


- 1 Laboratory Medicine, Department of Pathology, School of Medicine and Public Health, University of Wisconsin, Madison, WI, USA
- 2 Cellular and Molecular Pathology Training Program, School of Medicine and Public Health, University of Wisconsin, Madison, WI, USA
- 3 The Rockefeller University, New York, NY, USA
The host–pathogen biology during infection with Mycobacterium tuberculosis is incredibly complex and despite accelerating progress in research, remains poorly understood. Our limited understanding hinders the development of new drugs, next generation vaccines, and novel therapies. The granuloma is the site where mycobacteria are both controlled and allowed to persist, but it remains one of the least studied aspects of the host–pathogen relationship. Here, we review the development, application, potential uses, and limitations of a novel model of granuloma transplantation as a tool to study specific host–pathogen interactions that have been difficult to probe. Application of this new model has already contributed to our understanding of granuloma cell traffic, repopulation, and the relationship between systemic immunity and mycobacteria-containing granulomas. The data collected highlight the dynamic interaction between systemic and local immune processes and support a paradigm that defines the granuloma as a highly dynamic structure. Granuloma transplantation also has special potential as a novel latency model that can contribute to our understanding of host protection factors and bacterial mutants, and serve as a platform for drug testing.
Introduction
One-third of the world’s population is infected with Mycobacterium tuberculosis (Mtb; Barry et al., 2009), the bacterial cause of tuberculosis (TB) that killed approximately 1.7 million people in 2009 (2010). Over 90% of those infected harbor latent Mtb, a dormant state in which bacilli can persist for decades in the host without symptoms (Lin and Flynn, 2010). The HIV pandemic coupled with evolving drug resistance (Iseman and Sbarbaro, 1992; Cox et al., 2003; Gandhi et al., 2010; WHO Global Tuberculosis Control Report 2010, 2010) has dramatically increased the susceptibility of both infected and uninfected persons, especially those in developing countries and Sub-Saharan Africa (Corbett et al., 2003; Aaron et al., 2004). New antibiotics and improved vaccines are desperately needed to combat the evolving Mtb threat, but have been elusive in part because of a lack of basic understanding about the complex host–pathogen biology that occurs during infection.
At the heart of these complex interactions is the granuloma, the hallmark pathology of Mtb but also of other diseases, such as Crohn’s disease, leprosy, and histoplamosis to name a few (Chambers and Morson, 1979; Narayanan, 1988; Wheat et al., 2000; Wallis et al., 2004). The granuloma is a type IV hypersensitivity reaction formed by a special organization of immune cells that contains and kills bacilli, but also provides the long-term home needed for Mtb latency (Saunders and Cooper, 2000; Co et al., 2004; Russell, 2007; Davis and Ramakrishnan, 2008). The granuloma consists of macrophages, T-cells, B-cells, dendritic cells (DCs), natural killer (NK)-cells, and fibroblasts. It is initiated by resident macrophages that phagocytose bacilli and release proinflammatory cytokines, such as TNFα, to recruit additional cells (Flynn et al., 1995; Roach et al., 1999; Algood et al., 2005). CD4+ T-cells from the adaptive response are eventually recruited and enhance macrophage-mediated killing of bacilli by the release of IFNγ (Orme, 1987; Flynn et al., 1993; Stenger and Modlin, 1999). A fibrotic cuff encloses the granuloma after collagen deposition by fibroblasts (Dheda et al., 2005). Mtb have evolved special strategies to avoid cell-mediated killing during initial control of infection, and therefore a few bacilli persist in granulomas indefinitely (McCune et al., 1966; Armstrong and Hart, 1975; Sturgill-Koszycki et al., 1994; Xu et al., 1994; Via et al., 1997; Fortune et al., 2004; Deretic et al., 2006). A better understanding is needed about all parts of granuloma evolution – from its formation during acute infection, to its maintenance during chronic infection. The latter is especially important since any serious global attempt to combat TB will have to target the two billion latently infected people acting as Mtb reservoirs.
While Mtb was originally thought to have evolved around 10,000 years ago from Mycobacterium bovis (Sreevatsan et al., 1997), mounting evidence has suggested a much earlier emergence (Mostowy and Behr, 2005; Gibbons, 2008; Wirth et al., 2008; Smith et al., 2009). The host–pathogen biology that unfolds in the granuloma is one of the most complex of any host–pathogen pair, likely owing to this long history of co-evolution. Probably the ultimate puzzle of infection is that Mtb’s evolutionary strategy is built around the robust, acute granuloma responses that are required for host protection. Mtb hijacks the granuloma, leveraging both its formation and maintenance to allow a few bacilli to escape macrophage anti-microbial responses and persist (Hingley-Wilson et al., 2003; Volkman et al., 2004). Nevertheless, the granuloma as a host protective response prevents bacilli from disseminating to other tissues by constraining bacterial replication and survival. Bacterial persistence in the granuloma during peak anti-microbial responses is accompanied by changes in bacterial metabolism and changes in host cell metabolism that are in part driven by Mtb effector proteins and lipids (Hingley-Wilson et al., 2003; Russell, 2007). Up to decades after dormancy Mtb can again change its own metabolism, reactivate, and exert an entirely different pressure on the granuloma that leads to necrotic cell death (Gan et al., 2008; Porcelli and Jacobs, 2008; Divangahi et al., 2009). Necrosis of the granuloma center facilitates bacterial repopulation, dissemination, and finally transmission to other hosts after the granuloma cuff is ruptured during the physical stress of coughing (Manabe and Bishai, 2000; Lin and Flynn, 2010). The timing for reactivation of latent bacilli corresponds most closely with suppression or decay of important host immune factors which can impair granuloma integrity and set the stage for granuloma necrosis (Saunders and Cooper, 2000).
Many questions remain about the details of the complex granuloma–mycobacteria interactions in an infected host. For instance, what cells and at what rate do they traffic to and from granulomas? Cell traffic has important consequences on granuloma formation and maintenance, as well as the priming of systemic immunity by antigen-carrying cells that leave granulomas and migrate to secondary lymphoid organs. It can be technically challenging, however, to know if antigen-carrying cells identified in the lymph nodes have come directly from granulomas. Measuring rates of granuloma repopulation is also similarly difficult because it is almost impossible to know what cells have been there since the formation and which cells are newly arrived. We wanted to circumvent these conventional limitations and sought to develop a system with different color-coded or genetically marked cells to distinguish between cells of the local and systemic environment. We accomplished this goal by transplantation of infected tissue from one mouse to another, in which the fluorescent signature or cell allotype of donor and host tissue is different and can therefore be tracked.
In the same way that the fluorescent signature of donor and host tissues can be differentiated by transplantation, so also can the local and systemic immune environments during Mycobacterium infection. While granulomas may form during immune-competent conditions, systemic depletion of important factors, such as CD4+ T-cells, can lead to remodeling of the granuloma structure and reactivation of disease (Kaufmann, 2002). Simply infecting immune factor-deficient animals is often an inadequate approach in understanding the effects of systemic depletions on granuloma biology since those same factors may be required for granuloma formation. Transplantation can relocate granulomas from wild (WT) or genetically altered hosts into a recipient with WT or genetically altered characteristics, allowing us to measure the resulting response. This approach helps characterize local and systemic requirements for cells and molecules in the regulation of granulomas. It can also provide insight into the mechanisms of reactivation as well as what types of immune-deficient conditions put latently infected persons at risk.
Here, we highlight the development of this novel model of granuloma transplantation and how it has been used to probe cell trafficking, systemic immune cell priming, and the systemic vs. local requirements for host protection factors. We also outline other applications of the system that include its potential as a new latency model to test the effects of host protection factors, bacterial mutants, and new drugs. Ultimately, understanding parts of the mycobacterial host–pathogen biology discussed here will stem more targeted and thoughtful approaches toward new Mtb therapies, and this new tool can help in that endeavor.
Granuloma Transplantation: A New Tool
In this model, donor liver infected with M. bovis strain bacillus Calmette–Guerin (BCG) is transplanted into recipients by surgical insertion of an infected tissue piece underneath the recipient’s kidney capsule (Figure 1A). The capsule is rich in blood and oxygen, and so it is a permissive environment for transplanted organ survival. A small 1 mm incision through the capsule is made, and the capsule is pulled away from the kidney creating an open pocket for a piece of infected donor tissue. Approximately 1% of infected donor liver is transplanted. The tautness of the capsule holds the transplant piece in place and prevents it from being dislodged during or after surgery. Humanization of NOG/SCID mice is done by transplantation of human thymus in an almost identical fashion (McCune et al., 1988; Kaneshima et al., 1991).
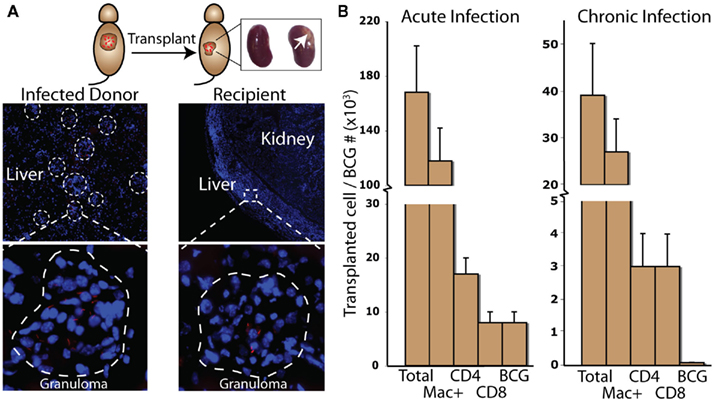
Figure 1. Intraperitoneal infection of BCG in mice results in mycobac-teria-containing liver granulomas [(A), left panels]. In this transplant model, approximately 1% of infected donor liver tissue is transplanted underneath the kidney capsule of recipient mice [(A), right panels]. The high oxygen/blood-rich environment maintains the viability of the tissue piece, and BCG-containing granulomas can be identified by microscopy 2 weeks after surgery. With this model, the total number of transplanted granuloma cells and bacterial CFU can be calculated for both acute and chronic transplant pieces (B).
Bacillus Calmette–Guerin-containing granulomas can be identified in donor-infected tissue both before and up to 2 weeks after transplantation, clearly demonstrating the viability of the tissue piece and survival of bacilli. Transplanted myeloid and lymphoid cells from the donor are almost exclusively from transplanted granulomas. Our experiments have taken advantage of the well-characterized murine BCG liver infection, but the model could also be used with any strain of Mycobacterium, including virulent Mtb. Intraperitoneal (IP) infection of BCG in mice results in the formation of liver granulomas that peak in size, bacterial load, and anti-microbial activity by 3 weeks post infection (Co et al., 2004). From 3 to 10 weeks post infection, bacterial load decreases 100- to 1,000-fold, and bacilli persist in a non-expanding state in smaller, more organized granulomas. Transplant pieces infected with BCG for 10 weeks mimic some of the latent conditions seen in humans infected with virulent Mtb. The resolution of acute infection and persistence of chronic infection mimics the progression of Mtb infection in humans that is considered a hallmark of the Mtb evolutionary strategy (Barry et al., 2009).
With this model both the location and approximate number of transplanted granuloma cells and bacilli can be estimated (Figure 1B). These calculations require knowing the total number of granuloma cells and bacterial CFU per organ, the proportion of transplanted organ to the whole, and the individual cell ratio out of the whole cell population. In our hands, IP injection of BCG in mice results in 1 × 107 (±2 × 106) granuloma cells and 5 × 105 CFU (±1 × 105) per gram of liver 3 weeks after infection. At this time granulomas are roughly 70% macrophage, 10% CD4, and 5% CD8. By 10 weeks, infection results in 3 × 106 (±3 × 105) granuloma cells and 5 × 104 CFU (±1 × 104) per liver, granulomas are roughly 70% macrophage, 8% CD4, and 8% CD8. The remaining cellular composition of 3 and 10 week granulomas consists of roughly 5–10% B-cells, 3–5% DCs, 1–2% NK-cells, and 1–2% γδ T-cells (not shown in Figure 1B). In our hands, per gram of liver there are approximately 5 × 105 (±1 × 105) CFU 3 weeks post infection and 5 × 103 (±1 × 103) CFU 10 weeks post infection. Since protocols, mice, and bacilli vary between labs, the number of transplanted cells and bacterial CFU will vary as well.
Cell Traffic
Granuloma cell traffic is difficult to study on an individual granuloma basis, but undoubtedly plays an important role in several components of granuloma biology. First, cell traffic is required not only in forming granulomas, but also in repopulating short-lived effector cells like CD4+ lymphocytes in existing granulomas. The susceptibility to Mtb reactivation in AIDS patients with ablated CD4+ T-cell numbers demonstrates the importance of this repopulation. Second, cell traffic is a key regulator of systemic immune priming/activation, since mycobacterial antigen from granulomas is captured by mobile cells such as DCs and carried to the lymph nodes (Reljic et al., 2005; Humphreys et al., 2006; Schreiber and Sandor, 2010). One important gap in our knowledge is how, if at all, antigen traffic regulates systemic immune responses during chronic infection and Mtb latency. Mtb’s persistence in the granuloma is reflective of its unique ability to influence its local environment and manipulate the processing, trafficking, and presentation of its own antigens (Gercken et al., 1994; Stenger et al., 1998; Hanekom et al., 2003). These processes help Mtb shape the overall immune response in a way that is favorable to its long-term survival in the granuloma. They may also help explain why the current BCG vaccine, while invaluable for protecting children from death by TB meningitis, does not actually prevent infection or provide long-term protection (Fine, 1988; Colditz et al., 1994, 1995). Understanding cell traffic and how it regulates antigen trafficking has important consequences on the development of next generation vaccines.
Rates of Repopulation
To better understand the dynamics and rates of cell traffic into acute and chronic granulomas we transplanted both acutely and chronically BCG-infected colorless liver into GFP-expressing recipients (Schreiber et al., 2011a). This system, when coupled with immunohistochemistry, allows a convenient way to measure both donor- and recipient-derived cells by comparing GFP+ (R) and GFP− (D) cells in transplanted granulomas (Figure 2A). It remains poorly understood how different cell subsets repopulate the granuloma over time, and how this repopulation, if any, differs between acute and chronic granulomas. The data show that 7 days after transplantation, approximately 35% (±4.7%) of CD4+ T-cells in acute granulomas are of recipient origin, and, surprisingly, 40% (±4.3) of CD4+ T-cells are of recipient origin after transplantation of chronic granulomas. Furthermore monoclonal, mycobacterial antigen-specific T-cells migrated into acutely and chronically infected transplanted tissue at a higher rate than of a non-specific polyclonal population. Together, these results show that both antigen-specific and non-specific CD4+ T-cells not only have access to, but also repopulate acute and chronic granulomas at a high rate. Measurements were also made of CD8+ T-cell, B-cell, and NK-cell migration. Seven days after transplant, CD8+ T-cells repopulate acute and chronic granulomas at comparable rates to CD4+ T-cells, while B-cells repopulate chronic granulomas more so than acute ones. Seven days after transplantation 2/3 of B-cells in chronic granulomas are of recipient origins, while only 1/3 are of recipient origin in acute granulomas (Figure 2B). NK-cells repopulate acute and chronic granulomas less than T-cells or B-cells. That many cell types have greater access to chronic granulomas than acute granulomas was an especially novel finding. Understanding these differences, as well as why certain cellular subsets such as B-cells have faster exchange rates, has yet to be investigated. Taken together, these results show a dynamic cell migration into both acute and chronic granulomas, and challenge the paradigm that defines chronic granulomas as static structures isolated from the systemic immune environment. These results also demonstrate the ability to measure cell-specific granuloma repopulation over time using granuloma transplantation.
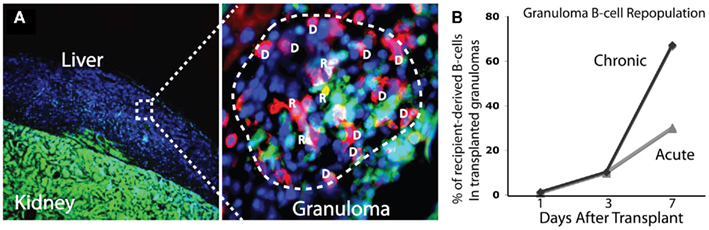
Figure 2. Transplantation of BCG-infected liver from colorless donor mice into GFP-expressing mice provides a simple way to distinguish donor and recipient tissues [(A), left]. When coupled with cell-specific antibodies, the proportion of donor (D) and recipient (R) cells in BCG-containing granulomas can be measured. Colocalization of cell-specific (shown here is CD4+ T-cells) and GFP fluorescent signals identifies recipient cells, while cell-specific signal alone identifies donor cells. In the image shown, donor cells are blue only (DAPI) or blue and red (donor CD4+ T-cell), while recipient cells are green (GFP) only or green and red (recipient CD4+ T-cell). Recipient B-cells repopulate chronic granulomas at a higher rate than acute granulomas (B). Seven days after transplant, 2/3 of B-cells in transplanted granulomas are of recipient origin.
This system not only allows measurements of cells that get into granulomas, but also cells that get out. We transplanted infected, GFP-expressing donors into colorless recipients and looked for GFP+ cells in recipient organs like spleen and lymph nodes. The results show that only a limited number of lymphocytes egress from the granuloma, which is not surprising considering most granuloma-resident T- and B-cells are effector cells that live only for a short time. Still, it is unknown if the few lymphocytes that do get out have any influence on the course of infection and anti-microbial immunity. Unlike for lymphocytes, we measured a high rate of DC egress, a cell type that is known to induce systemic immune responses and contribute to bacterial dissemination (Reljic et al., 2005; Humphreys et al., 2006; Schreiber and Sandor, 2010). The consequences of this DC egress is discussed in the next section.
Antigen Sampling
Dendritic cells in chronic granulomas (unlike in acute) express high levels of inhibitory molecules of T-cell activation like programmed death ligand 1 and 2 and low levels of costimulatory molecules (CD40 and B7), and therefore downregulate T-cell responses (Schreiber et al., 2010). In the same paper that describes these findings, the authors show that the same DCs that downregulate granuloma T-cell responses in the granuloma can also support systemic anti-microbial ones outside of the granuloma. A better understanding of systemic immune priming during latency will contribute to the development of next generation vaccines and novel therapies. Since the granuloma may be the only niche for mycobacteria during latency, any priming/upkeep of systemic responses likely requires antigen sampling (the removal of antigen from the granuloma by cells that transplant it to the lymph nodes to activate T-cells). Only recently has the fact that antigen sampling occurs during latency gained momentum, supported by studies highlighted in this review (below) and another report that identified antigen-specific T-cells in the peripheral blood of latently infected patients (Schuck et al., 2009). Still, the mechanisms and frequency of antigen sampling during Mtb infection remain poorly understood, as well as how these processes might explain the inefficacies of the BCG vaccine. Simply transferring antigen-specific T-cells into chronically infected mice is a limited approach because any measured priming could result from extra-granulomatous bacilli or newly formed lesions. Since the recipients of infected tissue are not infected themselves, the transplant model excludes these possibilities. The only mycobacteria in the system are in transplanted tissue in a known quantity. This makes it an excellent model to study the rate and mechanisms of systemic mycobacterial-specific T-cell priming during infection.
We transplanted infected CD11c-EYFP donors into WT recipients (and vice versa) and adoptively transferred mycobacterial-specific T-cells into recipients to measure T-cell priming that results directly from DC-mediated antigen sampling of acute and chronic granulomas (Schreiber et al., 2011b). The results show that not only do donor DCs migrate out of transplanted granulomas, but recipient ones also migrate in. Surprisingly, there was a higher rate of recipient DC migration into chronic granulomas than into acute ones. Seven days post-transplant, 35% of DCs in chronic granulomas have been repopulated with recipient DCs, while only 25% have been repopulated in acute granulomas. The data also show that DCs from acute and chronically infected transplants migrate out of granulomas into secondary lymph organs, but do not carry live bacilli, the absence of which was confirmed by fluorescent microscopy and CFU of recipient spleen and lymph nodes. Antigen-specific T-cells undergo proliferation after transfer into recipients of both acute and chronic granulomas. This T-cell priming requires recipient MHC II after transplantation of acute or chronic granulomas. It also requires donor CCR7 (responsible for migration of DCs to secondary lymphoid organs; Cyster, 1999; Randolph, 2001; Martin-Fontecha et al., 2003) from acute granulomas, but not chronic ones. These data demonstrate that after transplantation of acute granulomas, local, donor-derived DCs carry antigen to the lymph node, which is then transferred by an unknown mechanism to recipient DCs and presented to antigen-specific T-cells (Figure 3). Antigen sampling after transplantation of chronic granulomas also occurs despite the low bacteria load and restricted amount of antigen. Taken together, these data demonstrate several key points: that both acute and chronic granulomas are intensely surveyed by local and systemic antigen-presenting cells, and that chronic granulomas have previously unappreciated and dynamic interactions with the systemic immune environment.
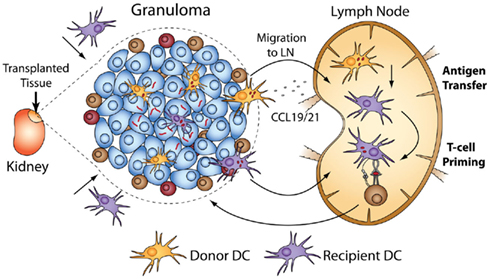
Figure 3. Transplantation of BCG-infected liver using CD11c-EYFP donors or recipients allows tracking of DC migration in and out of acute and chronic granulomas. After transplantation, donor DCs migrate out of granulomas, and recipient DCs also migrate in. The ability of DCs to prime systemic immune responses after surveying granulomas can be measured when coupled with adoptive transfer of antigen-specific T-cells into transplant recipients. Systemic T-cell priming after transplantation of acute granulomas results from CCR7-dependent migration to the lymph nodes of donor DCs that have BCG antigen. The antigen is transferred to recipient DCs and presented to mycobacterial-specific, transferred T-cells. Priming after transplantation of both acute and chronic granulomas was absolutely dependent on recipient MHC II.
Dissection of the Local and Systemic Requirements of Host Protection Factors
It is known that host protection factors such as T-cells, IFNγ, and TNFα play a vital role in the control of Mtb infection and also the maintenance of latent disease. Lymphocyte-deficient RAG knock-out (KO) mice, which are missing the genes required for T- and B-cell receptor recombination, have a limited ability to form granulomas in response to BCG infection. TNFαKO mice form large, unorganized lesions resulting from altered chemokine expression (Bean et al., 1999). IFNγKO mice are unable to induce potent anti-microbial response from mycobacteria-carrying macrophages (Cooper et al., 1993; Flynn et al., 1993). Unlike WT mice, all three KO animals ultimately die from uncontrolled bacterial replication, highlighting the absolute requirement for these host protection factors.
Many factors required for protection and granuloma formation have been described, but it is not always clear if these factors have to be produced in the granuloma or the systemic environment, or both. The transplant system allows us to better understand the local vs. systemic requirements for host protection factors. We transplanted acutely infected WT donor tissue into RAG KO recipients (Figure 4A, left) and did not detect bacterial dissemination from the transplants (Figure 4B, left). When we transplanted acutely infected RAG KO donor tissue into WT recipients (Figure 4A, right), however, bacteria disseminated into spleen and draining renal lymph nodes (Figure 4B, right). Similar results were obtained when chronically infected donors were transplanted. These results were especially surprising, since we expected conditions that mimic the ablated CD4+ T-cell numbers seen in AIDs patients to result in extensive bacterial dissemination. That we detected no dissemination suggests that the presence of some local, granuloma-contained host factors like lymphocytes can buffer, even if transiently, against systemic depletions (Figure 4C). This may explain why many types of stress on the immune system, such as infection with the common flu, do not usually result in reactivation of latent Mtb.
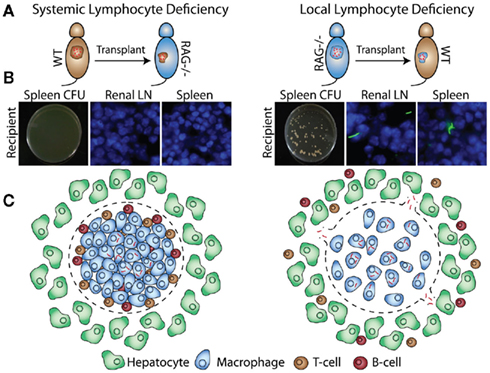
Figure 4. Bacillus Calmette–Guerin-infected WT or RAG KO donors are transplanted into WT or RAG KO recipients (A). Four weeks after transplantation, bacterial dissemination is measured by CFU and fluorescent microscopy of recipient spleen and renal lymph nodes (B). Transplantation of infected RAG KO donors into WT recipients, but not WT donors into RAG KO recipients, results in bacterial dissemination (C).
Transplantation of acutely and chronically BCG-infected WT donors into TNFαKO recipients, on the other hand, resulted in bacterial dissemination and expansion (data not shown). These show that, unlike lymphocytes, local TNFα cannot buffer against systemic depletions. Taken together with the lymphocyte-deficient transplants, the data reveal that not only can there be different local and systemic requirements for a single given protection factor, but also that this local/systemic balance is likely different between factors. The transplants we have done so far have allowed us to measure this requirement for only a handful of known important host factors (manuscript in preparation), but this transplant system can be used to measure any factor of interest for which there is a KO or over-expressing mouse.
Latency and Dissemination Model
Over 90% of the two billion people harboring Mtb are latently infected (Lin and Flynn, 2010). After immune stress such as HIV infection or anti-TNFα therapy, Mtb can reactivate and disseminate (Corbett et al., 2003). Dormant bacteria are incredibly difficult to detect, isolate, or grow during latency, and so the host–pathogen relationship during this stage remains especially poorly understood. For these same reasons the mechanisms and conditions needed for reactivation and dissemination of latent bacteria is also not well understood. One problem in unraveling Mtb latency and reactivation is the lack of animal latency models. Of the few, the Cornell model from the 1950s is the best described. In this model, Mtb-infected mice are treated with isoniazid and pyrazinamide for several months until there are no detectable bacilli in organ cultures (McCune and Tompsett, 1956; McCune et al., 1956). After ending treatment, Mtb in 1/3 of infected animals spontaneously reactivate; reactivation can also be induced after immunosuppressants. While this model mimics human disease in that it achieves low bacterial loads during latency, the artificial suppression of bacterial replication has made it controversial – antibiotics are started only 20 min after infection. The outcome of the Cornell model is also heavily dependent on the experimental parameters, including the dose of inoculum and dose of antibiotics (Scanga et al., 1999). New animal models of Mtb latency would be invaluable tools for the study of latent disease.
In the transplant model presented here, a small number of non-expanding mycobacteria at a known anatomical site can be maintained and then induced to disseminate and proliferate after transplantation into immune-deficient hosts such as TNFαKO recipients. Transplantation of chronically, BCG-infected donors into TNFαKO, but not WT recipients, results in dissemination (Figure 5). That granulomas with live, non-expanding bacilli disseminate when put into TNFαKO, but not WT recipients, highlights this model’s usefulness and relevance to human disease, as well as its potential to act as a latency and dissemination model. Additionally, granuloma transplantation can serve as a new platform for probing the effects of host protection factors, bacterial mutants, and the effects of new drugs during mycobacterial latency and dissemination. An advantage to the system is that the dissemination pattern and movement of bacteria can be tracked since transplant recipients have no pre-established granulomas or mycobacteria themselves.
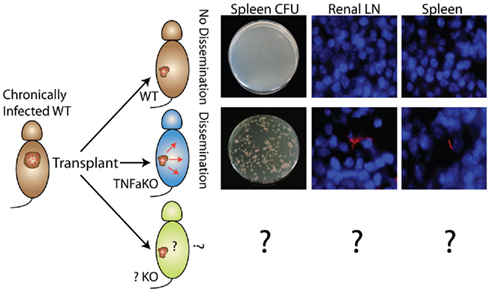
Figure 5. Non-expanding BCG from WT chronic granulomas can be induced to disseminate and expand after transplantation into TNFαKO, but not WT, recipients. Disseminated bacteria are measured by CFU and fluorescent microscopy of recipient target organs such as spleen and renal lymph node. These experiments highlight the system’s potential as a new model of bacterial dissemination to study host protection factors (bottom), bacteria mutants, and new drugs/therapies.
Host Factors Needed to Prevent Bacterial Dissemination
Our dissemination experiments show the model’s capacity to test how chronic granulomas containing non-expanding mycobacteria respond to systemic deficiencies of important immune factors. While systemic deficiencies are often the precursor to reactivation of latent Mtb, the extent to which and exactly what types of deficiencies matter is not well known. The importance of TNFα in maintaining latent disease was underlined by the observation that arthritis patients on anti-TNFα therapy had higher incidences of reactivation (Gardam et al., 2003; Wolfe et al., 2004; Keane, 2005). This model can test any host factor for which there is a KO animal – the unique advantage is that it tests systemic deficiencies against granulomas that evolved under immune-competent conditions.
Bacterial Factors Required for Dissemination
In addition to host factors, the bacterial proteins and pathways involved in Mtb reactivation and dissemination, while potentially important drug targets, remain poorly understood. Mtb transitions to and persists during latency by undergoing dramatic changes in its own metabolism – by shifting toward lipid-based food sources, for example – and by manipulating the cellular environment it occupies to create a niche for survival. Bacteria deficient in Mce4, a cholesterol transporter that facilitates cholesterol catabolism (with other enzymes like HsaC), have an impaired ability to survive during the chronic stages of infection (Pandey and Sassetti, 2008). Mice with defective isocitrate lyase 1, an enzyme that helps Mtb retain carbon instead of losing it to carbon dioxide during limited supply, have a similar inability to survive during chronic infection (Behr et al., 1999; Gordon et al., 1999; McKinney et al., 2000). While these strategies demonstrate Mtb’s metabolic adaptability during the transition into latency, they also imply an equivalent set of metabolic changes that drive Mtb out of latency to reactivate and disseminate.
The transplant model could help identify these lipid or protein factors by testing the influence of specific bacterial mutations on the dissemination of non-expanding mycobacteria (assuming the mutant reaches a non-expanding state). To do so would require transplantation of mutant BCG-infected donors into immune-deficient recipients, where any dissemination and expansion of mycobacteria can be measured. Admittedly, looking at factors driving the dissemination of non-dividing mycobacteria is only part of the total reactivation equation. One limitation in this approach is that IP infection of BCG in mice does not induce granuloma necrosis, which in humans is an important facilitator of bacterial dissemination (Tsai et al., 2006; Russell et al., 2009). Still, to uncover even a fraction of the total bacterial strategy related to reactivation and dissemination is an important contribution. The generation of any new “hits” on bacterial factors related to the dissemination of non-expanding BCG in mice could provide a more focused lens to examine the genome of virulent Mtb, which is 99.9% identical to that of BCG (Philipp et al., 1996). It is also likely that granuloma transplantation can be extended to include Mtb-infected donors, although at this point it is not clear if bacteria would be contained in the transplant (as in the case of BCG) or disseminate when put into WT recipients.
Drug Testing on Mycobacterial Latency and Dissemination
Most of the first-line anti-TB drugs preferentially target actively dividing mycobacteria. Some persisting mycobacteria escape the action of these drugs either through the development of drug resistance and/or the transition into dormancy (Wallis et al., 1999; Gomez and McKinney, 2004; Paramasivan et al., 2005; Zhang, 2005; Connolly et al., 2007). For this reason sterilization of an infected person by antibiotics is difficult. When drugs are administered during latent infection, the goal is to prevent reactivation and dissemination of latent bacilli (Blumberg et al., 2005). Mtb’s unique ability to transition into dormancy, coupled with the inefficacy of antibiotics against latent bacilli, has contributed to the huge reservoir of Mtb-infected people. Even a breakthrough vaccine preventing infection would still leave this group susceptible to reactivation and dissemination. Only recently have certain Mtb proteins – such as the unique prokaryotic proteosome – been identified that could have special potential as drug targets during bacterial latency (Darwin et al., 2003; Cerda-Maira and Darwin, 2009; Lin et al., 2009a; Gandotra et al., 2010). The development of drugs that target latent mycobacteria will require the identification of additional bacterial factors needed for dormancy and also a system to test the efficacy of new drugs on these targets. While in vitro assays on stationary mycobacteria can screen huge libraries of compounds to identify promising candidates, in vivo models more accurately reflect the biology of latency, which is needed to accurately measure the efficacy of latent bacteria-targeting drugs. The transplant system could be used as just this type of in vivo system. New drugs would be administered to chronically infected WT mice before transplantation into TNFαKO (or other KO) recipients to test if non-expanding mycobacteria can be eliminated by drugs.
Limitations
As with most surgeries, there is induced stress on recipients from incisions and displacement of transplanted tissues. The immune reaction to physical stress could be different from or augment the reaction resulting from mycobacteria-containing granulomas that are not transplanted. For this reason transplantation of uninfected tissue in any experiment is an important control in understanding the consequences of surgery alone and stress-induced immunity.
The length of survival of transplanted tissue itself is also a concern. While mycobacterial latency can persist indefinitely, it is unlikely that transplanted tissue would do so as well. We have detected viable bacilli in transplanted tissue up to 1 month after surgery. Two weeks after surgery viable BCG in transplanted tissue can be removed and retransplanted into an immune-deficient host to induce bacterial dissemination. Granuloma cells have a limited life-span, however, and our transplants using GFP+ recipients demonstrate the intense repopulation of recipient tissue and granulomas with donor cells. After a certain time transplanted tissue will likely be made up almost exclusively of immigrating host cells, and so there is a finite window to study mycobacterial–host interactions by granuloma transplantation.
Modeling latency and dissemination in the mouse has been particularly controversial. While virulent Mtb infection in humans usually results in control of infection and clinical latency, without antibiotic treatment Mtb in mice leads to a non-resolving acute infection (Sever and Youmans, 1957; Bloch et al., 1960; Orme, 1995). Mouse BCG infection shares some important features of human disease, such as the resolution of acute infection with high bacterial loads to chronic infection with a few persisting, non-expanding bacteria. There are, however, important differences. BCG lacks the RD1 region of the genome, which in Mtb encodes important virulence factors like ESAT6:CFP10, that drive/control much the host immune response (Hsu et al., 2003; Lewis et al., 2003; Guinn et al., 2004; Derrick and Morris, 2007; Smith et al., 2008). These virulence factors undoubtedly play a role in the overall shaping of the acute immune response required for the transition to latency. Non-expanding BCG from chronically infected mice can still be identified and cultured, but in humans latency is partly defined by the inability to find or culture bacteria. It is also unclear how closely the metabolic changes of BCG leading to a non-expanding state mirror the metabolic changes of Mtb in humans required for the transition to latency. The non-human primate model has gained recent attention for more closely resembling the spectrum of granulomas seen in humans, as well as the ability to transition into clinical latency after infection with virulent Mtb (Capuano et al., 2003; Lerche et al., 2008; Lin et al., 2009b). Transplantation in non-human primates, however, would be a costly and technically prohibitive procedure to almost any lab. In mice the surgery is inexpensive, requires no special instruments, and can be performed in less than 5 min per animal.
Technically the most feasible model for granuloma transplantation is mouse liver infected with BCG or Mtb. There are many important differences between mycobacterial granulomas in different hosts, different organs, and different mycobacterial species, all of which generate different granulomatous lesions. In humans, for example, some Mtb granulomas in the lung are necrotic and caseating; granuloma caseation occurs only in a few mouse strains (Tsai et al., 2006; Russell et al., 2009). There are, however, important similarities between liver granulomas in the mouse and other granulomas. The main cellular components of liver and lung granulomas are similar in most mycobacterial granulomas, as is the crucial role of cytokines such as TNFα and INFγ. Granuloma transplantation can be a useful tool as long as the limitations of the model of infection are accounted for during the interpretation of results. Many animal TB models, the differences of which are outside the scope of this review, have been described that differ in the strain of mycobacteria, the inoculation dose, the species of animal, and the route/tissue of infection (McMurray et al., 1996; Smith et al., 2000; Kaufmann, 2003; Gupta and Katoch, 2005), and the model described here will not reflect all of those differences.
Conclusion
The complex host–pathogen relationship during infection with mycobacteria, especially during bacterial latency, remains poorly understood. Dissecting this relationship will help the development of next generation vaccines and new drugs needed to combat the deadly TB-AIDS co-pandemic and Mtb drug resistance. Presented here is a new tool to help in this endeavor: granuloma transplantation. This unique system has been used to study parts of the host–pathogen biology that have been difficult to study with the available tools and animal models. First, we transplanted mycobacteria-containing granulomas into GFP+ recipients to study individual granuloma cell traffic and repopulation by host cells. The data show a complex and dynamic traffic of cells into pre-existing granulomas, a process that takes place not only in acute granulomas with high bacterial load and anti-microbial activity, but also in chronic granulomas with low bacterial load and low anti-microbial activity. Surprisingly, this repopulation takes place at a higher rate in chronic granulomas compared to acute ones for some cell types. These results challenge the idea that a chronic granuloma is a static structure, instead suggesting that it is repopulated with new cells and/or completely reformed at a new location. Next, we showed that recipient DCs migrate into, and donor DCs migrate out of, both acute and chronic granulomas and that this migration supports systemic priming of mycobacterial-specific CD4+ T-cells. The results suggest that even during times of low antigen load in granulomas with little anti-microbial activity, DC surveillance and transportation of bacterial antigen to the lymph nodes likely contribute to the maintenance and control of infection.
While these studies highlight the connectedness of local and systemic immune processes, we also studied the independence of local and systemic requirements for host protection factors by transplantation of immune-competent or -deficient hosts and donors. The results show that, for some host factors, the granuloma can protect against systemic deficiencies, but not for other host factors. Local lymphocytes can prevent bacterial dissemination in the absence of systemic lymphocytes, at least transiently, but local TNFα cannot prevent dissemination in the absence of systemic TNFα.
Finally, the transplant system has potential to serve as a novel latency and bacterial dissemination model. The best-known model of latency, the Cornell model, has remained relatively unchanged for over 50 years. Using our system, non-expanding mycobacteria from chronic granulomas can be maintained and then induced to disseminate by transplantation into TNFαKO mice. These conditions are analogous to the immune-deficient conditions that lead to reactivation of latent Mtb in humans. Our approach shows that the transplant system can be used to test the effects of any host protection factor on mycobacterial latency and dissemination. Additionally, the system can be used to test how specific bacterial mutants during chronic infection disseminate under immune-deficient conditions. These experiments could uncover new bacterial proteins and lipids involved in reactivation and dissemination, processes that in humans are required for Mtb transmission after latency. Finally, the system could serve as a useful in vivo platform to test the effects of new drugs on non-expanding bacilli.
New methods can provide new insights, and we have shown that granuloma transplantation is a useful approach to study key aspects of the host–mycobacterial relationship. Moving forward we hope the technology will grow in the diversity of its application and usefulness with other granuloma-inducing pathogens.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aaron, L., Saadoun, D., Calatroni, I., Launay, O., Memain, N., Vincent, V., Marchal, G., Dupont, B., Bouchaud, O., Valeyre, D., and Lortholary, O. (2004). Tuberculosis in HIV-infected patients: a comprehensive review. Clin. Microbiol. Infect. 10, 388–398.
Algood, H. M., Lin, P. L., and Flynn, J. L. (2005). Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin. Infect. Dis. 41(Suppl. 3), S189–S193.
Armstrong, J. A., and Hart, P. D. (1975). Phagosome-lysosome interactions in cultured macrophages infected with virulent tubercle-bacilli. Reversal of usual nonfusion pattern and observations on bacterial survival. J. Exp. Med. 142, 1–16.
Barry, C. E. III., Boshoff, H. I., Dartois, V., Dick, T., Ehrt, S., Flynn, J., Schnappinger, D., Wilkinson, R. J., and Young, D. (2009). The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7, 845–855.
Bean, A. G., Roach, D. R., Briscoe, H., France, M. P., Korner, H., Sedgwick, J. D., and Britton, W. J. (1999). Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol. 162, 3504–3511.
Behr, M. A., Wilson, M. A., Gill, W. P., Salamon, H., Schoolnik, G. K., Rane, S., and Small, P. M. (1999). Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284, 1520–1523.
Bloch, H., Yamamura, Y., and Walter, A. (1960). Bacterial populations in experimental murine tuberculosis. II. Studies in vaccinated mice. J. Infect. Dis. 106, 223–229.
Blumberg, H. M., Leonard, M. K. Jr., and Jasmer, R. M. (2005). Update on the treatment of tuberculosis and latent tuberculosis infection. JAMA 293, 2776–2784.
Capuano, S. V. III., Croix, D. A., Pawar, S., Zinovik, A., Myers, A., Lin, P. L., Bissel, S., Fuhrman, C., Klein, E., and Flynn, J. L. (2003). Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 71, 5831–5844.
Cerda-Maira, F., and Darwin, K. H. (2009). The Mycobacterium tuberculosis proteasome: more than just a barrel-shaped protease. Microbes Infect. 11, 1150–1155.
Co, D. O., Hogan, L. H., Kim, S. I., and Sandor, M. (2004). Mycobacterial granulomas: keys to a long-lasting host-pathogen relationship. Clin. Immunol. 113, 130–136.
Colditz, G. A., Berkey, C. S., Mosteller, F., Brewer, T. F., Wilson, M. E., Burdick, E., and Fineberg, H. V. (1995). The efficacy of bacillus Calmette-Guerin vaccination of newborns and infants in the prevention of tuberculosis: meta-analyses of the published literature. Pediatrics 96, 29–35.
Colditz, G. A., Brewer, T. F., Berkey, C. S., Wilson, M. E., Burdick, E., Fineberg, H. V., and Mosteller, F. (1994). Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA 271, 698–702.
Connolly, L. E., Edelstein, P. H., and Ramakrishnan, L. (2007). Why is long-term therapy required to cure tuberculosis? PLoS Med. 4, e120.
Cooper, A. M., Dalton, D. K., Stewart, T. A., Griffin, J. P., Russell, D. G., and Orme, I. M. (1993). Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 178, 2243–2247.
Corbett, E. L., Watt, C. J., Walker, N., Maher, D., Williams, B. G., Raviglione, M. C., and Dye, C. (2003). The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch. Intern. Med. 163, 1009–1021.
Cox, H., Hargreaves, S., and Ismailov, G. (2003). Effect of multidrug resistance on global tuberculosis control. Lancet 362, 1858–1859.
Cyster, J. G. (1999). Chemokines and cell migration in secondary lymphoid organs. Science 286, 2098–2102.
Darwin, K. H., Ehrt, S., Gutierrez-Ramos, J. C., Weich, N., and Nathan, C. F. (2003). The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302, 1963–1966.
Davis, J. M., and Ramakrishnan, L. (2008). “The very pulse of the machine”: the tuberculous granuloma in motion. Immunity 28, 146–148.
Deretic, V., Singh, S., Master, S., Harris, J., Roberts, E., Kyei, G., Davis, A., De Haro, S., Naylor, J., Lee, H. H., and Vergne, I. (2006). Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell. Microbiol. 8, 719–727.
Derrick, S. C., and Morris, S. L. (2007). The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell. Microbiol. 9, 1547–1555.
Dheda, K., Booth, H., Huggett, J. F., Johnson, M. A., Zumla, A., and Rook, G. A. (2005). Lung remodeling in pulmonary tuberculosis. J. Infect. Dis. 192, 1201–1209.
Divangahi, M., Chen, M., Gan, H., Desjardins, D., Hickman, T. T., Lee, D. M., Fortune, S., Behar, S. M., and Remold, H. G. (2009). Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 10, 899–906.
Flynn, J. L., Chan, J., Triebold, K. J., Dalton, D. K., Stewart, T. A., and Bloom, B. R. (1993). An essential role for interferon-gamma in resistance to Mycobacterium-tuberculosis infection. J. Exp. Med. 178, 2249–2254.
Flynn, J. L., Goldstein, M. M., Chan, J., Triebold, K. J., Pfeffer, K., Lowenstein, C. J., Schreiber, R., Mak, T. W., and Bloom, B. R. (1995). Tumor-necrosis-factor-alpha is required in the protective immune-response against Mycobacterium-tuberculosis in mice. Immunity 2, 561–572.
Fortune, S. M., Solache, A., Jaeger, A., Hill, P. J., Belisle, J. T., Bloom, B. R., Rubin, E. J., and Ernst, J. D. (2004). Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J. Immunol. 172, 6272–6280.
Gan, H., Lee, J., Ren, F., Chen, M., Kornfeld, H., and Remold, H. G. (2008). Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat. Immunol. 9, 1189–1197.
Gandhi, N. R., Nunn, P., Dheda, K., Schaaf, H. S., Zignol, M., Van Soolingen, D., Jensen, P., and Bayona, J. (2010). Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet 375, 1830–1843.
Gandotra, S., Lebron, M. B., and Ehrt, S. (2010). The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog. 6, e1001040.
Gardam, M. A., Keystone, E. C., Menzies, R., Manners, S., Skamene, E., Long, R., and Vinh, D. C. (2003). Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. Lancet Infect. Dis. 3, 148–155.
Gercken, J., Pryjma, J., Ernst, M., and Flad, H. D. (1994). Defective antigen presentation by Mycobacterium tuberculosis-infected monocytes. Infect. Immun. 62, 3472–3478.
Gibbons, A. (2008). American Association of Physical Anthropologists meeting. Tuberculosis jumped from humans to cows, not vice versa. Science 320, 608.
Gomez, J. E., and McKinney, J. D. (2004). M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis (Edinb.) 84, 29–44.
Gordon, S. V., Brosch, R., Billault, A., Garnier, T., Eiglmeier, K., and Cole, S. T. (1999). Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol. Microbiol. 32, 643–655.
Guinn, K. M., Hickey, M. J., Mathur, S. K., Zakel, K. L., Grotzke, J. E., Lewinsohn, D. M., Smith, S., and Sherman, D. R. (2004). Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol. Microbiol. 51, 359–370.
Gupta, U. D., and Katoch, V. M. (2005). Animal models of tuberculosis. Tuberculosis (Edinb.) 85, 277–293.
Hanekom, W. A., Mendillo, M., Manca, C., Haslett, P. A., Siddiqui, M. R., Barry, C. III., and Kaplan, G. (2003). Mycobacterium tuberculosis inhibits maturation of human monocyte-derived dendritic cells in vitro. J. Infect. Dis. 188, 257–266.
Hingley-Wilson, S. M., Sambandamurthy, V. K., and Jacobs, W. R. Jr. (2003). Survival perspectives from the world’s most successful pathogen, Mycobacterium tuberculosis. Nat. Immunol. 4, 949–955.
Hsu, T., Hingley-Wilson, S. M., Chen, B., Chen, M., Dai, A. Z., Morin, P. M., Marks, C. B., Padiyar, J., Goulding, C., Gingery, M., Eisenberg, D., Russell, R. G., Derrick, S. C., Collins, F. M., Morris, S. L., King, C. H., and Jacobs, W. R. Jr. (2003). The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. U.S.A. 100, 12420–12425.
Humphreys, I. R., Stewart, G. R., Turner, D. J., Patel, J., Karamanou, D., Snelgrove, R. J., and Young, D. B. (2006). A role for dendritic cells in the dissemination of mycobacterial infection. Microbes Infect. 8, 1339–1346.
Iseman, M. D., and Sbarbaro, J. A. (1992). The increasing prevalence of resistance to antituberculosis chemotherapeutic agents: implications for global tuberculosis control. Curr. Clin. Top. Infect. Dis. 12, 188–207.
Kaneshima, H., Shih, C. C., Namikawa, R., Rabin, L., Outzen, H., Machado, S. G., and Mccune, J. M. (1991). Human immunodeficiency virus infection of human lymph nodes in the SCID-hu mouse. Proc. Natl. Acad. Sci. U.S.A. 88, 4523–4527.
Kaufmann, S. H. (2002). Protection against tuberculosis: cytokines, T cells, and macrophages. Ann. Rheum. Dis. 61(Suppl. 2), ii54–ii58.
Kaufmann, S. H. (2003). Immune response to tuberculosis: experimental animal models. Tuberculosis (Edinb.) 83, 107–111.
Keane, J. (2005). TNF-blocking agents and tuberculosis: new drugs illuminate an old topic. Rheumatology (Oxford) 44, 714–720.
Lerche, N. W., Yee, J. L., Capuano, S. V., and Flynn, J. L. (2008). New approaches to tuberculosis surveillance in nonhuman primates. ILAR J. 49, 170–178.
Lewis, K. N., Liao, R., Guinn, K. M., Hickey, M. J., Smith, S., Behr, M. A., and Sherman, D. R. (2003). Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J. Infect. Dis. 187, 117–123.
Lin, G., Li, D., De Carvalho, L. P., Deng, H., Tao, H., Vogt, G., Wu, K., Schneider, J., Chidawanyika, T., Warren, J. D., Li, H., and Nathan, C. (2009a). Inhibitors selective for mycobacterial versus human proteasomes. Nature 461, 621–626.
Lin, P. L., Rodgers, M., Smith, L., Bigbee, M., Myers, A., Bigbee, C., Chiosea, I., Capuano, S. V., Fuhrman, C., Klein, E., and Flynn, J. L. (2009b). Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect. Immun. 77, 4631–4642.
Lin, P. L., and Flynn, J. L. (2010). Understanding latent tuberculosis: a moving target. J. Immunol. 185, 15–22.
Manabe, Y. C., and Bishai, W. R. (2000). Latent Mycobacterium tuberculosis-persistence, patience, and winning by waiting. Nat. Med. 6, 1327–1329.
Martin-Fontecha, A., Sebastiani, S., Hopken, U. E., Uguccioni, M., Lipp, M., Lanzavecchia, A., and Sallusto, F. (2003). Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J. Exp. Med. 198, 615–621.
McCune, J. M., Namikawa, R., Kaneshima, H., Shultz, L. D., Lieberman, M., and Weissman, I. L. (1988). The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science 241, 1632–1639.
McCune, R. M. Jr., Mcdermott, W., and Tompsett, R. (1956). The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J. Exp. Med. 104, 763–802.
McCune, R. M. Jr., and Tompsett, R. (1956). Fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. I. The persistence of drug-susceptible tubercle bacilli in the tissues despite prolonged antimicrobial therapy. J. Exp. Med. 104, 737–762.
McCune, R. M., Feldmann, F. M., Lambert, H. P., and Mcdermott, W. (1966). Microbial persistence. I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J. Exp. Med. 123, 445–468.
McKinney, J. D., Honer Zu Bentrup, K., Munoz-Elias, E. J., Miczak, A., Chen, B., Chan, W. T., Swenson, D., Sacchettini, J. C., Jacobs, W. R. Jr., and Russell, D. G. (2000). Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406, 735–738.
McMurray, D. N., Collins, F. M., Dannenberg, A. M. Jr., and Smith, D. W. (1996). Pathogenesis of experimental tuberculosis in animal models. Curr. Top. Microbiol. Immunol. 215, 157–179.
Mostowy, S., and Behr, M. A. (2005). The origin and evolution of Mycobacterium tuberculosis. Clin. Chest Med. 26, 207–216, v–vi.
Narayanan, R. B. (1988). Immunopathology of leprosy granulomas – current status: a review. Lepr. Rev. 59, 75–82.
Orme, I. (1995). Mechanisms underlying the increased susceptibility of aged mice to tuberculosis. Nutr. Rev. 53, S35–S38; discussion S38–S40.
Orme, I. M. (1987). The kinetics of emergence and loss of mediator T lymphocytes acquired in response to infection with Mycobacterium tuberculosis. J. Immunol. 138, 293–298.
Pandey, A. K., and Sassetti, C. M. (2008). Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U.S.A. 105, 4376–4380.
Paramasivan, C. N., Sulochana, S., Kubendiran, G., Venkatesan, P., and Mitchison, D. A. (2005). Bactericidal action of gatifloxacin, rifampin, and isoniazid on logarithmic- and stationary-phase cultures of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 49, 627–631.
Philipp, W. J., Nair, S., Guglielmi, G., Lagranderie, M., Gicquel, B., and Cole, S. T. (1996). Physical mapping of Mycobacterium bovis BCG pasteur reveals differences from the genome map of Mycobacterium tuberculosis H37Rv and from M. bovis. Microbiology 142(Pt 11), 3135–3145.
Porcelli, S. A., and Jacobs, W. R. Jr. (2008). Tuberculosis: unsealing the apoptotic envelope. Nat. Immunol. 9, 1101–1102.
Randolph, G. J. (2001). Dendritic cell migration to lymph nodes: cytokines, chemokines, and lipid mediators. Semin. Immunol. 13, 267–274.
Reljic, R., Di Sano, C., Crawford, C., Dieli, F., Challacombe, S., and Ivanyi, J. (2005). Time course of mycobacterial infection of dendritic cells in the lungs of intranasally infected mice. Tuberculosis (Edinb.) 85, 81–88.
Roach, D. R., Briscoe, H., Baumgart, K., Rathjen, D. A., and Britton, W. J. (1999). Tumor necrosis factor (TNF) and a TNF-mimetic peptide modulate the granulomatous response to Mycobacterium bovis BCG infection in vivo. Infect. Immun. 67, 5473–5476.
Russell, D. G., Cardona, P. J., Kim, M. J., Allain, S., and Altare, F. (2009). Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 10, 943–948.
Saunders, B. M., and Cooper, A. M. (2000). Restraining mycobacteria: role of granulomas in mycobacterial infections. Immunol. Cell Biol. 78, 334–341.
Scanga, C. A., Mohan, V. P., Joseph, H., Yu, K., Chan, J., and Flynn, J. L. (1999). Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect. Immun. 67, 4531–4538.
Schreiber, H. A., Harding, J. S., Altamirano, C. J., Hunt, O., Hulseberg, P. D., Fabry, Z., and Sandor, M. (2011a). Continuous repopulation of lymphocyte subsets in transplanted mycobacterial granulomas. Eur. J. Microbiol. Immunol. (Bp) 1, 59–69.
Schreiber, H. A., Harding, J. S., Hunt, O., Altamirano, C. J., Hulseberg, P. D., Stewart, D., Fabry, Z., and Sandor, M. (2011b). Inflammatory dendritic cells migrate in and out of transplanted chronic mycobacterial granulomas in mice. J. Clin. Invest. 121, 3902–3913.
Schreiber, H. A., Hulseberg, P. D., Lee, J., Prechl, J., Barta, P., Szlavik, N., Harding, J. S., Fabry, Z., and Sandor, M. (2010). Dendritic cells in chronic mycobacterial granulomas restrict local anti-bacterial T cell response in a murine model. PLoS ONE 5, e11453.
Schreiber, H. A., and Sandor, M. (2010). The role of dendritic cells in Mycobacterium-induced granulomas. Immunol. Lett. 130, 26–31.
Schuck, S. D., Mueller, H., Kunitz, F., Neher, A., Hoffmann, H., Franken, K. L., Repsilber, D., Ottenhoff, T. H., Kaufmann, S. H., and Jacobsen, M. (2009). Identification of T-cell antigens specific for latent Mycobacterium tuberculosis infection. PLoS ONE 4, e5590.
Sever, J. L., and Youmans, G. P. (1957). Enumeration of viable tubercle bacilli from the organs of nonimmunized and immunized mice. Am. Rev. Tuberc. 76, 616–635.
Smith, D., Wiegeshaus, E., and Balasubramanian, V. (2000). Animal models for experimental tuberculosis. Clin. Infect. Dis. 31(Suppl. 3), S68–S70.
Smith, J., Manoranjan, J., Pan, M., Bohsali, A., Xu, J., Liu, J., Mcdonald, K. L., Szyk, A., Laronde-Leblanc, N., and Gao, L. Y. (2008). Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect. Immun. 76, 5478–5487.
Smith, N. H., Hewinson, R. G., Kremer, K., Brosch, R., and Gordon, S. V. (2009). Myths and misconceptions: the origin and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 7, 537–544.
Sreevatsan, S., Pan, X., Stockbauer, K. E., Connell, N. D., Kreiswirth, B. N., Whittam, T. S., and Musser, J. M. (1997). Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc. Natl. Acad. Sci. U.S.A. 94, 9869–9874.
Stenger, S., and Modlin, R. L. (1999). T cell mediated immunity to Mycobacterium tuberculosis. Curr. Opin. Microbiol. 2, 89–93.
Stenger, S., Niazi, K. R., and Modlin, R. L. (1998). Down-regulation of CD1 on antigen-presenting cells by infection with Mycobacterium tuberculosis. J. Immunol. 161, 3582–3588.
Sturgill-Koszycki, S., Schlesinger, P. H., Chakraborty, P., Haddix, P. L., Collins, H. L., Fok, A. K., Allen, R. D., Gluck, S. L., Heuser, J., and Russell, D. G. (1994). Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263, 678–681.
Tsai, M. C., Chakravarty, S., Zhu, G., Xu, J., Tanaka, K., Koch, C., Tufariello, J., Flynn, J., and Chan, J. (2006). Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell. Microbiol. 8, 218–232.
Via, L. E., Deretic, D., Ulmer, R. J., Hibler, N. S., Huber, L. A., and Deretic, V. (1997). Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J. Biol. Chem. 272, 13326–13331.
Volkman, H. E., Clay, H., Beery, D., Chang, J. C., Sherman, D. R., and Ramakrishnan, L. (2004). Tuberculous granuloma formation is enhanced by a Mycobacterium virulence determinant. PLoS Biol. 2, e367.
Wallis, R. S., Broder, M. S., Wong, J. Y., Hanson, M. E., and Beenhouwer, D. O. (2004). Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin. Infect. Dis. 38, 1261–1265.
Wallis, R. S., Patil, S., Cheon, S. H., Edmonds, K., Phillips, M., Perkins, M. D., Joloba, M., Namale, A., Johnson, J. L., Teixeira, L., Dietze, R., Siddiqi, S., Mugerwa, R. D., Eisenach, K., and Ellner, J. J. (1999). Drug tolerance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 43, 2600–2606.
Wheat, J., Sarosi, G., Mckinsey, D., Hamill, R., Bradsher, R., Johnson, P., Loyd, J., and Kauffman, C. (2000). Practice guidelines for the management of patients with histoplasmosis. Infectious diseases society of America. Clin. Infect. Dis. 30, 688–695.
Wirth, T., Hildebrand, F., Allix-Beguec, C., Wolbeling, F., Kubica, T., Kremer, K., Van Soolingen, D., Rusch-Gerdes, S., Locht, C., Brisse, S., Meyer, A., Supply, P., and Niemann, S. (2008). Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 4, e1000160.
Wolfe, F., Michaud, K., Anderson, J., and Urbansky, K. (2004). Tuberculosis infection in patients with rheumatoid arthritis and the effect of infliximab therapy. Arthritis Rheum. 50, 372–379.
Xu, S., Cooper, A., Sturgill-Koszycki, S., Van Heyningen, T., Chatterjee, D., Orme, I., Allen, P., and Russell, D. G. (1994). Intracellular trafficking in Mycobacterium tuberculosis and Mycobacterium avium-infected macrophages. J. Immunol. 153, 2568–2578.
Keywords: granuloma, transplantation, mycobacteria, host–pathogen interactions
Citation: Harding JS, Schreiber HA and Sandor M (2011) Granuloma transplantation: an approach to study Mycobacterium–host interactions. Front. Microbio. 2:245. doi: 10.3389/fmicb.2011.00245
Received: 19 September 2011; Paper pending published: 10 October 2011;
Accepted: 23 November 2011; Published online: 12 December 2011.
Edited by:
Adel M. Talaat, University of Wisconsin Madison, USAReviewed by:
Jordi Torrelles, Ohio State University, USAMichael L. Vasil, University of Colorado Medical School, USA
Copyright: © 2011 Harding, Schreiber and Sandor. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Matyas Sandor, Laboratory Medicine, Department of Pathology, School of Medicine and Public Health, University of Wisconsin, 1300 University Avenue, Madison, WI 53706, USA. e-mail:bXNhbmRvckB3aXNjLmVkdQ==