 Sushrusha Nayak1
Sushrusha Nayak1 Debalina Sarkar1
Debalina Sarkar1 George Q. Perrin1
George Q. Perrin1 Babak Moghimi1
Babak Moghimi1 Brad E. Hoffman1 Shangzhen Zhou2 Barry J. Byrne1
Brad E. Hoffman1 Shangzhen Zhou2 Barry J. Byrne1 Roland W. Herzog1*
Roland W. Herzog1*- 1 Division of Cellular and Molecular Therapy, Department of Pediatrics, University of Florida, Gainesville, FL, USA
- 2 Division of Hematology, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
Intramuscular (IM) administration of an adeno-associated viral (AAV) vector represents a simple and safe method of gene transfer for treatment of the X-linked bleeding disorder hemophilia B (factor IX, F.IX, deficiency). However, the approach is hampered by an increased risk of immune responses against F.IX. Previously, we demonstrated that the drug cocktail of immune suppressants rapamycin, IL-10, and a specific peptide (encoding a dominant CD4+ T cell epitope) caused an induction of regulatory T cells (Treg) with a concomitant apoptosis of antigen-specific effector T cells (Nayak et al., 2009). This protocol was effective in preventing inhibitory antibody formation against human F.IX (hF.IX) in muscle gene transfer to C3H/HeJ hemophilia B mice (with targeted F9 gene deletion). Here, we show that this protocol can also be used to reverse inhibitor formation. IM injection of AAV1–hF.IX vector resulted in inhibitors of on average 8–10 BU within 1 month. Subsequent treatment with the tolerogenic cocktail accomplished a rapid reduction of hF.IX-specific antibodies to <2 BU, which lasted for >4.5 months. Systemic hF.IX expression increased from undetectable to >200 ng/ml, and coagulation times improved. In addition, we developed an alternative prophylactic protocol against inhibitor formation that did not require knowledge of T cell epitopes, consisting of daily oral administration of rapamycin for 1-month combined with frequent, low-dose intravenous injection of hF.IX protein. Experiments in T cell receptor transgenic mice showed that the route and dosing schedule of drug administration substantially affected Treg induction. When combined with intravenous antigen administration, oral delivery of rapamycin had to be performed daily in order to induce Treg, which were suppressive and phenotypically comparable to natural Treg.
Introduction
Gene therapy offers many advantages for treatment of the X-linked bleeding disorder hemophilia B, which is caused by mutations in coagulation factor IX (F.IX). In particular, adeno-associated viral (AAV) in vivo gene transfer to skeletal muscle or liver has been shown to direct long-term expression of functional F.IX in animal models, thereby reducing the incidence of spontaneous bleeding (Mingozzi and High, 2011b). A series of pioneering clinical trials on muscle- and liver-directed AAV gene transfer recently culminated in successful treatment patients with severe hemophilia B (Kay et al., 2000; Manno et al., 2003, 2006; Ponder, 2011). However, despite the low immunogenicity profile of AAV vectors, several concerns about immune responses to gene transfer remain. For example, prior natural infection with the parent or related virus may cause pre-existing immunity in humans, including neutralizing antibodies to viral particles (preventing gene transfer). Memory CD8+ T cell responses to capsid are also known to occur and may target transduced cells (Manno et al., 2006; Mingozzi et al., 2007b; Li et al., 2011; Mingozzi and High, 2011a). Immune responses to the transgene product are an additional concern. Large F9 gene deletions or other “null” mutations substantially increase the risk of immune responses, likely due to a lack of central tolerance (Cao et al., 2009a). A particular concern for gene therapy for inherited protein deficiencies such as hemophilia is the potential for antibody formation, which could then also negate conventional protein replacement therapy.
Muscle-directed gene transfer is an attractive treatment modality because of the safety and simplicity of vector administration, as demonstrated in clinical trials, but is hampered by an increased risk of immune responses against the transgene product (Herzog et al., 2001; Manno et al., 2003). For example, animals with null mutations are at high risk of formation of inhibitory antibodies (inhibitors) against F.IX (Herzog et al., 2001; Cao et al., 2009a). Previously, we demonstrated that the drug cocktail of immune suppressants rapamycin, IL-10, and a specific peptide (encoding a dominant CD4+ T cell epitope) caused an induction of CD4+CD25+FoxP3+ Treg with a concomitant antigen-specific apoptosis of effector T cells (Teff; Nayak et al., 2009). This protocol was effective in preventing inhibitor formation against human F.IX (hF.IX) upon subsequent IM administration of an AAV–hF.IX vector in hemophilia B mice with targeted F.IX gene deletion. Here, we used this model to test whether an inhibitor response to gene therapy can be reversed and whether a similar protocol can be developed that prevents inhibitor formation in a prophylactic manner without prior knowledge of T cell epitopes. Such epitopes may differ from one patient to another and may be difficult to map in a person who does not already have an immune response.
Materials and Methods
Animals
All mice were 6- to 12-week-old males at the onset of the experiments and housed in specific pathogen free conditions. Hemophilia B mice (F9 gene deletion) on C3H/HeJ background were as previously described (Cao et al., 2006, 2009a; Verma et al., 2010). DO11.10-tg Rag-2−/− BALB/c mice were obtained from Taconic (Germantown, NY, USA). This strain is transgenic for ovalbumin-specific DO11.10 CD4+ TCR and knock out for Rag-2 (DO11.10-tg Rag-2−/−; Cao et al., 2007a). Drug cocktails were administered via different routes in 200 μl of sterile PBS, three-times per week at the following doses: ovalbumin (ova) peptide (amino acid residues 323–339) or hF.IX-specific peptide (2A-54) were given at 100 μg/dose, rapamycin at 4 mg/kg/dose, and IL-10 at 50 ng/kg/dose (Nayak et al., 2009). In an alternative regimen, rapamycin was given by oral gavage at the same dose (but in a volume of only 100 μl) daily for 1 month. During that time, recombinant human F.IX (Benefix, Wyeth, Madison, NJ, USA) was given intravenous (IV) into the tail vein at 0.1 IU/mouse twice per week. Viral vector was administered at 1 × 1011 vector genomes (vg)/mouse by intramuscular (IM) injection into quadriceps and tibialis anterior muscles. Blood samples from hemophilia B mice were collected by tail-bleed into 3.8% sodium citrate buffer.
Reagents and Viral Vector
Ova and hF.IX peptides were synthesized by AnaSpec (San Jose, CA, USA). Murine IL-10 (Sigma Aldrich, St. Louis, MO, USA) and rapamycin (LC Laboratories, Woburn, MA, USA) stock solution were made in 0.02% carboxymethylcellulose and 0.25% Tween80. The previously published AAV-CMV-hF.IX vector contains the hF.IX cDNA (plus a 1.4-kb portion of the F9 gene) expressed from the cytomegalovirus immediate-early enhancer/promoter (Arruda et al., 2004). Vector genomes were packaged into serotype 1 capsid following triple transfection of HEK-293 cells, purified by PEG-precipitation and gradient centrifugation, and titers determined by quantitative slot–blot hybridization as published previously (Liu et al., 2003; Ayuso et al., 2010).
Assays on Plasma Samples
Levels of hF.IX were determined by ELISA as previously described (Cao et al., 2009a). Activated partial thromboplastin time (aPTT) was measured using a fibrometer (Fibrosystem; BBL, Cockeysville, MD, USA) and previously published protocols (Cao et al., 2009a). Bethesda assay to determine inhibitor titers was also performed as previously described. By definition, 1 Bethesda Unit (BU) represents a residual hF.IX activity of 50%.
Flow Cytometry
Splenocytes were isolated using standard protocols, and cell viability was measured using a hemocytometer and trypan blue staining. Antibody stains for surface and intracellular molecules were applied according to manufacturers’ protocols. Antibodies against mouse antigens CD4-FITC, CD25-PE, FoxP3-Alexa Fluor 647 were from eBioscience (San Diego, CA, USA). Isotype controls included Rat IgG2a-FITC, Rat IgG2b-PE, Rat IgG2a-Alexa Fluor 647, and other controls were unstained splenocytes and anti-Rat Ig Compensation Particles (BD Biosciences/Pharmingen). Data acquisition was carried out using the BD LSR II. Data analysis was done with FCS 3.0 software. Antibodies against mouse antigens CD4-PB, CD25-PE-Cy7, FoxP3-Alexa Fluor 647, GITR-PE, CD127-Alexa Flour 780 were from eBioscience (San Diego, CA, USA), and antibodies against mouse antigens CD25-PE, CTLA-4-PE were from BD Biosciences (San Jose, CA, USA).
In vitro Suppression Assay
Antigen presenting cells (APCs) were isolated from naïve D011.10-tg mice using MACS CD90.2 MicroBeads (Miltenyi Biotec, Auburn, CA, USA). CD4+CD25+ Treg were isolated from DO11.10-tg RAG-2−/− mice using mouse CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotech) and the CD4+CD25− Teff were isolated from DO11.10-tg RAG-2−/− mice using mouse CD4+ T Cell Isolation Kit II (Miltenyi Biotec). CD4+CD25− cells were labeled with 0.5 μM CFSE (carboxyfluorescein diacetate, succinimidyl ester, Invitrogen, Carlsbad, CA, USA) in PBS containing 2% FBS at a cell concentration of 3 × 106 cell/ml at room temperature for 5 min in the dark. These cells were then washed three times in PBS containing 2% FBS and re-suspended in DMEM containing 10% FBS and penicillin/streptomycin. CD4+CD25+ Treg were cultured with 2 × 105 CFSE-labeled Teff at different ratios as indicated and stimulated with 1 or 0.1 μg/ml ova peptide (AnaSpec), along with 2 × 104 APCs in DMEM supplemented with 10% FBS and penicillin/streptomycin in Thermo Scientific Nunclon D round bottom 96-well plates (Pittsburgh, PA, USA) and incubated at 37°C in 5% CO2 for 4 days.
Statistical Analysis
Results are reported as mean ± SD or ±SEM. Significant differences between an experimental group and control groups were determined with unpaired Student’s t-test. P values of <0.05 were considered significant. For comparison between multiple experimental groups, one-way ANOVA with variance was applied using Prism software (Irvine, CA, USA).
Results
Transient Immune Modulation with Rapamycin/IL-10/Specific Peptide Can Reverse Gene Therapy-Induced Inhibitor Formation
Hemophilia B mice with an F9 gene deletion on a C3H/HeJ genetic background have more robust immune responses to hF.IX than other strain backgrounds such as C57BL/6 or BALB/c (Mingozzi et al., 2003; Wang et al., 2005; Cao et al., 2006, 2009a; Verma et al., 2010). As expected, IM injection of the AAV1–hF.IX vector in these mice resulted in a high-titer inhibitory antibody response of on average 8–10 BU (up to 14 μg IgG1/ml plasma) within 1 month (Figures 1B–E). Coagulation times were not corrected (average aPTTs of ~80 s; an aPTT of 60 s corresponds to a coagulation activity of approximately 1%), and transduced mice lacked systemic hFIX expression (Figures 1F–I). Previously, we found that inhibitor formation in these mice, induced by muscle-directed hF.IX gene transfer, could be prevented by repeated with intraperitoneal (IP) administration of a cocktail comprised of rapamycin/IL-10/hFIX peptide; while cocktails lacking either IL-10 or the peptide were less effective (Nayak et al., 2009). Therefore, we decided to test whether the most optimal regimen could not only prevent but also reverse inhibitor formation in muscle gene transfer.
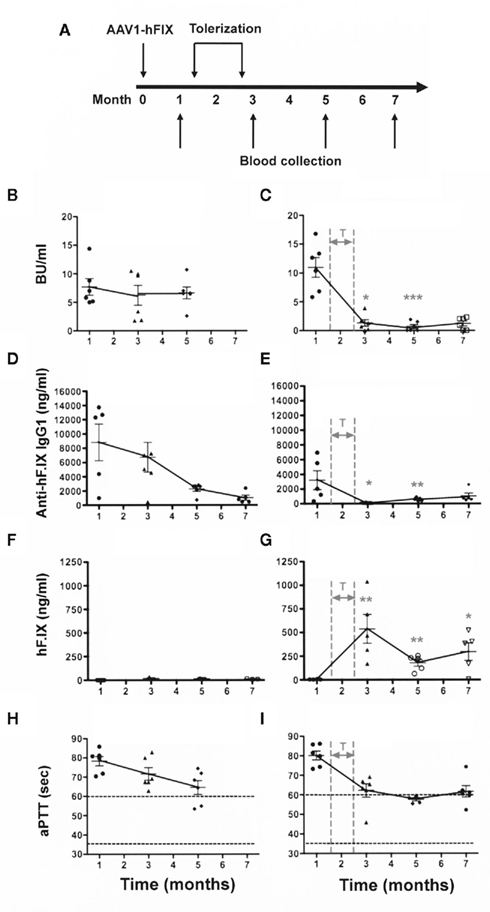
Figure 1. Elimination of a pre-existing/ongoing anti-hF.IX immune response. C3H/HeJ F9−/− mice (n = 12) were injected IM with AAV1-CMV-hF.IX (1 × 1011 vg/mouse) to generate an immune response. After demonstrating the existence of an immune response, half of the animals were given rapamycin/IL-10/specific peptide for 4 weeks [right panels: (C,E,G,I)]. Controls [left panels: (B,D,F,H)] only received gene transfer. (A) Experimental timeline. (B,C) Inhibitory antibody titers (in BU/ml). (D,E) Anti-hF.IX IgG1 antibody (ng/ml). (H,G) hF.IX plasma levels (ng/ml). (H,I) Coagulation times (aPTT in s). Data are shown as a function of time after vector administration in control mice (treated with vector only, left panels) or mice that received the immune modulation/tolerance regimen (starting 1.5 months after vector administration, indicated as “T” in right panels). Results are shown as averages at each time point (±SEM) and for individual animals. Statistically significant differences between the immune modulated and control groups (n = 6 per group) at a specific time point are indicated as gray asterisk in the panels of the immune modulated group with *P < 0.05, **P < 0.01, and ***P < 0.001.
Therefore, 1.5 months after vector administration, animals were randomly divided into the two groups (n = 6/group) graphed in the left and right columns of Figure 1. Animals of one group were not further manipulated (control group, Figures 1B,D,F,H), while the animals of the second group (Figures 1C,E,G,I) were treated with IP injections of rapamycin/IL-10/hFIX peptide, three-times per week for 4 weeks (Figure 1A). These treatments resulted in a substantial reduction of inhibitor titers to <2 BU by 1 month after the transient immune modulatory regimen had been stopped (Figure 1C). The difference between inhibitor titers at 1 month (i.e., prior to immune modulation) and all times points after immune modulation was highly significant (P < 0.001). Antibody titers to hF.IX were low to undetectable for the duration of the experiment (7 months after vector administration or 4.5 months after immune modulation; Figures 1C,E). The reduction in inhibitor/antibody titers was accompanied by an increase in systemic hF.IX antigen from 0 ng/ml before the tolerance regimen to an average level of ~250 ng/ml by 5–7 months (Figures 1C,D). Clotting times gradually declined to an average of 58–60 s (Figure 1E), representing a coagulation activity of approximately 1% of normal.
In contrast, transduced control mice that were not treated with the tolerance protocol maintained inhibitors for at least 5 months (Figure 1B). As seen in previously (Nayak et al., 2009), there was a gradual spontaneous decline in hF.IX-specific IgG titers over time and some improvement in the aPTT (Figures 1D,H). Nonetheless, no systemic hF.IX was detected in these mice for the duration of the experiment (i.e., 7 months after gene transfer, Figure 1F).
Prevention of Inhibitor Formation Using the HF.IX Protein as the Tolerizing Antigen
A problem with the prophylactic use of this protocol against inhibitor formation is that the specific T cell epitopes may not be known in a patient. However, when we used hF.IX protein (1 IU) instead of the peptide in an otherwise identical protocol, we failed to prevent inhibitor formation after subsequent administration of the AAV1–CMV–hF.IX vector (data not shown). Therefore, the protocol needed to be modified to use the entire hF.IX protein as the tolerizing antigen.
Keeping translatability in mind, subsequent experiments were adjusted for the fact that rapamycin is typically given orally in humans (IP administration is not a suitable route for humans) and that the cytokine IL-10 is not readily available for clinical application. Thus, a protocol that could realistically be used in humans would likely be based on oral administration of rapamycin and IV delivery of hF.IX protein in the absence of IL-10.
In a first attempt with such a modified protocol, rapamycin was given orally three-times per week for 4 weeks to hemophilia B mice (same C3H/HeJ strain described above). In addition, hF.IX was given IV at a reduced dose of 0.1 IU/mouse, twice per week. However, this protocol was only partially effective in preventing the immune response to hF.IX in gene therapy (Figures 2B,F). Two of five animals still developed IgG1 and >5 BU (Figure 2F). Next, we decided to test whether more frequent oral administration rapamycin could improve the protocol. Hence, rapamycin was given orally on a daily schedule for 1 month to hemophilia B mice, during which time a low-dose of hF.IX protein (0.1 IU) was also given IV twice per week (Figure 2A). AAV1-CMV-hF.IX was again injected IM 1 week before the end of the tolerance protocol. This regimen was highly effective and prevented antibody/inhibitor formation, resulting in systemic hF.IX (ranging from 50 to 300 ng/ml) and more robust correction of the aPTT to on average ~50 s (Figures 2B–I). Three of the initial five treated animals were still alive >8 months after gene transfer (>1 year old), at which time they still maintained their hF.IX protein levels with no detectable antibody (data not shown).
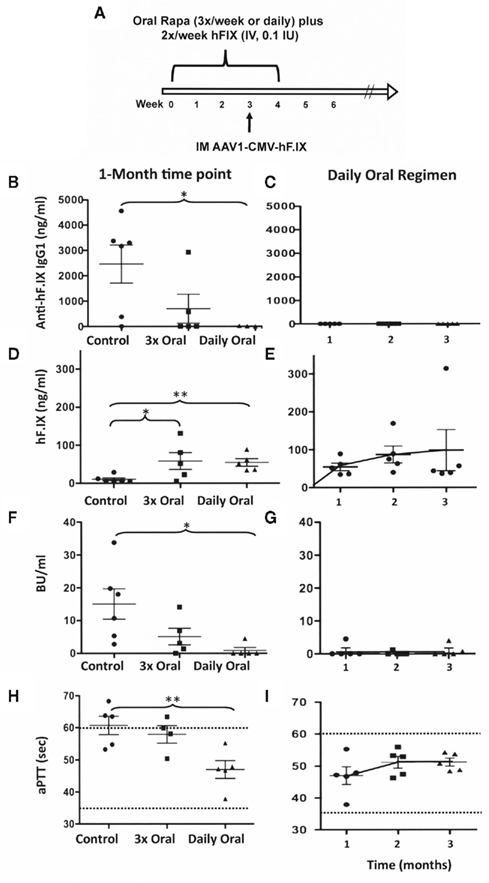
Figure 2. Optimization of a protocol that prevents inhibitor formation by administration of hF.IX protein and rapamycin. (A) Experimental timeline of the optimal protocol. For immune modulation, C3H/HeJ F9−/− mice received hF.IX IV injections (0.1 IU/dose) twice per week for 4 weeks. During that time, rapamycin was administered orally once per day. One week before the end of this 1-month regimen, AAV1-CMV-hF.IX (1 × 1011 vg/mouse) was given IM. Left panels: Comparison of IgG1 (B) and inhibitor (F) formation against hF.IX, of systemic hF.IX expression (D), and of coagulation times [aPTT (H)] at 1 month after vector administration in mice that had received vector only; or 2 IV doses of hF.IX (0.1 IU) plus 3 oral doses of rapamycin per week (with the first and last 30 min prior to hF.IX administration); or the optimal protocol (2 IV doses of F.IX plus daily rapamycin). Right panels: IgG1 (C) and inhibitor (G) formation against hF.IX, systemic hF.IX expression (E), and coagulation times [aPTT (I)] as a function of time after vector administration in mice that had received the optimal protocol (2 IV doses of F.IX plus daily rapamycin).
Effect of the Route of Drug Administration on the Effectiveness of the Protocol
Experimental results presented in Figure 2 suggested that the route of rapamycin administration and the dosing schedule had an impact on the effectiveness of the tolerance regimen. These questions are addressed in the following. In order to directly measure the ability to promote a shift from Teff to Treg as a function of the route of drug administration, we used T cell receptor (TCR) transgenic mice lacking endogenous Treg (DO11.10-tg Rag-2−/− BALB/c mice with a CD4+ TCR specific for ovalbumin). In these mice, IP, subcutaneous (SQ), intravenous (IV), and oral routes of drug administration were compared using our original cocktail. Rapamycin was either given as a cocktail with IL-10 and ova peptide, as published, three-times per week for 3 weeks via IP, SQ, or the IV route (Nayak et al., 2009). Alternatively, the IL-10/ova mix was given IP as published, while rapamycin was fed on the same day by oral gavage (within 30 min of administration of the other drugs).
All routes resulted in a substantial reduction in the frequency of ova-specific CD4+ T cells (Figure 3A). However, oral delivery of rapamycin was significantly less effective than the other routes (Figure 3A). Induction of ova-specific CD4+CD25+FoxP3+ Treg with a frequency of up to 20% of CD4+ T cells was achieved with all these routes (Figure 3B). Oral administration of rapamycin was again less effective in this regard, albeit not significantly different, from the other routes (Figure 3B).
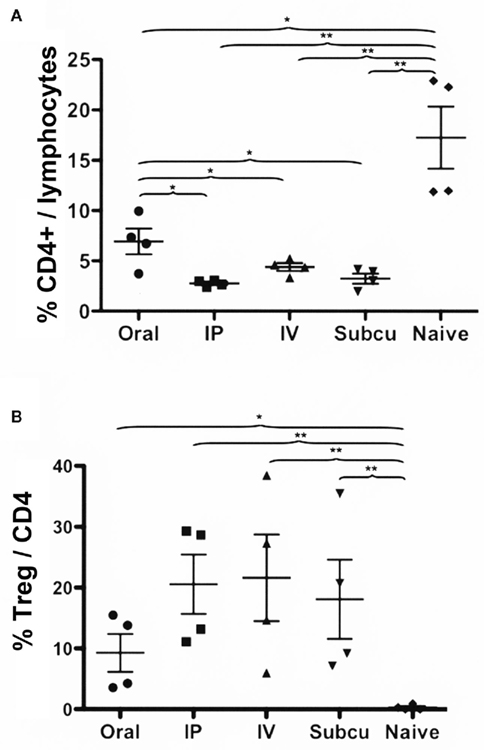
Figure 3. Comparison of Rapa/IL-10/specific peptide tolerization protocol using different routes of drug administration. DO.11.10-tg Rag−/− mice (n = 4/group) were treated for 3 weeks with Rapa/IL-10/ova-specific peptide, after which the spleens were harvested, and flow cytometry was used to determine: (A) percentage of CD4+ T cells of total gated splenocytes and (B) percentage of CD25+FoxP3+ cells of CD4+ T cells. Statistically significant differences between two groups are indicated as *P < 0.05 and **P < 0.01.
Data presented in Figure 2 showed that IV administration of antigen combined with daily gavage of rapamycin was superior in tolerance induction compared to performing gavage of rapamycin less frequently. To study this further, rapamycin was given orally to DO11.10-tg Rag-2−/− BALB/c mice either three-times per week or daily for 3 weeks. During this time, ova peptide was given IV three-times per week. For comparison, a rapamycin/ova peptide cocktail was injected IP three-times per week. As expected, reduction of Teff cells and Treg induction was not as effective for the IP method using this cocktail, which lacks IL-10 (Figures 4A,B). Both oral rapamycin regimens were less effective compared to the IP route (Figures 4A,B). Nonetheless, amongst the two oral rapamycin protocols, daily administration was significantly better for Treg induction (Figure 4B). At a frequency of three-times per week, nearly no Treg induction was observed. Reduction in total CD4+ T cell frequency was similar for both frequencies of oral delivery (Figure 4A).
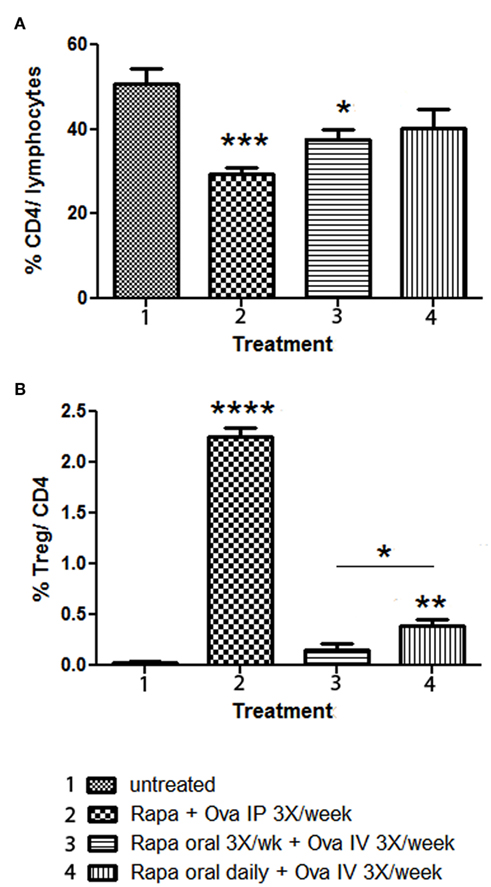
Figure 4. Comparison of Rapa/specific peptide tolerization protocol using different routes and schedules of drug administration. DO.11.10-tg Rag−/− mice (n = 4 per group) were treated for 3 weeks with Rapa/ova-specific peptide, after which the spleens were harvested, and flow cytometry was used to determine: (A) percentage of CD4+ T cells of total gated splenocytes, and (B) percentage of CD25+FoxP3+ cells of CD4+ T cells. Statistically significant differences between each group and untreated (naïve) control mice are indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. In (B), a statistically significant difference between mice that received rapamycin orally three-times per week or daily is also indicated.
Antigen/Rapamycin-Induced Treg Show an Immune Suppressive Phenotype
Further characterization of oral rapamycin/IV antigen induced Treg was carried out by extending treatment to 4 weeks. This resulted in higher Treg frequencies in the IP and oral protocols when performed in parallel for comparison (Figure 5E). The IP regimen was again superior in deletion of the ova-specific CD4+ T cells and in Treg induction (Figures 5D,E). Immunophenotyping, as shown in the form of examples in Figures 5A–C, suggested that FoxP3 expression was comparable for “natural,” IP-, and oral-induced Treg (Figure 5F; “natural” Treg refers to CD4+CD25+FoxP3+ cells isolated from spleens of naïve immune competent DO11.10-tg Rag-2+/+ BALB/c mice as opposed to Treg induced by exogenous antigen administration). Orally induced Treg were most consistently CD25hi. Similarly, all three types of Treg stained positive for surface expression of GITR (Figure 5G). Interestingly, oral-induced Treg showed a higher frequency of CTLA-4 expression compared to natural or IP-induced Treg (Figure 5H). Finally, ~97% of natural Treg and ~93% of IP-induced Treg were CD127−/low, respectively, while ~99% of oral-induced Treg were CD127−/low (data not shown).
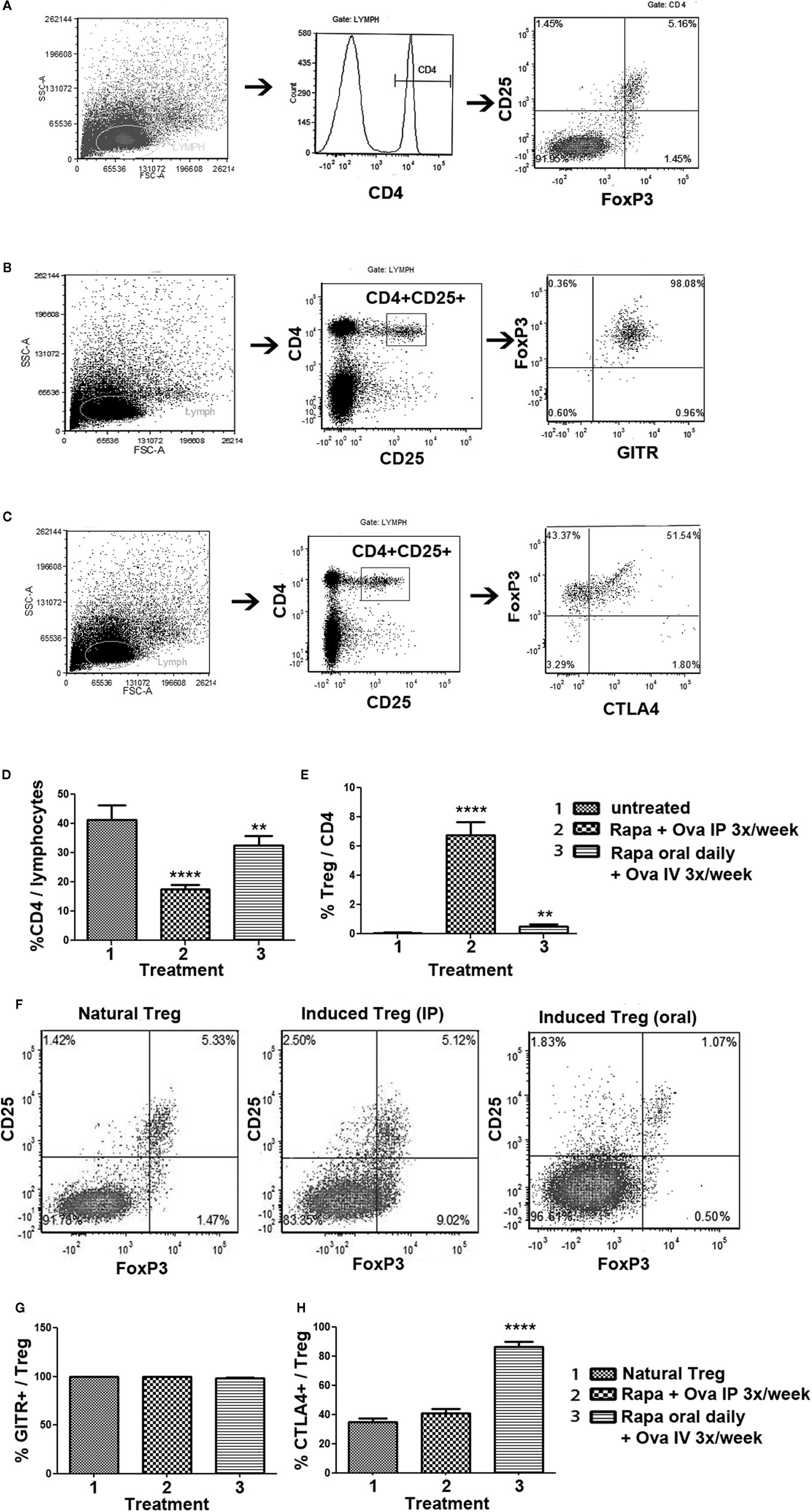
Figure 5. Phenotypic analysis of Treg induced by the rapa/specific peptide tolerization protocol using different routes and schedules of drug administration. DO11.10-tg Rag−/− BALB/c mice (n = 5 per group) were treated for 4 weeks with rapamycin/ova-specific peptide, after which splenocytes were analyzed by antibody stains and flow cytometry. (A–C) Examples for gating scheme and flow cytometric analysis of FoxP3 (A), GITR (B), and CTLA-4 (C) expression by induced CD4+CD25+ Treg. (D) Percentage of CD4+ T cells of total gated splenocytes, and (E) Percentage of CD25+FoxP3+ cells of CD4+ T cells as a function of treatment. Statistically significant differences between each group and untreated (naïve) control mice are indicated as **P < 0.01, and ****P < 0.0001. (F) Representative examples of CD25 and FoxP3 staining (gated on CD4+ splenocytes) for “natural” Treg (isolated from immune competent DO11.10-tg Rag+/+ mice), and induced Treg from DO11.10-tg Rag−/− mice treated with the IP or the oral protocol. (G) Percent GITR+ cells, and (H) percent CTLA-4+ cells amongst CD4+CD25+FoxP3+ Treg. All bar graphs are average ±SD.
In order to demonstrate that orally induced Treg were functional suppressors, proliferation of ova-specific CD4+ Teff was monitored by CFSE-dilution assay upon co-culture with increasing numbers of Treg isolated from spleens of DO11.10-tg Rag-2−/− BALB/c mice that had been treated for 4 weeks with the oral rapamycin/IV ova combination (Figure 6). Treg induced by daily oral rapamycin/IV ova suppressed Teff proliferation in response to stimulation with ova peptide in a Treg dose-dependent manner, resulting in more complete suppression at lower antigen doses (Figures 6A,B). Suppression was at least as effective for orally induced as for IP-induced Treg (compare Figures 6A,C).
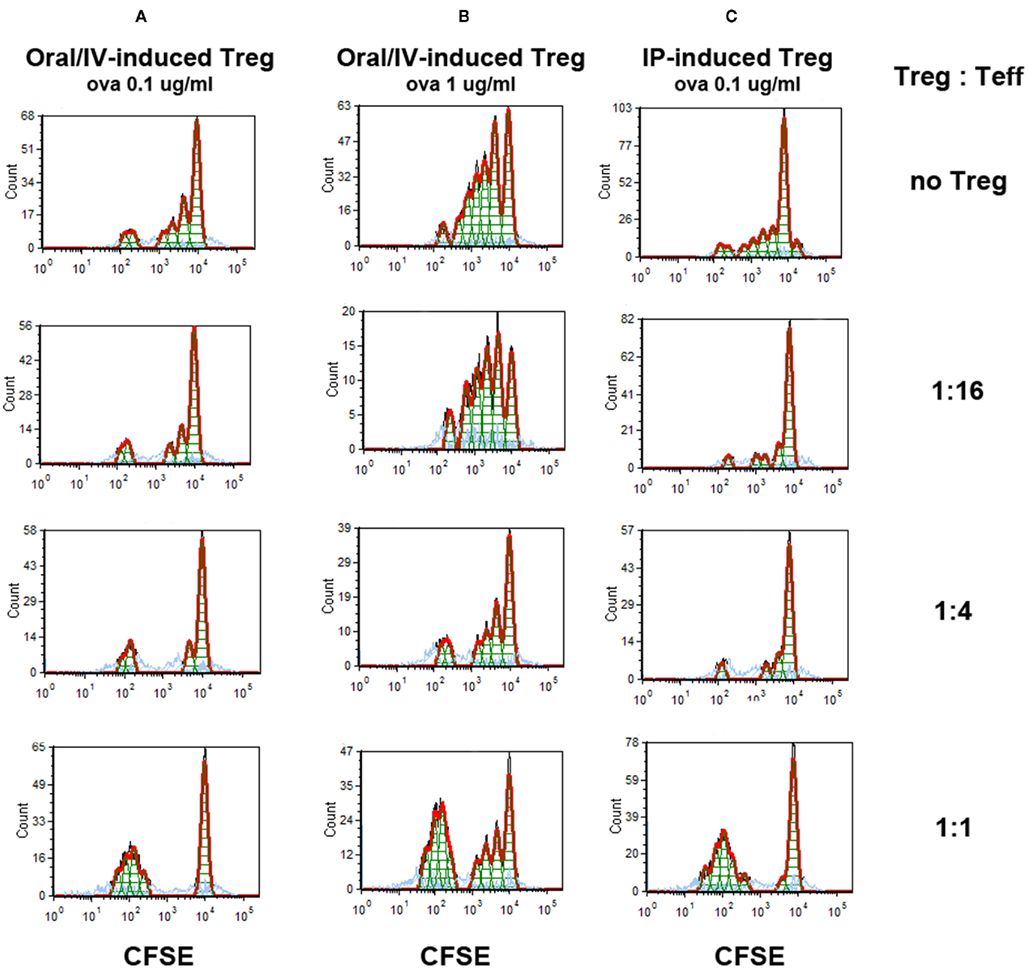
Figure 6. Suppression of antigen-specific Teff proliferation by induced Treg. Treg induced by 4 weeks of oral rapamycin/IV ova peptide-treated (A,B) or rapamycin/ova peptide IP-treated (C) DO11.10-tg Rag−/− BALB/c mice were co-cultured in the indicated ratios with Teff cells and APCs. Teff were CFSE-labeled ova-specific CD4+ T cell isolated from DO11.10-tg Rag−/− BALB/c mice. Conditioned media contained ova peptide at 0.1 μg/ml (A,C) or 1 μg/ml (B). CFSE-dilution was assayed by flow cytometry after 4 days of in vitro stimulation.
Discussion
Gene therapy with viral vectors holds a great deal of promise for the treatment of inherited protein deficiencies such as hemophilia. As with any protein or gene replacement therapy, there are inherent risks of immune responses to the therapeutic gene product. In the treatment of hemophilia, antibody (“inhibitor”) formation against the functional coagulation factor is of particular concern as a major complication of therapy. Our lab and others have used preclinical models to assess immune suppressive regimens for their ability to prevent this immune response when a drug or combination of drugs is given in conjunction with gene therapy (Sack and Herzog, 2009; Miao, 2010; Nayak and Herzog, 2010). However, the ability to reverse an immune response caused by gene transfer has not been studied thoroughly. Preliminary data suggested that monoclonal antibodies (anti-CD20/cyclosporine A combination or anti-CD3) might be useful (Nathwani et al., 2006; Peng et al., 2009; Sack and Herzog, 2009). Here, we clearly demonstrate that transient immune modulation using rapamycin can reverse antibody (“inhibitor”) formation.
Effects of Antigen/Rapamycin Combination
Previously, we found that MHC II antigen presentation to CD4+ T cells in the presence of the specific mTOR inhibitor rapamycin resulted in activation-induced cell death, primarily of Teff. In addition, CD4+CD25+FoxP3+ Tregs are induced in secondary lymphoid organs and expanded (Nayak et al., 2009). These Tregs are able to utilize alternative signaling pathways such as STAT5 to proliferate in response to cytokine growth factors, thereby bypassing the need for the mTOR pathway (Zeiser et al., 2008; Moghimi et al., 2011). The shift from an effector to a Treg response is further enhanced by the immunosuppressive cytokine IL-10, and the induced suppressive response can be adoptively transferred using CD4+CD25+ splenocytes (Battaglia et al., 2006a,b; Nayak et al., 2009; Moghimi et al., 2011).
While we previously showed prophylaxis against inhibitor formation in muscle-directed AAV–hF.IX gene transfer in hemophilia B C3H/HeJ mice, this new data demonstrates the enhanced reversal of a pre-existing response by our regimen. In this strain of hemophilia B mice, IM administration of AAV–hF.IX vector induces not only an inhibitor but also a CD8+ T cell response to hF.IX (Cao et al., 2009a). However, these infiltrating CD8+ T cells are not fully functional and fail to eliminate hFIX-expressing muscle fibers (Wang et al., 2005; Lin et al., 2007; Velazquez et al., 2009). Therefore, production of hF.IX protein persists in the muscle, which is critical for the restoration of systemic expression after antibody elimination. Consequently, we achieved systemic hF.IX expression and partial correction of the coagulation defect. Although the data demonstrate elimination of inhibitors (as measured by Bethesda assay) and restoration of coagulation following the 1-month immune modulatory protocol, circulating antigen levels (on average ~5% of normal) were higher than the level of coagulation activity (1–2% of normal). Low-titer antibodies against hF.IX were detectable by ELISA in rapamycin/IL-10/peptide-treated mice at late time points and may have limited coagulation activity.
Use of Peptide vs. Protein Antigen for Tolerance Induction
We have now shown in two different models (F.VIII replacement therapy in hemophilia A mice and F.IX muscle gene transfer in hemophilia B mice) that IV delivery of protein combined with daily feeding of rapamycin prevents inhibitor formation (Nayak et al., 2009; Moghimi et al., 2011). Use of the protein antigen for tolerance induction has the advantage that knowledge of specific epitopes is not required. Reduced protein doses appear better suited for tolerance induction with rapamycin (Moghimi et al., 2011). However, in the context of an ongoing immune response, the protein antigen may further stimulate B cell responses. In some cases (such as enzyme replacement therapy for hemophilia B or Pompe disease), anaphylactic reactions against the protein are a concern (Dimichele, 2007; Nayak et al., 2009; Sun et al., 2010; Verma et al., 2010). In this case, T cell epitope mapping followed by co-administration of rapamycin and specific peptides may represent a more effective means for targeting antigen-specific effector CD4+ T cells and expansion of Treg. This would promote hypo-responsiveness to the transgene product/therapeutic protein and reversal of the T help-dependent antibody response. Treg appear critical for tolerance to coagulation factors and other transgene products (Cao et al., 2007a,b, 2009b; Mingozzi et al., 2007a; Miao et al., 2009; Miao, 2010; Hoffman et al., 2011).
Impact of the Route of Drug Administration on Tolerance Induction
The dose of rapamycin in our experiments was chosen based on prior published animal studies (Teachey et al., 2006; Nayak et al., 2009; Moghimi et al., 2011). This dose has been determined to maintain a trough level of hF.IX in the blood (10–15 ng/ml) that is known to cause a tolerogenic effect (Teachey et al., 2009). Using direct injections such as IP, IV, or subcutaneous (rather than delivery via oral gavage), it is likely that maintenance of such a trough level requires less frequent administration. In addition, co-administration of a mixture of the antigen and rapamycin may have induced a more effective antigen presentation to T cells with a blocked mTOR pathway. Interestingly, there are now also IV preparations of rapamycin available for patients and could be considered for use in this protocol.
Nonetheless, our data demonstrate that hemophilic mice can be effectively tolerized to F.VIII (Moghimi et al., 2011) or F.IX (this study) using a combination of oral delivery of rapamycin and IV delivery of low-dose protein. Daily rapamycin was required for Treg induction. Orally induced CD4+CD25+ Treg expressed transcription factor FoxP3 and the co-stimulatory molecule GITR at levels comparable to IP-induced or natural Treg, while the inhibitory signaling molecule CTLA-4 was even more frequently expressed, and CD127 expression was low. Therefore, orally induced Treg had all the hallmark features of suppressor cells. A suppressive phenotype was confirmed by inhibition of Teff proliferation. Although not tested here, we previously found no evidence of antigen/rapamycin-induced splenic Treg to express IL-10, but provided evidence for TGF-β expression (Nayak et al., 2009; Moghimi et al., 2011). Experiments in the DO11.10-tg model as well as our published adoptive transfer studies (Nayak et al., 2009; Moghimi et al., 2011) demonstrate induction of antigen-specific Treg. However, we had also found that daily rapamycin administration for 1 month caused a general increase in frequency of circulating Treg (to 27–38% of CD4+ cells) in immune competent BALB/c mice, which within 6 weeks returned to the baseline level of approximately 10% (Moghimi, unpublished observations). We had also previously found general transient decreases in B and T cell frequencies by repeated rapamycin administration (Nayak et al., 2009). Therefore, the intravenously introduced antigen is likely presented in a generally immune suppressive context, while at the same time inducing antigen-specific deletion of Teff and induction of Treg. The combination of these effects likely facilitates tolerance induction. However, results from oral rapamycin scheduling strongly suggest that Treg induction is required for tolerance.
Advantages and Potential Complications of Rapamycin Treatment
There is ample clinical experience with rapamycin, including use in pediatric patients. Rapamycin is administered in combination with other drugs in solid organ transplantation and has recently been successfully tested for the treatment of autoimmune lymphoproliferative syndrome (ALPS; Teachey et al., 2009). It is also a promising drug for the treatment of autoimmune diabetes and can be combined with cytokines such as IL-2 or IL-10 for optimal results (Wells et al., 1999; Zheng et al., 2003; Battaglia et al., 2006a; Koulmanda et al., 2007; Nayak et al., 2009). Conceptually, the mode of action through which Teff are deleted and Treg are induced and expanded make rapamycin ideal for tolerance induction protocols. Rapamycin also has anti-inflammatory properties such as interfering with DC mobilization and function and increasing levels of hemeoxygenase-1 (HO), a stress-inducible, anti-inflammatory enzyme (Hackstein et al., 2003; Visner et al., 2003; Dimitrov et al., 2010). Furthermore, mTOR inhibition can reduce IFN I expression in response to TLR signaling (Manicassamy and Pulendran, 2009). Thus, there are multiple effects of rapamycin that could contribute to tolerance induction and it will be of great interest to study the effects of rapamycin on innate immune responses to AAV vectors, which are largely TLR9-dependent.
On the other hand, prolonged use of rapamycin may actually increase Th1/Th17/inflammatory responses, likely because blockage of mTOR can increase the use of the NF-κB pathway. This, however, may be countered by addition of IL-10 (Weichhart et al., 2008; Huber et al., 2011). In addition, rapamycin counteracts the immunosuppressive effects of glucocorticoids (Weichhart et al., 2011).
In general, immune suppressive regimens pose an increased risk for opportunistic infection and other possible side effects. Although we can achieve coagulation factor-specific tolerance with the 1-month rapamycin-based regimens, there are also transient effects on total B and T cell frequencies. In our initial rapamycin/IL-10 protocol, we found that B and T cell frequencies returned to normal with 2.5 months after treatment (Nayak et al., 2009). With the oral rapamycin protocol, these cell frequencies return to normal even faster (5 weeks), and subsequently, mice respond properly to viral immunization illustrating the highly transient nature of the immune suppressive protocol (Moghimi et al., 2011).
Neutropenia and dysregulation of lipid metabolism are additional side effects of rapamycin. In addition, rapamycin can also delay wound healing, suggesting that the drug should not be used during or early after surgery or other extensive tissue damage (Stallone et al., 2009). Many other side effects, such as kidney problems or increased cancer risk, are more linked to long-term use of the drug than to a very transient protocol.
In summary, we propose that alternative protocols, based on co-administration of rapamycin with the complete antigen or a known T cell epitope (which can be mapped during the course of an immune response), can be applied to prevent or reverse inhibitor responses in gene therapy for hemophilia. This approach could also be useful in gene therapy for other genetic diseases.
Author Contribution
Sushrusha Nayak, Debalina Sarkar, George Q. Perrin, Babak Moghimi, and Brad E. Hoffman performed experiments; Shangzhen Zhou provided AAV vector; Barry J. Byrne and Roland W. Herzog supervised the study; Sushrusha Nayak, Debalina Sarkar, George Q. Perrin, and Roland W. Herzog wrote the manuscript. Roland W. Herzog has been receiving royalty payments by Genzyme Corp. for license of AAV-FIX technology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grants R01 AI/HL51390 (to Roland W. Herzog), PO1 HD078810 (to Roland W. Herzog and Shangzhen Zhou), P01 HL59412 and P01 DK58327 (to Barry J. Byrne), T32DK074367 (support for Sushrusha Nayak), and by a Bayer Hemophilia Award (to Roland W. Herzog).
References
Arruda, V. R., Schuettrumpf, J., Herzog, R. W., Nichols, T. C., Robinson, N., Lotfi, Y., Mingozzi, F., Xiao, W., Couto, L. B., and High, K. A. (2004). Safety and efficacy of factor IX gene transfer to skeletal muscle in murine and canine hemophilia B models by adeno-associated viral vector serotype 1. Blood 103, 85–92.
Ayuso, E., Mingozzi, F., Montane, J., Leon, X., Anguela, X. M., Haurigot, V., Edmonson, S. A., Africa, L., Zhou, S., High, K. A., Bosch, F., and Wright, J. F. (2010). High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 17, 503–510.
Battaglia, M., Stabilini, A., Draghici, E., Gregori, S., Mocchetti, C., Bonifacio, E., and Roncarolo, M. G. (2006a). Rapamycin and interleukin-10 treatment induces T regulatory type 1 cells that mediate antigen-specific transplantation tolerance. Diabetes 55, 40–49.
Battaglia, M., Stabilini, A., Draghici, E., Migliavacca, B., Gregori, S., Bonifacio, E., and Roncarolo, M. G. (2006b). Induction of tolerance in type 1 diabetes via both CD4+CD25+ T regulatory cells and T regulatory type 1 cells. Diabetes 55, 1571–1580.
Cao, O., Armstrong, E., Schlachterman, A., Wang, L., Okita, D. K., Conti-Fine, B., High, K. A., and Herzog, R. W. (2006). Immune deviation by mucosal antigen administration suppresses gene-transfer-induced inhibitor formation to factor IX. Blood 108, 480–486.
Cao, O., Dobrzynski, E., Wang, L., Nayak, S., Mingle, B., Terhorst, C., and Herzog, R. W. (2007a). Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood 110, 1132–1140.
Cao, O., Furlan-Freguia, C., Arruda, V. R., and Herzog, R. W. (2007b). Emerging role of regulatory T cells in gene transfer. Curr. Gene Ther. 7, 381–390.
Cao, O., Hoffman, B. E., Moghimi, B., Nayak, S., Cooper, M., Zhou, S., Ertl, H. C., High, K. A., and Herzog, R. W. (2009a). Impact of the underlying mutation and the route of vector administration on immune responses to factor IX in gene therapy for hemophilia B. Mol. Ther. 17, 1733–1742.
Cao, O., Loduca, P. A., and Herzog, R. W. (2009b). Role of regulatory T cells in tolerance to coagulation factors. J. Thromb. Haemost. 7(Suppl. 1), 88–91.
Dimichele, D. (2007). Inhibitor development in haemophilia B: an orphan disease in need of attention. Br. J. Haematol. 138, 305–315.
Dimitrov, J. D., Dasgupta, S., Navarrete, A. M., Delignat, S., Repesse, Y., Meslier, Y., Planchais, C., Teyssandier, M., Motterlini, R., Bayry, J., Kaveri, S. V., and Lacroix-Desmazes, S. (2010). Induction of heme oxygenase-1 in factor VIII-deficient mice reduces the immune response to therapeutic factor VIII. Blood 115, 2682–2685.
Hackstein, H., Taner, T., Zahorchak, A. F., Morelli, A. E., Logar, A. J., Gessner, A., and Thomson, A. W. (2003). Rapamycin inhibits IL-4 – induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 101, 4457–4463.
Herzog, R. W., Mount, J. D., Arruda, V. R., High, K. A., and Lothrop, C. D. Jr. (2001). Muscle-directed gene transfer and transient immune suppression result in sustained partial correction of canine hemophilia B caused by a null mutation. Mol. Ther. 4, 192–200.
Hoffman, B. E., Martino, A. T., Sack, B. K., Cao, O., Liao, G., Terhorst, C., and Herzog, R. W. (2011). Nonredundant roles of IL-10 and TGF-beta in suppression of immune responses to hepatic AAV-factor IX gene transfer. Mol. Ther. 19, 1263–1272.
Huber, S., Gagliani, N., Esplugues, E., O’Connor, W. Jr., Huber, F. J., Chaudhry, A., Kamanaka, M., Kobayashi, Y., Booth, C. J., Rudensky, A. Y., Roncarolo, M. G., Battaglia, M., and Flavell, R. A. (2011). Th17 cells express interleukin-10 receptor and are controlled by Foxp3(−) and Foxp3(+) regulatory CD4(+) T cells in an interleukin-10-dependent manner. Immunity 34, 554–565.
Kay, M. A., Manno, C. S., Ragni, M. V., Larson, P. J., Couto, L. B., Mcclelland, A., Glader, B., Chew, A. J., Tai, S. J., Herzog, R. W., Arruda, V., Johnson, F., Scallan, C., Skarsgard, E., Flake, A. W., and High, K. A. (2000). Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat. Genet. 24, 257–261.
Koulmanda, M., Budo, E., Bonner-Weir, S., Qipo, A., Putheti, P., Degauque, N., Shi, H., Fan, Z., Flier, J. S., Auchincloss, H. Jr., Zheng, X. X., and Strom, T. B. (2007). Modification of adverse inflammation is required to cure new-onset type 1 diabetic hosts. Proc. Natl. Acad. Sci. U.S.A. 104, 13074–13079.
Li, H., Lasaro, M. O., Jia, B., Lin, S. W., Haut, L. H., High, K. A., and Ertl, H. C. (2011). Capsid-specific T-cell responses to natural infections with adeno-associated viruses in humans differ from those of nonhuman primates. Mol. Ther. 19, 2021–2030.
Lin, S. W., Hensley, S. E., Tatsis, N., Lasaro, M. O., and Ertl, H. C. (2007). Recombinant adeno-associated virus vectors induce functionally impaired transgene product-specific CD8+ T cells in mice. J. Clin. Invest. 117, 3958–3970.
Liu, Y. L., Wagner, K., Robinson, N., Sabatino, D., Margaritis, P., Xiao, W., and Herzog, R. W. (2003). Optimized production of high-titer recombinant adeno-associated virus in roller bottles. BioTechniques 34, 184–189.
Manicassamy, S., and Pulendran, B. (2009). Modulation of adaptive immunity with Toll-like receptors. Semin. Immunol. 21, 185–193.
Manno, C. S., Chew, A. J., Hutchison, S., Larson, P. J., Herzog, R. W., Arruda, V. R., Tai, S. J., Ragni, M. V., Thompson, A., Ozelo, M., Couto, L. B., Leonard, D. G., Johnson, F. A., Mcclelland, A., Scallan, C., Skarsgard, E., Flake, A. W., Kay, M. A., High, K. A., and Glader, B. (2003). AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 101, 2963–2972.
Manno, C. S., Pierce, G. F., Arruda, V. R., Glader, B., Ragni, M., Rasko, J. J., Ozelo, M. C., Hoots, K., Blatt, P., Konkle, B., Dake, M., Kaye, R., Razavi, M., Zajko, A., Zehnder, J., Rustagi, P. K., Nakai, H., Chew, A., Leonard, D., Wright, J. F., Lessard, R. R., Sommer, J. M., Tigges, M., Sabatino, D., Luk, A., Jiang, H., Mingozzi, F., Couto, L., Ertl, H. C., High, K. A., and Kay, M. A. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 12, 342–347.
Miao, C. H. (2010). Immunomodulation for inhibitors in hemophilia A: the important role of Treg cells. Expert Rev. Hematol. 3, 469–483.
Miao, C. H., Harmeling, B. R., Ziegler, S. F., Yen, B. C., Torgerson, T., Chen, L., Yau, R. J., Peng, B., Thompson, A. R., Ochs, H. D., and Rawlings, D. J. (2009). CD4+FOXP3+ regulatory T cells confer long-term regulation of factor VIII-specific immune responses in plasmid-mediated gene therapy-treated hemophilia mice. Blood 114, 4034–4044.
Mingozzi, F., Hasbrouck, N. C., Basner-Tschakarjan, E., Edmonson, S. A., Hui, D. J., Sabatino, D. E., Zhou, S., Wright, J. F., Jiang, H., Pierce, G. F., Arruda, V. R., and High, K. A. (2007a). Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood 110, 2334–2341.
Mingozzi, F., Maus, M. V., Hui, D. J., Sabatino, D. E., Murphy, S. L., Rasko, J. E., Ragni, M. V., Manno, C. S., Sommer, J., Jiang, H., Pierce, G. F., Ertl, H. C., and High, K. A. (2007b). CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13, 419–422.
Mingozzi, F., and High, K. A. (2011a). Immune responses to AAV in clinical trials. Curr. Gene Ther. 11, 321–330.
Mingozzi, F., and High, K. A. (2011b). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 12, 341–355.
Mingozzi, F., Liu, Y. L., Dobrzynski, E., Kaufhold, A., Liu, J. H., Wang, Y., Arruda, V. R., High, K. A., and Herzog, R. W. (2003). Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J. Clin. Invest. 111, 1347–1356.
Moghimi, B., Sack, B. K., Nayak, S., Markusic, D. M., Mah, C. S., and Herzog, R. W. (2011). Induction of tolerance to factor VIII by transient co-administration with rapamycin. J. Thromb. Haemost. 9, 1524–1533.
Nathwani, A. C., Gray, J. T., Ng, C. Y., Zhou, J., Spence, Y., Waddington, S. N., Tuddenham, E. G., Kemball-Cook, G., Mcintosh, J., Boon-Spijker, M., Mertens, K., and Davidoff, A. M. (2006). Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 107, 2653–2661.
Nayak, S., Cao, O., Hoffman, B. E., Cooper, M., Zhou, S., Atkinson, M. A., and Herzog, R. W. (2009). Prophylactic immune tolerance induced by changing the ratio of antigen-specific effector to regulatory T cells. J. Thromb. Haemost. 7, 1523–1532.
Nayak, S., and Herzog, R. W. (2010). Progress and prospects: immune responses to viral vectors. Gene Ther. 17, 295–304.
Peng, B., Ye, P., Rawlings, D. J., Ochs, H. D., and Miao, C. H. (2009). Anti-CD3 antibodies modulate anti-factor VIII immune responses in hemophilia A mice after factor VIII plasmid-mediated gene therapy. Blood 114, 4373–4382.
Sack, B. K., and Herzog, R. W. (2009). Evading the immune response upon in vivo gene therapy with viral vectors. Curr. Opin. Mol. Ther. 11, 493–503.
Stallone, G., Infante, B., Grandaliano, G., and Gesualdo, L. (2009). Management of side effects of sirolimus therapy. Transplantation 87, S23–S26.
Sun, B., Kulis, M. D., Young, S. P., Hobeika, A. C., Li, S., Bird, A., Zhang, H., Li, Y., Clay, T. M., Burks, W., Kishnani, P. S., and Koeberl, D. D. (2010). Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine Pompe disease. Mol. Ther. 18, 353–360.
Teachey, D. T., Greiner, R., Seif, A., Attiyeh, E., Bleesing, J., Choi, J., Manno, C., Rappaport, E., Schwabe, D., Sheen, C., Sullivan, K. E., Zhuang, H., Wechsler, D. S., and Grupp, S. A. (2009). Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br. J. Haematol. 145, 101–106.
Teachey, D. T., Obzut, D. A., Axsom, K., Choi, J. K., Goldsmith, K. C., Hall, J., Hulitt, J., Manno, C. S., Maris, J. M., Rhodin, N., Sullivan, K. E., Brown, V. I., and Grupp, S. A. (2006). Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS). Blood 108, 1965–1971.
Velazquez, V. M., Bowen, D. G., and Walker, C. M. (2009). Silencing of T lymphocytes by antigen-driven programmed death in recombinant adeno-associated virus vector-mediated gene therapy. Blood 113, 538–545.
Verma, D., Moghimi, B., Loduca, P. A., Singh, H. D., Hoffman, B. E., Herzog, R. W., and Daniell, H. (2010). Oral delivery of bioencapsulated coagulation factor IX prevents inhibitor formation and fatal anaphylaxis in hemophilia B mice. Proc. Natl. Acad. Sci. U.S.A. 107, 7101–7106.
Visner, G. A., Lu, F., Zhou, H., Liu, J., Kazemfar, K., and Agarwal, A. (2003). Rapamycin induces heme oxygenase-1 in human pulmonary vascular cells: implications in the antiproliferative response to rapamycin. Circulation 107, 911–916.
Wang, L., Cao, O., Swalm, B., Dobrzynski, E., Mingozzi, F., and Herzog, R. W. (2005). Major role of local immune responses in antibody formation to factor IX in AAV gene transfer. Gene Ther. 12, 1453–1464.
Weichhart, T., Costantino, G., Poglitsch, M., Rosner, M., Zeyda, M., Stuhlmeier, K. M., Kolbe, T., Stulnig, T. M., Horl, W. H., Hengstschlager, M., Muller, M., and Saemann, M. D. (2008). The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 29, 565–577.
Weichhart, T., Haidinger, M., Katholnig, K., Kopecky, C., Poglitsch, M., Lassnig, C., Rosner, M., Zlabinger, G. J., Hengstschlager, M., Muller, M., Horl, W. H., and Saemann, M. D. (2011). Inhibition of mTOR blocks the anti-inflammatory effects of glucocorticoids in myeloid immune cells. Blood 117, 4273–4283.
Wells, A. D., Li, X. C., Li, Y., Walsh, M. C., Zheng, X. X., Wu, Z., Nunez, G., Tang, A., Sayegh, M., Hancock, W. W., Strom, T. B., and Turka, L. A. (1999). Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat. Med. 5, 1303–1307.
Zeiser, R., Leveson-Gower, D. B., Zambricki, E. A., Kambham, N., Beilhack, A., Loh, J., Hou, J. Z., and Negrin, R. S. (2008). Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood 111, 453–462.
Keywords: adeno-associated virus, factor IX, hemophilia, tolerance, antibody, gene therapy, muscle, rapamycin
Citation: Nayak S, Sarkar D, Perrin GQ, Moghimi B, Hoffman BE, Zhou S, Byrne BJ and Herzog RW (2011) Prevention and reversal of antibody responses against factor IX in gene therapy for hemophilia B. Front. Microbio. 2:244. doi: 10.3389/fmicb.2011.00244
Received: 20 June 2011;
Accepted: 22 November 2011;
Published online: 07 December 2011.
Edited by:
Katherine High, Children’s Hospital of Philadelphia, USAReviewed by:
Dirk Dittmer, University of North Carolina at Chapel Hill, USASing Sing Way, University of Minnesota, USA
Copyright: © 2011 Nayak, Sarkar, Perrin, Moghimi, Hoffman, Zhou, Byrne and Herzog. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Roland W. Herzog, Cancer and Genetics Research Center, University of Florida, Room 203, 2033 Mowry Road, Gainesville, FL 32610, USA. e-mail:cmhlcnpvZ0B1ZmwuZWR1