 Anastasia Bragina1†
Anastasia Bragina1† Stefanie Maier1† Christian Berg2 Henry Müller1 Vladimir Chobot3 Franz Hadacek3
Stefanie Maier1† Christian Berg2 Henry Müller1 Vladimir Chobot3 Franz Hadacek3 Gabriele Berg1*
Gabriele Berg1*- 1 Institute of Environmental Biotechnology, Graz University of Technology, Graz, Austria
- 2 Institute of Plant Sciences, Karl-Franzens University of Graz, Graz, Austria
- 3 Department of Chemical Ecology and Ecosystem Research, University of Vienna, Vienna, Austria
Sphagnum mosses represent a main vegetation component in ombrotrophic wetlands. They harbor a specific and diverse microbial community with essential functions for the host. To understand the extend of host specificity and impact of environment, Sphagnum fallax and Sphagnum angustifolium, two phylogenetically closely related species, which show distinct habitat preference with respect to the nutrient level, were analyzed by a multifaceted approach. Microbial fingerprints obtained by PCR-single-strand conformation polymorphism of 16S rRNA and nitrogenase-encoding (nifH) genes were highly similar for both Sphagnum species. Similarity was confirmed for colonization patterns obtained by fluorescence in situ hybridization (FISH) coupled with confocal laser scanning microscopy (CLSM): Alphaproteobacteria were the main colonizers inside the hyaline cells of Sphagnum leaves. A deeper survey of Alphaproteobacteria by 16S rRNA gene amplicon sequencing reveals a high diversity with Acidocella, Acidisphaera, Rhodopila, and Phenylobacterium as major genera for both mosses. Nitrogen fixation is an important function of Sphagnum-associated bacteria, which is fulfilled by microbial communities of Sphagna in a similar way. NifH libraries of Sphagnum-associated microbial communities were characterized by high diversity and abundance of Alphaproteobacteria but contained also diverse amplicons of other taxa, e.g., Cyanobacteria and Deltaproteobacteria. Statistically significant differences between the microbial communities of both Sphagnum species could not be discovered in any of the experimental approach. Our results show that the same close relationship, which exists between the physical, morphological, and chemical characteristics of Sphagnum mosses and the ecology and function of bog ecosystems, also connects moss plantlets with their associated bacterial communities.
Introduction
Northern wetlands belong to the oldest vegetation forms with more or less constant conditions for thousands of years. Sphagnum-dominated peatlands represent one of the most extensive types of Northern wetlands (Dedysh, 2011). They cover with 4 million km2 approx. 3% of the Earth surface and have a high value for biodiversity–conservation, as reservoir of fresh water, for human welfare and our world climate due to its extraordinary role in carbon sequestration (Gorham, 1991; Clymo et al., 1998). Despite their age, these long-existing ecosystems are extremely sensitive to changing a-biotic factors connected with climate change (Belyea and Malmer, 2004; Dise, 2009). As the dominant vegetation component of the peatlands, Sphagnum moss has been used globally as an indicator of climate change (Gignac and Vitt, 1994; Whinam and Copson, 2006; Granath et al., 2009). The ecological significance of bogs is directly related to the physical, morphological, and chemical characteristics of Sphagnum peat mosses; which set Sphagnum apart from other mosses to practically every stage of the life cycle (Shaw et al., 2003). Moreover, Sphagnum mosses are able to change their environments: living Sphagna have extraordinarily high cation exchange capacity and therefore acidify their environment by exchanging tissue-bound protons for basic cations in surrounding water (Soudzilovskaia et al., 2010). Interestingly, Sphagnum leaves are highly specialized: they form a network of living, chlorophyll-containing chlorophytes and dead, cell content-free hyalocytes, which are responsible for their high water holding capacity. Sphagnum species also produce species specific bioactive secondary metabolites influencing microbial colonization (Opelt et al., 2007a).
Sphagnum mosses are colonized by diverse bacterial communities. Microbial populations involved in CH4 cycling, i.e., methanotrophic bacteria (Dedysh et al., 1998; Dedysh, 2002; Raghoebarsing et al., 2005; Larmola et al., 2010; rev. in Dedysh, 2011) as well as methanogens including archaea (Horn et al., 2003; Freitag et al., 2010) have attracted research interest due to their important function for methane emission. Recently, we could show that living Sphagnum mosses are colonized in high abundances with specific microorganisms, which fulfill other important functions like nutrient supply and pathogen defense for moss growth and health (Opelt et al., 2007a,b). An extremely high impact of the Sphagnum species was found on the structure of the microbial diversity, and this diversity is transferred directly from the sporophyte (within the sporangium capsule) to the gametophyte and vice versa (Bragina et al., 2011). In the latter, we analyzed bacterial communities of two ubiquitous Sphagnum species, S. magellanicum, and Sphagnum fallax, in three Alpine bogs in Austria. Extremely high differences between bacterial communities of both Sphagna were found by a combination of methods independently from the site. For example, a discriminative spectrum of bacteria was identified: while Alpha- and Gamma-proteobacteria dominated S. magellanicum, S. fallax was mainly colonized by Verrucomicrobia, Planctomycetes, and Alphaproteobacteria. In addition, bacterial communities were strongly driven by a-biotic factors (nutrient richness and pH), and correlated strongly with the composition of higher plant communities. The specific microbial diversity associated with the highly diverse Sphagnum genus (Daniels and Eddy, 1985) is largely unknown but important to understand and protect Sphagnum in bog ecosystems.
The objective of this work was to study the structure and function of Sphagnum-associated bacteria to understand extend and degree of host specificity. Therefore, two phylogenetically closely related and widely distributed species with overlapping micro-niches but varying trophic specialization were selected: Sphagnum angustifolium (Warnst.) C. E. O. Jensen and S. fallax H. Klinggr. (Daniels and Eddy, 1985; Flatberg, 1992; Sastad et al., 1999). Both Sphagnum species were first characterized by their secondary metabolite profile. A polyphasic approach was applied to study bacterial communities with a special focus on Alphaproteobacteria and nitrogen-fixing bacteria: (i) microbial fingerprints by PCR-single-strand-conformation polymorphism (SSCP) applying universal and group-specific 16S rRNA gene-targeting primers and nitrogenase (nifH) gene-specific primers, (ii) fluorescent in situ hybridization with universal and group-specific probes coupled with fluorescence in situ hybridization confocal laser scanning microscopy (FISH–CLSM) and image analysis, (iii) deep-sequencing of Alphaproteobacteria, and (iv) a functional approach to analyses the potential for nitrogen fixation by nitrogenase (nifH) genes in amplicon libraries.
Materials and Methods
Sampling Procedure
Adult gametophytes of S. angustifolium (section Cuspidata) and S. fallax (section Cuspidata) were sampled from the bog “Pürgschachen Moor” (Liezen, 1.7 km SW Ardning, N47° 34.789′E14°2017′) in Austria in July and November 2010. In this bog, S. angustifolium has broader ecological amplitude and grow in mesotrophic wet hollows with some ground water influence as well as in more ombrotrophic hummocks over ground water level. S. fallax grow only in wet mesotrophic conditions in which we collected the samples of both species in comparable ecological situations. The pH of the surrounding peat water was measured at all sampling points and showed mean values of 4.00 (SD, 0.15) for S. fallax and 4.04 (SD, 0.31) for S. angustifolium. Altogether eight independent replicates per Sphagnum species consisting of composite samplings of 15–20 plantlets were collected and stored separately. The approximate length of the sampled plants was 14.5 cm (SD, 2.3 cm) for S. fallax and 15.1 cm (SD, 2.5 cm) for S. angustifolium. The eight sampling points were situated in a distance of at least 10 m, while both species had a distance of not more than 1 m at each single point. The living green parts of the plantlets were placed into sterile plastic bags and transported cooled to the laboratory.
Chemical Analysis of Sphagnum Secondary Metabolites
Spectra of secondary metabolites of Sphagnum samples were analyzed using high-performance liquid chromatography with UV photodiode array detection (HPLC-PDA) as described previously (Opelt et al., 2007a).
Total-Community DNA Isolation
The bacterial fraction associated with gametophytes was extracted according to the modified protocol of Opelt and Berg (2004). Briefly, 5 g of plant material were physically disrupted with sterile pestle and mortar and re-suspended in 10 ml of 0.85% NaCl. Two milliliter of the suspension was centrifuged at 13000 rpm for 20 min at 4°C and the pellet was used for isolation of the total-community DNA. For mechanical lysis, the cells were homogenized twice in a FastPrep® FP120 Instrument (MP Biomedicals) for 30 s at speed 5.0. The obtained DNA was purified using the FastDNA® SPIN Kit for Soil (MP Biomedicals) according to the manufacturer’s protocol. Final aliquots of the total-community DNA were further applied in PCR-based approaches.
Microbial Fingerprinting by PCR-SSCP
Fingerprinting of the moss-associated bacterial communities was carried out by PCR-based SSCP described by Schwieger and Tebbe (1998). 16S rRNA genes of Bacteria were amplified with universal bacterial primers Com1/Unibac-II-927rP (Schwieger and Tebbe, 1998; Zachow et al., 2008). A set of Alphaproteobacteria-specific primers ADF681F/1492r, followed by ADF681F/927rP, was applied using a semi-nested protocol (Blackwood et al., 2005). Bacterial nitrogenase gene (nifH) fragments were amplified in a nested approach with nifH3/19F, nifH11/nifH22P primers (Yeager et al., 2004). The amplicons were separated using the TGGE Maxi system (Biometra) at 400 V and 26°C in acrylamide gel followed by silver staining.
Strand conformation polymorphism is based on the differences in the conformation of single-stranded DNA fragments. The electrophoretic mobility of the single-stranded DNA fragments depends on their three-dimensional conformation. Each of the amplification products was identified by its electrophoretic distance on SSCP gel and the number of DNA fragments. According to the distance of the bands, the SSCP gels were virtually divided into operational taxonomic units (OTUs). The presence or absence of individual amplified product DNA bands in each group was scored. The obtained matrix was used to compare statistically (see statistics).
Fluorescent in situ Hybridization and Confocal Laser Scanning Microscopy
Single gametophytes of S. angustifolium and S. fallax were fixed with 4% paraformaldehyde/phosphate buffered salt (3:1, v/v). Separated leaves were stained by in-tube FISH (Grube et al., 2009). Fluorescently labeled rRNA-targeting probe ALF968 specific for Alphaproteobacteria (Loy et al., 2007) was applied in combination with equimolar mixture of universal bacterial probes EUB338, EUB338II, EUB338III (Amann et al., 1990; Daims et al., 1999). Sphagnum samples were consequently hybridized with ALF968 (41°C, 45% formamide) followed by EUB338/EUB338II/EUB338III (41°C, 15% formamide). Negative control was hybridized with non-target NON-EUB probe (Amann et al., 1990) at the same stringency conditions applied for the positive FISH probes.
Confocal laser scanning microscopy was performed using a confocal microscope Leica TCS SPE (Leica Microsystems). Fluorescent dyes Cy3 and Cy5 labeled to the FISH probes were sequentially excited with 532 and 635 nm laser beams, respectively; the emitted light was detected in the range of 556–607 and 657–709 nm, respectively. An additional channel (excitation at 488 nm; emission range 508–556 nm) was used for acquiring the autofluorescence of the moss cells. Photomultiplier gain and offset were individually optimized for every channel and every field of view, in order to improve the signal/noise ratio. Confocal stacks were acquired with a Leica ACS APO 40X OIL CS objective (NA: 1.15) and a Leica ACS APO 63X OIL CS objective (NA: 1.30) by applying a Z-step of 0.4–0.8 μm. Three-dimensional reconstructions were created with the software Imaris 7.0 (Bitplane).
Deep-Sequencing and Bioinformatic Analysis
Diversity of Alphaproteobacteria and nitrogen-fixing bacteria associated with Sphagnum species was deeply investigated by barcoded pyrosequencing approach. The total-community DNA was amplified with the set of Alphaproteobacteria-specific primers ADF681F/Unibac-II-927r (Blackwood et al., 2005; Zachow et al., 2008) and nifH gene-specific primers nifH3/nifH4, nifH1/nifH2 in the nested approach (Zehr and Turner, 2001) using Taq-&Go™ Ready-to-use PCR Mix (MP Biomedicals). Duplicate PCR products from all templates were purified with Wizard® SV Gel and PCR Clean-Up System (Promega). Amplicons of each Sphagnum sp. were pooled together and subjected to the pyrosequencing using the Roche/454 GS FLX+ Titanium platform executed by GATC Biotech (Konstanz, Germany).
The 16S rRNA gene amplicon libraries specific for Alphaproteobacteria were analyzed as specified by Bragina et al. (2011). Shortly, raw sequencing reads were quality and length filtered (≥150 bp). Rarefaction analysis was performed for phylotype clusters of 97, 95, and 90% similarity by using the tools of the RDP’s Pyrosequencing Pipeline (Cole et al., 2009). Datasets were normalized to the same number of sequences. Richness estimates and diversity indices were calculated in the open source software package QIIME (Caporaso et al., 2010). Classification of the reads was performed using the BLAT pipeline within the web interface SnoWMAn version 1.111 with 80% confidence threshold.
Amplicon libraries of the nitrogenase gene (nifH) were explored using the FunGene Pipeline of RDP server2 with parameters stated by Farnelid et al. (2011). Primer sequences were trimmed and reads of a low quality and shorter 200 bp were removed. Filtered reads were translated into amino acid sequences and clipped at 60 aa. Further analyses were carried out on amino acid sequences. For the rarefaction, datasets were clustered with 100, 96, and 92% similarity cut-offs. Richness estimates and diversity indices were calculated for the subsets normalized to the same number of sequences by QIIME software. Compositional diversity was compared by Sørensen (Cs) and Shannon (H′) indices at 96% similarity level. Phylogenetic analysis was performed for the clusters of 92% similarity with ≥10 sequences. Reference sequences were obtained using the NCBI algorithm TBLASTN and a phylogenetic tree was constructed as described previously (Bragina et al., 2011).
Statistics
Computer-assisted analysis of SSCP profiles was performed using the GelCompare II version 5.1 software package (Applied Maths). Similarity matrices were constructed based on Pearson’s correlation coefficients (r) and cluster analyses were done by the unweighted pair group method with average linkages (UPGMA). SSCP profiles of the microbial communities generated with universal bacterial and nifH gene-specific primers were further applied for the multivariate analysis. Single DNA bands, characterized by the relative position and abundance on the gel, were defined as OTUs and used as response variables for detrended correspondence analyses (DCA) by Canoco 4.5 for Windows (Lepš and Smilauer, 2003).
Results
Profiling of the Sphagnum Secondary Metabolites
High-performance liquid chromatography-PDA profiling of plant extracts yielded notably similar chromatograms and spectra of the prevailing peaks of the secondary metabolites for both Sphagnum species. The UV spectra suggested various phenols and indol derivatives including sphagnic acid (peak 7; Figure 1). The only noteworthy difference was the more pronounced accumulation of flavonoid conjugates (naringenin and apigenin) in S. angustifolium. The late retention time precluded glycosides but more lipophilic molecules. The result testified common chemical basis for establishment of the similar microbial diversity of the compared moss species.
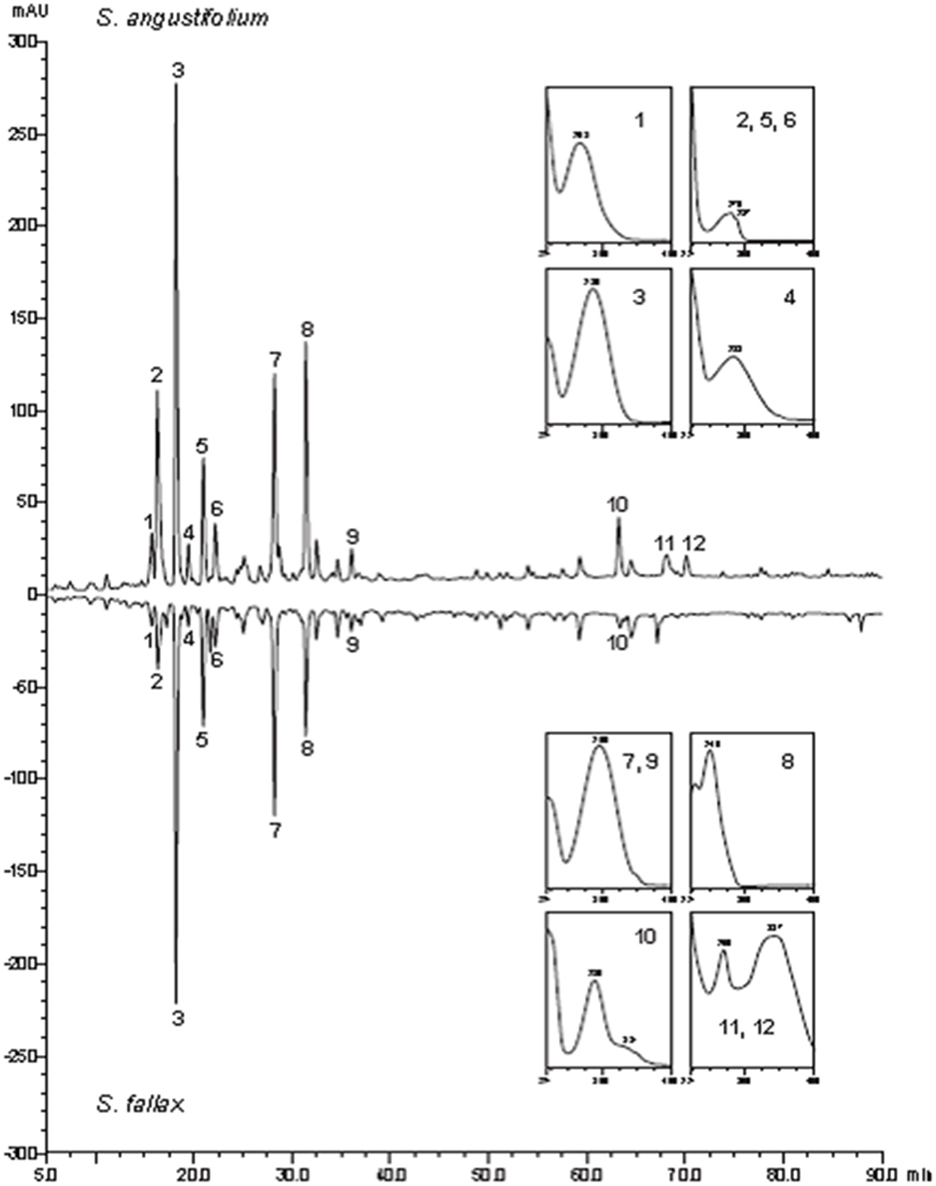
Figure 1. High-performance liquid chromatography -UV secondary metabolite profiles of S. angustifolium and S. fallax at 229 nm (UV spectra (200–400 nm; 1, phenol; 2, 5, 6, indole derivative; 3, 4, phenols; 7, 9, sphagnic acid; 8, phenol; 10, naringenin type flavonoid; 11, 12, apigenin flavonoids.
Molecular Fingerprinting of Microbial Communities
In the molecular fingerprinting approach we used a specific set of primers: universal primers to get an overview about the whole bacterial community associated with Sphagnum, primers for Alphaproteobacteria because this is a ubiquitous and cosmopolitan phylogenetic class associated with Sphagnum (Bragina et al., 2011), and nifH primers to detect nitrogen-fixing bacteria as important functional group (Opelt et al., 2007a). All SSCP fingerprints obtained with universal and group-specific primers revealed high similarity of the microbial communities associated with S. fallax and S. angustifolium (Figure 2). UPGMA analysis of the Bacteria profiles resulted in a minimum similarity level of 95.5% of bryophyte-associated communities. Alphaproteobacteria patterns formed a common cluster at 87.1% similarity. Fingerprints of the nifH gene clustered into two groups at 41.2% similarity. Within each cluster, samples of both Sphagna grouped together. This overlap of the microbial communities was confirmed statistically by a detrended correspondence analysis (Figure 3).
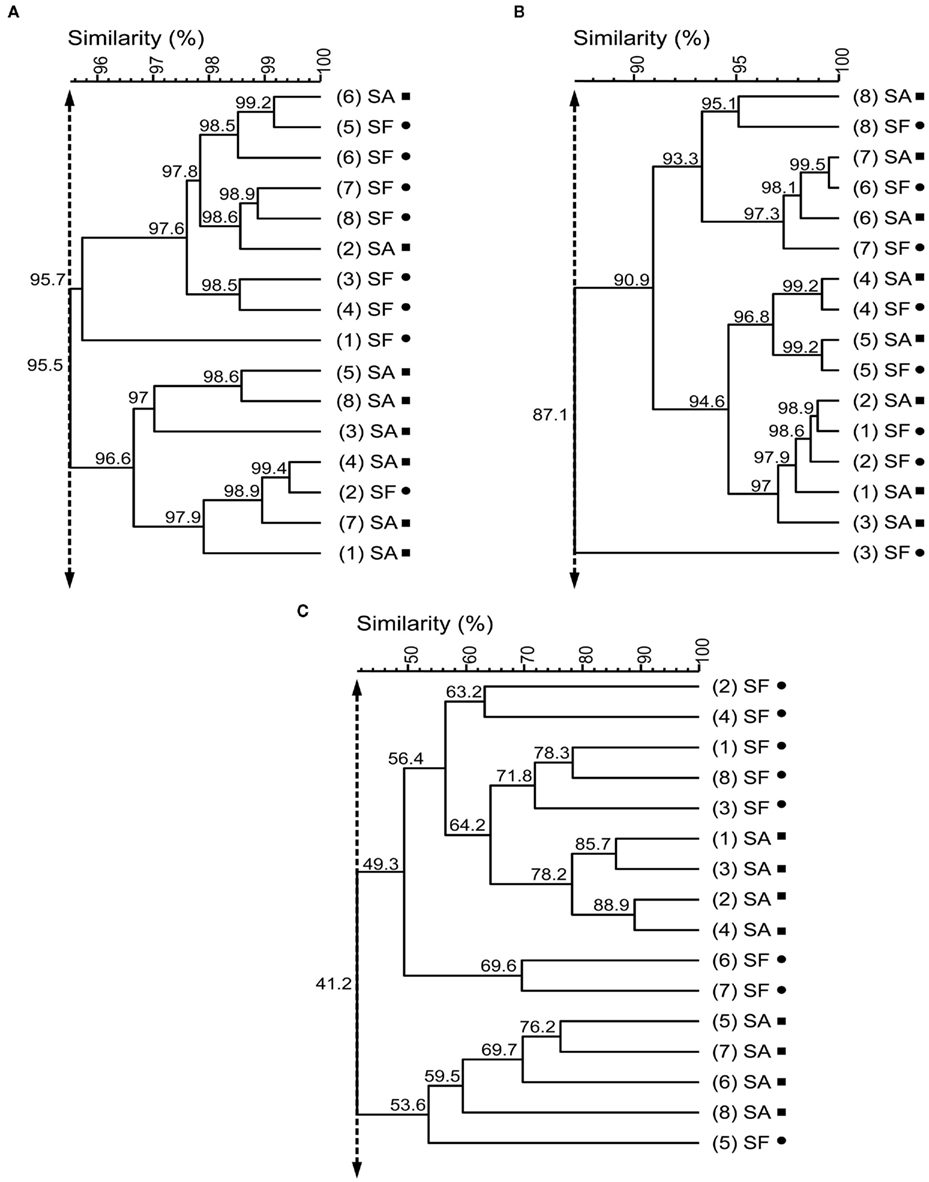
Figure 2. Unweighted pair group method with average linkages dendrograms of bacterial communities associated with S. angustifolium (SA, squares) and S. fallax (SF, circles). The dendrograms were generated from the PCR-SSCP profiles of Bacteria(A), Alphaproteobacteria (B), and bacterial nifH genes (C) using unweighted pair group method with average linkages (UPGMA). Numbers in round brackets indicate replicates. Double-headed vertical arrows indicate the similarity for the groupings.
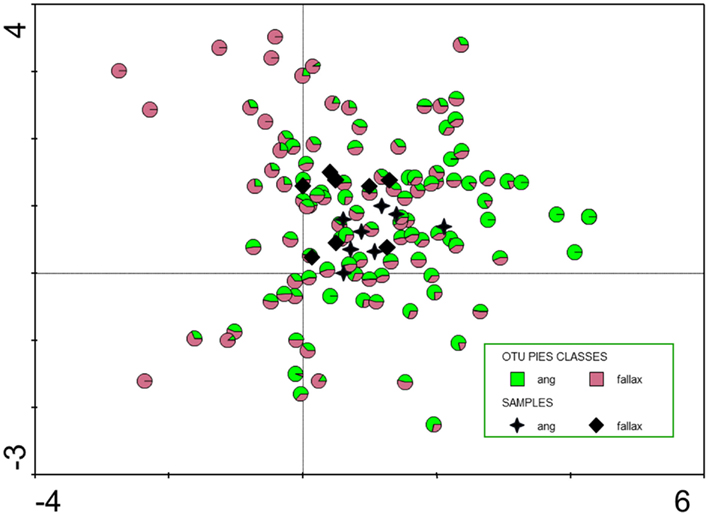
Figure 3. Detrended correspondence analysis (DCA, indirect unimodal gradient analysis) of operational taxonomic units (OTUs) identified by SSCP community fingerprints. Eigenvalues of first and second axis are 0.244 and 0.141, respectively; sum of all eigenvalues 1.715. Black stars and diamonds show the location of the 16 samples, colored circles the location of the 111 OTUs in the biplot. The colors indicate its preference to Sphagnum angustifolium (ang) or S. fallax, respectively.
Microbial Colonization Patterns
Sphagnum gametophytes characterized by the unique morphology were studied for microbial colonization patterns by FISH. Again, we applied universal and Alphaproteobacteria-specific probes according to the above mentioned reasons. The stem and branch leaves are usually differentiated in size and shape, but equally are formed by dimorphic leaf cells in which large, empty hyaline cells perforated by pores are enclosed in a network of narrower, chlorophyllose cells (Figures 4A,D). CLSM observation of both leave types showed dense colonization by bacterial colonies of the internal space of the gametophytes as shown for branch leaves in Figures 4B,E. Alphaproteobacterial cells presented up to 50% of the detected bacterial colonies. Three-dimensional reconstruction of the acquired images supported that bacteria primarily occupied dead hyaline cells Figures 4C,F. In conclusion, FISH–CLSM approach displayed similar colonization patterns for S. angustifolium and S. fallax by the bacterial communities.
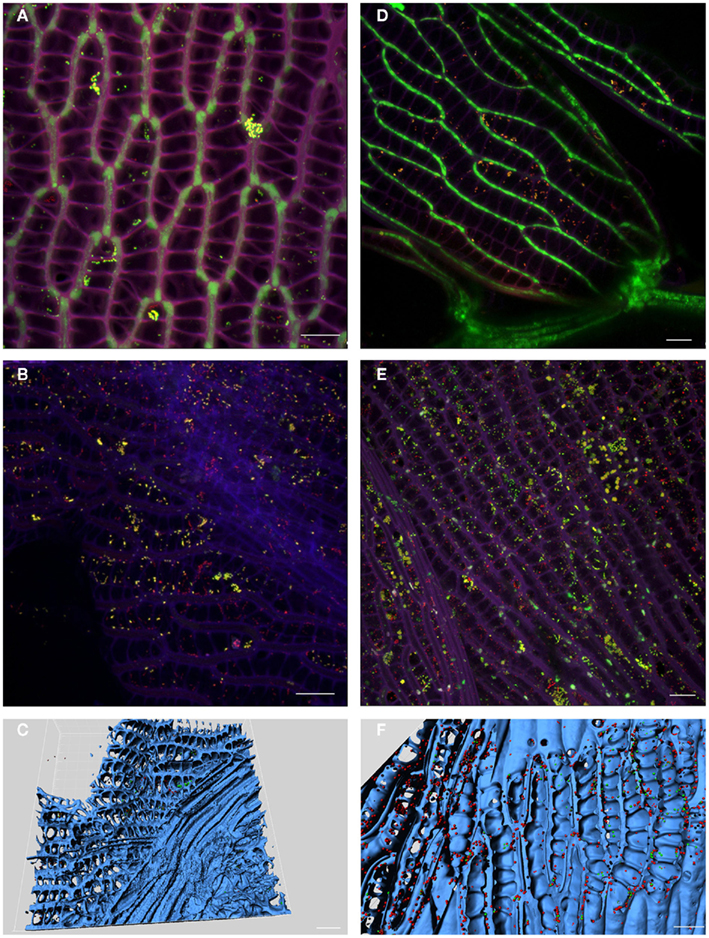
Figure 4. Localization of bacteria in moss gametophytes. Fluorescent in situ hybridization of S. angustifolium(A–C) and S. fallax (D–F) leaves showed colonization of hyaline cells by Alphaproteobacteria. Images acquired by confocal laser scanning microscopy (CLSM) (A,B,D,E): violet – cell walls of Sphagnum cells; green – chlorophyll-containing Sphagnum chlorocytes; yellow – Alphaproteobacteria; red – other bacteria. 3D computer reconstructions of CLSM images using Imaris7.0 (C,F): blue – moss tissue; red: Alphaproteobacteria; green: other bacteria. Scale bar = 20 μm.
Deep-Sequencing of Alphaproteobacteria and NIFH Genes
Alphaproteobacteria and nitrogen-fixing bacteria were selected to get a deeper insight by a pyrosequencing approach. The 16S rRNA gene amplicon libraries specific for Alphaproteobacteria were rarefied as shown in Figure 5. Richness estimation of the normalized datasets revealed that pyrosequencing effort reached 66.0–74.2% of estimated richness for the clusters of 90% similarity (Table 1). The clusters of 95 and 97% similarity reflected 49.3–51.2 and 45.4–46.2% of estimated richness, respectively.
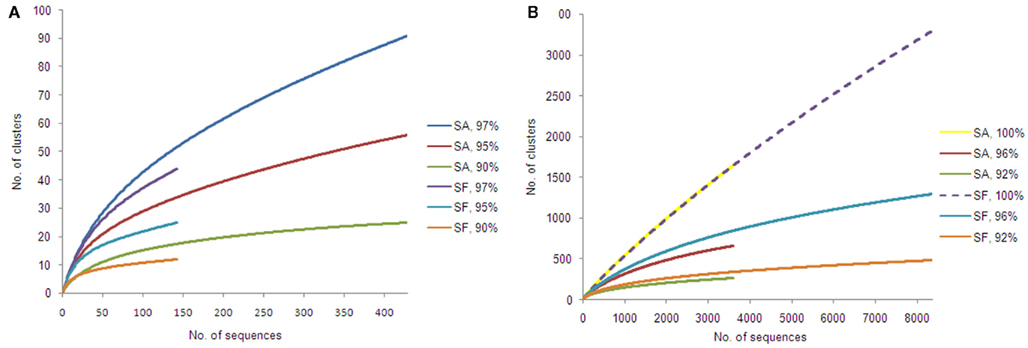
Figure 5. Rarefaction curves for amplicon libraries of Sphagnum samples. Saturation curves are presented for samples of S. angustifolium (SA) and S. fallax (SF). Alphaproteobacteria (A) and nifH datasets (B) were clustered with similarity cut-offs defined.
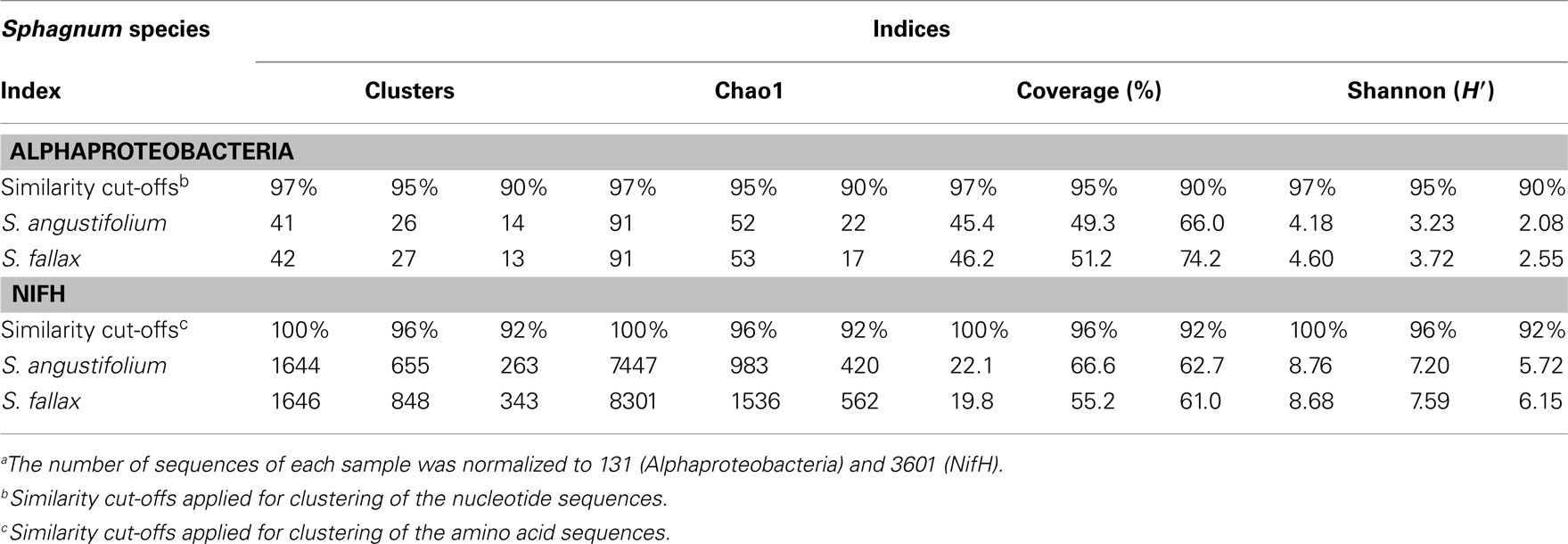
Table 1. Richness estimates and diversity indices for amplicon libraries of Sphagnum samplesa.
Taxonomic composition of alphaproteobacterial populations, compared at the ranks of families and genera, was substantially similar among Sphagnum spp. (Figure 6). Dominant Acetobacteraceae family was prevailed by genera Acidocella, Acidisphaera, and Rhodopila. Within families Sphingomonadaceae and Rhodospirillaceae, the most members belonged to Novosphingobium spp. and Magnetospirillum spp., correspondingly. Withal, composition and ratio of subdominant Caulobacteraceae varied between mosses. The family was more abundant in S. fallax sample and consisted of genus Phenylobacterium (detected all over) and genus Caulobacter (unique for S. fallax). Diversity of species was accessed by Shannon diversity index (H′) for clusters of 97% similarity. Comparison of the index values revealed a slightly higher diversity of Alphaproteobacteria for S. fallax (4.60) than for S. angustifolium (4.18).
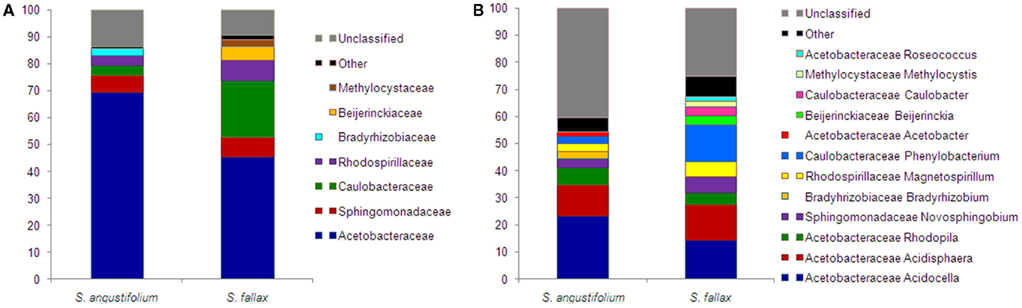
Figure 6. Taxonomic classification of Alphaproteobacteria associated with S. angustifolium and S. fallax. Pyrosequencing reads are classified at family (A) and genus level (B) with a confidence threshold of 80%. Groups below 1% of relative abundance are included in Other. Multi-colored charts at the legend are shown for each sample correspondingly.
According to the NCBI database, identified alphaproteobacterial genera comprise bacteria known for the nitrogen fixation. Particularly, genera Bradyrhizobium, Acetobacter, and Beijerinckia were found in both libraries, while genera Gluconacetobacter, Methylocystis, Methylosinus, and Rhizobium were solely detected in the S. fallax library.
Rarefaction analysis of the nitrogenase gene libraries resulted in similar saturation profiles of the Sphagnum samples (Figure 5). Normalized datasets represented 61.0–62.7% of estimated richness at 92% similarity (Table 1). The clusters of 96% similarity covered 55.2–66.6%, while unique clusters reflected 19.8–22.1% of estimated richness, correspondingly. Compositional diversity was assessed applying 96% similarity cut-off to confine the clusters. The Sørensen similarity index indicated that samples featured 53% identity. Nitrogenase diversity estimated by Shannon was again slightly higher for S. fallax (7.59), than for S. angustifolium (7.20; Table 1).
Phylogenetic analysis of the NifH composition revealed that retrieved sequences, 60 amino acids in length, were distributed among canonical Clusters I, III, and Sub-cluster IA (Figure 7). The most abundant Cluster I, comprising sequences of Proteobacteria and Cyanobacteria, was dominated by Alphaproteobacteria. According to the BLAST analysis, the most prevalent alphaproteobacterial amplicons were ≥95% similar to Bradyrhizobium, Azorhizobium, Rhizobium, Methylobacterium, Rhodocista, and Acetobacter species. Considerable proportion of amplicons showed 96–100% identity with Methylocella, Methylocapsa, and Beijerinckia reference sequences. Detected Betaproteobacteria were prevailed by Burkholderia spp. (≥95% similarity). Minor portion of Cluster I amplicons was affiliated with cyanobacterial genera Anabaena and Tolypotrix (100% similarity). Sub-cluster IA contained sequences 96–100% similar to Geobacter sp. Within the Cluster III amplicons grouped with reference Spirochaeta and Thermincola species (82–93% similarity). In conclusion, nifH amplicon libraries of Sphagnum-associated microbial communities were characterized by high diversity and abundance of alphaproteobacterial amplicons.
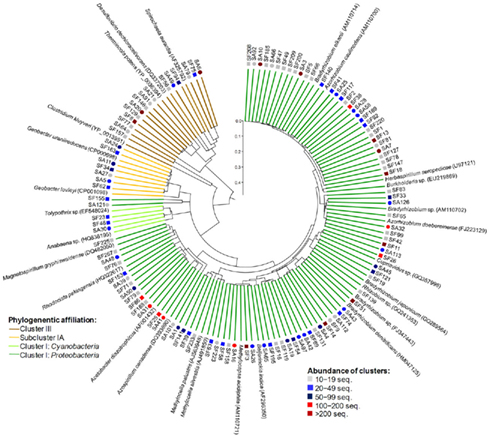
Figure 7. Phylogenetic composition of the nitrogenase gene (nifH) amplicon libraries of S. angustifolium (SA, circles) and S. fallax (SF, squares). Neighbor-joining phylogenetic tree was constructed with both one representative sequence per NifH cluster (92% similarity) and the nearest reference sequences (accession numbers in brackets). A partial sequence of the light-independent photochlorophyllide reductase subunit L (BchL) from Chlorobaculum tepidum (Acc. Nr. AAG12203) was used as out-group. Reliability of the tree topology was evaluated by 100 bootstrap resamplings (bootstrap values not shown). Abundance and phylogenetic affiliation of the clusters are indicated at the legend. Scale bar = 0.1 substitutions per site.
Discussion
Sphagnum mosses form an outstanding group of Bryophyta; they are unique in their morphological and developmental features at every stage of the life cycle (Shaw et al., 2003). More than for other plants, physical and chemical characteristics of Sphagnum mosses are related to the ecology and function of Northern peatlands. In this study we have shown the same deep relationship between moss plantlets and their associated bacterial communities. In the multifaceted approach applied to study the structure and function of bacteria, only minor but not statistically significant differences were found between S. angustifolium and S. fallax, two peat mosses which shared similar ecological conditions inside the bog ecosystem.
This high similarity is in contrast to former studies of Sphagnum-associated bacteria of different ecological amplitudes (Opelt et al., 2007c; Bragina et al., 2011). The main difference between the former studies and the present study is the close taxonomic and ecological relationship of the investigated S. angustifolium and S. fallax unlike S. magellanicum. S. magellanicum belongs to another section within the genus Sphagnum (section Sphagnum) and is typical for strong acidic, oligotrophic, and ombrotrophic habitats, whereas S. angustifolium and S. fallax (section Cuspidata) grow in weakly acid, more mesotrophic situations influenced by minerotrophic groundwater (Daniels and Eddy, 1985). Also from the morphological point of view, S. angustifolium and S. fallax are difficult to distinguish, and in former times both taxa were considered as varieties of one species S. recurvum P. Beauv (Smith, 1978). In contrast to S. magellanicum (Opelt et al., 2007a), S. angustifolium, and S. fallax are characterized by similar secondary metabolites. The overlap of common properties for the mosses was also found for the bacterial community. Figure 3 not only shows a high degree of similarity between the microbial communities also a differentiation in more S. fallax (negative along the first axis) and more S. angustifolium (positive along the first axis) preferring bacteria. So, the situation shown exemplarily in Figure 3 could be interpreted as an early state of specification of bacterial communities correspondent with an early state of host species differentiation. Interestingly, our results also explain differences between the theory of Sphagnum species specific communities established by Opelt et al. (2007c) and Bragina et al. (2011) and results obtained by Larmola et al. (2010) who identified only a-biotic drivers. For methanotrophs, they found for transplanted Sphagnum species bacterial pattern and activity typical for the a-biotic parameters of the destination site. However, this was an artificial experiment; the majority of the approx. 300 Sphagnum species has very narrow ecological amplitudes and would not grow in nature under different conditions (Daniels and Eddy, 1985). In Northern wetlands, which belong to an old vegetation type with more or less extreme but constant conditions for thousands of years, Sphagnum mosses have established a highly specific and adapted symbiosis with their associated microbes.
What are the ecological consequences of this specificity of the microbial community? Well-adapted to a-biotic parameters to the place where they live, Sphagnum mosses together with their microbiome as so called “meta-organisms” fulfill important functions for ecosystem services. These functions can only fulfilled in cooperation with the associated microbial community. The latter is responsible to fix nitrogen for the host plant, to solubilize phosphor but also to provide carbon from peat-delivered methane (Raghoebarsing et al., 2005; Opelt et al., 2007a). Studies have shown that oxidation of CH4 by methanotrophic microbes residing in the Sphagnum layer is controlled by environmental factors, i.e., water table and temperature (Larmola et al., 2010) but beside this the rate was specific for Sphagnum species (Gifford et al., 2011). Taken together, the specificity of the microbial community is essential to live under the extreme and highly varying ecological gradients within the bog ecosystem and to fulfill the ecological functions. The bog ecosystem is more complex than previously thought but this is important to know to maintain bog ecosystems in Northern wetlands. The high specificity, narrow ecological amplitude and closed relationship can be one reason that Sphagnum is highly sensitive to changing a-biotic parameters connected with climate change.
Little is known about the specific ecology of Sphagnum-associated bacteria which are to a high degree still not culturable (Dedysh, 2011). Dominant alphaproteobacterial taxa associated with Sphagna are known as acidophilic or acidotolerant bacteria able to grow chemo-organotrophically or phototrophically and to survive under oligotrophic conditions. Sphagnum-associated microbial communities should be included in biodiversity–conservation agenda and used for predictive microbial ecology as requested by Bodelier (2011).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was funded by the Austrian Science Foundation FWF by a grant to Gabriele Berg.
Footnotes
References
Amann, R. I., Binder, B. J., Olson, R. J., Chisholm, S. W., Devereux, R., and Stahl, D. A. (1990). Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925.
Belyea, L. R., and Malmer, N. (2004). Carbon sequestration in peatland: patterns and mechanisms of response to climate change. Glob. Chang. Biol. 10, 1043–1052.
Blackwood, C. B., Adam, O., and Buyer, J. S. (2005). Phylum- and class-specific PCR primers for general microbial community analysis. Appl. Environ. Microbiol. 71, 6193–6198.
Bodelier, P. L. (2011). Toward understanding, managing, and protecting microbial ecosystems. Front. Microbiol. 2:80. doi:10.3389/fmcib.2011.00080
Bragina, A., Berg, C., Cardinale, M., Shcherbakov, A., Chebotar, W., and Berg, G. (2011). Sphagnum mosses harbor highly specific bacterial diversity during their whole life cycle. ISME J. doi:10.1038/ismej.2011.151
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., Fierer, A., Pena, A. G., Goodrich, J. K., Gordon, J. I., Huttley, G. A., Kelley, S. T., Knights, D., Koenig, J. E., Ley, R. E., Lozupone, C. A., McDonald, D., Muegge, B. D., Pirrung, M., Reeder, J., Sevinsky, J. R., Tumbaugh, P. J., Walters, W. A., Widmann, J., Yatsunenko, T., Zaneveld, J., and Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336.
Clymo, R. S., Turunen, J., and Tolonen, K. (1998). Carbon accumulation in peatland. Oikos 81, 368–388.
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., Marsh, T., Garrity, G. M., and Tiedje, J. M. (2009). The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145.
Daims, H., Brühl, A., Amann, R., Schleifer, K. H., and Wagner, M. (1999). The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444.
Dedysh, S. N. (2011). Cultivating uncultured bacteria from northern wetlands: knowledge gained and remaining gaps. Front. Microbiol. 2:184. doi: 10.3389/fmcib.2011.00184
Dedysh, S. N., Panikov, N. S., and Tiedje, J. M. (1998). Acidophilic methanotrophic communities from Sphagnum peat bogs. Appl. Environ. Microbiol. 64, 922–929.
Farnelid, H., Andersson, A. F., Bertilsson, S., Al-Soud, W. A., Hansen, L. H., Sørensen, S., Steward, G. H., Hägstrom, Å., and Riemann, L. (2011). Nitrogenase gene amplicons from global marine surface waters are dominated by genes of non-cyanobacteria. PLoS ONE 6, e19223. doi:10.1371/journal.pone.0019223
Flatberg, K. I. (1992). The European taxa in the Sphagnum recurvum complex. 2. Amended descriptions of Sphagnum brevifolium and S. fallax. Lindbergia 17, 96–110.
Freitag, T. E., Toet, S., Ineson, P., and Prosser, J. I. (2010). Links between methane flux and transcriptional activities of methanogens and methane oxidizers in a blanket peat bog. FEMS Microbiol. Ecol. 73, 157–165.
Gifford, J., Hoopes, M., Larmola, T., Varner, R., Bubier, J., and Young, B. (2011). The Role of Sphagnum Mosses in Methane Oxidation in a Temperate Fen. Thesis, Mount Holyoke College, South Hadley, MA.
Gignac, L. D., and Vitt, D. H. (1994). Responses of Northern peatlands to climate change: effects on bryophytes. J. Hattori Bot. Lab. 75, 119–132.
Gorham, E. (1991). Nothern peatlands: role in the carbon cycle and probable responses to climatic warming. Ecol. Appl. 1, 182–195.
Granath, G., Wiedermann, M. M., and Strengbom, J. (2009). Physiological responses to nitrogen and sulphur addition and raised temperature in Sphagnum balticum. Oecologia 161, 481–490.
Grube, M., Cardinale, M., Vieira de Castro, J., Müller, H., and Berg, G. (2009). Species-specific structural and functional diversity of bacterial communities in lichen symbioses. ISME J. 3, 1105–1115.
Horn, M. A., Matthies, C., Küsel, K., Schramm, A., and Drake, H. L. (2003). Hydrogenotrophic methanogenesis by moderately acid-tolerant methanogens of a methane-emitting acidic peat. Appl. Environ. Microbiol. 69, 74–83.
Larmola, T., Tuittila, E. S., Tiirola, M., Nykänen, H., Martikainen, P. J., Yrjälä, K., Tuomivirta, T., and Fritze, H. (2010). The role of Sphagnum mosses in the methane cycling of a boreal mire. Ecology 91, 2356–2365.
Lepš, J., and Smilauer, P. (2003). Multivariate Analysis of Ecological Data Using Canoco. Cambridge: Cambridge University Press.
Loy, A., Maixner, F., Wagner, M., and Horn, M. (2007). probeBase-an online resource for rRNA-targeted oligonucleotide probes: new features 2007. Nucleic Acids Res. 35, D800–D804.
Opelt, K., and Berg, G. (2004). Diversity and antagonistic potential of bacteria associated with bryophytes from nutrient-poor habitats of the Baltic Sea Coast. Appl. Environ. Microbiol. 70, 6569–6579.
Opelt, K., Chobot, V., Hadacek, F., Schönmann, S., Eberl, L., and Berg, G. (2007a). Investigations of the structure and function of bacterial communities associated with Sphagnum mosses. Environ. Microbiol. 91, 2795–2809.
Opelt, K., Berg, C., and Berg, G. (2007b). The bryophyte genus Sphagnum is a reservoir for powerful and extraordinary antagonists and potentially facultative human pathogens. FEMS Microbiol. Ecol. 61, 38–53.
Opelt, K., Berg, C., Schönmann, S., Eberl, L., and Berg, G. (2007c). High specificity but contrasting biodiversity of Sphagnum-associated bacterial and plant communities in bog ecosystems independent of the geographical region. ISME J. 1, 502–516.
Raghoebarsing, A. A., Smolders, A. J. P., Schmid, M. C., Rijpstra, W. I. C., Wolters-Arts, Derksen, J. M., Jetten, M. S. M., Schouten, S., Damsté, J. S. S., Lamers, L. P. M., Roelofs, J. G. M., Op den Camp, H. J. M., and Strous, M. (2005). Methanotrophic symbionts provide carbon for photosynthesis in peat bogs. Nature 436, 1153–1156.
Sastad, S. M., Bard, P., and Kjetil, D. (1999). Habitat-specific genetic effects on growth rate and morphology across pH and water level gradients within a population of the moss Sphagnum angustifolium (Sphagnaceae). Am. J. Bot. 86, 1687–1698.
Schwieger, F., and Tebbe, C. C. (1998). A new approach to utilize PCR-singe-strand-conformation polymorphism for 16S rRNA gene-based microbial community analysis. Appl. Environ. Microbiol. 64, 4870–4876.
Shaw, A. J., Cox, C. J., and Boles, S. B. (2003). Global patterns in peatmoss biodiversity. Mol. Ecol. 12, 2553–2570.
Smith, A. J. E. (1978). The Moss Flora of Britain and Ireland. Cambridge: Cambridge University Press.
Soudzilovskaia, N. A., Cornelissen, J. H. C., During, H. J., van Logtestijn, R. S. P., Lang, S. I., and Aerts, R. (2010). Similar cation exchange capacities among bryophyte species refute a presumed mechanism of peatland acidification. Ecology 91, 2716–2726.
Whinam, J., and Copson, G. (2006). Sphagnum moss: an indicator of climate change in the sub-Antarctic. Polar Rec. 42, 43–49.
Yeager, C. M., Kornosky, L., Housman, C., Grote, E. E., Belnap, J., and Kuske, C. R. (2004). Diazotrophic community structure and function in two successional stages of biological soil crusts from the Colorado Plateau and Chihuahuan Desert. Appl. Environ. Microbiol. 70, 973–983.
Keywords: Sphagnum fallax, Sphagnum angustifolium, SSCP fingerprints, FISH–CLSM, amplicon library, Alphaproteobacteria, nitrogenase
Citation: Bragina A, Maier S, Berg C, Müller H, Chobot V, Hadacek F and Berg G (2012) Similar diversity of Alphaproteobacteria and nitrogenase gene amplicons on two related Sphagnum mosses. Front. Microbio. 2:275. doi: 10.3389/fmicb.2011.00275
Received: 11 November 2011;
Accepted: 22 December 2011;
Published online: 10 January 2012.
Edited by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology, Russian Academy of Sciences, RussiaReviewed by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology, Russian Academy of Sciences, RussiaSteffen Kolb, University of Bayreuth, Germany
Copyright: © 2012 Bragina, Maier, Berg, Müller, Chobot, Hadacek and Berg. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Gabriele Berg, Institute of Environmental Biotechnology, Graz University of Technology, Petersgasse 12, 8010 Graz, Austria. e-mail:Z2FicmllbGUuYmVyZ0B0dWdyYXouYXQ=
†Anastasia Bragina and Stefanie Maier have contributed equally to this work.