. A. (2008). Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther. 15, 808–816.
Keywords: AAV, muscle, immunity, tolerance, parvovirus, vaccination, canine, immune response
Citation: Arnett ALH, Garikipati D, Wang Z, Tapscott S and Chamberlain JS (2011) Immune responses to rAAV6: the influence of canine parvovirus vaccination and neonatal administration of viral vector. Front. Microbio. 2:220. doi: 10.3389/fmicb.2011.00220
Received: 12 July 2011;
Paper pending published: 03 August 2011;
Accepted: 16 October 2011;
Published online: 03 November 2011.
Edited by:
Roland W. Herzog, University of Florida, USAReviewed by:
Dongsheng Duan, University of Missouri, USATim Nichols, University of North Carolina at Chapel Hill, USA
Copyright: © 2011 Arnett, Garikipati, Wang, Tapscott and Chamberlain. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jeffrey S. Chamberlain, Department of Neurology, University of Washington School of Medicine, 1959 NE Pacific Street, Box 357720, Seattle, WA 98195-7720, USA. e-mail:anNjNUB1dy5lZHU=
†Present address: Dilip Garikipati, Department of Animal Sciences, Washington State University, Pullman, WA, USA.