 YuShuan Lai1
YuShuan Lai1


- 1 Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA, USA
- 2 Graduate Program in Cellular and Molecular Physiology, Sackler School of Graduate Biomedical Sciences, Tufts University School of Medicine, Boston, MA, USA
- 3 Center for Innovations in Wound Healing Research, Tufts University School of Medicine, Boston, MA, USA
- 4 Department of Molecular Biology and Microbiology, Tufts University School of Medicine, Boston, MA, USA
A member of the attaching and effacing (AE) family of pathogens, enterohemorrhagic Escherichia coli (EHEC) induces dramatic changes to the intestinal cell cytoskeleton, including effacement of microvilli. Effacement by the related pathogen enteropathogenic E. coli (EPEC) requires the activity of the Ca+2-dependent host protease, calpain, which participates in a variety of cellular processes, including cell adhesion and motility. We found that EHEC infection results in an increase in epithelial (CaCo-2a) cell calpain activity and that EHEC-induced microvillar effacement was blocked by ectopic expression of calpastatin, an endogenous calpain inhibitor, or by pretreatment of intestinal cells with a cell-penetrating version of calpastatin. In addition, ezrin, a known calpain substrate that links the plasma membrane to axial actin filaments in microvilli, was cleaved in a calpain-dependent manner during EHEC infection and lost from its normal locale within microvilli. Calpain may be a central conduit through which EHEC and other AE pathogens induce enterocyte cytoskeletal remodeling and exert their pathogenic effects.
Introduction
Enterohemorrhagic Escherichia coli (EHEC) causes serious diarrheal illnesses worldwide. A bacterium that can be found in a variety of ruminants, EHEC can be transmitted to humans by ingestion of contaminated foods. Amongst the various EHEC strains, EHEC serotype O157:H7 has caused most of the serious outbreaks (for review, see Croxen and Finlay, 2010 and Kaper et al., 2004). Symptoms of the disease include severe abdominal cramping, watery diarrhea, hemorrhagic colitis, and in rare cases, hemolytic uremic syndrome (HUS), a triad of hemolysis, thrombocytopenia, and renal failure. HUS, the leading cause of renal failure in children in the US, is caused by systemic absorption of the EHEC toxin, Shiga toxin (Stx), which inhibits protein synthesis (for review, see Tarr et al., 2005).
Enterohemorrhagic E. coli is a member of the attaching and effacing (AE) pathogen family, which also includes enteropathogenic E. coli (EPEC), an important cause of infantile diarrhea in developing countries, and the mouse pathogen Citrobacter rodentium (Mundy et al., 2005; Borenshtein et al., 2008; Frankel and Phillips, 2008). During infection of intestinal epithelial cells, these extracellular pathogens induce dramatic changes in the host cell membrane and cytoskeleton, collectively referred to as AE lesions. At sites of bacterial attachment, surface microvilli are effaced, and bacteria intimately adhere to the host cell surface, appearing to partially “sink” into the mammalian cell. Also characteristic of these lesions is the assembly of striking “pedestals” of filamentous (F-) actin beneath bound bacteria (Caron et al., 2006; Campellone, 2010).
Microvilli are highly organized structures that not only allow for a ∼30-fold increase in the apical surface area of intestinal epithelial, but also serve as sites of robust specialized transport, thereby enhancing enterocytes’ ability to absorb water and nutrients (Tyska and Mooseker, 2002; Brown and Mcknight, 2010; Lange, 2010). Loss of microvilli would therefore severely impair absorptive capacity and facilitate diarrheal disease. In addition, AE pathogens that are incapable of generating AE lesions display severe colonization defects and reduced disease phenotypes (Donnenberg et al., 1993; Tzipori et al., 1995; Marches et al., 2000; Tacket et al., 2000; Ritchie et al., 2003), and EHEC mutants defective in stimulating actin pedestal formation fail to expand their initial infectious niche (Ritchie et al., 2008; Crepin et al., 2010).
To generate AE lesions, these pathogens inject effectors into host cells via a contact-dependent type III secretion system (T3SS; Kaper et al., 2004; Croxen and Finlay, 2010). An essential effector is the translocated intimin receptor (Tir), which, after insertion into the host cell apical membrane, binds the bacterial surface protein intimin, thus promoting an intimate connection to the host cell (Kenny et al., 1997). The cytosolic domains of Tir then initiate a signaling cascade that ultimately hijacks a host cell signaling cascade to form filamentous actin pedestals beneath the bound bacteria (Caron et al., 2006; Hayward et al., 2006; Campellone, 2010).
Though both EHEC and EPEC translocate highly related Tir molecules that are required for the formation of morphologically indistinguishable pedestals, the two pathogens trigger F-actin assembly by different signaling pathways (Caron et al., 2006; Hayward et al., 2006; Campellone, 2010). Pedestal formation by EHEC requires a host adaptor, insulin receptor tyrosine kinase substrate (IRTKS) or insulin receptor tyrosine kinase substrate p53 (IRSp53; Vingadassalom et al., 2009; Weiss et al., 2009), and an additional type III-secreted bacterial effector, EspFU, also know as Tir-cytoskeleton coupling protein (TccP), which stimulates the actin nucleation factor neural Wiskott–Aldrich syndrome protein (N-WASP; Campellone et al., 2004; Garmendia et al., 2004). In contrast, EPEC pedestals require recruitment of the host adaptor protein Nck, which in turn binds and activates N-WASP (Gruenheid et al., 2001; Campellone et al., 2002).
While pedestal formation by AE pathogens has been well characterized, much less is known about the mechanisms promoting microvillar effacement. The core bundle of F-actin in a single microvillus is stabilized internally by villin and fimbrin and tethered laterally to adjacent plasma membrane by myosin1A:calmodulin cross-bridges. The bundle is anchored at the base to the terminal web via conventional acto-myosin interactions that ultimately associate with the basolateral membrane domain terminating in adherens and tight junctions where adjacent epithelial cells are tethered to one another (Tyska and Mooseker, 2002; Brown and Mcknight, 2010). In addition, ezrin, an ERM family protein required for microvillar development, is found at this apical cytoskeletal–membrane interface of polarized intestinal epithelia, and is thought to bridge the apical plasma membrane to microvillar F-actin core (Bretscher et al., 2002; Fehon et al., 2010). Core microvillar components are also in constant turnover, making microvilli highly dynamic structures (Tyska and Mooseker, 2002; Brown and Mcknight, 2010). Because of this, there are several potential host proteins that could be targeted by EHEC to alter the cytoskeleton and yield AE lesions. Given the complexity and inherent dynamic nature of microvilli, AE pathogen-induced microvilli effacement likely involves participation of key host adaptor proteins, just as pedestal formation does.
An important class of eukaryotic cytoskeletal regulators are the calpains, Ca+2-dependent proteases that cleave a variety of enzymes and regulatory proteins to modulate cellular function. Calpains are ubiquitously expressed in vertebrates and have been implicated in many important cellular processes, such as regulation of signal transduction, cell spreading and motility, membrane repair, cell death, embryogenesis, and tumor suppression (Potter et al., 1998; Croall and Ersfeld, 2007; Sorimachi et al., 2010). Many reported calpain substrates are involved in regulating the actin dynamics, especially during cellular adhesion and migration (Shuster and Herman, 1995; Potter et al., 1998; Franco and Huttenlocher, 2005; Lebart and Benyamin, 2006; Chan et al., 2010; Kotecki et al., 2010). Several microbial pathogens, promote disease through the inappropriate activation of calpain (Fettucciari et al., 2006; Wang et al., 2008; Goldmann et al., 2009; Dean et al., 2010; Perry et al., 2010; Sumitomo et al., 2010; Zhang et al., 2010). EPEC infection results in an increase intracellular Ca+2 levels in mammalian cells (Baldwin et al., 1991), and induces calpain activity in a manner dependent on T3SS (Hardwidge et al., 2004; Dean et al., 2010). We previously showed that calpain controls EPEC-induced enterocyte effacement in vitro (Potter et al., 2003).
While calpain’s roles in effacement and intestinal barrier disruption have been documented for EPEC, its involvement in EHEC infection has not been characterized. Since comparative analysis of pedestal formation by EPEC and EHEC have revealed that these related pathogens generate actin pedestals by fundamentally different means (Caron et al., 2006; Hayward et al., 2006), we sought to determine if calpain plays an important role in EHEC effacement, as it does in EPEC (Potter et al., 2003). We show, here, that infection of polarized CaCo-2a monolayers by EHEC increased calpain activity, and inhibition of calpain blocked effacement and reduced other morphological manifestations of cell damage. Calpain-dependent ezrin disruption was also observed upon EHEC infection. These results indicate that calpain, perhaps by cleaving ezrin, likely plays a central role in EHEC-induced microvillar effacement. An understanding of the calpain-regulated cytoskeletal remodeling that controls host cell adaptive responses to bacterial challenge may offer innovative therapeutic strategies aimed at preventing EPEC and EHEC pathogenesis.
Materials and Methods
Antibodies and Phalloidin
Alexa 488- and 546-conjugated phalloidin, were obtained from Invitrogen. Primary antibodies used were mouse anti-ezrin (Zymed # 357300), rabbit anti-O157 (Gibco), and goat anti-O157 (Fitzgerald Industries #70-XG13). Secondary antibodies are species-specific Alexa 350-, 488-, and 546-labeled anti-IgG (Invitrogen).
Cell Culture
CaCo-2a enterocytes, stably transfected with the calpastatin high over-expression plasmid pRC/CMV-3ΔCSN (HOX) or the pRC/CMV empty vector (CON), have been previously described (Potter et al., 2003). Cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 1% each penicillin–streptomycin–fungizone (PSF) and L-glutamine. Sub-confluent cultures were maintained in T175 flasks. For experiments, cultures were washed twice with HBSS, released with trypsin–EDTA, and seeded at confluence (approximately 1.5 × 105 cells/cm2) in tissue cultures vessels precoated with collagen (BD #354236). CaCo-2a cells were then allowed to differentiate for 7–14 days, and fed every 2 or 3 days. For electron microscopy, cells were seeded onto standard plastic six-well, tissue culture plates. For immunofluorescence staining, cells were seeded into 24-well glass bottom tissue culture plates (Mattek #P24G-1.5-13F). For calpain activity, cells were seeded into plastic 96-well black walled tissue culture plates with optically clear bottoms (Potter et al., 2003).
Bacterial Strains
Table 1 lists bacterial strains used in this study. The wild type, Stx-deficient EHEC strain of serotype O157:H7, strain TUV-93, a derivative of the prototypic E. coli O157:H7 strain EDL933, has been previously described. The lack of Shigatoxin production by TUV-93-0 permits the prolonged infection of a wide variety of cell lines without the induction of Stx-mediated cytotoxicity. TUV-93ΔescN was generated by lambda Red recombineering as previously described (Murphy and Campellone, 2003).

Table 1. Bacterial strains used in this study.
Infection
In preparation for infection, individual colonies from freshly streaked plates were grown in 1 mL of LB with appropriate antibiotics for up to 8 h. Ten microliter of this day culture was transferred to DMEM/100 mM HEPES/antibiotics and incubated overnight at 37°C with 5% CO2 to induce T3SS expression. For infection, bacteria were resuspended in DMEM/2% FBS/20 mM HEPES/2 mM glutamine to the following MOI’s: EHEC at 500:1, EPEC at 100:1. Higher MOI’s were necessary for EHEC infections due to less efficient binding of EHEC to cultured cells; this is a commonly observed phenotype that was noted at least two decades ago (Cantey and Moseley, 1991). Strain TUV-93 also does not adhere to in vitro cultured cells as well as EHEC harvested from infected piglets (Brady et al., 2011). The latter observation suggests that the poor cell attachment by in vitro-grown EHEC, as were analyzed here (and in virtually all published cell binding studies), reflect the relative lack of a putative adhesin (or adhesins) that is expressed at higher levels when EHEC is growing in the mammalian host.
Quantification of Calpain Activity
Calpain activity was assayed by either one of two fluorogenic calpain substrate kits. For assays using AnaSpec’s Calpain Activity Kit (#72150), CaCo-2a cell monolayers are seeded in 96-well TC treated plates with clear bottoms and black walls (Ibidi #89626) 10–14 days prior to use. Cells were infected as described above. Infected monolayers were washed twice with PBS. Fifty microliter of assay buffer was added to each well and incubated at room temperature (RT) for 5–10 min. Cells were scraped with a pipette tip and 50 mL of fluorogenic substrate in assay buffer was added to each well. Plates were spun at 1000 rpm on a tabletop centrifuge (700 × g) for 5 min to reduce bubbles and incubated at RT in the dark for up to 60 min. For assays using Calbiochem’s InnoZyme™ Calpain Activity Assay Kit, (#CBA054), CaCo-2a cell monolayers are seeded in six-well TC treated plates 10–14 days prior to use. Cells were infected as described above. Infected monolayers were washed twice with PBS, and lysed with CytoBusterTM (EMD #71009) on ice for 5 min. Cell lysates were scraped, transferred into 1.5 mL tubes and spun at 12,000 g at 4°C for 15 min. The supernatants were collected and used immediately for measuring calpain activity per kit instructions or stored at −80°C. Fluorescence was measured using a Molecular Devices SpectraMax Gemini XS at 354 nm excitation and 442 nm emission for the Anaspec kit and 320 nm excitation and 480 nm emission for the Calbiochem kit, and corrected for background fluorescence (i.e., that of wells with substrate only).
Calpastat Treatment
Calpastat, a cell-penetrating calpastatin peptide (Croce et al., 1999) was synthesized at the Tufts Peptide Core Facility using solid phase Fmoc chemistry. Peptides were purified by high performance liquid chromatography; molecular mass and purity were confirmed by mass spectrometry. A 25 mM peptide stock was prepared in 100 mM HEPES, pH 7.4 and stored at −20°C. For treatments, cell monolayers were washed once with HBSS and cell culture media containing Calpastat at the specified concentrations was added. Cells were incubated at 37°C in 5% CO2 for 1 h.
Scanning Electron Microscopy
Cell monolayers were washed twice with HBSS, then fixed by immersion in 2.5% glutaraldehyde in 100 mM sodium phosphate buffer (pH 7.2) for a minimum of 2 h at RT. The fixed samples were then washed three times in the same buffer. Following the third wash, monolayers were dehydrated through a graded series of ethanol to 100% and then critical point dried in liquid CO2. The bottoms of the dishes were cut off and using silver conductive paste, the plastic disks with the cells attached on the surface were affixed to aluminum scanning electron microscopy (SEM) stubs and sputter coated with Au/Pd (80/20). The specimens were then examined using an FEI Quanta 200 FEG MK II scanning electron microscope at 10 kV accelerating voltage. Each specimen was systematically observed at 1000×, 2500×, and 5000× at five separate areas spaced throughout the disk. Images were assessed by four individuals, one of whom was blinded and another of whom was not involved in the study. Assessments were unanimous concerning the presence or absence of effacement. Representative images at 10,000× were taken with disks tilted about 30°.
Immunofluorescence Microscopy
For indirect immunofluorescence imaging studies, CaCo-2a cells were seeded at confluence and allowed to differentiate in 24-well glass bottom culture plates (Mattek #P24G-1.5-13F). At the indicated time points following infection, cells were washed with warm DMEM, then fixed in 4% formaldehyde/DMEM for 5 min at RT. Cells were permeabilized with 0.1% Triton buffer for 90 s at RT, washed three times with PBS, and then incubated with the specified primary antibodies for 1 h at RT. Labeled secondary antibodies and/or Alexa 488- or 544-conjugated phalloidin (Invitrogen) were applied for 45 min at RT. Experiments were repeated three times. For each experiment, greater than 5 fields were examined per condition; roughly 20–50 cells per field were viewed/scored. Images were acquired on a Zeiss 200 M inverted microscope with a Hamamatsu cooled-CCD digital camera (ER) and MetaMorph 7.0 imaging software (Molecular Devices Corp, PA, USA).
Subcellular Fractionation and Western Blotting
Infected and uninfected cells were washed with warm TBS to remove media and unattached bacteria. Extraction buffer containing 40 mM HEPES pH 7.2, 50 mM PIPES, 75 mM NaCl, 1 mM MgCl2, 0.5 mM EGTA, Protease Inhibitor Cocktail (1:1000, Sigma), 1 mM sodium orthovanadate, and 0.1% Triton detergent was applied to the cultures, 0.5 ml per well of a six-well plate, and plates were placed on an orbital shaker at 50 rpm for 10 min at RT. The extraction buffer was then removed to tubes held on ice. The remaining cellular residue was collected with 200 μL boiling hot Laemmli sample buffer (Sambrook and Russell, 2001). All extracts were dialyzed against 4 L distilled water at 4°C for 4 h with one water change at 2 h, using SnakeSkin dialysis tubing, 10 K MWCO (Pierce). Dialyzed samples were snap frozen in liquid nitrogen, lyophilized overnight, and brought up in 200 μL each Laemmli sample buffer. All samples were boiled for 3 min and loaded onto 10% acrylamide gels for SDS-PAGE, followed by transfer to nitrocellulose membranes. Membranes were stained for ponceau to confirm equal protein loading, then probed for ezrin using monoclonal mouse anti-ezrin (Zymed #357300).
Results
Calpain Activity is Increased Upon EHEC Infection
To investigate whether calpain activity is affected by EHEC, we used two different commercially available kits that utilized calpain-specific fluorogenic substrates. Compared to uninfected cell monolayers, calpain activity increased after 3 h of infection by EHEC. As reported (Dean et al., 2010), EPEC also induced an increase in calpain activity, while the T3SS deficient EHEC mutant Δescn did not. Figure 1 shows the results of one representative experiment. Pretreatment of polarized cell monolayers with a cell-penetrating calpastatin inhibitor for 1 h prior to EHEC infection reduced EHEC-induced calpain activation to near uninfected levels (Data not shown).
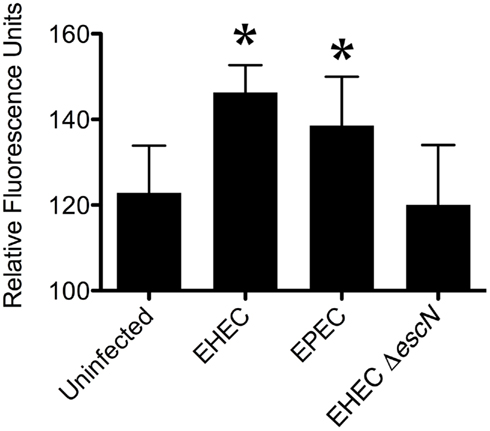
Figure 1. Enterohemorrhagic Escherichia coli increased calpain activity during infection. Calpain activity was measured with a fluorogenic, calpain-specific substrates in lysates of uninfected CaCo-2a cells, or of cells infected with EHEC, EPEC and EHEC ΔescN for 3 h. Shown are the means (±SE) with (*) indicating significance of p < 0.05 by student’s T-test compared against the uninfected group.
Calpastat, a Cell-Penetrating Calpain Inhibitor Prevents EHEC-Induced Effacement
To assess the role of calpain in EHEC-induced effacement, we infected polarized CaCo-2a CON cells with or without pretreatment with Calpastat, a cell-penetrating version of the endogenous calpain inhibitor, calpastatin (Croce et al., 1999; Carragher, 2006). SEM of uninfected control cells revealed distinct microvilli and cellular borders, although some variation in the density and arrangement of microvilli was observed (Figure 2A, and data not shown).

Figure 2. Enterohemorrhagic Escherichia coli infection produced additional gross monolayer damage that was diminished by inhibition of calpain. Uninfected CaCo-2a CON monolayers (A), or monolayers infected for 2 h with EPEC (C) or EHEC, with (D) or without (B) a 1-h pretreatment with Calpastat (2.5 μM) were visualized by SEM under 1000× magnification. Scale bars are 100 μm.
When CON cells were infected with EHEC and assessed by SEM at low (1000×) magnification (Figure 2) or TEM (data not shown), CON cells suffered severe “blebbing” and rounding, indicating cellular damage (Figure 2B). This apparent toxicity was not observed after infection with EPEC (Figure 2C), in which cell monolayers looked nearly identical to uninfected monolayers (Figure 2A). Pretreatment with 2.5 mM Calpastat for 1 h prior to EHEC infection diminished these effects (Figure 2D), suggesting that calpain activity is required for this cellular damage and raising the possibility that calpain may be critical for EHEC-induced cellular pathogenesis.
Higher magnification assessment of monolayers at 5000× to 10,000× revealed that a 2-h infection by EHEC resulted in the predicted microvillar effacement (Figure 3B) as compared to uninfected controls (Figure 3A). When CON cells were subjected to a 1-h pretreatment with 2.5 mM of Calpastat prior to EHEC infection and assessed by 10,000× magnification SEM, most EHEC-bound cells retained largely intact microvilli (Figure 3C), similar in quality to uninfected CaCo-2a CON monolayers (Figure 3A). As expected, the control infection by EPEC in the absence of Calpastat treatment also revealed effacement (Figure 3D).
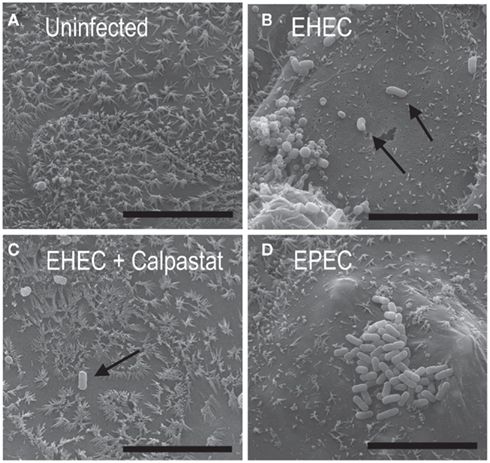
Figure 3. A cell-penetrating calpain inhibitor prevents microvilli effacement by EHEC. Uninfected CaCo-2a CON monolayers (A) and monolayers infected for 2 h with EHEC without (B) or with (C) 1 h pretreatment with 2.5 μM Calpastat, or infected with EPEC (D) were visualized by SEM at 10,000× magnification with a 30° tilt. Scale bars are 10 μm.
Ectopic Expression of Calpastatin Inhibits Effacement by EHEC
We previously showed that the calpastatin-overexpressing CaCo-2a cell line, HOX, for which the pRC/CMV vector-transfected CaCo-2a CON line serves as an appropriate “wildtype” control, resists effacement by EPEC (Potter et al., 2003). Like the parent CaCo-2a and vector control CON line, these cells polarize and appear to differentiate, in that they develop trans-epithelial resistance and, by SEM, generate microvilli (albeit morphologically distinct from those on CON cells; Potter et al., 2003). Like CON cells, some cell-to-cell variability with respect to microvillar length and density (data not shown) were observed. HOX microvilli were, intact and in general, shorter than those observed in CON cells, (Figure 4A). As reported; these phenotypic differences compared to wild type CaCo-2 cells are likely due to the altered cytoskeletal architecture resulting from disrupted calpain-driven cytoskeletal remodeling (Potter et al., 2003).
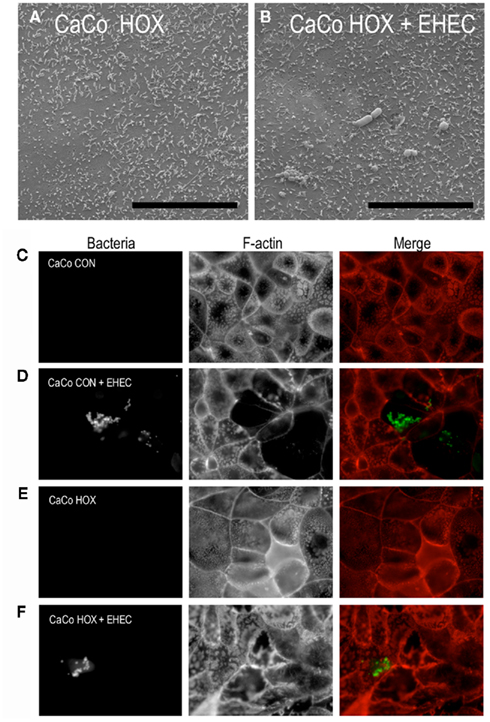
Figure 4. The calpastatin overexpressing cell line, HOX, resisted microvillar effacement by AE pathogens. Uninfected polarized CaCo-2a HOX monolayers (A) and polarized HOX monolayers infected for 2 h with EHEC (B) were visualized by SEM at 10,000× magnification and with a 30° tilt. Scale bars for SEM’s are 10 μm. Polarized control CaCo-2a CON (C,D) and calpastatin-overexpressing CaCo-2a HOX cells (E,F) were stained for F-actin (red) and EHEC (green) without (C,E) or with infection (D,F) by EHEC. F-actin was stained using Alexa 568-conjugated phalloidin; EHEC were stained with anti-O157 antibody.
When stained for F-actin using fluorescent phalloidin and imaged near the apical surface of the monolayers, the microvillar F-actin core bundles of polarized CON and HOX cells appeared as a punctate fluorescent pattern on the apical surface (Figures 4C,E) when viewed “end-on,” while the circumferential “belt” of F-actin encircling the apical cytoskeletal–membrane domain could also be readily distinguished. This apical F-actin staining pattern was consistent with the pattern of microvilli observed by SEM (Figures 3D and 4A). In addition, infection of polarized CON cells by EHEC induced the loss of this punctate fluorescent staining (Figure 4D), consistent with the loss of microvilli observed during EHEC-induced effacement as revealed by high magnification SEM (Figure 3A). (Note that the EHEC strain used in this study requires 4–6 h of infection to form actin pedestals, and were not detected in the timeframe of the current study.)
In stark contrast to the effacement of CON cells by EHEC, infected HOX cells retained their microvillar-associated punctate F-actin fluorescence staining (Figure 4F). The retention of microvillar integrity by HOX cells after 2 h (Figure 4F) or 3 h (data not shown) was confirmed by SEM (Figure 4B and data not shown). These results indicate that ectopic expression of calpastatin and the concomitant inhibition of calpain renders HOX cells resistant to effacement by EHEC infection.
The Calpain Substrate Ezrin is Cleaved in Response to EHEC Infection and Lost from Microvilli
We have previously shown that calpain-mediated cleavage of ezrin occurs during cell spreading and migration, a process that, like effacement, involves extensive actin remodeling (Shuster and Herman, 1995; Potter et al., 1998, 2003). Given that EHEC induces calpain activity upon infection of mammalian cells, we tested for ezrin cleavage upon infection by EHEC. Immunoblotting of subcellular fractions of EHEC-infected CaCo-2a CON cells revealed ezrin cleavage (Figure 5A). The full-length 80 kDa species was detected in both soluble and insoluble fractions. The cleaved 55 kDa ezrin fragment, which is not associated with the plasma membrane, was detected at very slight levels in the soluble fraction of uninfected CON cells. Endogenous calpain activity is present in all cells, supporting various functions in situ such as cellular differentiation, membrane polarization, or cell cycle progression. Therefore a small amount of cleaved ezrin should exist, even in uninfected cells. Infection with EHEC increased this pool of cleaved ezrin. A significant amount of full-length ezrin was still detected in EHEC-infected CON monolayers likely because, as can be seen in Figures 4D,F, EHEC does not infect cells uniformly, but rather heavily infects a fraction of cells. Thus, a mixture of infected and uninfected cells contributes to the lysate collected for western blotting. The 55 kDa ezrin cleavage product was not observed in uninfected or EHEC-infected HOX cells, indicating that calpain is required for its generation (Figure 5A).
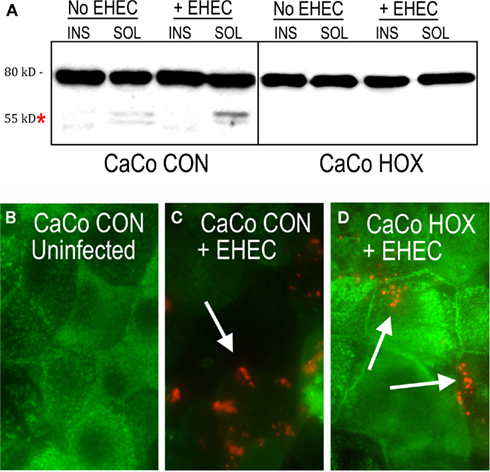
Figure 5. Enterohemorrhagic Escherichia coli infection caused calpain-dependent loss of mirovillar ezrin and ezrin cleavage. Lysates from subcellular fractions of polarized control CaCo-2a CON and HOX monolayers with and without 6 h EHEC infection by were probed for ezrin by western blot. Asterisk (or arrow) indicates the ∼55 kDa ezrin cleavage product (A). Polarized CaCo-2a CON cells without (B) and with 6 h infection by EHEC (C) and EHEC-infected polarized HOX monolayers (D) were stained for ezrin (green) and EHEC (red). EHEC was detected by anti-O157 antibody.
Immunofluorescence staining of ezrin in polarized CaCo-2a CON cells revealed a punctate pattern at the apical surface almost identical to that of microvilli staining by phalloidin (Figure 5B). Images taken at the apical surface showed a staining pattern that is consistent with its role in linking the plasma membrane to core microvillar actin bundles (Fehon et al., 2010). To determine if EHEC-mediated cleavage of ezrin correlated with a change in the cellular distribution of ezrin, we stained CON cells for ezrin after EHEC infection. Interestingly, the apical punctate ezrin pattern disappeared throughout the entire infected cell (Figure 5C). Cleavage of ezrin would result from its release from the apical surface, thus appearing as a loss of staining in the focal plane imaged. This change in localization was dependent on calpain activity, because calpastatin-overexpressing HOX cells retained their punctate ezrin staining pattern during infection (Figure 5D).
Discussion
In response to challenge by AE pathogens, intestinal epithelial cells undergo a coordinated and robust remodeling of the apical membrane–cytoskeletal domain (Goosney et al., 2000; Hardwidge et al., 2004; Caron et al., 2006). Highly organized core bundles of actin in the microvilli are disassembled while the apical membrane conforms to newly synthesized actin pedestals. These structural alterations likely disrupt overall epithelial cell function and integrity and contribute to EHEC- and EPEC-induced gastroenteritis and diarrhea. While the process of pedestal formation is well characterized, the mechanisms of microvillar effacement are poorly understood. Previous work indicated that EPEC infection triggers a rise intracellular Ca+2 in mammalian cells (Baldwin et al., 1991), and EPEC-mediated effacement requires the Ca+2-regulated host protease, calpain (Potter et al., 2003). In this study, we showed that EHEC infection of CaCo-2a cells induces an increase in calpain activity that is required for EHEC-mediated effacement. By analogy to EPEC, we postulate that type III-secreted effectors play a role in both calpain activation and microvillar effacement (Hardwidge et al., 2004; Dean et al., 2010). In fact, an EHECΔescN mutant, which is defective in type III secretion, did not trigger an increase in calpain activity.
Effacement likely involves disruption of protein–protein and protein–plasma membrane interactions that contribute to microvillar integrity. Calpain is known to target a number of cytoskeletal elements, including the ERM family member ezrin (Shuster and Herman, 1995; Potter et al., 1998; Frame et al., 2002; Franco and Huttenlocher, 2005; Lebart and Benyamin, 2006), which contributes to microvillar integrity by indirectly linking the plasma membrane to the axial actin microfilamentous bundles (Shuster et al., 1996; Bretscher et al., 2002; Saotome et al., 2004). Another gut pathogen, H. pylori, triggers calpain-dependent cleavage of ezrin in gastric parietal cells, resulting in redistribution of the protein, distortion of microvillar structure and disruption of apical secretory function (Wang et al., 2008). We showed here that EHEC-mediated effacement of microvilli is accompanied by calpain-dependent cleavage of ezrin and loss of its apical localization.
The targeting of ezrin by AE pathogens is likely to induce widespread changes in the structure and function of intestinal epithelial cells. EPEC triggers the transient formation of filopodia that are significantly destabilized by the expression of a dominant negative ezrin mutant (Berger et al., 2009). Ezrin has also has been implicated in the assembly of junctional complexes (Pujuguet et al., 2003), and we found that EHEC infection results in a calpain-dependent redistribution away from cell–cell junctions. This relocalization may well have functional consequences on epithelial barrier function, given the observations that the addition of calpain-inhibitory peptides (Dean et al., 2010) and or the expression of a dominant negative ezrin mutant (Simonovic et al., 2001) diminish EPEC-induced disruption of tight junctions and trans-epithelial resistance.
More generally, apical and junctional cytoskeletal domains are functionally integrated: F-actin rich microvillar rootlet structures penetrate the terminal web, making intimate contact with the circumferentially disposed array of filamentous actin, conventional myosins and intermediate filaments, which all join together in the terminal web at adherens and tight, occluding junctions (Drenckhahn and Dermietzel, 1988; Bretscher, 1991; Nelson, 2003). Thus, the ability of AE pathogens to dramatically alter the apical cytoskeleton is likely to induce changes in the basolateral and terminal web-associated cytoskeleton including the membrane–cytoskeletal interface. Consistent with this notion, EPEC infection is known to cause the redistribution of many junctional and basolateral domain proteins (Goosney et al., 2001; Muza-Moons et al., 2003, 2004; Guttman et al., 2006, 2007), some of which are recruited to actin pedestals (Peralta-Ramirez et al., 2008; Huett et al., 2009). It will be of interest to determine whether the requirement for calpain in the redistribution of ezrin is reflected in a similar requirement for the redistribution of some or all of these other molecules. Finally, we observed EHEC infection of confluent epithelial cell monolayers resulted in striking cell rounding and “blebbing”, which to our knowledge has not been previously reported in response to infection by AE pathogens but is consistent with a general disruption of membrane–cytoskeletal interactions and cell adhesion. Although this damage was calpain-dependent, similar cellular damage was not induced by EPEC, indicating that this manifestation is separable from effacement and is specific to EHEC. This difference could reflect the fact that EHEC encodes a larger number of injected T3SS effectors than does EPEC (Tobe et al., 2006; Wong et al., 2011). Although EHEC-mediated cell rounding could be specifically linked to ezrin cleavage, other actin-associated calpain substrates, such as the focal adhesion protein talin and actin assembly factor and cortactin might also or alternatively be involved (Frame et al., 2002; Franco and Huttenlocher, 2005; Lebart and Benyamin, 2006).
In summary, AE pathogens such as EHEC induce a broad range of cytoskeletal disruption, among them pedestal formation, tight junction disruption and microvillar effacement. These alterations are predicted to diminish both the absorptive capacity and the barrier function of intestinal epithelium, thereby contributing to diarrhea, a major and potentially life-threatening manifestation of disease. The manifold cellular consequences of infection are undoubtedly in part due to diversity of effectors translocated to the cell (Tobe et al., 2006). In addition, an emerging picture is that a central modulator such as calpain, which has the potential to alter many cytoskeletal structures, may be a conduit through which AE family members, as well as other infectious agents, exert pleomorphic pathogenic effects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to David Potter for the longstanding collaborations focused on calpain-dependent signaling in metazoan cells, and to Lara Strittmatter and Greg Hendricks of the UMass Electron Microscopy Core for dedicated and skilled work. This work is supported in part by NIH EY 15125 to Ira M. Herman, NIH-R01-AI46454 to John M. Leong, and a Summer Intern Fellowship provided to Kathleen Riley through NIH Training Grant T35 DK07635.
References
Baldwin, T. J., Ward, W., Aitken, A., Knutton, S., and Williams, P. H. (1991). Elevation of intracellular free calcium levels in HEp-2 cells infected with enteropathogenic Escherichia coli. Infect. Immun. 59, 1599–1604.
Berger, C. N., Crepin, V. F., Jepson, M. A., Arbeloa, A., and Frankel, G. (2009). The mechanisms used by enteropathogenic Escherichia coli to control filopodia dynamics. Cell. Microbiol. 11, 309–322.
Borenshtein, D., Mcbee, M. E., and Schauer, D. B. (2008). Utility of the Citrobacter rodentium infection model in laboratory mice. Curr. Opin. Gastroenterol. 24, 32–37.
Brady, M. J., Radhakrishnan, P., Liu, H., Magoun, L., Murphy, K. C., Mukherjee, J., Donohue-Rolfe, A., Tzipori, S., and Leong, J. M. (2011). Enhanced actin pedestal formation by enterohemorrhagic Escherichia coli O157:H7 adapted to the mammalian host. Front. Microbio. 2:226. doi: 10.3389/fmicb.2011.00226
Bretscher, A. (1991). Microfilament structure and function in the cortical cytoskeleton. Annu. Rev. Cell Biol. 7, 337–374.
Bretscher, A., Edwards, K., and Fehon, R. G. (2002). ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3, 586–599.
Brown, J. W., and Mcknight, C. J. (2010). Molecular model of the microvillar cytoskeleton and organization of the brush border. PLoS ONE 5, e9406. doi:10.1371/journal.pone.0009406
Campellone, K. G. (2010). Cytoskeleton-modulating effectors of enteropathogenic and enterohaemorrhagic Escherichia coli: Tir, EspFU and actin pedestal assembly. FEBS J. 277, 2390–2402.
Campellone, K. G., Giese, N., Tipper, O. J., and Leong, J. M. (2002). A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol. Microbiol. 43, 1227–1241.
Campellone, K. G., Robbins, D., and Leong, J. M. (2004). EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev. Cell 7, 217–228.
Cantey, J. R., and Moseley, S. L. (1991). HeLa cell adherence, actin aggregation, and invasion by nonenteropathogenic Escherichia coli possessing the eae gene. Infect. Immun. 59, 3924–3929.
Caron, E., Crepin, V. F., Simpson, N., Knutton, S., Garmendia, J., and Frankel, G. (2006). Subversion of actin dynamics by EPEC and EHEC. Curr. Opin. Microbiol. 9, 40–45.
Carragher, N. O. (2006). Calpain inhibition: a therapeutic strategy targeting multiple disease states. Curr. Pharm. Des. 12, 615–638.
Chan, K. T., Bennin, D. A., and Huttenlocher, A. (2010). Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 285, 11418–11426.
Crepin, V. F., Girard, F., Schüller, S., Phillips, A. D., Mousnier, A., and Frankel, G. (2010). Dissecting the role of the Tir:Nck and Tir:IRTKS/IRSp53 signalling pathways in vivo. Mol. Microbiol. 75, 308–323.
Croall, D., and Ersfeld, K. (2007). The calpains: modular designs and functional diversity. Genome Biol. 8, 218.
Croce, K., Flaumenhaft, R., Rivers, M., Furie, B., Furie, B. C., Herman, I. M., and Potter, D. A. (1999). Inhibition of calpain blocks platelet secretion, aggregation, and spreading. J. Biol. Chem. 274, 36321–36327.
Croxen, M. A., and Finlay, B. B. (2010). Molecular mechanisms of Escherichia coli pathogenicity. (Report). Nat. Rev. Microbiol. 8, 26.
Dean, P., Mühlen, S., Quitard, S., and Kenny, B. (2010). The bacterial effectors EspG and EspG2 induce a destructive calpain activity that is kept in check by the co-delivered Tir effector. Cell. Microbiol. 12, 1308–1321.
Donnenberg, M. S., Tzipori, S., Mckee, M. L., O’brien, A. D., Alroy, J., and Kaper, J. B. (1993). The role of the eae gene of enterohemorrhagic Escherichia coli in intimate attachment in vitro and in a porcine model. J. Clin. Invest. 92, 1418–1424.
Drenckhahn, D., and Dermietzel, R. (1988). Organization of the actin filament cytoskeleton in the intestinal brush border: a quantitative and qualitative immunoelectron microscope study. J. Cell Biol. 107, 1037–1048.
Fehon, R. G., Mcclatchey, A. I., and Bretscher, A. (2010). Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287.
Fettucciari, K., Fetriconi, I., Mannucci, R., Nicoletti, I., Bartoli, A., Coaccioli, S., and Marconi, P. (2006). Group B Streptococcus induces macrophage apoptosis by calpain activation. J. Immunol. 176, 7542–7556.
Frame, M. C., Fincham, V. J., Carragher, N. O., and Wyke, J. A. (2002). v-SRC’S hold over actin and cell adhesions. Nat. Rev. Mol. Cell Biol. 3, 233–245.
Franco, S. J., and Huttenlocher, A. (2005). Regulating cell migration: calpains make the cut. J. Cell. Sci. 118, 3829–3838.
Frankel, G., and Phillips, A. D. (2008). Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: getting off the pedestal. Cell. Microbiol. 10, 549–556.
Garmendia, J., Phillips, A. D., Carlier, M.-F., Chong, Y., Schüller, S., Marches, O., Dahan, S., Oswald, E., Shaw, R. K., Knutton, S., and Frankel, G. (2004). TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton†. Cell. Microbiol. 6, 1167–1183.
Goldmann, O., Sastalla, I., Wos-Oxley, M., Rohde, M., and Medina, E. (2009). Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell. Microbiol. 11, 138–155.
Goosney, D. L., Devinney, R., and Finlay, B. B. (2001). Recruitment of cytoskeletal and signaling proteins to enteropathogenic and enterohemorrhagic Escherichia coli pedestals. Infect. Immun. 69, 3315–3322.
Goosney, D. L., Gruenheid, S., and Finlay, B. B. (2000). GUT FEELINGS: enteropathogenic E. coli (EPEC) Interactions with the host. Annu. Rev. Cell Dev. Biol. 16, 173–189.
Gruenheid, S., Devinney, R., Bladt, F., Goosney, D., Gelkop, S., Gish, G. D., Pawson, T., and Finlay, B. B. (2001). Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat. Cell Biol. 3, 856–859.
Guttman, J. A., Li, Y., Wickham, M. E., Deng, W., Vogl, A. W., and Finlay, B. B. (2006). Attaching and effacing pathogen-induced tight junction disruption in vivo. Cell. Microbiol. 8, 634–645.
Guttman, J. A., Samji, F. N., Li, Y., Deng, W., Lin, A., and Finlay, B. B. (2007). Aquaporins contribute to diarrhoea caused by attaching and effacing bacterial pathogens. Cell. Microbiol. 9, 131–141.
Hardwidge, P. R., Rodriguez-Escudero, I., Goode, D., Donohoe, S., Eng, J., Goodlett, D. R., Aebersold, R., and Finlay, B. B. (2004). Proteomic analysis of the intestinal epithelial cell response to enteropathogenic Escherichia coli. J. Biol. Chem. 279, 20127–20136.
Hayward, R. D., Leong, J. M., Koronakis, V., and Campellone, K. G. (2006). Exploiting pathogenic Escherichia coli to model transmembrane receptor signalling. Nat. Rev. Microbiol. 4, 358–370.
Huett, A., Leong, J. M., Podolsky, D. K., and Xavier, R. J. (2009). The cytoskeletal scaffold Shank3 is recruited to pathogen-induced actin rearrangements. Exp. Cell Res. 315, 2001–2011.
Jerse, A. E., Yu, J., Tall, B. D., and Kaper, J. B. (1990). A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. U.S.A. 87, 7839–7843.
Kaper, J. B., Nataro, J. P., and Mobley, H. L. T. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140.
Kenny, B., Devinney, R., Stein, M., Reinscheid, D. J., Frey, E. A., and Finlay, B. B. (1997). Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91, 511–520.
Kotecki, M., Zeiger, A. S., Van Vliet, K. J., and Herman, I. M. (2010). Calpain- and talin-dependent control of microvascular pericyte contractility and cellular stiffness. Microvasc. Res. 80, 339–348.
Lange, K. (2010). Fundamental role of microvilli in the main functions of differentiated cells: outline of an universal regulating and signaling system at the cell periphery. J. Cell. Physiol. 226, 896–927.
Lebart, M.-C., and Benyamin, Y. (2006). Calpain involvement in the remodeling of cytoskeletal anchorage complexes. FEBS J. 273, 3415–3426.
Marches, O., Nougayrede, J.-P., Boullier, S., Mainil, J., Charlier, G., Raymond, I., Pohl, P., Boury, M., De Rycke, J., Milon, A., and Oswald, E. (2000). Role of Tir and intimin in the virulence of rabbit enteropathogenic Escherichia coli serotype O103:H2. Infect. Immun. 68, 2171–2182.
Mundy, R., Macdonald, T. T., Dougan, G., Frankel, G., and Wiles, S. (2005). Citrobacter rodentium of mice and man. Cell. Microbiol. 7, 1697–1706.
Murphy, K., and Campellone, K. (2003). Lambda red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4, 11. doi:10.1186/1471-2199-4-11
Muza-Moons, M. M., Koutsouris, A., and Hecht, G. (2003). Disruption of cell polarity by enteropathogenic Escherichia coli enables basolateral membrane proteins to migrate apically and to potentiate physiological consequences. Infect. Immun. 71, 7069–7078.
Muza-Moons, M. M., Schneeberger, E. E., and Hecht, G. A. (2004). Enteropathogenic Escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cells. Cell. Microbiol. 6, 783–793.
Peralta-Ramirez, J., Hernandez, J. M., Manning-Cela, R., Luna-Munoz, J., Garcia-Tovar, C., Nougayrede, J.-P., Oswald, E., and Navarro-Garcia, F. (2008). EspF interacts with nucleation-promoting factors to recruit junctional proteins into pedestals for pedestal maturation and disruption of paracellular permeability. Infect. Immun. 76, 3854–3868.
Perry, S. W., Barbieri, J., Tong, N., Polesskaya, O., Pudasaini, S., Stout, A., Lu, R., Kiebala, M., Maggirwar, S. B., and Gelbard, H. A. (2010). Human immunodeficiency virus-1 tat activates calpain proteases via the ryanodine receptor to enhance surface dopamine transporter levels and increase transporter-specific uptake and Vmax. J. Neurosci. 30, 14153–14164.
Potter, D. A., Srirangam, A., Fiacco, K. A., Brocks, D., Hawes, J., Herndon, C., Maki, M., Acheson, D., and Herman, I. M. (2003). Calpain regulates enterocyte brush border actin assembly and pathogenic Escherichia coli-mediated effacement. J. Biol. Chem. 278, 30403–30412.
Potter, D. A., Tirnauer, J. S., Janssen, R., Croall, D. E., Hughes, C. N., Fiacco, K. A., Mier, J. W., Maki, M., and Herman, I. M. (1998). Calpain regulates actin remodeling during cell spreading. J. Cell Biol. 141, 647–662.
Pujuguet, P., Del Maestro, L., Gautreau, A., Louvard, D., and Arpin, M. (2003). Ezrin regulates E-cadherin-dependent adherens junction assembly through Rac1 activation. Mol. Biol. Cell 14, 2181–2191.
Ritchie, J. M., Brady, M. J., Riley, K. N., Ho, T. D., Campellone, K. G., Herman, I. M., Donohue-Rolfe, A., Tzipori, S., Waldor, M. K., and Leong, J. M. (2008). EspFU, a type III-translocated effector of actin assembly, fosters epithelial association and late-stage intestinal colonization by E. coli O157:H7. Cell. Microbiol. 10, 836–847.
Ritchie, J. M., Thorpe, C. M., Rogers, A. B., and Waldor, M. K. (2003). Critical roles for stx2, eae, and tir in enterohemorrhagic Escherichia coli-induced diarrhea and intestinal inflammation in infant rabbits. Infect. Immun. 71, 7129–7139.
Sambrook, J., and Russell, D. (eds). (2001). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.
Saotome, I., Curto, M., and Mcclatchey, A. I. (2004). Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev. Cell 6, 855–864.
Shuster, C. B., and Herman, I. M. (1995). Indirect association of ezrin with F-actin: isoform specificity and calcium sensitivity. J. Cell Biol. 128, 837–848.
Shuster, C. B., Lin, A. Y., Nayak, R., and Herman, I. M. (1996). βCAP73: a novel β actin-specific binding protein. Cell Motil. Cytoskeleton 35, 175–187.
Simonovic, I., Arpin, M., Koutsouris, A., Falk-Krzesinski, H. J., and Hecht, G. (2001). Enteropathogenic Escherichia coli activates Ezrin, which participates in disruption of tight junction barrier function. Infect. Immun. 69, 5679–5688.
Sorimachi, H., Hata, S., and Ono, Y. (2010). Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp. Animals 59, 549–566.
Sumitomo, T., Nakata, M., Higashino, M., Jin, Y., Terao, Y., Fujinaga, Y., and Kawabata, S. (2010). Streptolysin S contributes to group A streptococcal translocation across an epithelial barrier. J. Biol. Chem. 286, 2750–2761.
Tacket, C. O., Sztein, M. B., Losonsky, G., Abe, A., Finlay, B. B., Mcnamara, B. P., Fantry, G. T., James, S. P., Nataro, J. P., Levine, M. M., and Donnenberg, M. S. (2000). Role of EspB in experimental human enteropathogenic Escherichia coli infection. Infect. Immun. 68, 3689–3695.
Tarr, P. I., Gordon, C. A., and Chandler, W. L. (2005). Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365, 1073–1086.
Tobe, T., Beatson, S. A., Taniguchi, H., Abe, H., Bailey, C. M., Fivian, A., Younis, R., Matthews, S., Marches, O., Frankel, G., Hayashi, T., and Pallen, M. J. (2006). An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. 103, 14941–14946.
Tyska, M. J., and Mooseker, M. S. (2002). MYO1A (brush border myosin I) dynamics in the brush border of LLC-PK1-CL4 cells. Biophys. J. 82, 1869–1883.
Tzipori, S., Gunzer, F., Donnenberg, M. S., De Montigny, L., Kaper, J. B., and Donohue-Rolfe, A. (1995). The role of the eaeA gene in diarrhea and neurological complications in a gnotobiotic piglet model of enterohemorrhagic Escherichia coli infection. Infect. Immun. 63, 3621–3627.
Vingadassalom, D., Kazlauskas, A., Skehan, B., Cheng, H.-C., Magoun, L., Robbins, D., Rosen, M. K., Saksela, K., and Leong, J. M. (2009). Insulin receptor tyrosine kinase substrate links the E. coli O157:H7 actin assembly effectors Tir and EspFU during pedestal formation. Proc. Natl. Acad. Sci. U.S.A. 106, 6754–6759.
Wang, F., Xia, P., Wu, F., Wang, D., Wang, W., Ward, T., Liu, Y., Aikhionbare, F., Guo, Z., Powell, M., Liu, B., Bi, F., Shaw, A., Zhu, Z., Elmoselhi, A., Fan, D., Cover, T. L., Ding, X., and Yao, X. (2008). Helicobacter pylori VacA disrupts apical membrane-cytoskeletal interactions in gastric parietal cells. J. Biol. Chem. 283, 26714–26725.
Weiss, S. M., Ladwein, M., Schmidt, D., Ehinger, J., Lommel, S., Städing, K., Beutling, U., Disanza, A., Frank, R., JäNsch, L., Scita, G., Gunzer, F., Rottner, K., and Stradal, T. E. B. (2009). IRSp53 links the enterohemorrhagic E. coli effectors Tir and EspFU for actin pedestal formation . Cell Host Microbe 5, 244–258.
Wong, A. R. C., Pearson, J. S., Bright, M. D., Munera, D., Robinson, K. S., Lee, S. F., Frankel, G., and Hartland, E. L. (2011). Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol. Microbiol. 80, 1420–1438.
Keywords: CaCo-2, calpastat, calpastatin, ezrin, attaching and effacing lesion, microvilli
Citation: Lai Y, Riley K, Cai A, Leong JM and Herman IM (2011) Calpain mediates epithelial cell microvillar effacement by enterohemorrhagic Escherichia coli. Front. Microbio. 2:222. doi: 10.3389/fmicb.2011.00222
Received: 11 July 2011;
Paper pending published: 27 July 2011;
Accepted: 20 October 2011;
Published online: 08 November 2011.
Edited by:
Elizabeth L. Hartland, The University of Melbourne, AustraliaReviewed by:
Carolyn J. Hovde, University of Idaho, USASabrina Mühlen, University of Melbourne, Australia
Copyright: © 2011 Lai, Riley, Cai, Leong and Herman. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: John M. Leong, Department of Molecular Biology and Microbiology, Tufts University School of Medicine, 136 Harrison Avenue, Boston, MA, 02111, USA. e-mail:am9obi5sZW9uZ0B0dWZ0cy5lZHU=; Ira M. Herman, Department of Molecular Physiology and Pharmacology, Tufts University School of Medicine, 136 Harrison Avenue, Boston, MA 02111, USA. e-mail:aXJhLmhlcm1hbkB0dWZ0cy5lZHU=