 Michael John Brady1†
Michael John Brady1† Padhma Radhakrishnan1†
Padhma Radhakrishnan1† Hui Liu1 Loranne Magoun1 Kenan C. Murphy1
Hui Liu1 Loranne Magoun1 Kenan C. Murphy1 Jean Mukherjee2 Arthur Donohue-Rolfe2 Saul Tzipori2
Jean Mukherjee2 Arthur Donohue-Rolfe2 Saul Tzipori2 John M. Leong1,3*
John M. Leong1,3*- 1 Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA, USA
- 2 Division of Infectious Disease, Department of Biomedical Sciences, Cummings School of Veterinary Medicine, Tufts University, North Grafton, MA, USA
- 3 Department of Molecular Biology and Microbiology, Tufts University School of Medicine, Boston, MA, USA
Upon intestinal colonization, enterohemorrhagic Escherichia coli (EHEC) induces epithelial cells to generate actin “pedestals” beneath bound bacteria, lesions that promote colonization. To induce pedestals, EHEC utilizes a type III secretion system to translocate into the mammalian cell bacterial effectors such as translocated intimin receptor (Tir), which localizes in the mammalian cell membrane and functions as a receptor for the bacterial outer membrane protein intimin. Whereas EHEC triggers efficient pedestal formation during mammalian infection, EHEC cultured in vitro induces pedestals on cell monolayers with relatively low efficiency. To determine whether growth within the mammalian host enhances EHEC pedestal formation, we compared in vitro-cultivated bacteria with EHEC directly isolated from infected piglets. Mammalian adaptation by EHEC was associated with a dramatic increase in the efficiency of cell attachment and pedestal formation. The amounts of intimin and Tir were significantly higher in host-adapted than in in vitro-cultivated bacteria, but increasing intimin or Tir expression, or artificially increasing the level of bacterial attachment to mammalian cells, did not enhance pedestal formation by in vitro-cultivated EHEC. Instead, a functional assay suggested that host-adapted EHEC translocate Tir much more efficiently than does in vitro-cultivated bacteria. These data suggest that adaptation of EHEC to the mammalian intestine enhances bacterial cell attachment, expression of intimin and Tir, and translocation of effectors that promote actin signaling.
Introduction
Enterohemorrhagic Escherichia coli (EHEC) serotype O157:H7 is the leading cause of outbreaks of bloody diarrhea and is often associated with the triad of hemorrhagic colitis, thrombocytopenia, and renal failure in the United States (Karmali, 1989). The life threatening sequelae of EHEC infections are due to the production of Shiga toxins (Karmali, 1989; Noel and Boedeker, 1997; Teel et al., 2003). EHEC belongs to a unique subset of intestinal pathogens that cause attaching and effacing (AE) lesions on the intestinal epithelium during infection. AE lesions are histo-pathological alterations of the intestinal epithelial surface that are characterized by loss of brush border microvilli and formation of actin rich pedestals beneath bound bacteria (Pai et al., 1986). Enteropathogenic E. coli (EPEC) is a related intestinal pathogen that causes infantile diarrhea that also generates AE lesions in the host (Frankel et al., 1998; Nataro and Kaper, 1998; Celli et al., 2000; Campellone and Leong, 2003).
An ~35 kB pathogenicity island in E. coli O157:H7 termed the locus of enterocyte effacement (LEE) is required for AE lesion formation (McDaniel et al., 1995; McDaniel and Kaper, 1997). Some of the genes on the LEE required for AE lesion formation encode a type III secretion apparatus that injects bacterial effectors into host cells. The best-characterized secreted effector is translocated intimin receptor (Tir) which, when localized in the host cell membrane, serves as a receptor for intimin, a LEE encoded outer membrane adhesin. Intimin is necessary for intimate attachment to epithelial cells and its interaction with Tir is required for production of AE lesions at the enterocyte–bacteria interface. AE lesion formation can be conceptually divided into multiple stages: initial attachment of the bacteria to the host epithelia, type III secretion during which Tir and other E. coli secreted proteins (Esps) are translocated into host cells, and finally Intimin-mediated Tir ligation at the plasma membrane, which triggers host cell signaling events that lead to actin assembly (Donnenberg et al., 1997; Hayward et al., 2006; Frankel and Phillips, 2008; Campellone, 2010). Consistent with this model, mammalian cells injected with Esps delivered by a pre-infecting intimin-deficient EPEC mutant are capable of initiating robust actin focusing upon challenge with another strain or particle expressing intimin (Rosenshine et al., 1996; Liu et al., 1999b).
Enterohemorrhagic Escherichia coli generates far fewer pedestals than EPEC on cultured mammalian cells. EHEC also exhibits relatively poor mammalian cell binding and actin pedestal formation on cultured monolayers after in vitro cultivation, in contrast to the robust AE lesion formation on intestinal epithelia observed during mammalian infection (Karch et al., 1987; Tzipori et al., 1987; Cantey and Moseley, 1991). This apparent paradox raises the possibility that growth in the mammalian host environment enhances the ability of EHEC to form AE lesions. To directly determine the relative efficiencies of virulence-associated phenotypes, we compared in vitro-cultured bacteria to EHEC isolated directly from infected piglets. We found that adaptation of EHEC to the mammalian host is associated with enhanced mammalian cell binding, increased amounts of intimin and Tir, better translocation of functional Tir, and dramatically more efficient pedestal formation on cultured mammalian cells.
Materials and Methods
In vitro Bacterial and Tissue Culture
The strains and plasmids used in this study are listed (Table 1). To enhance biosafety, we and other labs routinely utilized an Stx-deficient derivative of EHEC EDL933 (Riley et al., 1983) when examining virulence features unrelated to toxin production. TUV93-0, lacking Stx, is predicted to be incapable of causing hemorrhagic colitis, but for simplicity, here we nevertheless refer to this as an “EHEC” strain. For infections, EHEC strains were cultured in LB, Miller (BD Difco) broth at 37°C with shaking for approximately 7 h, diluted 1:250 into DMEM with high-glucose (Gibco-BRL) supplemented with 100 mM HEPES (pH 7.4), and incubated at 37°C overnight without shaking (in vitro-cultured). It has been shown that there is maximal secretion of Esps by EHEC under these conditions (Ebel et al., 1996). E. coli K-12 strains were cultured in LB broth at 37°C with shaking. When appropriate, strains were cultured in media supplemented with 100 μg/mL of ampicillin (pInt or pIL22) or 35 μg/mL of Kanamycin A (pTir).
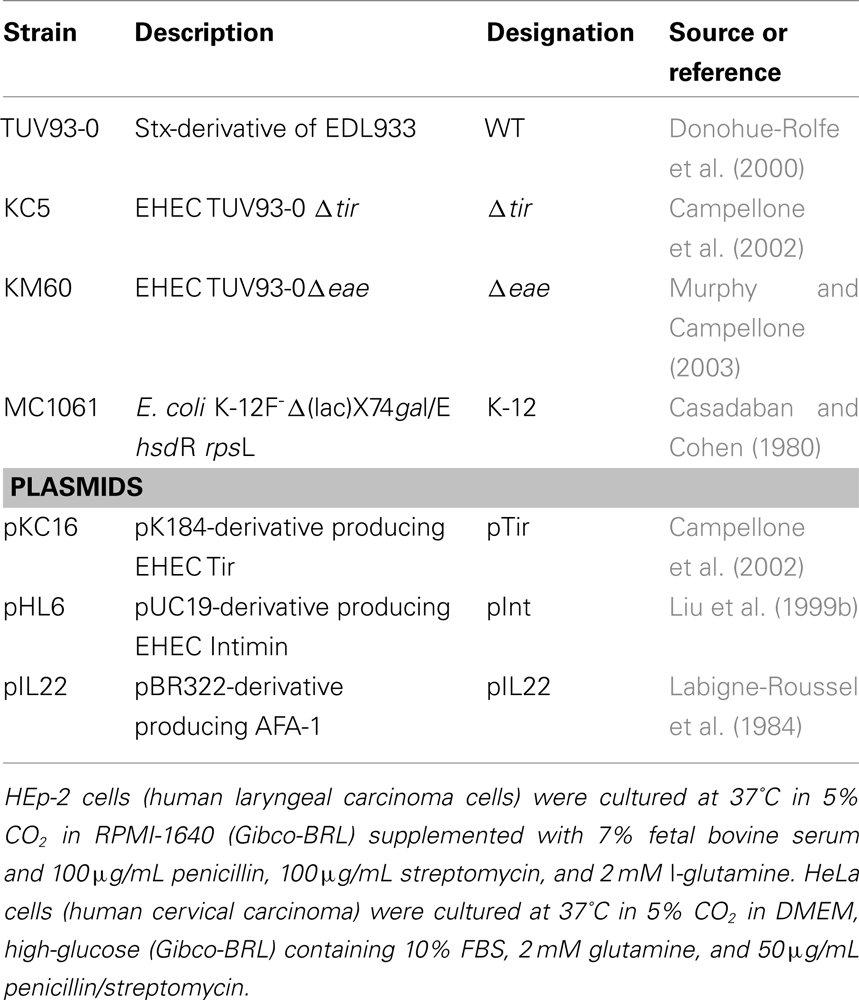
Table 1. Strains and plasmids used in this study.
Isolation of EHEC from Infected Gnotobiotic Piglets
Piglets were delivered by cesarean section and maintained in microbiologic isolation (gnotobiotic) for 24 h and infected with an oral challenge ~5 × 109 EHEC as described previously (Tzipori et al., 1992, 1995). All animals were monitored daily for signs of clinical disease and euthanized 48–72 h after oral challenge.
To harvest bacteria from the intestines, both ileal and spiral colon were collected from each animal and flushed with chilled sterile PBS to remove the majority of gut contents and pooled. To obtain additional adherent EHEC, intestine sections were dissected and the mucosal surfaces were gently scraped with a sterile spatula, flushed with sterile PBS and combined with the initial washings. To separate EHEC from crude intestinal contents, the pooled preparation was centrifuged and washed five to six times with PBS (host-adapted EHEC). To quantitate EHEC density, cells were enumerated using a hemacytometer, re-suspended at 1 × 108 bacteria/mL and confirmed by standard spread plating. The intestinal contents (“mucus” for use in control experiments) from uninfected piglets was obtained similarly.
In one set of control experiments, 20% “mucus” was supplemented to EHEC cultures grown in DMEM with high-glucose to determine whether in vitro growth in the presence of intestinal contents altered cell binding or pedestal formation. In a second set of control experiments, 20% “mucus” was added to in vitro-cultured EHEC immediately prior to cell binding and pedestal formation assays to determine whether mucus itself promoted mammalian cell binding or pedestal formation. In a third set of control experiments to differentiate between genetic (stable) and phenotypic (transient) adaptation, host-adapted EHEC were passaged in vitro as described previously and characterized for cell binding and pedestal formation.
Quantification of Bacterial Adherence
Bacterial adherence assays were performed as previously described (Liu et al., 1999a). Briefly, HEp-2 cells were seeded in RPMI-1640 supplemented with 20 mM HEPES (pH 7.0), 2% fetal bovine serum, and 0.5% D-mannose (RHFM medium), infected with approximately 5 × 105 bacteria for an MOI of approximately 10, and incubated for 3 h. Infected monolayers were washed six times in PBS to remove unbound bacteria and lysed with 0.5% TritonX-100. The cell lysates were serially diluted and plated on LB agar to determine the percent of bound bacteria. Significant differences from binding by in vitro-cultured EHEC was determined using a two-tailed, paired, Student’s t-test and defined as a probability of P ≤ 0.05.
Assessment of Actin Pedestal Formation
To quantitate the ability of EHEC strains to form pedestals on mammalian cells, coverslips that had been seeded with HEp-2 cells in RHFM (as described above) were infected with 5 × 105 EHEC. After 3 h incubation at 37°C, the monolayers were washed with PBS and further incubated for 3 h after addition of fresh RHFM. The cells were washed, fixed with 2.5% para-formaldehyde, stained for bacteria with DAPI and filamentous actin with TRITC-conjugated phalloidin (Sigma; Knutton et al., 1989; Liu et al., 1999a, 2002). To quantify pedestal formation, bacteria-associated with actin pedestals was determined as a function of 100 HEp-2 cells examined. One hundred eighty mammalian cells on three coverslips per bacterial preparation were examined in three independent experiments. Significance was determined as previously described (Vingadassalom et al., 2010).
Preparation of Cell Lysates and Immunoblotting
For Intimin and Tir expression, 108 bacteria were re-suspended in 20 μL of 1× Laemmli buffer, lysed by heating to 100°C for 10 min and subjected to 10% SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes. Membranes were blocked in PBS + 5% milk (PBSM) before treatment with sheep anti-Intimin (antiserum from a sheep immunized with a C-terminal 300-residues fragment of EHEC 86–24 intimin, gift of A. O’Brien) diluted 1:5000 or rabbit anti-Tir (diluted 1:10,000) antibodies. Following washes, membranes were treated with anti-sheep or anti-rabbit secondary IgG antiserum conjugated to alkaline phosphatase (diluted 1:5000) and developed (Acheson et al., 1995).
For Tir translocation, HEp-2 or HeLa cells were seeded into six well tissue culture plates (Becton–Dickinson Labware) and infected with bacterial strains at an MOI of approximately 10 for 6 h. Following infection, the monolayer were washed with PBS to remove unbound bacteria, lifted with Trypsin, centrifuged for 2 min and the pellet was re-suspended in 1× Laemmli buffer and boiled for 10 min. The samples were analyzed by 10% SDS-PAGE and western blotted for Tubulin and Tir (as described above).
Quantification of Surface Expression of Intimin
An enzyme-linked immunosorbent assay (ELISA) was used to quantify the levels of intimin expression on the bacterial surface (Liu et al., 2002). In brief, equivalent numbers of indicated strains (100 μL aliquots of 1 × 108 bacteria/mL) were seeded on poly-L-lysine-treated 96-well cell culture dishes (Becton–Dickinson Labware), centrifuged for 10 min at 1225 × g and incubated at room temperature for 20 min. The bacteria were then fixed with 3% para-formaldehyde for 1 h at room temperature, washed in PBS and blocked with 5% milk in PBS (PBSM) at room temperature for 1 h. Fixed bacteria were probed with Intimin anti-sera for 1 h at room temperature (diluted 1:500). The plates were washed with PBS and PBS + 0.5% Tween-20, followed by addition of the secondary horseradish peroxidase (HRP)-conjugated anti-sheep IgG antibody (diluted 1:10,000 in PBSM; Sigma). Parallel plates were subjected to anti-O157 antiserum (Difco) diluted 1:1000 in PBSM followed by probing with HRP-conjugated anti-rabbit IgG (Sigma) diluted in PBSM to determine the relative number of bound bacteria. The LPS signal obtained for the different bacterial strains within each experiment were within 5%. The mean OD600 values (±SD) of quadruplicate samples after subtraction of background (i.e., signal when the primary antibody was omitted) and normalized to bacterial number are shown.
Bacterial Binding and Pedestal Formation in Prime and Challenge Assays
To assess functional Tir delivery, a “prime and challenge” assay was used (Vingadassalom et al., 2010). HEp-2 monolayers were infected with 5 × 105 of either host-adapted or in vitro-cultivated EHECΔeae or EHECΔtir with or without plasmid pKC16 (aka pTir) for 3 h, gentamicin (100 μg/ml) treated for 30 min to kill priming bacteria, and challenged with 5 × 105 either native E. coli K-12 (vector or un-transformed controls) or E. coli K-12 expressing intimin from plasmid pHL6 (aka pInt) and incubated for an additional 3 h at 37°C. To assess bacterial binding, monolayers were lysed and serially plated as described above. To assess actin pedestal formation, infected monolayers were examined microscopically as described above. Significance was determined as previously described (Vingadassalom et al., 2010).
Results
Host-Adapted EHEC Bind and Generate Actin Pedestals on Cultured Mammalian Cells More Efficiently Than in vitro-Cultivated EHEC
To test whether EHEC adapts to the mammalian host environment by altering its ability to interact with mammalian cells, we cultivated EHEC O157:H7 strain EDL933 under conditions that have been previously developed to maximize host cell interaction and pedestal formation (“in vitro-cultured”; Ebel et al., 1996), or isolated them from the large bowel of gnotobiotic piglets 24–48 h following oral infection (“host-adapted”; see Materials and Methods). Mammalian cell monolayers were infected with either of these two preparations of EHEC and actin pedestal formation was quantified. Whereas less than 5 actin pedestals were observed per 100 mammalian cells for in vitro-cultured EHEC, almost 50 pedestals were observed for the same strain after host adaptation (Figure 1A). Similar results were observed under identical conditions using HeLa instead of HEp-2 cells (data not shown). This change does not appear to be hereditable because growth of host-adapted bacteria overnight in vitro resulted in a complete reversal of enhanced cell attachment activity (data not shown).
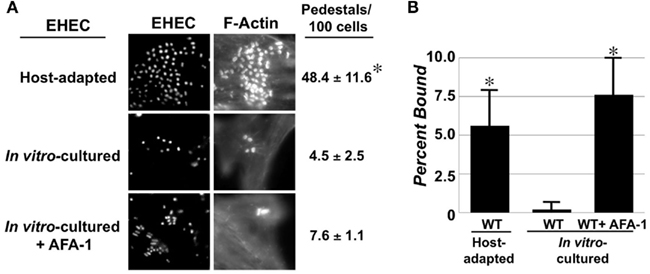
Figure 1. Enterohemorrhagic Escherichia coli actin pedestal formation and adhesion are enhanced upon host adaptation. (A) Mammalian cell monolayers were infected with host-adapted EHEC (top), in vitro-cultured EHEC (middle) or in vitro-cultured EHEC ectopically expressing the afimbrial adhesin AFA-1 (bottom). Bacteria were stained with DAPI (“EHEC”) and actin pedestals with Alexa568-phalloidin (”F-actin”). The percentage of actin pedestals (±SD) per 100 mammalian cells was determined visually (right). (B) Binding of the same three strains to mammalian cell monolayers was determined by plating for viable counts. Data represent the mean ± SD from three independent experiments. *P ≤ 0.05.
Host-adapted EHEC also appeared to bind to mammalian cells much more efficiently than did in vitro-cultivated EHEC (Figure 1A, “EHEC,” compare top two rows). When we quantified viable bacterial counts after lysis of infected monolayers, we found that whereas only 0.2% of in vitro-cultivated EHEC bound to monolayers, more than 5% of host-adapted bacteria bound to cells, representing a more than 25-fold increase (Figure 1B). Thus, adaptation to the mammalian host results in dramatic increases in both cell attachment and pedestal formation by EHEC.
To determine whether the high efficiency of actin pedestal formation upon host adaptation was solely a consequence of its enhanced ability to bind to monolayers, we required a means to dramatically increase mammalian cell binding of in vitro-cultured EHEC. Accordingly, we ectopically expressed the afimbrial adhesion AFA-1 from uropathogenic E. coli, encoded on pIL22 to artificially increased mammalian cell binding (Labigne-Roussel et al., 1984). Indeed, AFA-1 increased cell attachment by in vitro-cultivated EHEC to a level comparable to that observed for host-adapted EHEC (Figure 1B). Nevertheless, this enhanced cell binding was not associated with a significant increase in the frequency of actin pedestals (Figure 1A). Thus, the higher efficiency of pedestal formation by host-adapted EHEC is not simply a consequence of increased cell binding, suggesting that host adaptation results in expression of EHEC products that specifically contribute to actin signaling.
Intimin Expression is Increased during Host Adaptation but is not Solely Responsible for Enhanced Pedestal Formation
Given that intimin participates in cell attachment and pedestal formation, we assessed the relative levels of this adhesin expressed by in vitro-cultivated and host-adapted EHEC by Western blotting. As expected, no intimin was detected in extracts of in vitro-cultivated EHECΔeae (Figure 2A, rightmost lane). Greater amounts of intimin were produced by host-adapted than by in vitro-cultivated EHEC (Figure 2A, first two lanes). Densitometric analysis suggested a fourfold increase, but was complicated by the multitude of intimin-related species that are typically produced (data not shown; Donnenberg et al., 1993; McKee and O’Brien, 1996; Agin and Wolf, 1997; Liu et al., 2002). The apparent increase in expression of intimin in whole cell lysates by host-adapted bacteria was reflected in the level of surface protein detected by ELISA of intact bacteria using anti-intimin antiserum, which revealed that host-adapted bacteria expressed more than threefold higher levels of the adhesin on their surface than their in vitro-cultivated counterparts (Figure 2B).
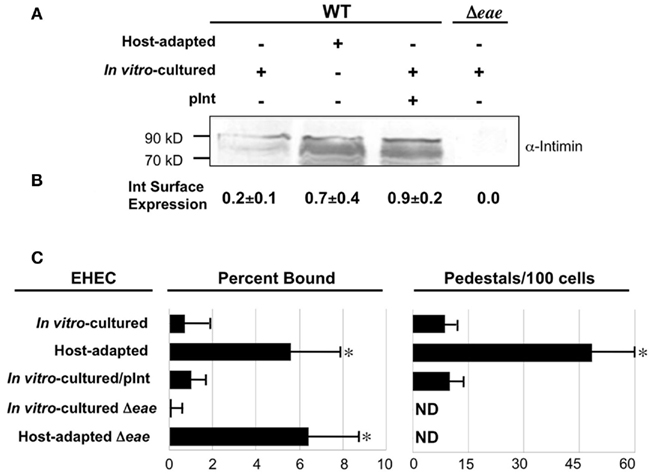
Figure 2. Intimin expression is increased during host adaptation but is not solely responsible for enhanced pedestal formation. (A) Equivalent numbers of bacteria were utilized to generate bacterial lysates of host-adapted or in vitro-cultivated EHEC that were subjected to immunoblotting using anti-intimin antibody. The in vitro-cultivated EHEC included wild type bacteria ±pInt, a plasmid that encodes intimin (pInt, lane 3), and EHECΔeae, which does not express intimin (lane 4). Molecular weight markers are indicated at left. (B) Equivalent numbers of the indicated strains were added to microtiter wells, fixed, and the relative number of bound bacteria was normalized by ELISA using anti-O157 antiserum. Parallel microtiter wells were subjected to anti-intimin ELISA to determine relative intimin surface expression. Shown are the mean OD600 values (±SD) of quadruplicate samples after subtraction of background (i.e., signal when the primary antibody was omitted) and normalized to bacterial number. (C) The percent of bacteria bound, determined by plating for viable counts, and the frequency of pedestal formation was determined as described in Figure 1. ND, not detected. *P ≤ 0.05.
In addition to promoting pedestal formation by binding to Tir, intimin may enhance binding by recognizing mammalian proteins, such as β1-chain integrins (Frankel et al., 1996) and nucleolin (Sinclair and O’Brien, 2002). To determine whether intimin overexpression by host-adapted EHEC can account for the observed high efficiency of actin pedestal formation, we artificially increased intimin expression of in vitro-cultured EHEC by introducing a plasmid (“pInt”) that encodes intimin. When evaluated by immunoblotting for total intimin and ELISA for surface expression, EHEC harboring pInt expressed levels of intimin comparable to those achieved upon host adaptation (Figures 2A,B, middle lanes), while EHEC harboring a control vector expressed levels of intimin comparable to in vitro-cultured WT (data not shown). However, the overexpression of intimin by in vitro-cultivated EHEC did not result in a concomitant increase in either host cell binding or actin pedestal formation (Figure 2C).
To determine whether the increased production of intimin by host-adapted bacteria is responsible the enhanced mammalian binding we observed, we tested cell binding of an EHEC eae mutant, which cannot produce intimin. We found that although host-adapted EHEC eae was, as predicted, completely incapable of generating actin pedestals (Figure 2C, right), this strain bound to mammalian cells indistinguishably from host-adapted wild type EHEC. Thus, the high level of mammalian cell binding observed for host-adapted EHEC does not reflect intimin-mediated attachment. Clearly, other bacterial factors are required to account for the high efficiency of cell interaction observed upon host adaptation of EHEC.
Tir Expression is Increased during Host Adaptation but is not Solely Responsible for Enhanced Pedestal Formation
Translocated intimin receptor and intimin are expressed from a co-transcribed mRNA (Sanchez-SanMartin et al., 2001) so we also compared Tir expression by host-adapted or in vitro-cultivated bacteria. Immunoblot analysis of cell lysates revealed that an ~80 kDa species that Tir production by host-adapted bacteria was considerably greater than by in vitro-cultured EHEC (Figure 3A, lanes 1 and 2); densitometric analyses suggested an approximately fivefold increase (data not shown).
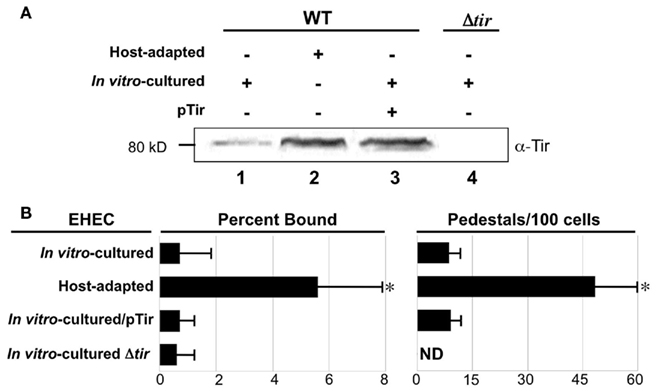
Figure 3. Translocated intimin receptor expression is increased during host adaptation but is not solely responsible for enhanced pedestal formation. (A) Equivalent numbers of bacteria were utilized to generate bacterial lysates of host-adapted or in vitro-cultivated EHEC that were subjected to immunoblotting using anti-intimin antibody. The in vitro-cultivated EHEC included wild type bacteria ± pTir a plasmid that encodes Tir (pTir, lane 3), and EHECΔtir (lane 4). Molecular weight is indicated at left. (B) Percent bacteria bound and the frequency of pedestal formation was determined as described in Figure 1. ND, not detected.*P ≤ 0.05.
To determine if the increase in Tir expression was responsible for the enhanced adherence and pedestal formation by host-adapted bacteria, we introduced a high-copy plasmid expressing Tir (pTir) into EHEC that resulted in the levels of Tir expression comparable to that of host-adapted EHEC (Figure 3A, lane 3). Consistent with the observation that adherence to monolayers was not influenced by the absence or overexpression of intimin (Figure 2), the absence or overexpression of Tir also had no effect on binding of in vitro-cultivated EHEC to mammalian cells (Figure 3B). In addition, overexpression of Tir by in vitro-cultivated EHEC did not result in an increase in pedestal formation (Figure 3B, right). These data indicate that although Tir production is increased upon host adaptation, this increase is not sufficient to account for the dramatic increase in the ability to generate actin pedestals upon growth in the mammalian host.
Host Adaptation Results in Increased Tir Translocation
Tir functions to promote cell attachment and pedestal formation only after insertion into the mammalian cell plasma membrane
After translocation into the mammalian host, EHEC Tir becomes serine phosphorylated by PKA, a modification that can be detected by an increase in apparent molecular mass upon SDS-PAGE (DeVinney et al., 1999; Warawa and Kenny, 2001). To assess the relative Tir translocation efficiencies, we infected HeLa cell monolayers with equivalent numbers of either host-adapted or in vitro-cultivated EHEC, and characterized the amount of modified Tir by Western blotting (Kenny, 1999). Analysis of replicate samples revealed that host-adapted EHEC translocates a significantly greater amount of Tir into monolayers than in vitro grown EHEC (Figure 4, compare lanes 1 and 2 with lanes 3 and 4). Similar results were observed under identical conditions using HeLa instead of HEp-2 cells (data not shown). Enhanced Tir translocation by host-adapted EHEC is not a simple consequence of enhanced host cell binding, because an increase in modified Tir was not observed when HeLa cells were infected with in vitro-cultivated EHEC that is highly adhesive by virtue of expressing AFA-1 (Figure 4, lane 5). Importantly, since there is approximately equivalent amounts of modified Tir, Tir translocation appears to be independent of AFA-1-mediated enhanced cell binding, suggesting that ectopic expression of AFA-1 neither increased nor decreased the efficiency of type III secretion (Figure 4, compare lanes 4 and 5).
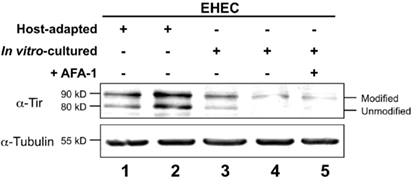
Figure 4. Translocated intimin receptor translocation is enhanced upon host adaptation. Mammalian cell monolayers were infected for 6 h with equivalent numbers of host-adapted EHEC isolated from the intestine of two different gnotobiotic piglets (lanes 1–2) or in vitro-cultured EHEC strains alone (lanes 3–4) or in vitro-cultured EHEC harboring a plasmid encoding AFA-1 (lane 5). Lysates of infected monolayers were resolved by SDS-PAGE and immunoblotted for Tir and tubulin. Western blots from a representative experiment are shown. The migration of unmodified and modified Tir is indicated (right), and numbers on the left indicate apparent molecular mass.
To determine if the apparent increased translocation of Tir by host-adapted bacteria resulted in enhanced Tir function, we utilized a “prime and challenge” assay
We first infected monolayers with either host-adapted or in vitro-cultured EHECΔeae, which translocates Tir but is not capable of intimin-mediated binding. Bacteria were removed by gentamicin treatment and washing. These “primed” monolayers were then “challenged” with either an E. coli K-12 strain (“K-12”) or an E. coli strain harboring a plasmid expressing EHEC intimin (“K-12/pInt”), and after washing, bound bacteria were quantified by plating for viable counts.
Binding of E. coli K-12/pInt to unprimed mammalian cell monolayers (data not shown), or monolayers primed with in vitro-cultured EHECΔtir, was indistinguishable from binding of E. coli K-12 (i.e., ~1% of inoculum), indicating that in this experimental system, intimin does not promote binding to an endogenous host cell receptor (Table 2, top row). E. coli K-12/pInt also did not demonstrate significant binding to monolayers primed with in vitro-cultured EHECΔeae, indicating that, upon in vitro growth, EHEC does not have the ability to translocate sufficient levels of Tir to allow detection in this functional assay (Table 2, row 2). Artificial overexpression of Tir in this strain, by introduction of pTir, was also not sufficient to promote translocation of functional amounts of Tir (Table 2, row 3). In contrast, E. coli K-12/pInt bound at levels 10-fold above background to monolayers that had been primed with host-adapted EHECΔeae (Table 2, bottom row). Microscopic visualization of the monolayers after staining bound bacteria and F-actin revealed that E. coli K-12/pInt also generated pedestals on these cells, but not on cells primed with in vitro-cultured EHECΔeae. These data indicate that growth in the mammalian host results in enhanced levels of translocation.
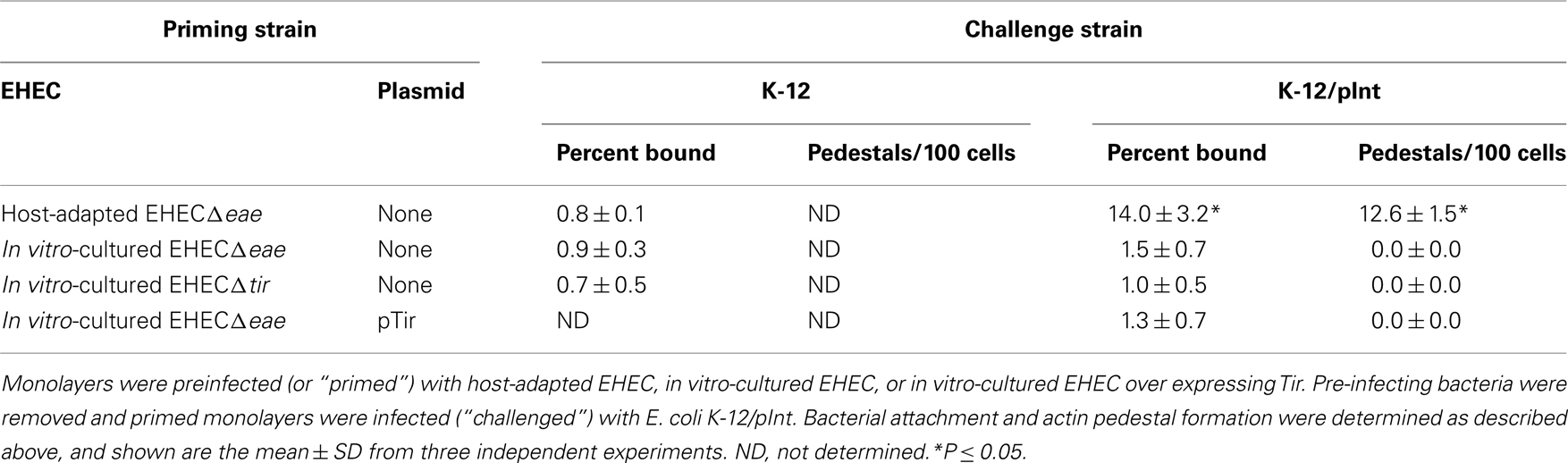
Table 2. Translocation of functional Tir is enhanced upon host adaptation.
Discussion
Bacteria are capable of remarkable phenotypic adaptation during mammalian infection and several bacterial pathogens, including the AE pathogen C. rodentium, exhibit enhanced infectivity upon passage through mammalian hosts (Merrell et al., 2002; Wiles et al., 2005). We hypothesized that host adaptation by EHEC might explain the longstanding paradox that in vitro-cultured EHEC typically attach to and generate actin pedestals on mammalian cell monolayers with low efficiency (Karch et al., 1987; Cantey and Moseley, 1991) yet generate extensive AE lesions on intestinal epithelial cells during mammalian infection (Tzipori et al., 1987). In fact, we found that EHEC isolated from the piglet intestine bound to HEp-2 cell monolayers ~25-fold more efficiently and generated actin pedestals with ~10-fold more efficiently than in vitro-cultivated bacteria. AE lesion formation is associated with enhanced colonization (Vlisidou et al., 2006; Ritchie et al., 2008; Crepin et al., 2010), and it is tempting to speculate that the increased efficiency of interaction with cultured mammalian cells observed upon host adaptation is associated with the increased colonization efficiency. Indeed, the hyperinfectious phenotype attained by C. rodentium following passage through mice is associated with greater host cell binding and pedestal formation, similar to that observed in this study (Wiles et al., 2005; Bishop et al., 2007).
Host-adapted bacteria produced significantly higher levels of intimin and Tir, two proteins documented to promote both bacterial adhesion and localized actin assembly (Jerse et al., 1990; Kenny et al., 1997). However, the enhanced cell attachment phenotype by host-adapted EHEC did not require intimin expression, and this intimin-independence suggests that Tir is also unlikely to be required for this phenotype. In addition, artificially increasing levels of these proteins by in vitro-cultivated bacteria was not sufficient to phenocopy host-adapted EHEC. Several EHEC surface proteins have been demonstrated or postulated to promote attachment to mammalian cells (i.e., AidA, Iha, Lpf, ToxB, HCP; Ebel et al., 1998; Spears et al., 2006; Xicohtencatl-Cortes et al., 2009; Yin et al., 2009a,b, 2011), but the role of any of these adhesins in binding of host-adapted EHEC remains to be demonstrated.
The high efficiency of cell attachment and pedestal formation by host-adapted EHEC appeared to be at least in part a manifestation of enhanced type III translocation. Immunoblotting of extracts of infected monolayers suggested that the translocated effector Tir was delivered more efficiently to mammalian cells by host-adapted than by in vitro-cultivated EHEC. In addition, in an assessment of Tir translocation that depends on the detection of two critical Tir functions, we found that preinfection of monolayers with host-adapted but not in vitro-cultured bacteria resulted in significantly greater Tir-mediated bacterial binding and actin pedestal formation. Given that actin pedestal formation is largely dependent on a second translocated effector, EspFU (TccP; Campellone et al., 2004; Garmendia et al., 2004) it appears likely that translocation of at least some other type III effectors is also increased during host adaptation.
The AE pathogens EPEC and C. rodentium each encode a type IV pilus capable of retraction that is required for colonization (Donnenberg et al., 1992; Mundy et al., 2003), and the ability of the EPEC type IV pilus to retract has recently been linked to increased type III translocation and actin pedestal formation (Zahavi et al., 2011). Thus, it is tempting to speculate that the EHEC retractable type IV pilus HCP, which appears to be expressed during human infection (Xicohtencatl-Cortes et al., 2007), may play a role in promoting cell attachment and type III translocation by host-adapted EHEC. We showed here that artificial enhancement of bacterial binding by the ectopic expression of the afimbrial adhesin AFA-1, which is not capable of retraction, did not result in a concomitant increase in Tir translocation or pedestal formation by in vitro-cultured EHEC. Although AFA-1 is not normally expressed in EHEC, it does not appear to interfere with (or enhance) Tir translocation and importantly, and its ectopic expression allowed us to test whether increased pedestal formation was solely a function of increased (albeit artificial) adherence. We cannot exclude the possibility that host-adapted bacteria express one or more adhesin(s) that, by co-ordinated regulation or function with the type III system, facilitate(s) both cell binding and subsequent Tir translocation. Studies to identify the bacterial factor(s) responsible for enhanced cell attachment and pedestal formation by EHEC are ongoing.
Enterohemorrhagic Escherichia coli dam and hfq mutants, which each suffer pleomorphic regulatory alterations (Murphy et al., 2008; Bhatt et al., 2011), exhibit increased cell binding, effector translocation, and pedestal formation, similar to that observed here for host-adapted EHEC (Campellone et al., 2007; Hansen and Kaper, 2009; Shakhnovich et al., 2009). It is possible that loss of dam or hfq function mimics some aspects of host adaptation. The induction of cell attachment and pedestal forming capacities by host-adapted EHEC were lost upon overnight in vitro culture (P. Radhakrishnan, M. J. Brady, data not shown), indicating that these phenotypes are due to a reversible alteration by EHEC growing in the host, similar to the transient nature of enhanced host cell interaction by C. rodentium (Bishop et al., 2007). Although the regulation of the LEE pathogenicity island that encodes the type III translocation system of AE pathogens has been studied extensively (Ando et al., 2007; Musken et al., 2008; House et al., 2009; for review, see Mellies et al., 2007), it is not known what environmental signal(s) results in host adaptation by EHEC. Quorum sensing and growth phase have been shown to regulate virulence factors of AE or other intestinal pathogens (Puente et al., 1996; Sperandio et al., 1999; Antunes et al., 2010). However, manipulation of the growth phase of in vitro-cultivated EHEC did not result in efficient host cell attachment or actin pedestal formation (P. Radhakrishnan, unpublished observations) and broth cultures of EHEC that achieved approximately the same density of bacteria as was attained in the piglet intestine (~109 bacteria/ml) bound to and generated pedestals on mammalian cell monolayers inefficiently, indicating that quorum sensing per se is not a sufficient trigger for host adaptation in binding and type III translocation. In addition, mammalian factors such as catecholamines have been implicated in the regulation of EHEC virulence, but in vitro culture of EHEC in the presence of epinephrine or porcine intestinal mucus did not increase mammalian cell binding or pedestal formation (M. J. Brady and P. Radhakrishnan, unpublished observation). Thus, future studies are required to understand the regulatory pathway(s) responsible for host adaptation by EHEC. Such investigations, as well as studies to identify the microbial products that promote enhanced cell interaction of bacteria growing within the mammalian host, would provide both insight into the pathogenesis of EHEC infection and potential targets for therapeutic intervention.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Alison O’Brien for EHEC Intimin anti-sera, Carol Kumamoto for OmpA anti-sera, Jon Goguen, Matt Waldor, Donald Tipper, and Martin Marinus for helpful discussions. This study was funded by NIH Grant R01 AI46454 awarded to John Leong.
References
Acheson, D. W., De Breucker, S. A., Jacewicz, M., Lincicome, L. L., Donohue-Rolfe, A., Kane, A. V., and Keusch, G. T. (1995). Expression and purification of Shiga-like toxin II B subunits. Infect. Immun. 63, 301–308.
Agin, T. S., and Wolf, M. K. (1997). Identification of a family of intimins common to Escherichia coli causing attaching-effacing lesions in rabbits, humans, and swine. Infect. Immun. 65, 320–326.
Ando, H., Abe, H., Sugimoto, N., and Tobe, T. (2007). Maturation of functional type III secretion machinery by activation of anaerobic respiration in enterohaemorrhagic Escherichia coli. Microbiology 153, 464–473.
Antunes, L. C., Ferreira, R. B., Buckner, M. M., and Finlay, B. B. (2010). Quorum sensing in bacterial virulence. Microbiology 156, 2271–2282.
Bhatt, S., Romeo, T., and Kalman, D. (2011). Honing the message: post-transcriptional and post-translational control in attaching and effacing pathogens. Trends Microbiol. 19, 217–224.
Bishop, A. L., Wiles, S., Dougan, G., and Frankel, G. (2007). Cell attachment properties and infectivity of host-adapted and environmentally adapted Citrobacter rodentium. Microbes Infect. 9, 1316–1324.
Campellone, K. G. (2010). Cytoskeleton-modulating effectors of enteropathogenic, and enterohaemorrhagic Escherichia coli: Tir, EspFU, and actin pedestal assembly. FEBS J. 277, 2390–2402.
Campellone, K. G., Giese, A., Tipper, D. J., and Leong, J. M. (2002). A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol. Microbiol. 43, 1227–1241.
Campellone, K. G., and Leong, J. M. (2003). Tails of two Tirs: actin pedestal formation by enteropathogenic E. coli and enterohemorrhagic E. coli O157:H7. Curr. Opin. Microbiol. 6, 82–90.
Campellone, K. G., Robbins, D., and Leong, J. M. (2004). EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev. Cell 7, 217–228.
Campellone, K. G., Roe, A. J., Lobner-Olesen, A., Murphy, K. C., Magoun, L., Brady, M. J., Donohue-Rolfe, A., Tzipori, S., Gally, D. L., Leong, J. M., and Marinus, M. G. (2007). Increased adherence and actin pedestal formation by dam-deficient enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 63, 1468–1481.
Cantey, R. J., and Moseley, S. L. (1991). HeLa cell adherence, actin aggregation, and invasion by noneteropathogenic Escherichia coli possessing the eae gene. Infect. Immun. 59, 3924–3929.
Casadaban, M. J., and Cohen, S. N. (1980). Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138, 179–207.
Celli, J., Deng, W., and Finlay, B. B. (2000). Enteropathogenic Escherichia coli (EPEC) attachment to epithelial cells: exploiting the host cell cytoskeleton from the outside. Cell. Microbiol. 2, 1–9.
Crepin, V. F., Girard, F., Schüller, S., Phillips, A. D., Mousnier, A., and Frankel, G. (2010). Dissecting the role of the Tir:Nck and Tir:IRTKS/IRSp53 signalling pathways in vivo. Mol. Microbiol. 75, 308–323.
DeVinney, R., Stein, M., Reinscheid, D., Abe, A., Ruschkowski, S., and Finlay, B. B. (1999). Enterohemorrhagic Escherichia coli O157:H7 produces Tir, which is translocated to the host cell membrane but is not tyrosine phosphorylated. Infect. Immun. 67, 2389–2398.
Donnenberg, M. S., Giron, J. A., Nataro, J. P., and Kaper, J. B. (1992). A plasmid-encoded type IV fimbrial gene of enteropathogenic Escherichia coli associated with localized adherence. Mol. Microbiol. 6, 3427–3437.
Donnenberg ,M. S., Lai, L. C., Taylor, K. A. (1997). The locus of enterocyte effacement pathogenicity island of enteropathogenic Escherichia coli encodes secretion functions and remnants of transposons at its extreme right end. Gene. 184, 107–114.
Donnenberg, M. S., Yu, J., and Kaper, J. B. (1993). A second chromosomal gene necessary for intimate attachment of enteropathogenic Escherichia coli to epithelial cells. J. Bacteriol. 175, 4670–4680.
Donohue-Rolfe, A., Kondova, I., Oswald, S., Hutto, D., and Tzipori, S. (2000). Escherichia coli O157:H7 strains that express Shiga toxin (Stx) 2 alone are more neurotropic for gnotobiotic piglets than are isotypes producing only Stx1 or both Stx1 and Stx2. J. Infect. Dis. 181, 1825–1829.
Ebel, F., Deibel, C., Kresse, A. U., Guzman, C. A., and Chakraborty, T. (1996). Temperature- and medium-dependent secretion of proteins by Shiga toxin- producing Escherichia coli. Infect. Immun. 64, 4472–4479.
Ebel, F., Podzadel, T., Rohde, M., Kresse, A. U., Kramer, S., Deibel, C., Guzman, C. A., and Chakraborty, T. (1998). Initial binding of Shiga toxin-producing Escherichia coli to host cells and subsequent induction of actin rearrangements depend on filamentous EspA-containing surface appendages. Mol. Microbiol. 30, 147–161.
Frankel, G., Lider, O., Hershkoviz, R., Mould, A. P., Kachalsky, S. G., Candy, D., Cahalon, L., Humphries, M. J., and Dougan, G. (1996). The cell-binding domain of intimin from enteropathogenic Escherichia coli binds to beta1 integrins. J. Biol. Chem. 271, 20359–20364.
Frankel, G., Philips, A. D., Novakova, M., Batchelor, M., Hicks, S., and Dougan, G. (1998). Generation of Escherichia coli intimin derivatives with differing biological activities using site-directed mutagenesis of the intimin C-terminus domain. Mol. Microbiol. 29, 559–570.
Frankel, G., and Phillips, A. D. (2008). Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: getting off the pedestal. Cell. Microbiol. 10, 549–556.
Garmendia, J., Phillips, A. D., Carlier, M. F., Chong, Y., Schuller, S., Marches, O., Dahan, S., Oswald, E., Shaw, R. K., Knutton, S., and Frankel, G. (2004). TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell. Microbiol. 6, 1167–1183.
Hansen, A. M., and Kaper, J. B. (2009). Hfq affects the expression of the LEE pathogenicity island in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 73, 446–465.
Hayward, R. D., Leong, J. M., Koronakis, V., and Campellone, K. G. (2006). Exploiting pathogenic Escherichia coli to model transmembrane receptor signalling. Nat. Rev. Microbiol. 4, 358–370.
House, B., Kus, J. V., Prayitno, N., Mair, R., Que, L., Chingcuanco, F., Gannon, V., Cvitkovitch, D. G., and Barnett Foster, D. (2009). Acid-stress-induced changes in enterohaemorrhagic Escherichia coli O157: H7 virulence. Microbiology 155, 2907–2918.
Jerse, A. E., Yu, J., Tall, B. D., and Kaper, J. B. (1990). A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. USA 87, 7839–7843.
Karch, H., Heesemann, J., Laufs, R., O’Brien, A. D., Tacket, C. O., and Levine, M. M. (1987). A plasmid of enterohemorrhagic Escherichia coli O157:H7 is required for expression of a new fimbrial antigen and for adhesion to epithelial cells. Infect. Immun. 55, 455–461.
Karmali, M. A. (1989). Infection by verocytotoxin-producing Escherichia coli. Clin. Microbiol. Rev. 2, 15–38.
Kenny, B. (1999). Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol. Microbiol. 31, 1229–1241.
Kenny, B., Devinney, R., Stein, M., Reinscheid, D. J., Frey, E. A., and Finlay, B. B. (1997). Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91, 511–520.
Knutton, S., Baldwin, T., Williams, P. H., and Mcneish, A. S. (1989). Actin accumulation at sites of bacterial adhesion to tissue culture cells: basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 57, 1290–1298.
Labigne-Roussel, A. F., Lark, D., Schoolnik, G., and Falkow, S. (1984). Cloning and expression of an afimbrial adhesin (AFA-I) responsible for P blood group-independent, mannose-resistant hemagglutination from a pyelonephritic Escherichia coli strain. Infect. Immun. 46, 251–259.
Liu, H., Magoun, L., and Leong, J. M. (1999a). β1-Chain integrins are not essential for intimin-mediated host cell attachment and enteropathogenic Escherichia coli-mediated actin polymerization. Infect. Immun. 67, 2045–2049.
Liu, H., Magoun, L., Luperchio, S., Schauer, D. B., and Leong, J. M. (1999b). The Tir-binding region of enterohaemorrhagic Escherichia coli intimin is sufficient to trigger actin condensation after bacterial-induced host cell signalling. Mol. Microbiol. 34, 67–81.
Liu, H., Radhakrishnan, P., Magoun, L., Prabu, M., Campellone, K. G., Savage, P., He, F., Schiffer, C. A., and Leong, J. M. (2002). Point mutants of EHEC intimin that diminish Tir recognition and actin pedestal formation highlight a putative Tir binding pocket. Mol. Microbiol. 45, 1557–1573.
McDaniel, T. K., Jarvis, K. G., Donnenberg, M. S., and Kaper, J. B. (1995). A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 92, 1664–1668.
McDaniel, T. K., and Kaper, J. B. (1997). A cloned pathogenicity island from enteropathogenic Escherichia coli confers the attaching and effacing phenotype on E. coli K-12. Mol. Microbiol. 23, 399–407.
McKee, M. L., and O’Brien, A. D. (1996). Truncated enterohemorrhagic Escherichia coli (EHEC) O157:H7 intimin (EaeA) fusion proteins promote adherence of EHEC strains to HEp-2 cells. Infect. Immun. 64, 2225–2233.
Mellies, J. L., Barron, A. M., and Carmona, A. M. (2007). Enteropathogenic and enterohemorrhagic Escherichia coli virulence gene regulation. Infect. Immun. 75, 4199–4210.
Merrell, D. S., Butler, S. M., Qadri, F., Dolganov, N. A., Alam, A., Cohen, M. B., Calderwood, S. B., Schoolnik, G. K., and Camilli, A. (2002). Host-induced epidemic spread of the cholera bacterium. Nature 417, 642–645.
Mundy, R., Pickard, D., Wilson, R. K., Simmons, C. P., Dougan, G., and Frankel, G. (2003). Identification of a novel type IV pilus gene cluster required for gastrointestinal colonization of Citrobacter rodentium. Mol. Microbiol. 48, 795–809.
Murphy, K. C., and Campellone, K. G. (2003). Lambda red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol. Biol. 4, 11. doi: 10.1186/1471-2199-4-11
Murphy, K. C., Ritchie, J. M., Waldor, M. K., Lobner-Olesen, A., and Marinus, M. G. (2008). Dam methyltransferase is required for stable lysogeny of the Shiga toxin (Stx2)-encoding bacteriophage 933W of enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol. 190, 438–441.
Musken, A., Bielaszewska, M., Greune, L., Schweppe, C. H., Muthing, J., Schmidt, H., Schmidt, M. A., Karch, H., and Zhang, W. (2008). Anaerobic conditions promote expression of Sfp fimbriae and adherence of sorbitol-fermenting enterohemorrhagic Escherichia coli O157:NM to human intestinal epithelial cells. Appl. Environ. Microbiol. 74, 1087–1093.
Nataro, J. P., and Kaper, J. B. (1998). Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201.
Noel, J. M., and Boedeker, E. C. (1997). Enterohemorrhagic Escherichia coli: a family of emerging pathogens. Dig. Dis. 15, 67–91. [Review].
Pai, C. H., Kelly, J. K., and Meyers, G. L. (1986). Experimental infection of infant rabbits with verotoxin-producing Escherichia coli. Infect. Immun. 51, 16–23.
Puente, J. L., Bieber, D., Ramer, S. W., Murray, W., and Schoolnik, G. K. (1996). The bundle-forming pili of enteropathogenic Escherichia coli: transcriptional regulation by environmental signals. Mol. Microbiol. 20, 87–100.
Riley, L. W., Remis, R. S., Helgerson, S. D., Mcgee, H. B., Wells, J. G., Davis, B. R., Hebert, R. J., Olcott, E. S., Johnson, L. M., Hargrett, N. T., Blake, P. A., and Cohen, M. L. (1983). Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308, 681–685.
Ritchie, J. M., Brady, M. J., Riley, K. N., Ho, T. D., Campellone, K. G., Herman, I. M., Donohue-Rolfe, A., Tzipori, S., Waldor, M. K., and Leong, J. M. (2008). EspFU, a type III-translocated effector of actin assembly, fosters epithelial association and late-stage intestinal colonization by E. coli O157:H7. Cell. Microbiol. 10, 836–847.
Rosenshine, I., Ruschkowski, S., and Finlay, B. B. (1996). Expression of attaching/effacing activity by enteropathogenic Escherichia coli depends on growth phase, temperature, and protein synthesis upon contact with epithelial cells. Infect. Immun. 64, 966–973.
Sanchez-SanMartin, C., Bustamante, V. H., Calva, E., and Puente, J. L. (2001). Transcriptional regulation of the orf19 gene and the tir-cesT-eae operon of enteropathogenic Escherichia coli. J. Bacteriol. 183, 2823–2833.
Shakhnovich, E. A., Davis, B. M., and Waldor, M. K. (2009). Hfq negatively regulates type III secretion in EHEC and several other pathogens. Mol. Microbiol. 74, 347–363.
Sinclair, J. F., and O’Brien, A. D. (2002). Cell surface-localized nucleolin is a eukaryotic receptor for the adhesin intimin-gamma of enterohemorrhagic Escherichia coli O157:H7. J. Biol. Chem. 277, 2876–2885.
Spears, K. J., Roe, A. J., and Gally, D. L. (2006). A comparison of enteropathogenic and enterohaemorrhagic Escherichia coli pathogenesis. FEMS Microbiol. Lett. 255, 187–202.
Sperandio, V., Mellies, J. L., Nguyen, W., Shin, S., and Kaper, J. B. (1999). Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 96, 15196–15201.
Teel, L. D., Steinberg, B. R., Aronson, N. E., and O’Brien, A. D. (2003). Shiga toxin-producing Escherichia coli-associated kidney failure in a 40-year-old patient and late diagnosis by novel bacteriologic and toxin detection methods. J. Clin. Microbiol. 41, 3438–3440.
Tzipori, S., Gunzer, F., Donnenberg, M. S., De Montigny, L., Kaper, J. B., and Donohue-Rolfe, A. (1995). The role of the eaeA gene in diarrhea and neurological complications in a gnotobiotic piglet model of enterohemorrhagic Escherichia coli infection. Infect. Immun. 63, 3621–3627.
Tzipori, S., Karch, H., Wachsmuth, K. I., Robins-Browne, R. M., O’Brien, A. D., Lior, H., Cohen, M. L., Smithers, J., and Levine, M. M. (1987). Role of a 60-megadalton plasmid and Shiga-like toxins in the pathogenesis of infection caused by enterohemorrhagic Escherichia coli O157:H7 in gnotobiotic piglets. Infect. Immun. 55, 3117–3125.
Tzipori, S., Montanaro, J., Robins-Browne, R. M., Vial, P., Gibson, R., and Levine, M. M. (1992). Studies with enteroaggregative Escherichia coli in the gnotobiotic piglet gastroenteritis model. Infect. Immun. 60, 5302–5306.
Vingadassalom, D., Campellone, K. G., Brady, M. J., Skehan, B., Battle, S. E., Robbins, D., Kapoor, A., Hecht, G., Snapper, S. B., and Leong, J. M. (2010). Enterohemorrhagic E. coli requires N-WASP for efficient type III translocation but not for EspFU-mediated actin pedestal formation. PLoS Pathog. 6, e1001056. doi: 10.1371/journal.ppat.1001056
Vlisidou, I., Dziva, F., La Ragione, R. M., Best, A., Garmendia, J., Hawes, P., Monaghan, P., Cawthraw, S. A., Frankel, G., Woodward, M. J., and Stevens, M. P. (2006). Role of intimin-tir interactions and the tir-cytoskeleton coupling protein in the colonization of calves and lambs by Escherichia coli O157:H7. Infect. Immun. 74, 758–764.
Warawa, J., and Kenny, B. (2001). Phosphoserine modification of the enteropathogenic Escherichia coli Tir molecule is required to trigger conformational changes in Tir and efficient pedestal elongation. Mol. Microbiol. 42, 1269–1280.
Wiles, S., Dougan, G., and Frankel, G. (2005). Emergence of a “hyperinfectious” bacterial state after passage of Citrobacter rodentium through the host gastrointestinal tract. Cell. Microbiol. 7, 1163–1172.
Xicohtencatl-Cortes, J., Monteiro-Neto, V., Ledesma, M. A., Jordan, D. M., Francetic, O., Kaper, J. B., Puente, J. L., and Giron, J. A. (2007). Intestinal adherence associated with type IV pili of enterohemorrhagic Escherichia coli O157:H7. J. Clin. Invest. 117, 3519–3529.
Xicohtencatl-Cortes, J., Monteiro-Neto, V., Saldana, Z., Ledesma, M. A., Puente, J. L., and Giron, J. A. (2009). The type 4 pili of enterohemorrhagic Escherichia coli O157:H7 are multipurpose structures with pathogenic attributes. J. Bacteriol. 191, 411–421.
Yin, X., Chambers, J. R., Wheatcroft, R., Johnson, R. P., Zhu, J., Liu, B., and Gyles, C. L. (2009a). Adherence of Escherichia coli O157:H7 mutants in vitro and in ligated pig intestines. Appl. Environ. Microbiol. 75, 4975–4983.
Yin, X., Wheatcroft, R., Chambers, J. R., Liu, B., Zhu, J., and Gyles, C. L. (2009b). Contributions of O island 48 to adherence of enterohemorrhagic Escherichia coli O157:H7 to epithelial cells in vitro and in ligated pig ileal loops. Appl. Environ. Microbiol. 75, 5779–5786.
Yin, X., Zhu, J., Feng, Y., Chambers, J. R., Gong, J., and Gyles, C. L. (2011). Differential gene expression and adherence of Escherichia coli O157:H7 in vitro and in ligated pig intestines. PLoS ONE 6, e17424. doi: 10.1371/journal.pone.0017424
Zahavi, E. E., Lieberman, J. A., Donnenberg, M. S., Nitzan, M., Baruch, K., Rosenshine, I., Turner, J. R., Melamed-Book, N., Feinstein, N., Zlotkin-Rivkin, E., and Aroeti, B. (2011). Bundle forming pilus retraction enhances enteropathogenic Escherichia coli infectivity. Mol. Biol. Cell. 22, 2436–2447.
Keywords: host adaptation, actin assembly, translocation, EHEC, intimin, Tir
Citation: Brady MJ, Radhakrishnan P, Liu H, Magoun L, Murphy KC, Mukherjee J, Donohue-Rolfe A, Tzipori S and Leong JM (2011) Enhanced actin pedestal formation by enterohemorrhagic Escherichia coli O157:H7 adapted to the mammalian host. Front. Microbio. 2:226. doi: 10.3389/fmicb.2011.00226
Received: 19 July 2011; Accepted: 25 October 2011;
Published online: 15 November 2011.
Edited by:
Elizabeth L. Hartland, The University of Melbourne, AustraliaReviewed by:
Steffen Backert, University College Dublin, IrelandMichael L. Vasil, University of Colorado Medical School, USA
Charles Martin Dozois, Institut National de la Recherche Scientifique, Canada
Andrew Roe, University of Glasgow, UK
Copyright: © 2011 Brady, Radhakrishnan, Liu, Magoun, Murphy, Mukherjee, Donohue-Rolfe, Tzipori and Leong. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: John M. Leong, Department of Molecular Biology and Microbiology, Tufts University School of Medicine, 136 Harrison Avenue, Boston, MA, 02111, USA. e-mail:am9obi5sZW9uZ0B1bWFzc21lZC5lZHU=
†Michael John Brady and Padhma Radhakrishnan have contributed equally to this work.