 Shirajum Monira1
Shirajum Monira1 Shota Nakamura2
Shota Nakamura2 Kazuyoshi Gotoh2 Kaori Izutsu2 Haruo Watanabe3 Nur Haque Alam1
Kazuyoshi Gotoh2 Kaori Izutsu2 Haruo Watanabe3 Nur Haque Alam1 Hubert Ph. Endtz1
Hubert Ph. Endtz1 Alejandro Cravioto1 Sk. Imran Ali1
Alejandro Cravioto1 Sk. Imran Ali1 Takaaki Nakaya2 Toshihiro Horii2
Takaaki Nakaya2 Toshihiro Horii2 Tetsuya Iida2 and Munirul Alam1*
Tetsuya Iida2 and Munirul Alam1*- 1 International Center for Diarrhoeal Disease Research, Dhaka, Bangladesh
- 2 Research Institute for Microbial Diseases, Osaka University, Osaka, Japan
- 3 National Institute of Infectious Diseases, Shinjuku-ku, Tokyo, Japan
Poor health and malnutrition in preschool children are longstanding problems in Bangladesh. Gut microbiota plays a tremendous role in nutrient absorption and determining the state of health. In this study, metagenomic tool was employed to assess the gut microbiota composition of healthy and malnourished children. DNA was extracted from fecal samples of seven healthy and seven malnourished children (n = 14; age 2–3 years) were analyzed for the variable region of 16S rRNA genes by universal primer PCR followed by high-throughput 454 parallel sequencing to identify the bacterial phyla and genera. Our results reveal that the healthy children had a significantly higher number of operational taxonomic unit in their gut than that of the malnourished children (healthy vs. malnourished: 546 vs. 310). In malnourished children, bacterial population of the phyla Proteobacteria and Bacteroidetes accounted for 46 and 18%, respectively. Conversely, in healthy children, Proteobacteria and Bacteroidetes accounted for 5% and 44, respectively (p < 0.001). In malnourished children, the phylum Proteobacteria included pathogenic genera, namely Klebsiella and Escherichia, which were 174-fold and 9-fold higher, respectively, than their healthy counterpart. The predominance of potentially pathogenic Proteobacteria and minimal level of Bacteroidetes as commensal microbiota might be associated to the ill health of malnourished children in Bangladesh.
Introduction
The human gastrointestinal tract harbors a diverse microbial community throughout its extent, which support their hosts mostly for healthy living. It has been estimated that the adult human colonic bacteria reaches approximately 1014 organisms throughout the GI tract, which is 10 times the total number of somatic and germ cells within the body. The colon of a healthy human adult harbors around 400–500 different species of bacteria belonging to 190 different genera, although a few genera together comprise the major cultivable flora of the feces (Ramakrishna, 2007). These bacteria have important metabolic roles, e.g., production of short-chain fatty acids and vitamins such as vitamin k and biotin, structural functions like immune system development and protective functions, e.g., pathogen displacement by colonization competition (Ann and O’Hara, 2006).
Colonization of commensal bacteria in the gut of newborns has profound implications in weaning period. For example, Bifidobacterium spp. were found as dominant microbiota in infants that were exclusively breastfed and these infants were less likely to suffer from diarrhea (Harmsen et al., 2000). Indigenous microbiota play an important role in developing homeostatic immunity by suppressive mechanisms to avoid local and peripheral hypersensitivity to innocuous antigens particularly food proteins and components of commensal bacteria by dendritic cells (DCs) and regulatory T cells in mesenteric lymph nodes (Brandtzaeg, 2010). Pathogenic bacteria can alter the gut microbiota proportion significantly. Namely, colonization of Vibrio cholerae in the gut epithelial cells causes profuse loss of water and electrolytes which results in expulsion of Bifidobacteria and invites increasing proportion of Enterobacteriaceae (Monira et al., 2010). Role of gut microbiota in immune diseases has been demonstrated and bacterial imbalance has been associated with pathologies such as inflammatory bowel disease and obesity (Ley et al., 2006; Marteau et al., 2004). Microbial ecology of the gastrointestinal tract also plays a key role in nutrition research into the relation between colonic microbiota signatures and age (Mariat et al., 2009), diet (De Filippo et al., 2010), dietary allergies (Lay et al., 2005), or diseases (Frank et al., 2007). These observations have stimulated renewed interests in research on the GI tract microbiota (biomarkers) of healthy and diseased individuals (Tap et al., 2009). Diet and nutritional status are specifically most important factors which are known as the modifiable determinants of human health. As gut microbiota play an important role in nutrient extraction from food in the lower part of GI tract, the nutritional value of food is influenced partially by a person’s gut microbial community (microbiota) and its component genes (microbiome; Turnbaugh et al., 2009).
Malnutrition continues to remain a persistent problem for children under 5 years of age in developing countries. The concept of malnutrition is changing rapidly and it is not always related to lack of food, but can also be related to the set of gut flora that can play both positively and negatively by influencing the intestinal absorption of nutrients. It has been hypothesized that changes in intestinal microbial ecology and the microbiome because of pathogenic infection or treatment by antibiotics can also disrupt the intricate balance between the gut flora and thus affect the harvest of nutrients from the diet. Additionally, human genetic polymorphisms that alter host genes can also have impact on nutrient absorption and metabolism (Ahmed et al., 2009). In Bangladesh, which is one of the most densely populated developing country where, about half of the children under 5 years of age suffer from acute malnutrition and growth stunting, although data of gut microbiota of children of different nutritional status are lacking. The present study was designed with the aim to assess the composition of gut microbiota in malnourished and healthy children of Bangladesh.
Materials and Methods
Study Subjects
This study was conducted on seven healthy and seven malnourished children in the International Centre for Diarrheal Disease Research, Bangladesh (ICDDR,B). These subjects were selected as controls in our previous study (Monira et al., 2009). The age of the children was between 2 and 3 year and their weight for height was ≥100% for healthy children and ≤70% for malnourished children according to the National Center for Health Statistics median (WHO, 1983). The malnourished children were enrolled from a nearby slum area in the Mohakhali area of Dhaka, close to the Dhaka Hospital of ICDDR,B. Healthy children of the ICDDR,B staff members were enrolled as healthy subjects in this study. The socio-economic status of the parents of healthy children is moderate to high and their monthly income ranges from US$ 600 to 900. On the other hand, the parents of malnourished children are from poor socio-economic status and their monthly income ranges from US$ 100 to 150. We did not record the specific dietary history of these children however, they received usual Bangladeshi foods, e.g., rice, meat, milk, fish, egg, fruits, and vegetables. As the poor families cannot afford protein-rich foods, the malnourished children are unlikely to have access to foods namely meat, milk, fish, and egg. Their baseline characteristics are presented in Table 1. The ethical review committee of the ICDDR,B had approved both the clinical study and the present study, and informed consent was obtained from the parents or legal guardians of the child before enrollment. Also, the ethical review committee of the Research Institute for Microbial Disease, Osaka University, had approved the present study. These children residing at their home did not present with any disease and did not take antibiotics during the last 2 months. Fecal samples collected from children at their home in the morning and maintaining cold chain brought to the laboratories. Health workers visited the children and their mothers the day before sampling and after taking their consent gave the stool collection containers and taught the collection procedure. Health workers collected the sample on the following day. Samples were immediately preserved at −20°C freeze after coming to laboratory and stored until processing for DNA extraction.
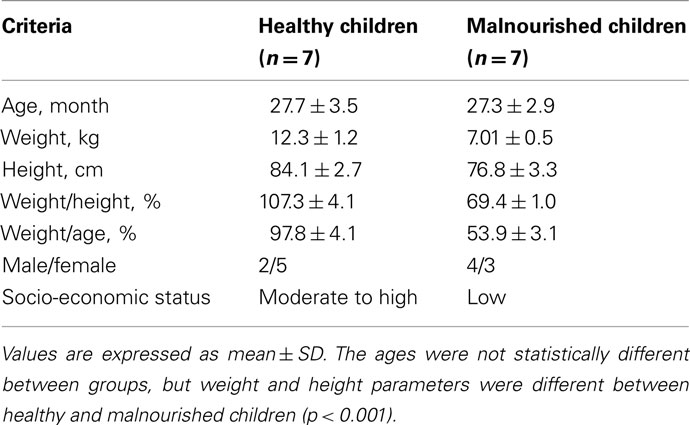
Table 1. Baseline characteristics of study children.
Extraction and Purification of Total DNA
DNA was extracted from fecal samples according to the method of Magne et al. (2006). 125 mg (wet weight) fecal sample was suspended in 625 μl breaking buffer [0.8 mol/L guanidinium isothiocyanate, 4% N-lauroyl sarcosine, 20 mmol/L Tris (pH 8.0), 80 mmol/L sodium phosphate buffer (pH 8.0)] and incubated for 1 h at 70°C. Afterward, 750 μl glass beads 0.1 mm in diameter (Sigma, St Louis, MO, USA) and 15 mg polyvinylpolypyrrolidone were added. Bacterial cells were lysed in a vortex mixer at high speed (10 cycles consisting of 1 min of vortexing and 1 min of storage in ice). The mixture was centrifuged at 20,000 g for 3 min at 4°C. After recovery of the supernatant, the pellet was washed three times with 200 μl TENP (50 mm Tris–HCl [pH 8.0], 20 mm EDTA [pH 8.0], 100 mm NaCl, and 1% [w/v] polyvinylpolypyrrolidone). The four obtained supernatants were pooled. Nucleic acids were extracted with one volume of phenol. The aqueous phase was washed twice by use of chloroform–isoamyl-alcohol (24:1). DNA was precipitated by use of 100% isopropanol, and the pellet was washed with 70% v/v isopropanol, dried, and resuspended in 50–100 μl of sterile water and stored at −20°C. The amount and integrity of DNA were estimated by use of 1% (w/v) agarose gel electrophoresis containing ethidium bromide (1 mg/ml) in 1× TBE (Tris Borate EDTA).
454 Sequencing and Data Analysis
Relative abundance of bacterial phyla in fecal specimens was estimated by sequencing the PCR amplicons targeting 16S rRNA gene for the DNA samples extracted from each fecal specimen. PCR was performed using a primer set (784F: 5′-AGGATTAGATACCCTGGTA-3′ and 1061R: 5′-CRRCACGAGCTGACGAC-3′). The primer set targets the V5-V6 region of the 16S rRNA genes (Andersson et al., 2008). To amplify the targeted region, 1 μl of extracted DNA was served as the template in 50-μl reactions using Prime STAR HS premix (Takara Bio Inc., Japan). Each reaction mixture contained 10 pmol of each primer. The PCR conditions was 30 cycles of 98°C for 10 s, 55°C for 15 s, and 72°C for 20 s. Two 100 μl of 3-cycle reconditioning PCR reactions were performed per sample to eliminate heteroduplexes (Thompson et al., 2002), with 10-μl aliquots of the initial PCR product mixture as the template and other PCR conditions unchanged. Products of the two reconditioning PCR reactions per sample were combined and purified using QIAquick PCR purification columns (Qiagen). The amplified PCR products were used as a template for pyrosequencing with the GS Junior platform (454 Life Sciences). Pyrosequencing was performed by following the manufacturer’s instruction using MID tags. Sequencing runs yielded 14,093 reads for one sample on the average. The obtained data were then subjected to a data analysis pipeline. Data analysis was performed on each read sequence by computational tools, as constructed previously (Nakamura et al., 2008) with some modifications. Bacterial rRNA typing was performed by BLASTN search against the comprehensive rRNA database “silva” release 94 (Pruesse et al., 2007) using a threshold of E-value <1E-40. All the bacterial species obtained from each fecal sample were classified into their phylogenetic groups and the proportion of different phyla was estimated. The estimation of operational taxonomic unit (OTU) was performed by the program ESPRIT using default settings (Sun et al., 2009). According to Sun et al., considering the sequencing error rate of the high-throughput DNA sequencers, using the d value below 0.10 may overestimate the OTU number. Also, the paper describes that it is possible to evaluate the diversity of microbiota with the d value of 0.10 appropriately and we followed this.
Statistical Analysis
Data were entered into a personal computer using a statistical package (SPSS version 11.5; LEAD Technologies Inc., Charlotte, NC, USA). Baseline characteristics of the study children were compared by analysis of variance. Differences between groups were analyzed for significance using t-test. Data are presented as mean ± SD.
Results
Fecal samples were collected from seven healthy children of ICDDR,B staff members and seven malnourished children of a nearby slum located close to the ICDDR,B Dhaka hospital. None of the children had any history of disease, and they did not have any antibiotic therapy during the last 2 months. To determine the bacterial lineages present in the fecal microbiota of these healthy and malnourished children, pyrosequencing technology was adopted targeting the PCR-amplified hyper variable region V5–V6 of the 16S rRNA gene with a GS Junior platform (454 Life Sciences). The average sequence length was mean ± SD: 292.3 ± 0.557 vs. 293.9 ± 1.25 bp in healthy vs. malnourished children, respectively. The sequencing generated a dataset consisting of 197,315 filtered high quality classifiable 16S rRNA gene sequences with a mean ± SD of 14,093 ± 6,234 sequences per sample. Interestingly, we observed a significantly higher number of OTU in healthy children by comparing between these data from healthy and malnourished children (no. of OTU: 546 in healthy vs. 310 in malnourished children, respectively, Figure 1). This indicates greater diversity of gut microbiota in healthy children than that in malnourished children.
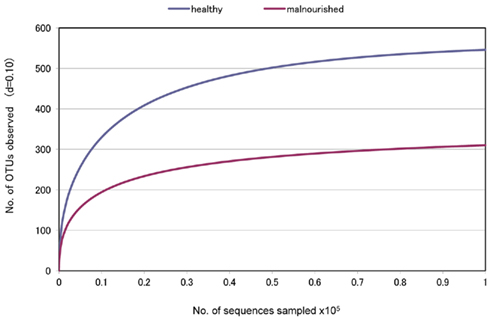
Figure 1. “Rarefaction Curve” showing differences in the divergence between the gut microbiota of healthy and malnourished children. d, distance cut off value.
Almost 98–99% of the sequences in all of the healthy and malnourished children samples were found to belong to the four most populated bacterial phyla, namely Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria. The healthy children differed from the malnourished children in relative proportion of all four phyla. The mean relative abundance of Bacteroidetes and Firmicutes in healthy children was 44% for each of the phylum, while in malnourished children; these two phyla were 18 and 32%, respectively. On the other hand, Proteobacteria was more abundant in malnourished children (46%) than their healthy counterparts (5%). The proportion of Actinobacteria in healthy and malnourished children was 6 and 1%, respectively. From this observation Bacteroidetes, Firmicutes, and Actinobacteria were 2.4, 1.4, and 6 times higher in healthy children than malnourished and Proteobacteria was 9.2 times higher in malnourished than healthy children. Statistical analysis indicates that Bacteroidetes and Proteobacteria differ significantly (p < 0.05 and p < 0.001, respectively) between these two groups of children (Figure 2). The distribution of four major bacterial phyla in all of the 14 children from the two defined health groups is presented in Figure 3. In the gut of healthy and malnourished children, the number of genera belonging to dominant bacterial phyla was strikingly different; 62 genera were observed in healthy, while only 37 genera in malnourished children (Figure 4). In addition, malnourished children had pathogenic genera belonging to the family Enterobacteriaceae, namely Klebsiella and Escherichia that were higher by 174-fold and 9-fold respectively, than that of healthy counterpart (Table 2).
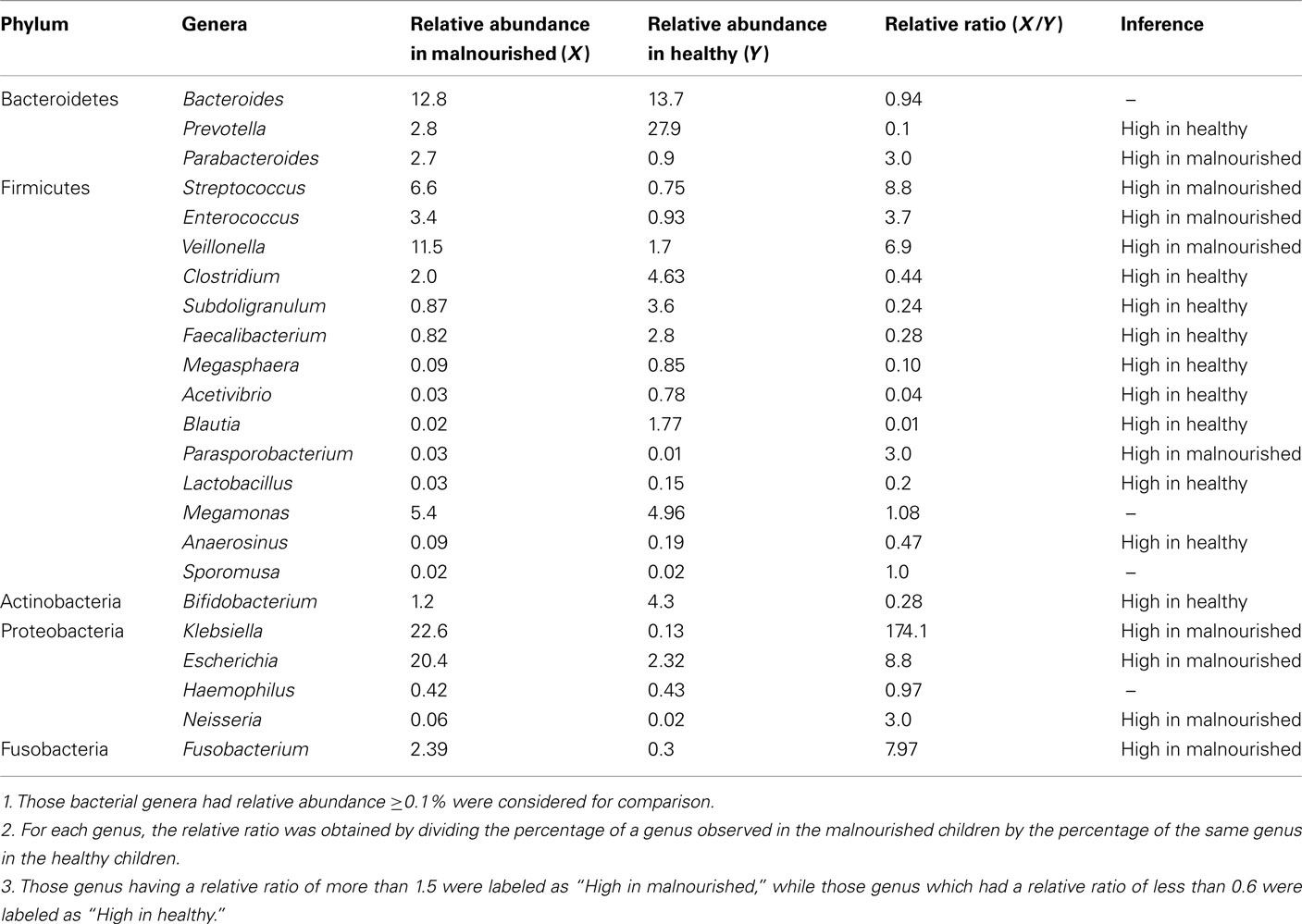
Table 2. Comparison of genera assignments obtained for malnourished and healthy children at the taxonomic level of phylum.
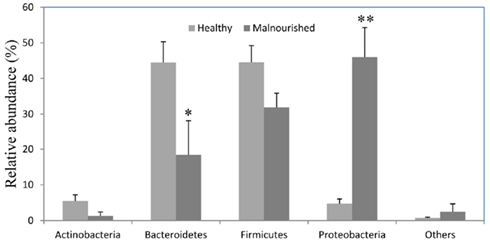
Figure 2. Relative abundance (percentage of sequences) of the dominant bacterial phyla in the gut of healthy and malnourished children. Values are expressed as mean ± SE. *Indicates significant difference (p < 0.05) and **(p < 0.001) between two groups of children. Light gray, healthy; dark gray, malnourished children.
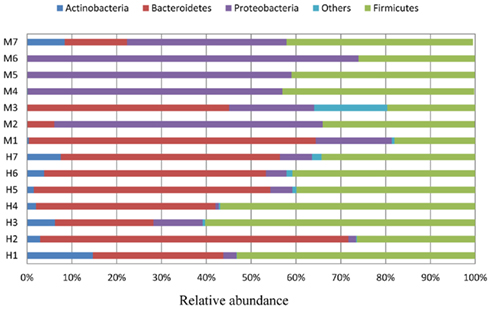
Figure 3. Each individual of two groups of children (healthy, H1 ∼ H7; malnourished, M1 ∼ M7) showing relative abundance (percentage of sequences) of the dominant gut bacteria.
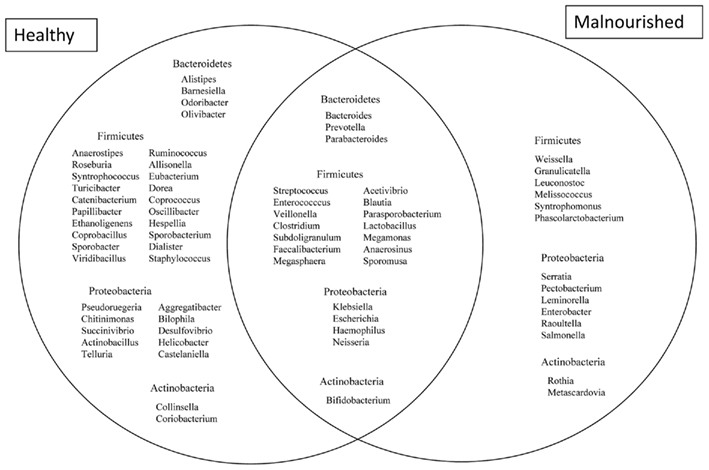
Figure 4. Venn diagram showing the distribution of genera belonging to the major phyla between healthy and malnourished children. Bacterial genera with the relative abundance of ≥0.1% were considered for comparison. A total of 39 genera are characteristic of healthy children, 23 genera common between healthy and malnourished children and 14 genera are characteristic of malnourished children.
Discussion
The data presented in this study provide important insights into the distinctive compositions of colonic microbiota, showing that the healthy children possessed significantly higher number of OTU in their gut than that of the malnourished children in Bangladesh, a developing country of 160 million people, where poor child health and malnutrition are recognized as a longstanding problem. We obtained four major bacterial phyla, e.g., Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria as most dominant bacterial groups in the gut of all 14 children irrespective of their health status. Our results appeared in line with the results presented in some other studies in which bacteria of these four phyla were shown to represent the largest part of the human gut microbiota (Backhed et al., 2005; Qin et al., 2010). Bacteroidetes bacteria represented one of the major phyla of colonic bacteria which are generally considered as a group of bacteria responsible for the synthesis of short chain fatty acids from dietary fiber (Xu et al., 2003). The observed lower relative abundance of the phylum Bacteroidetes in Bangladeshi malnourished children (healthy vs. malnourished; 44 vs. 18%) might explain their poor health because these bacteria are widely recognized for their ability to digest complex dietary material and help their hosts with better nutrition and energy extracted from various foods and dietary supplements. However, our findings differed significantly with the observation of a recently reported study showing gut microbiota of children in India in which Bacteroidetes bacteria were shown to be more abundant in malnourished than in healthy children (Gupta et al., 2011). This disparity of gut bacteria may presumably be due to low sample size of that study which analyzed only one malnourished child and compared with a healthy child, contrary to the present study in which gut microbiota of seven children in each group were analyzed and compared. Although Bacteroidetes phyla ranged from 10 to 30% in the healthy children of other countries (Bibiloni et al., 2006; Eckburg et al., 2005; Frank et al., 2007; Lay et al., 2005), we observed a relatively higher abundance of Bacteroidetes in the healthy children of Bangladesh. Interestingly, in the present study, those healthy children that had higher number of Bacteroidetes bacteria in their gut harbored lower number of Actinobacteria, namely Bifidobacterium sp. and vice versa (data not shown).
The Proteobacteria represented the fourth and the lowermost component (relative abundance 5%) of the fecal microbiota in healthy children of Bangladesh. This relative abundance appeared in agreement with that of healthy children of other countries where incidence of Proteobacteria ranged from 5 to 10% (Bibiloni et al., 2006; Eckburg et al., 2005; Frank et al., 2007; Lay et al., 2005). The predominance of Proteobacteria observed in the gut of malnourished children of Bangladesh was unique in the present study. This is interesting because three types of gut microbiota, termed “enterotypes,” were proposed (Arumugam et al., 2011) in which Bacteroides, Prevotella, and Ruminococcus were dominant respectively, and reported to be most common fecal microbiota of individuals irrespective of any nation or continent. Although the population included in the said study were adult individuals, and from developed countries (Arumugam et al., 2011), we studied malnourished children of Bangladesh as representatives of most populated developing countries where majority of the world population lives. So, the observed predominance of Proteobacteria in the gut of malnourished population of Bangladesh might be suggestive of more available enterotypes other than the proposed three on earth, depending on geographic location, age and health status of the individuals, and requires further studies.
In the present study, we observed some common components of Proteobacteria such as, Klebsiella, Escherichia, and Neisseria that are manifold higher in malnourished than the healthy children. Species of these genera are well established human pathogens; and thus higher relative abundance of pathogenic bacteria in malnourished children may be suggestive that such children often suffer from some subclinical infections in their gastrointestinal tract, which likely results in malabsorption of nutrients and chronic declination of health. It is presumable that poor hygiene and frequent illnesses are predisposing for the facultative gut bacteria to settle as commensal, which can cause illness during immune compromised conditions. This assumption may be further substantiated by the recent observation that the gastrointestinal infection mainly diarrhea is caused by not only a single pathogen, but also by a consortium of two or more pathogenic bacteria simultaneously (Lindsay et al., 2011). On the other hand, no defined etiology was found in more than one third of the diarrhea cases. The possibility of the association of as yet unknown pathogens in the gut microbiota cannot be ruled out, considering that existing sequence data in the literatures and databases, e.g., in NCBI GenBank, DDBJ, or in EMBL are scarce. We observed higher relative abundance of Klebsiella and Escherichia bacteria, which are member of Enterobacteriaceae family, in malnourished than healthy children. In sharp contrary to our observation in the present study, members of the Enterobacteriaceae family were shown recently to be 2.5 times higher in healthy children than that of their malnourished counterparts (Gupta et al., 2011). Moreover, unlike data presented in the current study, bacteria belonging to Campylobacteraceae and Helicobacteraceae family was shown in abundance in malnourished than in healthy children in that study, although we did not observe such results in our study except that Helicobacter sp. was found in only one healthy child (Figure 4), and also in very low abundance.
Recent development in our understanding is that the gut microbiota, namely Firmicutes bacteria can influence the energy absorption from food. It has been becoming increasingly evident that the gut bacteria belonging to the Firmicutes phyla are involved in energy resorption and obesity (Ley et al., 2005; Ley et al., 2006). In the present study, Firmicutes bacteria, which included mainly the Clostridium and Lactobacillus constituted a relatively less abundant group in both healthy (44%) and malnourished (32%) children of Bangladesh compared to the children of developed countries (range 50–70%; Bibiloni et al., 2006; Eckburg et al., 2005; Frank et al., 2007; Lay et al., 2005). The lower abundance of Firmicutes bacteria in Bangladeshi children might explain their poor health, especially for the malnourished children. However, the role of gut microbiota has been becoming increasingly evident in health and diseases. It has been shown that the colonization of infants gut by a group of Lactobacilli decreases the risk of allergy at 5 years of age despite these children have allergic heredity (Johansson et al., 2011). Conversely, changes in microbiota composition can facilitate pathogenic bacteria such as C. difficile to colonize the gut of infants (Rousseau et al., 2011).
Bangladesh is a developing country with about half of the children aged between 0.6 and <5 years suffering from severe malnutrition. The government of Bangladesh and many non-government organizations (NGOs) are working jointly to alleviate the malnutrition from children. Although sufficient and balanced food intake remains a big challenge for the poverty-stricken people world-wide, well digestion, and proper energy extraction from foods are dependent largely on the type of gut microbiota. As gut microbiota can play a crucial role in maximizing energy extraction from food in the lower part of gastrointestinal tract, a clear picture of the gut bacteria remains a key step toward designing intervention and proper management of malnutrition. This study revealed differences in the gut microbial community which were related to the health status, mostly nutritional. We chose arbitrarily a minimum of 2 months post antibiotic treatment as a prerequisite for sample collection. The idea is that, it is easier for mother to remember any event of illness of her child within this time period. Again, in our previous study we observed that malnourished children with cholera who received antibiotics, resume their gut microbiota proportion upto the level of malnourished children (without cholera) after 1 month (Monira et al., 2010). At the same time this study has limitations that it could not collect and analyze large number of samples covering a wide range of people of various socioeconomic status and areas of Bangladesh. Lack of meta data and specific dietary history are among the other limitations of this study. Nonetheless, to our knowledge, this study is the first of its kind in Bangladesh, and the differences that were observed in the composition and abundance of major bacterial phyla in the two defined groups of children, healthy and malnourished, were consistent. The distinct signature patterns of gut microbiota in healthy and malnourished children of Bangladesh may have profound implication in determining health in poor resource settings. Finally, future studies of gut microbiota among larger number of samples and from different populations might better explain the role of gut microbiota in malnourished children. In the light of these findings intervention studies to reestablish the normal gut flora in malnourished children might be designed.
Following is the URL for our sequence data: http://imet.gen-info.osaka-u.ac.jp/en/suppdata01.html
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was jointly supported by ICDDR,B, the National Institute of Infectious Diseases (NIID), Japan, and a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports Culture, and Technology, Japan (MEXT), the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases by the MEXT and the project for the International Research Center for Infectious Diseases, Research Institute for Microbial Diseases, Osaka University from the MEXT. ICDDR,B acknowledges with gratitude the commitment of NIID and MEXT, Japan to the Center’s research efforts. ICDDR,B also gratefully acknowledges the following donors which provide unrestricted support to the Centre’s research efforts: Australian Agency for International Development (AusAID), Government of the People’s Republic of Bangladesh, Canadian International Development Agency (CIDA), Embassy of the Kingdom of the Netherlands (EKN), Swedish International Development Cooperation Agency (SIDA), and the Department for International Development, UK (DFID).
References
Ahmed, T., Haque, R., Shamsir Ahmed, A. M., Petri, W. A. Jr., and Cravioto, A. (2009). Use of metagenomics to understand the genetic basis of malnutrition. Nutr. Rev. 67, 201–206.
Andersson, A. F., Lindberg, M., Jakobsson, H., Backhed, F., Nyren, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3, e2836. doi:10.1371/journal.pone.0002836
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., Fernandes, G. R., Tap, J., Bruls, T., Batto, J. M., Bertalan, M., Borruel, N., Casellas, F., Fernandez, L., Gautier, L., Hansen, T., Hattori, M., Hayashi, T., Kleerebezem, M., Kurokawa, K., Leclerc, M., Levenez, F., Manichanh, C., Nielsen, H. B., Nielsen, T., Pons, N., Poulain, J., Qin, J., Sicheritz-Ponten, T., Tims, S., Torrents, D., Ugarte, E., Zoetendal, E. G., Wang, J., Guarner, F., Pedersen, O., de Vos, W. M., Brunak, S., Doré, J., MetaHIT ConsortiumAntolín, M., Artiguenave, F., Blottiere, H. M., Almeida, M., Brechot, C., Cara, C., Chervaux, C., Cultrone, A., Delorme, C., Denariaz, G., Dervyn, R., Foerstner, K. U., Friss, C., van de Guchte, M., Guedon, E., Haimet, F., Huber, W., van Hylckama-Vlieg, J., Jamet, A., Juste, C., Kaci, G., Knol, J., Lakhdari, O., Layec, S., Le Roux, K., Maguin, E., Mérieux, A., Melo Minardi, R., M’rini, C., Muller, J., Oozeer, R., Parkhill, J., Renault, P., Rescigno, M., Sanchez, N., Sunagawa, S., Torrejon, A., Turner, K., Vandemeulebrouck, G., Varela, E., Winogradsky, Y., Zeller, G., Weissenbach, J., Ehrlich, S. D., and Bork, P. (2011). Enterotypes of the human gut microbiome. Nature 473, 174 –180.
Backhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A., and Gordon, J. I. (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920.
Bibiloni, R., Mangold, M., Madsen, K. L., Fedorak, R. N., and Tannock, G. W. (2006). The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J. Med. Microbiol. 55, 1141–1149.
Brandtzaeg, P. (2010). Homeostatic impact of indigenous microbiota and secretory immunity. Benef. Microbes 1, 211–227.
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., Collini, S., Pieraccini, G., and Lionetti, P. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696.
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., Gill, S. R., Nelson, K. E., and Relman, D. A. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638.
Frank, D. N., Amand, A. L. St, Feldman, R. A., Boedeker, E. C., Harpaz, N., and Pace, N. R. (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. U.S.A. 104, 13780–13785.
Gupta, S. S., Mohammed, M. H., Ghosh, T. S., Kanungo, S., Nair, G. B., and Mande, S. S. (2011). Metagenome of the gut of a malnourished child. Gut Pathog. 3, 7.
Harmsen, H. J., Wildeboer-Veloo, A. C., Raangs, G. C., Wagendorp, A. A., Klijn, N., Bindels, J. G., and Welling, G. W. (2000). Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J. Pediatr. Gastroenterol. Nutr. 30, 61–67.
Johansson, M. A., Sjogren, Y. M., Persson, J. O., Nilsson, C., and Sverremark-Ekstrom, E. (2011). Early colonization with a group of Lactobacilli decreases the risk for allergy at five years of age despite allergic heredity. PLoS ONE 6, e23031. doi: 10.1371/journal.pone.0023031
Lay, C., Rigottier-Gois, L., Holmstrom, K., Rajilic, M., Vaughan, E. E., de Vos, W. M., Collins, M. D., Thiel, R., Namsolleck, P., Blaut, M., and Doré, J. (2005). Colonic microbiota signatures across five northern European countries. Appl. Environ. Microbiol. 71, 4153–4155.
Ley, R. E., Backhed, F., Turnbaugh, P., Lozupone, C. A., Knight, R. D., and Gordon, J. I. (2005). Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U.S.A. 102, 11070–11075.
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023.
Lindsay, B., Ramamurthy, T., Sen Gupta, S., Takeda, Y., Rajendran, K., Nair, G. B., and Stine, O. C. (2011). Diarrheagenic pathogens in polymicrobial infections. Emerging Infect. Dis. 17, 606–611.
Magne, F., Abely, M., Boyer, F., Morville, P., Pochart, P., and Suau, A. (2006). Low species diversity and high interindividual variability in faeces of preterm infants as revealed by sequences of 16S rRNA genes and PCR-temporal temperature gradient gel electrophoresis profiles. FEMS Microbiol. Ecol. 57, 128–138.
Mariat, D., Firmesse, O., Levenez, F., Guimaraes, V., Sokol, H., Dore, J., Corthier, G., and Furet, J. P. (2009). The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 9, 123. doi:10.1186/1471-2180-9-123
Marteau, P., Lepage, P., Mangin, I., Suau, A., Dore, J., Pochart, P., and Seksik, P. (2004). Review article: gut flora and inflammatory bowel disease. Aliment. Pharmacol. Ther. 20(Suppl. 4), 18–23.
Monira, S., Alam, N. H., Suau, A., Magne, F., Nair, G. B., Karmakar, P. C., Rahman, M., Pochart, P., and Desjeux, J. F. (2009). Time course of bacterial diversity in stool samples of malnourished children with cholera receiving treatment. J. Pediatr. Gastroenterol. Nutr. 48, 571–578.
Monira, S., Hoq, M. M., Chowdhury, A. K., Suau, A., Magne, F., Endtz, H. P., Alam, M., Rahman, M., Pochart, P., Desjeux, J. F., and Alam, N. H. (2010). Short-chain fatty acids and commensal microbiota in the faeces of severely malnourished children with cholera rehydrated with three different carbohydrates. Eur. J. Clin. Nutr. 64, 1116–1124.
Nakamura, S., Maeda, N., Miron, I. M., Yoh, M., Izutsu, K., Kataoka, C., Honda, T., Yasunaga, T., Nakaya, T., Kawai, J., Hayashizaki, Y., Horii, T., and Iida, T. (2008). Metagenomic diagnosis of bacterial infections. Emerging Infect. Dis. 14, 1784–1786.
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., and Glöckner, F. O. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196.
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., Nielsen, T., Pons, N., Levenez, F., Yamada, T., Mende, D. R., Li, J., Xu, J., Li, S., Li, D., Cao, J., Wang, B., Liang, H., Zheng, H., Xie, Y., Tap, J., Lepage, P., Bertalan, M., Batto, J. M., Hansen, T., Le Paslier, D., Linneberg, A., Nielsen, H. B., Pelletier, E., Renault, P., Sicheritz-Ponten, T., Turner, K., Zhu, H., Yu, C., Li, S., Jian, M., Zhou, Y., Li, Y., Zhang, X., Li, S., Qin, N., Yang, H., Wang, J., Brunak, S., Doré, J., Guarner, F., Kristiansen, K., Pedersen, O., Parkhill, J., Weissenbach, J., MetaHIT ConsortiumBork, P., Ehrlich, S. D., and Wang, J. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65.
Ramakrishna, B. S. (2007). The normal bacterial flora of the human intestine and its regulation. J. Clin. Gastroenterol. 41, S2–S6.
Rousseau, C., Levenez, F., Fouqueray, C., Dore, J., Collignon, A., and Lepage, P. (2011). Clostridium difficile colonization in early infancy is accompanied by changes in intestinal microbiota composition. J. Clin. Microbiol. 49, 858–865.
Sun, Y., Cai, Y., Liu, L., Yu, F., Farrell, M. L., McKendree, W., and Farmerie, W. (2009). ESPRIT: estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 37, e76.
Tap, J., Mondot, S., Levenez, F., Pelletier, E., Caron, C., Furet, J. P., Ugarte, E., Muñoz-Tamayo, R., Paslier, D. L., Nalin, R., Dore, J., and Leclerc, M. (2009). Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 11, 2574–2584.
Thompson, J. R., Marcelino, L. A., and Polz, M. F. (2002). Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by “reconditioning PCR.” Nucleic Acids Res. 30, 2083–2088.
Turnbaugh, P. J., Ridaura, V. K., Faith, J. J., Rey, F. E., Knight, R., and Gordon, J. I. (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 1, 6ra14.
WHO. (1983). Measuring Change in Nutritional Status. Guidelines for Assessing the Nutritional Impact of Supplementary Feeding Programmes for Vulnerable Groups. Geneva: World Health Organization.
Keywords: microbiota, gut, 16S rDNA, children, nutrition
Citation: Monira S, Nakamura S, Gotoh K, Izutsu K, Watanabe H, Alam NH, Endtz HP, Cravioto A, Ali SI, Nakaya T, Horii T, Iida T and Alam M (2011) Gut microbiota of healthy and malnourished children in bangladesh. Front. Microbio. 2:228. doi: 10.3389/fmicb.2011.00228
Received: 22 August 2011;
Accepted: 26 October 2011;
Published online: 21 November 2011.
Edited by:
Awdhesh Kalia, The University of Texas MD Anderson Cancer Center, USAReviewed by:
Loren Hauser, Oak Ridge National Laboratory, USAEvelyne Forano, Institut National de la Recherche Agronomique, France
Copyright: © 2011 Monira, Nakamura, Gotoh, Izutsu, Watanabe, Alam, Endtz, Cravioto, Ali, Nakaya, Horii, Iida and Alam. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Munirul Alam, Enteric and Food Microbiology Laboratory, Laboratory Sciences Division, International Center for Diarrhoeal Diseases Research, Bangladesh, 68 Shaheed Tajuddin Ahmed Sarani, Mohakhali, Dhaka 1212, Bangladesh. e-mail:bXVuaXJ1bEBpY2RkcmIub3Jn