 Sayuri Seki1,2
Sayuri Seki1,2 Tetsuro Matano1,2*
Tetsuro Matano1,2*- 1 AIDS Research Center, National Institute of Infectious Diseases, Tokyo, Japan
- 2 The Institute of Medical Science, The University of Tokyo, Tokyo, Japan
Cytotoxic T lymphocyte (CTL) responses exert a suppressive effect on HIV and simian immunodeficiency virus (SIV) replication. Under the CTL pressure, viral CTL escape mutations are frequently selected with viral fitness costs. Viruses with such CTL escape mutations often need additional viral genome mutations for recovery of viral fitness. Persistent HIV/SIV infection sometimes shows replacement of a CTL escape mutation with an alternative escape mutation toward higher viral fitness. Thus, multiple viral genome changes under CTL pressure are observed in the chronic phase of HIV/SIV infection. HIV/SIV transmission to HLA/MHC-mismatched hosts drives further viral genome changes including additional CTL escape mutations and reversions under different CTL pressure. Understanding of viral structure/function and host CTL responses would contribute to prediction of HIV evolution and control of HIV prevalence.
Introduction
Virus-specific CD8+ cytotoxic T lymphocyte (CTL) responses play a central role in the control of HIV and simian immunodeficiency virus (SIV) replication (Borrow et al., 1994; Koup et al., 1994; Matano et al., 1998; Jin et al., 1999; Schmitz et al., 1999; Goulder and Watkins, 2008). CTLs recognize viral antigen-derived peptides (epitopes) presented by major histocompatibility class I (MHC-I) molecules on the surface of viral-infected cells. Under the CTL pressure, viral mutations in and around epitope-coding regions which result in viral escape from CTL recognition are frequently selected with the cost of viral fitness (Phillips et al., 1991; Borrow et al., 1997; Goulder et al., 1997; Price et al., 1997). Thus, analysis of structural and functional constraints in viral proteins could facilitate determination of effective CTLs that can limit viral escape options, contributing to immunogen design in development of CTL-inducing AIDS vaccines.
We previously developed an AIDS vaccine using a Sendai virus vector expressing Gag (SeV-Gag), which induces Gag-specific CTL responses efficiently. Our analysis showed vaccine-based control of a SIVmac239 challenge in a group of Burmese rhesus macaques possessing the MHC-I haplotype 90-120-Ia (Matano et al., 2004; Kawada et al., 2008). Gag206–216 (IINEEAADWDL) epitope-specific CTL responses exert a suppressive effect on SIV replication and select for a CTL escape mutation, GagL216S, leading to a leucine (L)-to-serine (S) substitution at the 216th amino acid (aa) in Gag capsid (CA) with viral fitness costs (Kobayashi et al., 2005). Our studies starting with this finding revealed viral genome changes in persistent SIV infection, providing insights into HIV/SIV evolution.
Loss of Viral Fitness by Escape Mutations and its Recovery by Compensatory Mutations
In contrast to the SIVmac239 challenge experiment, 90-120-Ia-positive vaccinees failed to control a challenge with another pathogenic SIV strain, SIVsmE543-3 (Hirsch et al., 1997), which has the same Gag206–216 amino acid sequence with SIVmac239. SIVsmE543-3 has a different amino acid (glutamate [E]) from SIVmac239 (aspartate [D]) at Gag residue 205, and this GagD205E change resulted in escape from Gag206–216-specific CTL recognition, leading to failure in control of SIVsmE543-3 replication in 90-120-Ia-positive vaccinees (Moriya et al., 2008).
Theoretically, Gag206–216-specific CTL responses can select for either GagD205E or GagL216S mutation. SIVmac239-infected 90-120-Ia-positive macaques, however, select the latter GagL216S mutation but not GagD205E in a year postchallenge. This suggests a possibility that the GagD205E substitution in SIVmac239 results in larger reduction of viral fitness than GagL216S. Indeed, our analysis in vitro revealed much lower replicative ability of the virus with this GagD205E substitution, SIVmac239Gag205E, compared to the wild-type SIVmac239 (Inagaki et al., 2010). On LuSIV cells, which contain a luciferase indicator gene under the control of the SIVmac239 long terminal repeat, SIVmac239Gag205E infection showed significantly lower luciferase activity compared to wild-type SIVmac239, indicating suppression of the early phase of this mutant virus replication.
Further passage of SIVmac239Gag205E-infected culture supernatants in vitro found an additional mutation, GagV340M, resulting in a valine (V)-to-methionine (M) substitution at the 340th aa in Gag. Interestingly, SIVmac239 has V while SIVsmE543-3 has M at the Gag residue 340. SIVmac239Gag205E340M showed similar replication kinetics with wild-type SIVmac239, indicating compensation for loss of viral fitness in SIVmac239Gag205E by addition of the GagV340M substitution. Thus, CTL escape mutations resulting in loss of viral fitness could be selected with compensatory mutations. Figure 1 is a schema indicating the interaction between escape and compensatory mutations.
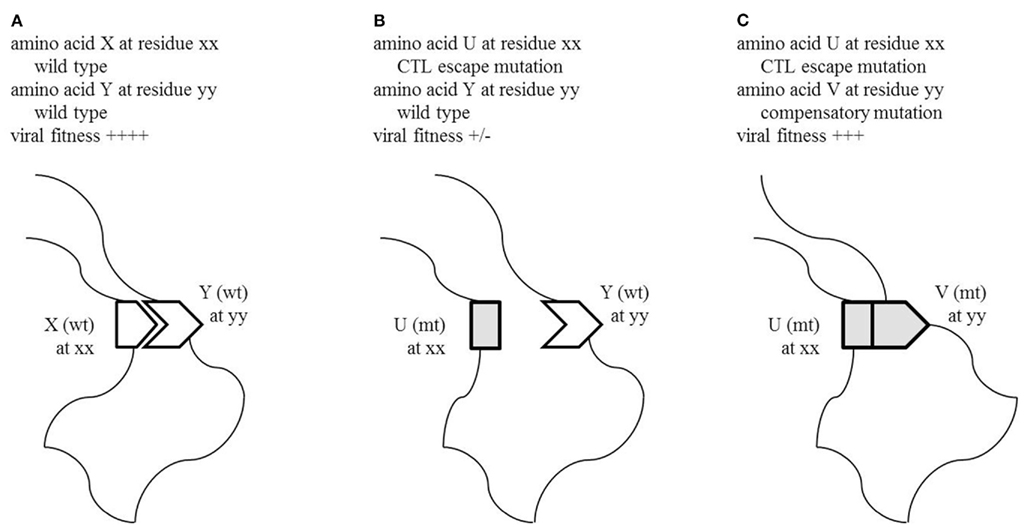
Figure 1. Schema of recovery of viral fitness by a compensatory mutation. (A) Functional interaction between amino acid X at residue xx and Y at residue yy in wild-type viral protein is critical for viral replication. (B) A CTL escape mutation leading to an amino acid change from X to U at residue xx results in loss of viral fitness. (C) An additional compensatory mutation leading to an amino acid change from Y to V partially or fully restores viral function and replication.
Gag CA Intermolecular Interaction
The Gag CA is comprised of the N-terminal (NTD) and the C-terminal domains (CTD) (Momany et al., 1996; Gamble et al., 1997; Berthet-Colominas et al., 1999). Modeling of CA monomer structure showed that the Gag 205th residue is located in the helix 4 of CA NTD and the 340th is in the loop between helices 10 and 11 of CTD. A possibility of intramolecular contact between Gag residues 205 and 340 is not supported by this modeling. However, CA molecules are known to form hexamer lattice in mature virions (Ganser et al., 1999; Li et al., 2000; Ganser-Pornillos et al., 2007, 2008; Pornillos et al., 2009). Modeling of CA hexamer structure revealed that the Gag 205th residue is located in close proximity to the 340th of the adjacent CA molecule. The molecular model of CA hexamers incorporating the GagD205E substitution suggested shortening of the distance between Gag205 and Gag340 residues, which appeared compensated by GagV340M substitution. Thus, there may be intermolecular interaction between Gag residues 205 and 340 in CA hexamers. This is consistent with our results obtained by viral core stability assay. The core stability was reduced by the GagD205E substitution but recovered by the GagV340M substitution. Loss of viral fitness by GagD205E and its recovery by GagV340M implies a structural constraint for functional interaction between CA NTD and CTD involved in the formation of CA hexamers. In addition to previous reports on intramolecular compensation for loss of viral fitness by CTL escape mutations (Friedrich et al., 2004a; Crawford et al., 2007), our results present evidence indicating intermolecular compensation.
Replacement of a CTL Escape Mutation with an Alternative Escape Mutation Toward Higher Viral Fitness
As stated above, SIVmac239-infected 90-120-Ia-positive macaques usually select the Gag206–216-specific CTL escape mutation, GagL216S, but not GagD205E in a year postchallenge. After that, however, we found that the GagD205E mutation together with GagV340M became dominant instead of GagL216S in a 90-120-Ia-positive macaque (Inagaki et al., 2010). In this macaque, neither GagD205E nor GagV340M was detected until week 123 after SIVmac239 challenge, but both became detectable at week 137 and were dominant at week 150. In contrast, the GagL216S mutation dominant until week 123 was undetectable at week 150. Thus, in this animal, SIVmac239Gag216S, whose replicative ability is lower than wild-type SIVmac239 but higher than SIVmac239Gag205E, became dominant under Gag206–216-specific CTL pressure in the early phase, while in the later phase, this mutant virus was replaced with SIVmac239Gag205E340M, whose replicative ability is similar with the wild-type. This indicates replacement of a CTL escape mutation with an alternative escape mutation toward higher viral fitness in the chronic phase, implying persistent Gag206–216-specific CTL pressure for more than 2 years after selection of the CTL escape mutation.
Multiple Viral Genome Changes under CTL Pressure
In another study (Kawada et al., 2006), we observed accumulation of multiple CTL escape mutations in viral genomes in SIV-infected macaques. SeV-Gag-vaccinated animals possessing MHC-I haplotype 90-120-Ia elicited Gag206–216-specific CTL responses and controlled viral replication with rapid selection of the GagL216S mutation after SIVmac239 challenge. Among these SIV controllers, two animals (V3 and V5) accumulated additional gag mutations and showed reappearance of plasma viremia around week 60 postchallenge. Both animals first selected a Gag241–249 epitope-specific CTL escape mutation leading to a GagD244E (aspartic acid [D] to glutamic acid [E] at the 244th aa in Gag) substitution, and then, a Gag373–380 epitope-specific CTL escape mutation leading to a GagA373T (alanine [A] to threonine [T] at the 373rd) or GagP376S (proline [P] to S at the 376th) substitution during the period of viral control. At the viremia reappearance, SIVmac239Gag216S244E247L312V373T with five gag mutations, L216S, D244E, I247L (isoleucine [I] to L at the 247th), A312V (A to V at the 312th), and A373T, became dominant in one of them (V5), and SIVmac239Gag145A216S244E376S with four gag mutations leading to V145A (V to A at the 145th), L216S, D244E, and P376S became dominant in the other (V3). These viruses with multiple gag mutations showed lower replicative ability in vitro than SIVmac239Gag216S carrying single GagL216S mutation. Indeed, SIVmac239Gag216S244E247L312V373T carrying five gag mutations had lower replicative ability in vitro compared to SIVmac239Gag216S244E373T carrying three gag mutations. These results suggest that selection of CTL escape mutations even with viral fitness costs could be advantageous for viral replication in vivo under CTL pressure.
SIV Transmission into MHC-Mismatched Hosts Drives Further Viral Genome Changes
Previous studies (Friedrich et al., 2004b; Kobayashi et al., 2005; Loh et al., 2007) reported reversion of CTL escape mutations in the absence of CTL pressure by transmission of SIVs carrying single escape mutations between MHC-mismatched hosts. SIVs carrying CTL escape gag mutations selected in 90-120-Ia-positive macaques showed lower replicative ability in vitro. We then examined in vivo replicative ability of those SIVs carrying CTL escape mutations in 90-120-Ia-negative macaques (Seki et al., 2008). Coinoculation of macaques with SIVmac239GagL216S and SIVmac239Gag216S244E373T resulted in rapid selection of the former; i.e., D244E and A373T mutations were undetectable even in the acute phase, indicating lower replicative ability in vivo of the latter carrying three escape mutations than the former. Reversion of L216S was observed in a few months, confirming lower replicative ability in vivo of SIVmac239Gag216S than wild-type SIVmac239. Further competition indicated lower replicative ability in vivo of SIVmac239Gag216S244E247L312V373T carrying five gag mutations than SIVmac239Gag216S244E373T carrying three.
We next examined viral genome changes after challenge of 90-120-Ia-negative macaques with SIVs carrying multiple CTL escape mutations selected in 90-120-Ia-positive macaques. Challenge with SIVs carrying five gag mutations, L216S, D244E, I247L, A312V, and A373T, resulted in persistent viremia in all four 90-120-Ia-negative macaques. Two animals exhibited higher viral loads. One of them rapidly developed AIDS at week 18 while the other developed AIDS 2 years postchallenge. The former showed reversion of I247L and A312V but still had three CTL escape mutations, L216S, D244E, and A373T at AIDS onset. The latter showed reversion of four mutations in a year postchallenge, but the A373T mutation remained dominant without reversion until AIDS onset. In the remaining two animals that exhibited lower viral loads, multiple gag mutations including L216S and D244E were still dominant without reversion 1 year after challenge.
Thus, in the experiment of challenge with SIVs carrying multiple CTL escape mutations, the reversion of all the mutations was not required for AIDS onset, while transmission with SIVs carrying single CTL escape mutations showed their rapid reversion. This suggests that even HIVs accumulating multiple CTL escape mutations with viral fitness costs can induce persistent viral infection leading to AIDS progression after their transmission into HLA/MHC-mismatched individuals.
The reversion of the L216S mutation was delayed or not observed after challenge with SIVs carrying multiple gag mutations, whereas challenge with SIVmac239Gag216S resulted in its reversion in a few months. This may be due to the predominant selection of the reversion of other mutations, compensatory mutations, or to lower viral replication efficiency in the former case. Our results suggest that CTL escape mutations resulting in viral fitness costs may not always revert rapidly after their transmission into MHC-mismatched hosts and can be transmitted further to other hosts, driving further viral genome changes with accumulation of mutations (Figure 2). These results provide an important insight into HIV evolution in human individuals with divergent HLA/MHC polymorphisms.
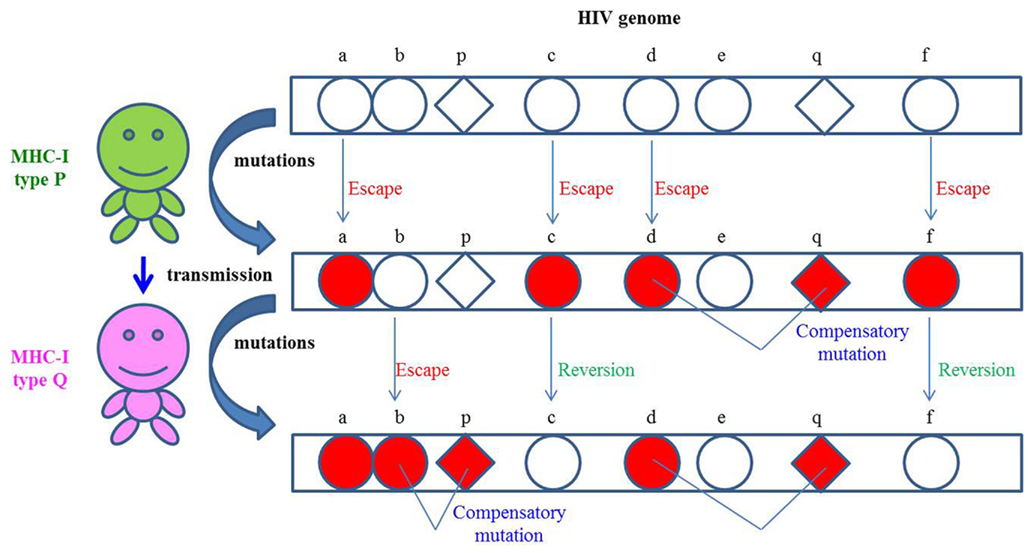
Figure 2. Schema of HIV/SIV transmission resulting in accumulation of multiple viral mutations. Multiple CTL escape mutations resulting in viral fitness costs do not always revert rapidly even in the absence of CTL pressure after their transmission into HLA/MHC-mismatched hosts and such mutants can be transmitted further to other hosts. New escape mutations and compensatory mutations are also observed with transmissions. Thus, CTL affects HIV/SIV evolution in individuals with divergent HLA/MHC polymorphisms.
Concluding Remarks
Cytotoxic T lymphocyte responses exert strong selective pressure on HIV and play a central role in viral evolution (Kaslow et al., 1996; Brander and Walker, 2003; Kiepiela et al., 2004; O’Connor et al., 2004). Correlation of frequencies of viral epitope variants with prevalence of restricting HLA alleles has been shown, indicating HIV adaptation to HLA polymorphisms at a population level (Kawashima et al., 2009). Loss of viral fitness by CTL escape mutations may contribute to HIV control (Martinez-Picado et al., 2006; Schneidewind et al., 2007), but our results indicate the potential of even such HIVs with lower viral fitness to induce AIDS progression. Elucidation of structural constraints of viral antigens for viral function would lead to determination of conserved, escape-resistant epitopes whose mutations largely diminish viral replicative ability (Dahirel et al. 2011), contributing to immunogen design in development of CTL-inducing AIDS vaccines.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Berthet-Colominas, C., Monaco, S., Novelli, A., Sibai, G., Mallet, F., and Cusack, S. (1999). Head-to-tail dimers and interdomain flexibility revealed by the crystal structure of HIV-1 capsid protein (p24) complexed with a monoclonal antibody Fab. EMBO J. 18, 1124–1136.
Borrow, P., Lewicki, H., Hahn, B. H., Shaw, G. M., and Oldstone, M. B. (1994). Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68, 6103–6110.
Borrow, P., Lewicki, H., Wei, X., Horwitz, M. S., Peffer, N., Meyers, H., Nelson, J. A., Gairin, J. E., Hahn, B. H., Oldstone, M. B., and Shaw, G. M. (1997). Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTL) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3, 205–211.
Brander, C., and Walker, B. D. (2003). Gradual adaptation of HIV to human host populations: good or bad news? Nat. Med. 9, 1359–1362.
Crawford, H., Prado, J. G., Leslie, A., Hué, S., Honeyborne, I., Reddy, S., van der Stok, M., Mncube, Z., Brander, C., Rousseau, C., Mullins, J. I., Kaslow, R., Goepfert, P., Allen, S., Hunter, E., Mulenga, J., Kiepiela, P., Walker, B. D., and Goulder, P. J. R. (2007). Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81, 8346–8351.
Dahirel, V., Shekhar, K., Pereyra, F., Miura, T., Artyomov, M., Talsania, S., Allen, T. M., Altfeld, M., Carrington, M., Irvine, D. J., Walker, B. D., and Chakraborty, A. K. (2011). Coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc. Natl. Acad. Sci. U.S.A. 108, 11530–11535.
Friedrich, T. C., Frye, C. A., Yant, L. J., O’Connor, D. H., Kriewaldt, N. A., Benson, M., Vojnov, L., Dodds, E. J., Cullen, C., Rudersdorf, R., Hughes, A. L., Wilson, N., and Watkins, D. I. (2004a). Extra-epitopic compensatory substitutions partially restore fitness to simian immunodeficiency virus variants that escape from an immunodominant cytotoxic T-lymphocyte response. J. Virol. 78, 2581–2585.
Friedrich, T. C., Dodds, E. J., Yant, L. J., Vojnov, L., Rudersdorf, R., Cullen, C., Evans, D. T., Desrosiers, R. C., Mothé, B. R., Sidney, J., Sette, A., Kunstman, K., Wolinsky, S., Piatak, M., Lifson, J., Hughes, A. L., Wilson, N., O’Connor, D. H., and Watkins, D. I. (2004b). Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat. Med. 10, 275–281.
Gamble, T. R., Yoo, S., Vajdos, F. F., von Schwedler, U. K., Worthylake, D. K., Wang, H., McCutcheon, J. P., Sundquist, W. I., and Hill, C. P. (1997). Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278, 849–853.
Ganser, B. K., Li, S., Klishko, V. Y., Finch, J. T., and Sundquist, W. I. (1999). Assembly and analysis of conical models for the HIV-1 core. Science 283, 80–83.
Ganser-Pornillos, B. K., Cheng, A., and Yeager, M. (2007). Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131, 70–79.
Ganser-Pornillos, B. K., Yeager, M., and Sundquist, W. I. (2008). The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 18, 203–217.
Goulder, P. J., Phillips, R. E., Colbert, R. A., McAdam, S., Ogg, G., Nowak, M. A., Giangrande, P., Luzzi, G., Morgana, B., Edwards, A., McMichael, A. J., and Rowland-Jones, S. (1997). Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3, 212–217.
Goulder, P. J. R., and Watkins, D. I. (2008). Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat. Rev. Immunol. 8, 619–630.
Hirsch, V., Adger-Johnson, D., Campbell, B., Goldstein, S., Brown, C., Elkins, W., and Montefiori, D. (1997). A molecularly cloned, pathogenic, neutralization-resistant simian immunodeficiency virus, SIVsmE543-3. J. Virol. 71, 1608–1620.
Inagaki, N., Takeuchi, H., Yokoyama, M., Sato, H., Ryo, A., Yamamoto, H., Kawada, M., and Matano, T. (2010). A structural constraint for functional interaction between N-terminal and C-terminal domains in simian immunodeficiency virus capsid proteins. Retrovirology 7, 90.
Jin, X., Bauer, D. E., Tuttleton, S. E., Lewin, S., Gettie, A., Blanchard, J., Irwin, C. E., Safrit, J. T., Mittler, J., Weinberger, L., Kostrikis, L. G., Zhang, L., Perelson, A. S., and Ho, D. D. (1999). Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189, 991–998.
Kaslow, R. A., Carrington, M., Apple, R., Park, L., Muñoz, A., Saah, A. J., Goedert, J. J., Winkler, C., O’Brien, S. J., Rinaldo, C., Detels, R., Blattner, W., Phair, J., Erlich, H., and Mann, D. L. (1996). Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2, 405–411.
Kawada, M., Igarashi, H., Takeda, A., Tsukamoto, T., Yamamoto, H., Dohki, S., Takiguchi, M., and Matano, T. (2006). Involvement of multiple epitope-specific cytotoxic T lymphocyte responses in vaccine-based control of simian immunodeficiency virus replication in rhesus macaques. J. Virol. 80, 1949–1958.
Kawada, M., Tsukamoto, T., Yamamoto, H., Iwamoto, N., Kurihara, K., Takeda, A., Moriya, C., Takeuchi, H., Akari, H., and Matano, T. (2008). Gag-specific cytotoxic T lymphocyte-based control of primary simian immunodeficiency virus replication in a vaccine trial. J. Virol. 82, 10199–10206.
Kawashima, Y., Pfafferott, K., Frater, J., Matthews, P., Payne, R., Addo, M., Gatanaga, H., Fujiwara, M., Hachiya, A., Koizumi, H., Kuse, N., Oka, S., Duda, A., Prendergast, A., Crawford, H., Leslie, A., Brumme, Z., Brumme, C., Allen, T., Brander, C., Kaslow, R., Tang, J., Hunter, E., Allen, S., Mulenga, J., Branch, S., Roach, T., John, M., Mallal, S., Ogwu, A., Shapiro, R., Prado, J. G., Fidler, S., Weber, J., Pybus, O. G., Klenerman, P., Ndung’u, T., Phillips, R., Heckerman, D., Harrigan, P. R., Walker, B. D., Takiguchi, M., and Goulder, P. (2009). Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458, 641–645.
Kiepiela, P., Leslie, A. J., Honeyborne, I., Ramduth, D., Thobakgale, C., Chetty, S., Rathnavalu, P., Moore, C., Pfafferott, K. J., Hilton, L., Zimbwa, P., Moore, S., Allen, T., Brander, C., Addo, M. M., Altfeld, M., James, I., Mallal, S., Bunce, M., Barber, L. D., Szinger, J., Day, C., Klenerman, P., Mullins, J., Korber, B., Coovadia, H. M., Walker, B. D., and Goulder, P. J. (2004). Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432, 769–775.
Kobayashi, M., Igarashi, H., Takeda, A., Kato, M., and Matano, T. (2005). Reversion in vivo after inoculation of a molecular proviral DNA clone of simian immunodeficiency virus with a cytotoxic-T-lymphocyte escape mutation. J. Virol. 79, 11529–11532.
Koup, R. A., Safrit, J. T., Cao, Y., Andrews, C. A., McLeod, G., Borkowsky, W., Farthing, C., and Ho, D. D. (1994). Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68, 4650–4655.
Li, S., Hill, C. P., Sundquist, W. I., and Finch, J. T. (2000). Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407, 409–413.
Loh, L., Batten, C. J., Petravic, J., Davenport, M. P., and Kent, S. J. (2007). In vivo fitness costs of different Gag CD8 T-cell escape mutant simian-human immunodeficiency viruses for macaques. J. Virol. 81, 5418–5422.
Martinez-Picado, J., Prado, J. G., Fry, E. E., Pfafferott, K., Leslie, A., Chetty, S., Thobakgale, C., Honeyborne, I., Crawford, H., Matthews, P., Pillay, T., Rousseau, C., Mullins, J. I., Brander, C., Walker, B. D., Stuart, D. I., Kiepiela, P., and Goulder, P. (2006). Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80, 3617–3623.
Matano, T., Kobayashi, M., Igarashi, H., Takeda, A., Nakamura, H., Kano, M., Sugimoto, C., Mori, K., Iida, A., Hirata, T., Hasegawa, M., Yuasa, T., Miyazawa, M., Takahashi, Y., Yasunami, M., Kimura, A., O’Connor, D. H., Watkins, D. I., and Nagai, Y. (2004). Cytotoxic T lymphocyte-based control of simian immunodeficiency virus replication in a preclinical AIDS vaccine trial. J. Exp. Med. 199, 1709–1718.
Matano, T., Shibata, R., Siemon, C., Connors, M., Lane, H. C., and Martin, M. A. (1998). Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J. Virol. 72, 164–169.
Momany, C., Kovari, L. C., Prongay, A. J., Keller, W., Gitti, R. K., Lee, B. M., Gorbalenya, A. E., Tong, L., McClure, J., Ehrlich, L. S., Summers, M. F., Carter, C., and Rossmann, M. G. (1996). Crystal structure of dimeric HIV-1 capsid protein. Nat. Struct. Mol. Biol. 3, 763–770.
Moriya, C., Igarashi, H., Takeda, A., Tsukamoto, T., Kawada, M., Yamamoto, H., Inoue, M., Iida, A., Shu, T., Hasegawa, M., Nagai, Y., and Matano, T. (2008). Abrogation of AIDS vaccine-induced cytotoxic T lymphocyte efficacy in vivo due to a change in viral epitope flanking sequences. Microbes Infect. 10, 285–292.
O’Connor, D. H., McDermott, A. B., Krebs, K. C., Dodds, E. J., Miller, J. E., Gonzalez, E. J., Jacoby, T. J., Yant, L., Piontkivska, H., Pantophlet, R., Burton, D. R., Rehrauer, W. M., Wilson, N., Hughes, A. L., and Watkins, D. I. (2004). A dominant role for CD8-T-lymphocyte selection in simian immunodeficiency virus sequence variation. J. Virol. 78, 14012–14022.
Phillips, R. E., Rowland-Jones, S., Nixon, D. F., Gotch, F. M., Edwards, J. P., Ogunlesi, A. O., Elvin, J. G., Rothbard, J. A., Bangham, C. R., Rizza, C. R., and McMichael, A. J. (1991). Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354, 453–459.
Pornillos, O., Ganser-Pornillos, B. K., Kelly, B. N., Hua, Y., Whitby, F. G., Stout, C. D., Sundquist, W. I., Hill, C. P., and Yeager, M. (2009). X-Ray Structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292.
Price, D. A., Goulder, P. J., Klenerman, P., Sewell, A. K., Easterbrook, P. J., Troop, M., Bangham, C. R., and Phillips, R. E. (1997). Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. U.S.A. 94, 1890–1895.
Schmitz, J. E., Kuroda, M. J., Santra, S., Sasseville, V. G., Simon, M. A., Lifton, M. A., Racz, P., Tenner-Racz, K., Dalesandro, M., Scallon, B. J., Ghrayeb, J., Forman, M. A., Montefiori, D. C., Rieber, E. P., Letvin, N. L., and Reimann, K. A. (1999). Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283, 857–860.
Schneidewind, A., Brockman, M. A., Yang, R., Adam, R. I., Li, B., Le Gall, S., Rinaldo, C. R., Craggs, S. L., Allgaier, R. L., Power, K. A., Kuntzen, T., Tung, C. S., LaBute, M. X., Mueller, S. M., Harrer, T., McMichael, A. J., Goulder, P. J., Aiken, C., Brander, C., Kelleher, A. D., and Allen, T. M. (2007). Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 81, 12382–12393.
Keywords: HIV, SIV, MHC, cytotoxic T lymphocyte, escape mutation, viral fitness, capsid
Citation: Seki S and Matano T (2012) CTL escape and viral fitness in HIV/SIV infection. Front. Microbio. 2:267. doi: 10.3389/fmicb.2011.00267
Received: 15 November 2011;
Accepted: 16 December 2011;
Published online: 04 January 2012.
Edited by:
Akio Adachi, The University of Tokushima Graduate School, JapanReviewed by:
Hirofumi Akari, Kyoto University, JapanYasuko Yokota, National Institute of Infectious Diseases, Japan
Copyright: © 2012 Seki and Matano. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Tetsuro Matano, AIDS Research Center, National Institute of Infectious Diseases, 1-23-1 Toyama, Shinjuku-ku, Tokyo 162-8640, Japan. e-mail:dG1hdGFub0BuaWguZ28uanA=